

In this study, Bevacqua et al. set out to investigate the roles of SIX2 and SIX3 transcription factors in human β cells. Using genetic and genomic approaches, this study unveils SIX2 and SIX3 functions crucial for postnatal human islet and β-cell development and maturation and reveals how β-cell dysfunction might develop in diabetes.
Keywords: β cells, islet, transcription factor, diabetes mellitus, pancreas
Abstract
The physiological functions of many vital tissues and organs continue to mature after birth, but the genetic mechanisms governing this postnatal maturation remain an unsolved mystery. Human pancreatic β cells produce and secrete insulin in response to physiological cues like glucose, and these hallmark functions improve in the years after birth. This coincides with expression of the transcription factors SIX2 and SIX3, whose functions in native human β cells remain unknown. Here, we show that shRNA-mediated SIX2 or SIX3 suppression in human pancreatic adult islets impairs insulin secretion. However, transcriptome studies revealed that SIX2 and SIX3 regulate distinct targets. Loss of SIX2 markedly impaired expression of genes governing β-cell insulin processing and output, glucose sensing, and electrophysiology, while SIX3 loss led to inappropriate expression of genes normally expressed in fetal β cells, adult α cells, and other non-β cells. Chromatin accessibility studies identified genes directly regulated by SIX2. Moreover, β cells from diabetic humans with impaired insulin secretion also had reduced SIX2 transcript levels. Revealing how SIX2 and SIX3 govern functional maturation and maintain developmental fate in native human β cells should advance β-cell replacement and other therapeutic strategies for diabetes.
Development of vital organs, like the brain and pancreas, includes a prenatal stage when the embryonic organ anlage, specification, and expansion of major differentiated cell types, organ morphology, and anatomic position are established, followed by a postnatal stage when differentiated cell types acquire mature physiological functions and refine their cellular interactions (Reinert et al. 2014; Kim et al. 2020). Improved understanding of the mechanisms underlying postnatal “functional maturation” in cells like pancreatic islet cells could accelerate efforts to improve therapies for diabetes, including production of replacement human β cells from renewable sources (Arda et al. 2016, 2018; Bakken et al. 2016; Sneddon et al. 2018).
During prenatal and neonatal stages, islet β cells transiently proliferate and expand. In childhood and thereafter, β cells reduce proliferation (Teta et al. 2005; Meier et al. 2008; Wang et al. 2016a), increase insulin production, and enhance glucose-dependent insulin secretion, all hallmarks of mature β-cell function (Artner et al. 2007, 2010; Aguayo-Mazzucato et al. 2011; Arda et al. 2016). The genetic and molecular mechanisms governing this age-dependent β-cell functional maturation are intensely sought. Prior studies in mice suggest that transcription factors (TFs) regulate functional maturation of β cells (Aguayo-Mazzucato et al. 2011; Lantz et al. 2004; Schaffer et al. 2013; Yoshihara et al. 2016). However, less is known about the role of TFs in human β-cell maturation, reflecting the challenges of studying postnatal development in native human β cells. Mutations in genes encoding TFs including PDX1, NEUROD1, MAFA, GATA6, and RFX6 have been linked to monogenic forms of diabetes mellitus and impaired β-cell function in humans, suggesting roles in β-cell maturation (for reviews, see Sellick et al. 2004; Allen et al. 2012; Patel et al. 2017; Iacovazzo et al. 2018; Urakami 2019). Thus, TFs may represent a class of factors governing age-dependent postnatal β-cell functional maturation in humans.
We and others have found that SIX2 and SIX3, members of the sine oculis homeobox family of TFs, are first expressed in the β cells of children by 9–10 yr of age (Blodgett et al. 2015; Arda et al. 2016; Wang et al. 2016b; Cyphert et al. 2019), followed by increased expression in adulthood. Moreover, neither SIX2 nor SIX3 mRNA are detectable in human α cells (Blodgett et al. 2015; Arda et al. 2016; Wang et al. 2016b). SIX2 and SIX3 are encoded by linked genes on human chromosome 2 (OMIM: 604994 and 603714). While they show high homology in their N-terminal SIX domain and their central homeodomain (HD), other portions of SIX2 and SIX3 differ (Kawakami et al. 2000) and could mediate distinct interactions. For example, SIX3, but not SIX2, interacts with members of the groucho family of corepressors (Kobayashi et al. 2001; López-Ríos et al. 2003). In addition, SIX2 has roles in development of kidneys, skull, stomach, and other organs (Kobayashi et al. 2008; Self et al. 2009; He et al. 2010; Park et al. 2012), while SIX3 has roles in forebrain and eye development (Jeong et al. 2008; Liu et al. 2010).
SIX2 and SIX3 have largely nonoverlapping tissue expression patterns that correspond with their distinct roles in human organogenesis. In the pancreas, however, both SIX2 and SIX3 are expressed in human β cells, with coincident onset of postnatal expression and a shared cis-regulatory element that coregulates islet SIX2 and SIX3 expression (Spracklen et al. 2018). This element encompasses single nucleotide polymorphisms (SNPs) associated with type 2 diabetes (T2D) and fasting hyperglycemia (Kim et al. 2011; Spracklen et al. 2020). Nevertheless, it remains unknown whether SIX2 and SIX3 are required for normal adult β-cell function, what β-cell genes these TFs regulate, and whether their expression is altered in diabetes.
Prior studies suggest that SIX2 or SIX3 might regulate genes with roles in restricting native β-cell proliferation and enhancing insulin production and secretion. The onset of SIX2 and SIX3 expression in postnatal human β cells temporally coincides with age-dependent enhancement of insulin production and secretion (Blodgett et al. 2015; Arda et al. 2016). Moreover, misexpression of SIX3 in immature islets from children stimulated glucose-dependent insulin secretion (Arda et al. 2016). Neither SIX2 nor SIX3 is expressed in mouse islets (Benner et al. 2014; Baron et al. 2016); thus, loss-of-function studies that clearly identify requirements for SIX2 or SIX3 in β-cell function will require studies in native human islets or alternative experimental systems. Attempts to generate insulin-secreting β cells from human stem cells have only produced immature progeny that express SIX2, often at low levels, and fail to express SIX3 (Sneddon et al. 2018; Nair et al. 2019; Veres et al. 2019; Velazco-Cruz et al. 2020). To overcome these limitations, here we investigated SIX2 and SIX3 in adult human islets, using recently described genetic methods permitting targeted loss of function in primary human islets (Peiris et al. 2018). Controlled islet cell dispersion and reaggregation to develop human “pseudoislets” (Scharp et al. 1980; Tze and Tai 1982; Arda et al. 2016) allowed us to achieve shRNA-mediated SIX2 or SIX3 suppression in human adult β cells. Collectively, these studies demonstrate that SIX2 and SIX3 coordinately regulate distinct genetic programs in human β cells, including expression of target genes governing mature β-cell function and maintaining β-cell fate. Moreover, we show that SIX2 expression is reduced in β cells purified from human donors with T2D and impaired islet insulin secretion, suggesting roles for SIX2 in the pathogenesis of β-cell defects in T2D.
Results
Reduced SIX2 or SIX3 expression impairs regulated insulin secretion by human islets
We used RNAi-based strategies to reduce SIX2 or SIX3 mRNA levels in primary human adult islets. After dispersion of primary islets, lentiviral transduction was used to achieve shRNA-mediated suppression of SIX2 or SIX3 (hereafter, SIX2kd or SIX3kd) and to simultaneously express a GFP transgene (Peiris et al. 2018), followed by reaggregation into pseudoislets (Fig. 1A,B; Materials and Methods). We used immunostaining to detect β cells (insulin [INS]), α cells (glucagon [GCG]), and δ cells (somatostatin [SST]) in pseudoislets, and verified reaggregation of these principal islet cell types in appropriate proportions (Supplemental Fig. S1). Five days after lentiviral infection, we confirmed significant reduction of SIX2 or SIX3 in pseudoislets by qRT-PCR (Fig. 1C,D; Materials and Methods). In contrast, we did not detect altered SIX3 mRNA levels after SIX2kd or altered SIX2 mRNA levels after SIX3kd (Fig. 1E,F), providing evidence that SIX2 and SIX3 expression are not mutually cross-regulated.
Figure 1.

shRNA-mediated suppression of SIX2 and SIX3 in primary human islets results in impaired glucose-stimulated insulin secretion. (A) Schematics of the lentiviral constructs coding for a short hairpin RNA (shRNA) and GFP. (B) Schematic detailing the pseudoislet technique. (C) SIX2 mRNA expression in primary human islets. (Gray bar) Control, (green bar) SIX2kd. n = 9 independent donor repetitions. (D) SIX3 mRNA expression in primary human islets. (Gray bar) Control, (blue bar) SIX3kd. n = 11 independent donor repetitions. SIX3 mRNA were not altered by SIX2kd (n = 8 independent donor repetitions) (E), and SIX2 mRNA levels were not affected by SIX3kd (n = 11 independent donor repetitions) (F). (G,I) In vitro glucose-stimulated insulin secretion from human pseudoislets control, SIX2kd (n = 9 independent donor repetitions) (G), or SIX3kd (n = 12 independent donor repetitions) (I). Secreted insulin normalized to insulin content. (Black lines) Significant differences within the control, (red lines) significant differences within the KD groups, (green lines) significant differences between control and KD conditions. (H,J) Total insulin from human pseudoislets after transduction with SIX2kd (n = 9 independent donor repetitions) (H) or SIX3kd (n = 12 independent donor repetitions) (J). Data presented as mean; error bars represent the standard error. Two-tailed t-tests used to generate P-values: (*) P < 0.05, (**) P < 0.01, (***) P < 0.0001.
We subsequently assessed glucose-stimulated insulin secretion (GSIS) following SIX2kd or SIX3kd in primary human pseudoislets. Like in our prior studies (Peiris et al. 2018), control pseudoislets infected with lentivirus expressing nontargeting shRNA (“control”) showed a significant increase in insulin secretion after a glucose step increase from 2.8 to 5.6 mM, 16.7 mM, 25 mM, or 25 mM glucose supplemented with the secretion potentiator IBMX (Fig. 1G,I). By comparison, insulin secretion by pseudoislets after SIX2kd was significantly blunted in 2.8 and 16.7 mM glucose and trended toward reduction at 5.6 mM (Fig 1G). Likewise, after SIX3kd, there was significant blunting of insulin secretion in 2.8, 5.6, 16.7, and 25 mM glucose (Fig. 1I). GSIS data were normalized to total pseudoislet insulin content, which was not significantly altered after SIX2kd (Fig. 1H) or SIX3kd (Fig. 1J). This suggests that reduced insulin release from islets after SIX2kd or SIX3kd reflects impaired secretion. Together, our findings provide index evidence that reduced SIX2 or SIX3 function impairs human adult islet β-cell function.
Elucidating the SIX2-dependent adult β-cell transcriptome
SIX2 is an established transcriptional regulator (Kobayashi et al. 2008; Self et al. 2009; He et al. 2010; Park et al. 2012), but SIX2-dependent genetic targets in adult islet β cells are unknown. To identify genes regulated by SIX2, we purified β cells from pseudoislets after SIX2kd (Fig. 2A–C; Materials and Methods). Intracellular labeling with antibodies against insulin and glucagon followed by flow cytometry and GFP gating (Fig. 2C; Supplemental Fig. S2A; Peiris et al. 2018) enriched for INS+ GFP+ β cells; qRT-PCR analysis of this cell subset confirmed enrichment of mRNA encoding INS, and depletion of GCG, or of the acinar and ductal cell markers, CPA1 and KRT19 (Supplemental Fig. S2B). We verified efficient shRNA-mediated suppression of SIX2 in INS+ GFP+ β cells compared with control INS+ GFP+ β cells (Supplemental Fig. S2C). We then produced and sequenced RNA-seq libraries from SIX2kd and control β cells (n = 4 independent donors) (Materials and Methods). Pearson correlation and hierarchical clustering analysis revealed clustering of SIX2kd and control samples from the same donor (Supplemental Fig. S2D,E), reflecting the expected inter-donor variability we and others have previously reported (Arda et al. 2016; Segerstolpe et al. 2016; Enge et al. 2017; Peiris et al. 2018). We used the DE-Seq2 algorithm (Love et al. 2014) to identify differentially expressed (DE) genes following SIX2kd. Expression of 1242 genes, including SIX2 itself, was significantly decreased after SIX2kd (Fig. 2D,G; Supplemental Table S2), whereas expression of 928 genes was significantly increased (P < 0.05) (Fig. 2I; Supplemental Table S3). In contrast, we did not detect changes in expression of β-cell SIX3 upon SIX2kd (Fig. 2E). Gene ontology (GO) term analysis (Fig. 2F) suggested unifying molecular functions in these enriched gene sets, including terms related to adult β-cell function, proliferation, and cell cycle regulation.
Figure 2.

RNA-seq of SIX2kd β cells reveals genes regulated by SIX2 in primary human islets. (A,B) SIX2kd human pseudoislets. (A) Bright field. (B) Blue light (488 nm). Scale bars, 500 µm. (C) FACS scheme used to sort GFP+ β cells. (D,E) Normalized transcript levels of SIX2 (D) and SIX3 (E) in GFP+ β−cells. (Gray bar) Control, (green bar) SIX2kd. n = 4. (F) GO term enrichment in genes deregulated in β cells post-SIX2kd. (G,H) KEGG pathway enrichment in genes up-regulated (G) or down-regulated (H) in β cells post-SIX2kd (n = 4). (I) Heat map showing all differentially expressed (DE) genes in β cells post-SIX2kd. The data are presented as mean; error bars represent the standard error. (*) P < 0.05
Genes with decreased expression in SIX2kd β cells included those encoding cardinal β-cell factors like INS, CHGA, CHGB, and IAPP; insulin processing enzymes like CPE and PCSK2; and transcription factors like PAX6, NEUROD1, NKX6.1, MLXIPL, TCF7L2, ESRRG, and MAFB (Fig. 2G,H; Supplemental Table S2). In addition, we noted severe reduction of mRNAs encoding glucokinase (GCK), the principal sensor of glucose flux in β cells (Matschinsky et al. 1993); glucagon receptor (GCGR); regulators of glycolysis and β-cell stimulus secretion coupling encoded by TPI1, ALDH2, ALDOA, ENO2, PGK1, and FBP1; and CAMK1D, a postulated type 2 diabetes risk gene (Fig. 2G; Thurner et al. 2018; Miguel-Escalada et al. 2019). In contrast, we observed increased levels of mRNAs encoding regulators of DNA replication and cell cycle factors in SIX2kd β cells, including NUSAP1, MAX, and SRF (Fig. 2G,I). This is consistent with prior findings (Arda et al. 2016) providing evidence that SIX2 expression may enforce β-cell cycle arrest and studies linking β-cell cycle exit to enhanced function (Helman et al. 2016). GO and KEGG pathway analysis of DE genes (Materials and Methods) revealed significant enrichment of terms including regulation of hormone secretion, glucose homeostasis, calcium signaling, and insulin signaling (Fig. 2F,H,I). Thus, these data suggest that SIX2 is required to maintain hallmark adult β-cell functions involved with insulin production and processing, glucose sensing, and proliferation, and support our finding of impaired GSIS after SIX2kd in adult human islets.
To validate our findings further, we assessed whether genes regulated by SIX2kd were enriched in gene sets whose expression changed in β cells expressing SIX2 (see the Materials and Methods; Blodgett et al. 2015; Arda et al. 2016). For example, we asked whether the set of genes with decreased expression after SIX2kd overlapped with DE genes whose mRNA normally increased with advancing age. In this case, we identified 287 genes with decreased expression after SIX2kd in β cells, whose expression was significantly increased in SIX2+ adult β cells compared with fetal or juvenile SIX2neg β cells (P < 3.873 × 10−30) (Fig. 3A,B; Supplemental Table S7). These included CDKN1C and CDKN1A, which encode inhibitors of cell proliferation; CACNA1C and CACNA1D, which encode calcium channels; GCGR, which encodes a receptor for glucagon and the incretin GLP-1 (Svendsen et al. 2018); and MLXIPL (also known as ChREBP), a glucose-activated transcription factor that regulates GCGR (Iizuka et al. 2012), GREM1, IAPP, and DLK1 (Fig. 3A,B; Supplemental Table S7). Based on similar logic, we identified 146 genes with significantly increased expression after SIX2kd, whose expression is normally decreased in SIX2+ adult β cells (P < 5.2 × 10−5) (Fig. 3B; Supplemental Table S7; Blodgett et al. 2015; Arda et al. 2016). These included NUSAP1 and SRF, postulated regulators of islet cell proliferation (Zeng et al. 2017), and RFX7, a marker of pancreatic progenitor cells (Kim-Muller et al. 2016). We also compared this data set with DE transcriptomic data recently reported after SIX2 loss in β-like cells (“SC-β”) generated from a human embryonic stem cell line (Velazco-Cruz et al. 2020). We found a general lack of concordance in DE genes (Materials and Methods): Specifically, we observed 30% (377/1242) overlap of genes with reduced expression and 14% (132/928) overlap of genes with increased expression after SIX2kd (Supplemental Fig. S3A,B; Supplemental Table S12). Many mRNAs that decreased after SIX2kd in native β cells, like MAFB, GIPR, and GREM1, did not change after SIX2kd in SC-β cells. Other mRNAs that decreased after SIX2kd in adult β cells, like CDKN1C, GCGR, TCF7L2, and NKX6.1, were found increased after SIX2kd in SC-β cells (Velazco-Cruz et al. 2020). These findings indicate that genetic programs in native human β cells and SC-β cells are distinct and demonstrate advantages of investigating SIX2-dependent gene expression in primary adult β cells.
Figure 3.
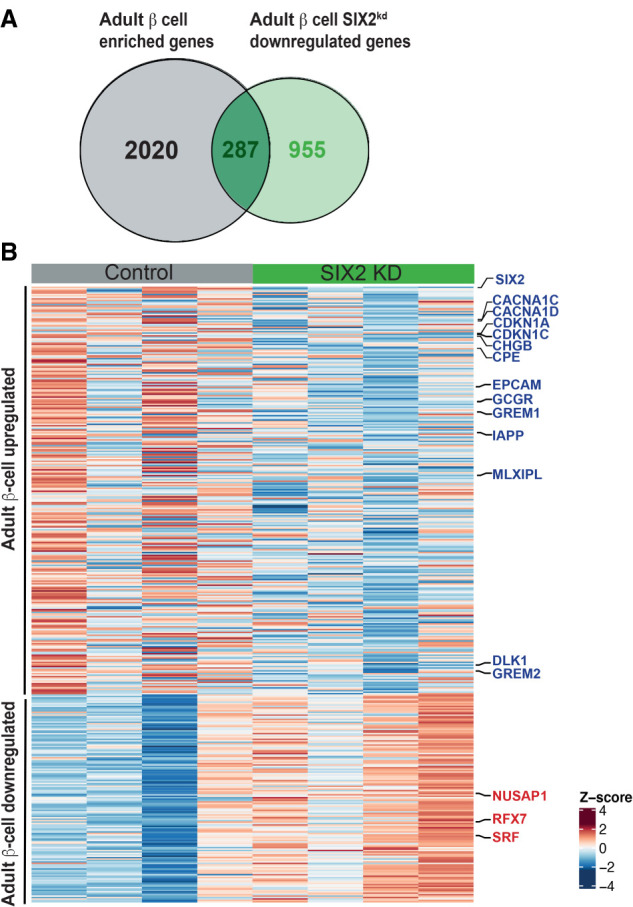
SIX2kd in primary β cells results in down-regulation of genes enriched in adult β-cell. (A) Venn diagram showing adult β-cell genes down-regulated post-SIX2kd. (B) Heat map of adult down-regulated genes and juvenile up-regulated genes in adult β cells post-SIX2kd.
Identifying direct genetic targets of SIX2 regulation in human β cells
To identify direct genetic targets of SIX2 in primary human islet cells, we performed cleavage under targets and release using nuclease (CUT&RUN) (Skene and Henikoff 2017; Hainer et al. 2019), which enables the sensitive detection of genomic loci bound by TFs (Fig. 4A; Materials and Methods). Because antibodies that detected native islet SIX2 for CUT&RUN were not available, we misexpressed in pseudoislets a transgene encoding human SIX2 tagged with the FLAG immuno-epitope (SIX2-FLAG) from a rat insulin promoter element (RIP) (Karlsson et al. 1987), then sequenced DNA bound by the SIX2-FLAG protein with an anti-FLAG antibody (Supplemental Fig. S4A). Thus, interpretation of CUT&RUN here is qualified by the possibility that SIX2-FLAG may bind sites not bound by native SIX2. We used the HOMER algorithm (Heinz et al. 2010) to identify genomic regions that were bound by SIX2-FLAG in samples from three islet donors (P ≤ 0.01 compared with IgG controls). Heat map visualization of independent peaks (Fig. 4B) as well as histogram plotting of averaged reads (Supplemental Fig. S4B) showed enrichment of read densities in the peak centers for the SIX2-FLAG libraries, whereas IgG controls showed minimal enrichment at these sites (Fig. 4B; Supplemental Fig. S4B). As further validation of the specificity of CUT&RUN, we found that SIX2-FLAG-bound genomic peaks were significantly enriched for the SIX2 DNA-binding motif, as well as the SIX1 DNA-binding motif, and motifs of other β-cell-enriched TFs, like MAFB (Fig. 4C). We used the GREAT algorithm (McLean et al. 2010) to associate SIX2-FLAG-bound genomic regions to 10,270 genes (Fig. 4D). Inadequate yields precluded analogous SIX3-FLAG studies (Materials and Methods).
Figure 4.

Identification of presumptive SIX2 genetic targets in primary human β cells using CUT&RUN. (A) Schematic of the CUT&RUN approach: Pseudoislets overexpressing SIX2-FLAG under the RIP promoter were used for CUT&RUN with anti-FLAG antibody (n = 3 independent donors). (B) Heat map showing enrichment of peak read densities at the center of the peak for the CUT&RUN libraries generated with FLAG antibody, but not for IgG. Peaks were called using HOMER. (C) Enriched motifs in the differential peaks were identified by HOMER. (D) Overlap of the SIX2-associated genomic regions and the SIX2kd DE genes. (E,F) Tracks showing SIX2-FLAG genomic regions associated with GCGR (E) or CHGA (F). Accessible chromatin regions in human islets are shown by ATAC-seq, H3K4me3, and H3K27ac ChiP-seq tracks. SIX2-FLAG CUT&RUN peaks are shown in pink boxes (note: for GCGR, two peaks are shown), and regulated genes are highlighted in green boxes.
To nominate candidate genes that might be directly regulated by SIX2, we then identified genes that (1) neighbored SIX2-FLAG binding sites and (2) had differential expression after SIX2kd. After intersection of CUT&RUN with 2170 DE genes after SIX2kd, we identified 1186 “overlapping genes” (Fig. 4D; Supplemental Table S4). Sixty-four percent of these genes (754/1186) had reduced mRNA levels after SIX2kd, consistent with a role for SIX2 in activating β-cell gene expression. Presumptive direct SIX2 targets identified by this approach included PAX6, IAPP, MAFB, CDKN1C, DLK1, PCSK2, GCGR, MLXIPL, and CHGA. As expected, SIX2-FLAG-bound genomic regions in presumptive SIX2 target genes were found in accessible chromatin and colocalized with activation-associated H3K4me3 and H3K27ac histone marks identified by prior islet ATAC-seq and ChIP-seq studies (Mularoni et al. 2017). This alignment further supports the conclusion that SIX2 binds active genomic regulatory elements governing hallmark β-cell genes (Fig. 4E,F; Supplemental Fig. S4C–E; Supplemental Table S4). Thus, our targeted nuclease-based analysis revealed hundreds of SIX2-associated candidate regulatory elements, including many that likely regulate expression of hallmark β-cell factors.
SIX3 and SIX2 regulate distinct gene sets in adult human β cells
Gain-of-function studies have linked SIX3 to adult human β-cell functional maturation (Arda et al. 2016). To identify genes whose expression is regulated by SIX3 in human β cells, we used flow cytometry to purify β cells from human pseudoislets after SIX3kd (Supplemental Fig. S5A–C; Materials and Methods). Like in our SIX2kd studies, we confirmed enrichment of INS+ GFP+ β cells—and depletion of non-β cells—with qRT-PCR analysis (Supplemental Fig. S5D), then generated, sequenced, and analyzed RNA-seq libraries (n = 3 independent donors). We achieved 50% reduction of SIX3 mRNA in purified INS+ GFP+ β cells (Fig. 5A; Supplemental Fig. S5E). In contrast, SIX2 mRNA levels were not detectably changed (Fig. 5A). As expected, analysis of RNA-seq libraries with Pearson correlation analysis and hierarchical clustering revealed close clustering of SIX3kd and control β-cell samples from the same donor (Fig. 5B; Supplemental Fig. S5F). With the DE-Seq2 algorithm (Materials and Methods), we identified 263 genes with significantly decreased mRNA levels (P < 0.05) (Supplemental Table S5), including SIX3 itself (Fig. 5A), and 372 genes with significantly increased mRNA levels in SIX3kd β cells (P < 0.05) (Fig. 5E; Supplemental Table S6). Thus, the number of DE genes with increased mRNA outnumbered those with decreased mRNA after SIX3kd, unlike DE genes after SIX2kd. Gene ontology (GO) analysis suggested molecular functions of the enriched gene sets, including regulation of DNA binding, response to glucose stimulus, and DNA replication (Fig. 5C; Supplemental Fig. S5G). Up-regulated DE genes associated with the latter term included established regulators of β-cell replication, like MYC, MAX, and INSM1 (Fig. 5C,E). Pathway analysis of the DE up-regulated genes (Materials and Methods) included TGF-β signaling, type 2 diabetes mellitus, and calcium signaling pathway (Fig. 5D).
Figure 5.

RNA-seq of SIX3kd β cells reveals a distinct gene set regulated by SIX3. (A) Normalized transcript levels of SIX3 and SIX2 in GFP-expressing β cells. (Gray bar) Control, (blue bar) SIX3kd . n = 3. (B) Heat map of the sample-to-sample distances for all the samples used in this experiment. (C) GO term enrichment in genes deregulated in β cells post-SIX3kd. (D) KEGG pathways enriched in genes up-regulated in β cells post-SIX3kd (n = 3). (E) Heat map showing all up-regulated genes upon SIX3kd in β cells. (F) Overlapped DE genes in adult β cells post SIX2kd and SIX3kd. (G) Fold transcript levels of non-β-cell genes significantly altered in β cells post-SIX3kd (n = 3). The data are presented as mean; error bars represent the standard error. (*) P < 0.05.
The DE genes after SIX3kd were largely distinct from DE genes after SIX2kd; only 133/2805 DE genes (<5%) overlapped between SIX2kd and SIX3kd (Fig. 5F; Supplemental Table S8; Supplemental Fig. S6A,B). Moreover, among these 133 DE genes, 112/133 (84%) changed in the opposite direction after SIX2kd compared with SIX3kd, such as KCNMB2, GREM2, OLIG1, and TLE2. After SIX3kd there was also a significant increase of mRNAs encoding genes not usually expressed in adult human β cells. This included genes encoding factors enriched or exclusively expressed in islet α cells or ε cells, like DPP4, NPNT, TMEM236, ADORA2A, and GHRL, which encodes the ε-cell hormone ghrelin, and MBOAT4 (which encodes an acetyl-transferase for ghrelin) (Fig. 5E,G). While not reaching statistical significance, we also detected an average 2.5-fold increase of mRNA encoding GCG in SIX3kd β cells (Fig. 5G). In SIX3kd β cells, we also identified increased average levels of 70 mRNAs that are typically expressed highly in fetal β cells, but attenuated or extinguished in adult β cells (Fig. 6A,B; Supplemental Table S9; Blodgett et al. 2015; Arda et al. 2016). This latter group of genes included MYC (Puri et al. 2018) and a “disallowed” gene (Pullen et al. 2010) that encodes hexokinase (HK2) (for review, see Lemaire et al. 2016). Moreover, none of these mRNAs increased in β cells after SIX2kd. Together, our findings support the view that SIX2 and SIX3 regulate distinct gene sets in human β cells.
Figure 6.

SIX3 represses non-β-cell programs in the adult β-cell. (A) Venn diagram showing juvenile β-cell genes up-regulated post-SIX3kd in adult β cells. (B) Heat map of adult down-regulated and juvenile up-regulated genes in adult β cells post-SIX3kd. (C) Schematic detailing the juvenile pseudoislet technique used to overexpress SIX3-FLAG (SIX3-ox) in juvenile pseudoislets (n = 2) and of the constructs used to overexpress SIX3 in juvenile pseudoislets: FACS was used to sort FLAG+ β cells. (D) SIX3, INS, and GCG mRNA expression in FLAG+-expressing β cells post FACS. (White bar) Control , (red bar) SIX3-ox. n = 2. (E) Schematics showing proposed coordinated regulation of maturation by SIX2 and SIX3 in the β-cell. The data are presented as mean; error bars represent the standard error. (*) P < 0.05.
We previously showed that SIX3 expression in pseudoislets from juvenile human donors aged 0.5–2 yr (which lack SIX3 expression) was sufficient to stimulate insulin secretion in vitro (Arda et al. 2016); however, transcriptome studies were not performed. To assess changes of gene expression stimulated by SIX3, we misexpressed a SIX3-FLAG transgene in pseudoislets from two juvenile donors (ages 1.5 and 3 yr) (Fig. 6C; Supplemental Table S1). Flow cytometry and western blotting verified and quantified expression of transgenic SIX3-FLAG (Supplemental Fig. S7A–C). qRT-PCR analysis of purified β cells showed a reduction in GCG levels and an average increase of INS mRNA levels following SIX3 misexpression (Fig. 6D), as previously reported (Arda et al. 2016). RNA-seq of purified SIX3-FLAG+ INS+ β cells (Materials and Methods) confirmed reduced mRNAs encoding GCG, HK2, MYC, TMEM236, MBOAT4, RUNX2, GREM2, and NPNT, mRNAs found increased after SIX3kd in primary β cells (Supplemental Fig. S7D; Supplemental Table S10). Thus, SIX3 gain- and loss-of-function studies here produced reciprocal changes in expression of multiple genes, supporting the view that SIX3 suppresses adult β-cell expression of gene sets expressed abundantly in fetal or neonatal β cells, or in adult α and ε cells. We conclude that SIX3 reinforces mature β-cell function, in part, by suppressing fetal gene expression programs and alternative islet cell fates (Fig. 6E).
Human β-cell expression of SIX2 is reduced in islets from T2D donors
It remains unknown whether the expression of SIX2 or SIX3 changes in islet β cells obtained from T2D patients. We found that both SIX2 and SIX3 mRNA appeared to be reduced in purified whole islets from subjects with established T2D (n = 4), compared with islets from nondiabetic controls (n = 7) (Fig. 7A; for donor information, see Supplemental Table S1). In contrast, expression of GPD2 and LEPROTL2, previously found up-regulated in prior studies (Segerstolpe et al. 2016), was unaltered in T2D islets (Fig. 7A).
Figure 7.

SIX2 expression is reduced in β cells from T2D donors. (A) Gene expression levels in whole islets from nondiabetic (ND; gray bars) (n = 5) or type 2 diabetic (T2D; red bar) (n = 3). (B) Heat map showing DE genes in nondiabetic versus T2 diabetic β cells. (C,D) Box plots displaying TPM counts of SIX2 (C) and SIX3 (D) in nondiabetic (ND) (n = 5) β cells (dark-gray bars) and α cells (light-gray bars) or type 2 diabetic (T2D) β cells (red bars) and α cells (dark-red bars) (n = 3). (E–G) Box plots displaying TPM GCGR (E), MAFB (F), and CDKN1C (G) in β-cells of ND (n = 5) and T2D (n = 3), the expression of which is DE in adult β cells post-SIX2kd. (Gray bars) Çontrol, (green bars) SIX2kd. (H) Insulin secretion of ND (n = 5) versus T2D (n = 3; see the Materials and Methods). (I) Plot of the total area under the curve of the released insulin. The data in A are presented as mean; error bars represent the standard error. Box plots show the mean. (Red *) P ≤ 0.01, (black *) P < 0.05.
Bulk RNA-seq from FACS-purified α and β cells was used to assess SIX2 and SIX3 expression in β cells (Fig. 7B–D; see the Materials and Methods). Transcriptome analysis confirmed significant reduction of SIX2 mRNA in purified β cells from T2D islets compared with β cells from control islets (P ≤ 0.01) (Fig. 7B–C; Supplemental Table S11), while SIX3 mRNA levels were not significantly changed (Fig. 7D). Thus, SIX2 expression was reduced in β cells in islets from cadaveric T2D donors. Moreover, β-cell regulation of SIX2 and SIX3 in these T2D islets was uncoupled. We also detected little to no expression of SIX2 and SIX3 in purified α cells from control or T2D islets (Fig. 7C,D). In addition to reduced SIX2 expression in β cells, we also noted impaired expression of SIX2 target genes, including MAFB, GREM1, GCGR, and CDKN1C, a candidate T2D risk gene (Fig. 7B,E–G; Supplemental Table S11). Consistent with our studies of impaired insulin secretion after SIX2kd (Fig. 1G), glucose-stimulated insulin secretion studies revealed impaired insulin secretion by islets from subjects with T2D compared with nondiabetic controls (Fig. 7H,I; see the Materials and Methods).
Although SIX3 mRNA levels were not detectably changed in our sampling of T2D β cells, a subset of SIX3 targets was differentially expressed in T2D β cells, including DPP4, NPNT, and GHRL; TGF-β signaling factors encoded by TGFB1 and THBS1; and the disallowed gene HK2 (Fig. 7B). This raises the possibility that factors collaborating with SIX3 to regulate these genes might be changed in T2D β cells.
Together, these findings suggest that dysregulation of SIX2- and SIX3-dependent genetic programs could contribute to impaired islet β-cell fate and function in T2D. These findings also support prior genome-wide association studies linking the locus encoding SIX2 and SIX3 to risk for T2D and diabetes-related traits (Kim et al. 2011; Hachiya et al. 2017; Varshney et al. 2017; Spracklen et al. 2018, 2020), raising the possibility that genetic influences might additionally modulate β-cell expression of SIX2 or SIX3.
Discussion
Here, we overcame inherent challenges facing postnatal human developmental studies to investigate the roles of SIX2 and SIX3 in human pancreatic β-cell maturation. Using genetic approaches, we show that SIX2 is required for expression of multiple hallmark genes in human β cells. In addition to regulation of genes governing β-cell function, we show that SIX3—unlike SIX2—suppresses expression of genes typically expressed in α cells or other non-β cells. Thus, our studies provide index evidence that SIX2 and SIX3 regulate distinct sets of genetic targets in adult human β cells. shRNA-mediated suppression of either SIX2 or SIX3 expression in primary human islets impaired regulated insulin secretion by β cells. Supporting these findings, we found evidence of reduced expression of SIX2 and downstream targets in islet β cells from human subjects with T2D, which coincided with significantly reduced insulin secretion by these islets. In sum, our study unveils a requirement for SIX2 and SIX3 in establishing and maintaining adult human β-cell function and fate (Fig. 6E).
Prior to our study, it was unclear what roles SIX2 and SIX3 had in adult human β-cell function. SIX2 and SIX3 are coexpressed in adult human β cells, and developmental studies of human islet cells have revealed coincident increases of SIX2 and SIX3 expression after the first decade of life (Arda et al. 2016, 2018; Blodgett et al. 2015). Moreover, studies of putative SIX2 and SIX3 cis-regulatory elements in humans and other systems have suggested these genes may be coregulated (Spracklen et al. 2018; Suh et al. 2010). A common set of SIX2 and SIX3 targets identified here includes regulators of cell cycle progression. However, the onset of SIX2 and SIX3 in human β cells occurs well after the period of neonatal expansion (Blodgett et al. 2015; Arda et al. 2016), suggesting that the post-mitotic state of β cells is established by other factors, and then reinforced by SIX2 and SIX3. Consistent with this possibility, we observed that reduction of SIX2 alone or SIX3 alone did not increase markers of β-cell S-phase like MKI67 (Supplemental Tables S2–S5). Moreover, we showed in our prior work that misexpression of either SIX2 or SIX3 was sufficient to suppress proliferation of the human β-cell line EndoCβH1 (Arda et al. 2016). Enforcement of the post-mitotic state in β cells has been linked to attainment of mature function (Helman et al. 2016; Puri et al. 2018; Mandelbaum et al. 2019).
The majority of differentially expressed genes after SIX2kd showed reduced expression, including those encoding crucial human β-cell factors like insulin and glucokinase, and essential TFs that coordinate pancreatic islet development and β-cell function in humans. Consistent with this, we observed reduced insulin secretion after SIX2kd. In contrast, after SIX3kd the majority of DE genes showed increased expression. These included transcripts encoding factors not normally expressed in healthy β cells, like the ε-cell hormone ghrelin, the α-cell-enriched protease DPP4, and the disallowed factor HK2 (Dhawan et al. 2015). These SIX3kd findings are consistent with previous studies suggesting that SIX3 can function as a transcriptional repressor (Kobayashi et al. 2001). Thus, our loss-of-function studies revealed SIX2- and SIX3-dependent mechanisms that regulate native maturation and fate of human β cells. Here, the degree of SIX2 mRNA reduction after SIX2kd was greater than the degree of SIX3 mRNA loss after SIX3kd; thus, future studies that achieve more complete SIX2 or SIX3 loss of function could identify additional β-cell genetic targets. Our study was also limited by the inherent variability of cadaveric human islet donors, as we and others have previously reported (Arda et al. 2016; Segerstolpe et al. 2016; Enge et al. 2017; Peiris et al. 2018). While SIX3 expression is restricted to the β cell, SIX2 is also expressed in islet δ cells (Baron et al. 2016; Muraro et al. 2016). Thus, changes observed after SIX2kd could reflect both β-cell autonomous and nonautonomous mechanisms. Studies here also revealed that SIX2 and SIX3 expression in β cells can be genetically uncoupled and correspond well with our data showing that SIX2 and SIX3 regulate distinct β-cell gene sets. Thus, distinct mechanisms likely govern expression and activity of SIX2 and SIX3 in β cells from healthy and diabetic cadaveric donors.
Elucidating how SIX2 and SIX3 expression are regulated should be aided by our identification of their targets in human β cells. Prior studies have shown that transcription factors like SIX2 and SIX3 expressed in human β cells show increased expression with age (Aguayo-Mazzucato et al. 2011; Blodgett et al. 2015; Arda et al. 2016; Wang et al. 2016b). For shRNA-based studies here we used islets from donors >22 yr of age, when adult levels of SIX2 and SIX3 have been established. Intense interest in SIX2 and SIX3 regulation also stems from association of the locus encoding these factors to T2D and related traits like fasting hyperglycemia (Kim et al. 2011; Hachiya et al. 2017; Varshney et al. 2017; Spracklen et al. 2018, 2020). However, studies of whole islet RNA, or prior single islet cell RNA-seq investigations by us and others (Segerstolpe et al. 2016; Camunas-Soler et al. 2020) were not sufficiently sensitive to detect changes of SIX2 or SIX3 mRNA in β cells isolated from donors with T2D. Studies here provide index evidence that (1) expression of SIX2, and a subset of SIX2-dependent genes like GCGR, are significantly reduced in β cells from T2D donors, and (2) islets from T2D donors with reduced β-cell expression of SIX2 had impaired insulin secretion (Fig. 7). While we did not detect changes in SIX3 expression in T2D islets here, additional studies are required to exclude the possibility that β-cell SIX3 dysregulation is a feature of T2D.
A recent report described phenotypes after SIX2 loss in hPSC-derived β-like cells (SC-β cells) (Velazco-Cruz et al. 2020). While 25% of these insulin+ SC-β cells express SIX2 mRNA, they lack other markers of mature β cells like MAFB or SIX3, thus precluding studies of SIX3 loss of function. After shRNA-mediated suppression of SIX2, Velazco-Cruz et al. (2020) reported reduced insulin protein content, loss of glucose-stimulated insulin secretion without effects on “basal” insulin secretion at low glucose concentration (2 mM), and significant changes in expression of >10,000 genes, assessed by RNA-seq of unsorted hESC progeny, with enrichment of gene sets related to insulin secretion and calcium signaling. This included significant increases of multiple transcripts encoding islet α cell or δ cell products, like SST, DPP4, MBOAT4, and FSTL1 (Velazco-Cruz et al. 2020). These findings support the conclusion that SIX2 is required for SC-β cells derived from hESCs to acquire some features of native human β cells.
In our study, we assessed the effects of SIX2 or SIX3 loss in native human β cells. After SIX2kd, we observed reduction of both basal and glucose-stimulated insulin secretion, without reduction of islet insulin content. FACS purification of Insulin+ β cells (and elimination of SIX2+ δ cells) and RNA-seq revealed significant changes in mRNA levels of 2100 genes, with gene set analysis revealing enrichment of terms related to proliferation, insulin secretion, calcium signaling, carbohydrate metabolism, and exocytosis. While some of these gene sets overlapped with those in SC-β cells (Velazco-Cruz et al. 2020), there was an overall lack of concordance between gene sets (Supplemental Fig. S3; Supplemental Table S12). For example, after SIX2kd we observed reduced mRNA encoding the calcium channel subunits CACNA1A and CACNA1D (“calcium signaling”), incretin receptors GIPR and GCGR (“insulin secretion”), and transcription factors with established roles in native β-cell regulation like MAFB, NEUROD1, NKX6.1, and TCF7L2; these changes were not noted in SC-β cells. Moreover, we did not observe increased expression of non-β-cell markers like DPP4, MBOAT4, and FSTL1 after SIX2kd. Instead, we observed increased expression of these genes, and other non-β-cell or disallowed genes after SIX3kd. These contrasts raise the possibility that SIX2 activity in SC-β cells includes ectopic functions normally fulfilled by SIX3 or other factors. Together, our findings clarify the importance of investigating SIX2 and SIX3 functions in bona fide adult β cells. While models of human β-cell development, like stem cell-derived insulin+ cells and immortalized β-cell lines have value (Sneddon et al. 2018), to date they remain fundamentally different from genuine pancreatic islet cells in gene regulation, function, proliferation, and cellular composition. Our studies further suggest that simultaneous expression of both SIX2 and SIX3 may be required to produce consummately functional replacement β cells from renewable sources, like human stem cell lines.
In summary, this study unveils SIX2 and SIX3 functions crucial for postnatal human islet and β-cell development and maturation and reveals how β-cell dysfunction might develop in diabetes. Our work demonstrates that SIX2 and SIX3 coordinately govern distinct genetic programs that increase insulin production and enhance mature β-cell physiological functions, enforce β-cell fate by suppressing alternative genetic programs, and suppress proliferation. Findings here also provide a unique developmental “roadmap” for achieving human β-cell replacement.
Materials and methods
Human islet procurement
Deidentified human islets were obtained from healthy, nondiabetic organ donors or type 2 diabetic donors procured through the Integrated Islet Distribution Network (IIDP), National Diabetes Research Institute (NDRI), International Institute for the Advancement of Medicine (IIAM), and the Alberta Diabetes Institute Islet Core. For T2D studies, data from the Human Pancreas Analysis Program (HPAP-RRID: SCR_016202) Database (https://hpap.pmacs.upenn.edu), a Human Islet Research Network (RRID: SCR_014393) consortium (UC4-DK-112217 and UC4-DK-112232) was used. See Supplemental Table S1 for details.
Constructs and lentivirus production
Lentiviral constructs coding for shRNAs targeting human SIX3 or SIX2 were obtained from Dharmacon. plenti-CMV-SIX3-cMyc-DDK was used in juvenile islet experiments (Origine). plenti-RIP-SIX2-cMyc-DDK was generated by replacing the CMV promoter of plenti-CMV-SIX2-cMyc-DDK (Origine) with the rat insulin promoter (RIP). Lentiviruses were produced by transfection of HEK293T cells with lentiviral constructs, pMD2.G (Addgene 12259) and psPAX2 (Addgene 12260) packaging constructs. Supernatants were collected and concentrated by PEG-it (System Biosciences).
Human pseudoislet generation and transduction
Human islets were dispersed into single cells by enzymatic digestion (Accumax, Invitrogen) and transduced with 1 × 109 viral units/1 mL lentivirus. Transduced islet cells were cultured in ultra-low attachment well plates for 5 d prior to further analysis.
RNA extraction and quantitative RT-PCR
RNA was isolated from whole pseudoislets using the PicoPure RNA isolation kit (Life Technologies). For sorted β and α cells, RNA was isolated using the RecoverALL isolation kit (Invitrogen by Thermo Fisher Scientific). cDNA was synthesized using the Maxima first strand cDNA synthesis kit (Thermo Scientific), and gene expression was assessed by PCR using TaqMan gene expression mix (Thermo Scientific) and the following probes ACTIN-B, Hs4352667_m1; SIX2, Hs00232731_m1; SIX3, Hs00193667_m1; insulin, Hs00355773_m1; glucagon, Hs00174967_m1; CPA-1, Hs00156992_m1; and KRT19, Hs01051611_gH.
In vitro insulin secretion assays
Batches of 25 pseudoislets were used for in vitro secretion assays as previously described (Peiris et al. 2018). Briefly, pseudoislets were incubated at 2.8, 5.6, 16.7, 25, and 25 mM + IBMX glucose concentrations for 60 min each, and supernatants were collected. Secreted human insulin in the supernatants and pseudoislet lysates were quantified using a human insulin ELISA kit (Mercodia). Secreted insulin levels are presented as a percentage of total insulin content. Perifusion data of T2 diabetic versus nondiabetic samples were acquired from the Human Pancreas Analysis Program (HPAP-RRID: SCR_016202).
Immunohistochemistry
Human pseudoislets were fixed for 1 h at 4°C and embedded in collagen (Wako Chemicals) and OCT before sectioning and staining as previously described (Arda et al. 2016). Primary antibodies used were guinea pig anti-Insulin (1:1000; DAKO A0564), mouse anti-glucagon (1:500; Sigma), and mouse anti-SST (1:500). Secondary antibodies were incubated for 2 h at room temperature. Images were obtained using a Leica SP2 confocal microscope.
Intracellular staining and FACS sorting of human islet cells
Detailed protocol can be found in Peiris et al. (2018). Briefly, pseudoislets were dispersed into single cells and stained with LIVE/DEAD Fixable Near-IR dead cell stain kit (Life Technologies) prior to fixation with 4% paraformaldehyde. After permeabilization, cells were stained with the following antibodies: guinea pig anti-insulin (1:100; Dako) followed by anti-guinea pig Alexa Fluor 555 (1:100; Sigma) and mouse anti-glucagon antibody Alexa Fluor 647 (1:100; Santa Cruz Biotechnology). Juvenile islet cells from SIX3-FLAG pseudoislets were stained with anti-FLAG antibody-555 (1:100; Biolegend). Labeled cells were sorted on a special order five-laser FACS Aria II (BD Biosciences) using a 100-µm nozzle, with appropriate compensation controls and doublet removal. Sorted cells were collected into low retention tubes containing 50 µL of FACS buffer.
RNA isolation and preparation of RNA-seq libraries
A total of 20,000 sorted, fixed β cells were used for each RNA-seq library construction of RNA with RIN number >7. SMART-seq v4 Ultra Low input RNA kit (Clontech) was used to amplify cDNA, which was subsequently sheared, resulting in 200- to 500-bp fragments. RNA-seq libraries were generated using the Low Input library preparation kit v2 (Clontech). Barcoded libraries were then multiplexed and sequenced as paired-end 150-bp reads on the Illumina HiSeq4000 platform. A total of eight libraries were generated from four different donors used for the SIX2kd (four libraries) and the respective control β cells (four libraries), while six libraries were generated from three different donor β cells used for SIX3kd and their respective controls.
Bioinformatic and statistical analysis
RNA-seq analysis was performed on SIX2kd and control β-cell libraries from four donors and on SIX3kd and control β-cell libraries from three donors. FastQC v0.11.4 was used for quality control. All libraries had >75 million reads, and barcodes were trimmed using Trimgalore_0.5.0. Reads were aligned to the human genome index (hg19) using STAR v2.6.1d (Dobin et al. 2013). Transcripts per million (TPM) were quantified using RSEM v1.3.0 (Li and Dewey 2011). Differentially expressed genes with fold change were detected using the DESeq2 R package (Love et al. 2014) for the two experimental conditions. The Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.7 was used (Huang et al. 2009) for gene set enrichment analysis. RNA-seq data sets of genes enriched in adult β cells versus juvenile β cells were obtained from Arda et al. (2016) (GEO: GSE79469) and Blodgett et al. (2015) (GEO: GSE67543). The probability of finding x overlapping genes was calculated using the hypergeometric probability formula that considers the total number of genes in the genome. RNA-seq data of sorted β and α cells of T2D versus nondiabetic donors (HPAP-RRID:SCR_016202) and of SIX2kd SC-β cells (GEO: GSE147737) (Velazco-Cruz et al. 2020) were analyzed per our data using DESeq2 R package (Love et al. 2014).
CUT&RUN and library preparation
Six-hundred-thousand redispersed islet cells were used as input material for each CUT&RUN, which was performed from three donors, as described (Skene and Henikoff 2017; Hainer et al. 2019). Briefly, nuclei were extracted with nuclear extraction buffer and added to concanavalin A bead slurries (Polysciences). After blocking, the nuclei/beads were washed in wash buffer and resuspended with rabbit anti-FLAG (Sigma-Millipore F7425) or IgG (Millipore) antibodies overnight at 4°C. Protein A-micrococcal nuclease (pA-MN; EpiCypher donation) was added to a concentration of 1:400 to nuclei. Cleavage was induced by 100 mM CaCl2 for 30 min at 0°C. DNA fragments were released for 20 min at 37°C and purified using phenol/chloroform/isoamyl alcohol followed by chloroform extraction and precipitated with glycogen and ethanol. DNA was resuspended in 0.1× TE and used for library construction with NEBnext Ultra II library kit. Libraries were sequenced as 2 × 75 on HiSeq4000.
CUT&RUN data analysis
Paired-end reads were trimmed and aligned as per CUT&RUNtools (Zhu et al. 2019). Briefly, Trimmomatic (Bolger et al. 2014) was used for trimming and Bowtie2 (Langmead and Salzberg 2012) for alignment. HOMER (Heinz et al. 2010) was used for peak calling. Genome browser tracks were generated from mapped reads using the “makeUCSCfile” command. Peaks were called using the “findPeaks” command. The GREAT algorithm was used for gene annotation (McLean et al. 2010). Motifs were identified using the “findMotifs” command. P-values for motif enrichment were performed by HOMER software, using a binomial test.
Statistical analysis
For qRT-PCR and GSIS, the number of biological or technical replicates (n), measure of central tendency (e.g., mean), standard deviation, and statistical analysis is detailed in each figure legend. Graphs and statistical analysis were produced and performed using GraphPad Prism (version 8) software.
Data visualization
Cytometery data were analyzed and graphed using FlowJo software (version 10.8, Beckton, Dickinson, and Company).
Heat maps were made with ComplexHeatmap. Browser tracks were made with the UCSC genome browser. The graphics were made with BioRender.
Data availability
The data discussed in this publication have been deposited in NCBI's GeneExpression Omnibus (Edgar et al. 2002) and are accessible under accession number GSE164628.
Supplementary Material
Acknowledgments
We thank past and current members of the Kim group for advice and encouragement, especially Dr. Y. Hang, Dr. S. Park, and Dr. A. Ibarra Urizar for technical guidance and advice; Dr. C. Chang, Dr. Y. Hang (Kim group), and Dr. R. Bottino (Allegheny Health Network) for assistance in tissue procurement; Dr. M. Angulo (K. Chua group, Stanford) for help with pull-downs and the CUT&RUN protocol; Dr. G. Oliver (Northwestern University Feinberg School of Medicine) for initial discussions about SIX2 and SIX3; N. Koska for help with antibody testing; and members of the Kim laboratory for comments on the manuscript. We thank Dr. R. Nair (Diabetes Genomics Analysis Core, Stanford Diabetes Research Center) for help with bioinformatics and programming. We thank Professor K. Loh and Professor A. Gloyn for advice and encouragement. We gratefully acknowledge organ donors and their families, and islet procurement through the Alberta Diabetes Institute Islet Core, Integrated Islet Distribution Program (National Institutes of Health UC4 DK098085), the National Disease Research Interchange, and the International Institute for the Advancement of Medicine. R.J.B. was supported by a postdoctoral fellowship from JDRF (3-PDF-2018-584-A-N) and is on leave from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET)-Universidad de Buenos Aires, Instituto de Investigaciones en Producción Animal (INPA), Buenos Aires, Argentina. H.P. was supported by fellowships from the Maternal and Child Health Research Institute (School of Medicine, Stanford University, UL1TR001085), the American Diabetes Association (1-16-PDF-086), and a Young Investigator Award from the Stanford Institute for Immunity, Transplantation, and Infection; R.L.W. was supported by fellowships from the Division of Endocrinology National Institutes of Health T32 training grant in the Department of Medicine, Stanford University (DK007217-41, to A. Hoffman and F. Kraemer) and JDRF (3-PDF-2020-931-A-N); and S.K. was supported by a fellowship from the Larry L. Hillblom Foundation (2017-D-008-FEL). Work in the Kim laboratory was supported by the National Institutes of Health (R01 DK107507, R01 DK108817, and U01 DK123743 to S.K.K., and U01 DK123716 to S.K.K. [MPI] and A. Powers [contact PI]), and JDRF Northern California Center of Excellence (to S.K.K. and M. Hebrok). Work here was also supported by National Institutes of Health grant P30 DK116074 (to S.K.K.), and by the Stanford Islet Research Core, and Diabetes Genomics and Analysis Core of the Stanford Diabetes Research Center.
Author contributions: R.J.B. and S.K.K. conceptualized the study and directed the work. R.J.B. and S.K.K. were responsible for the methodology. R.J.B., J.Y.L., H.P., R.L.W., S.K., M.S.H.F., and X.G. performed the investigations. R.J.B. and S.K.K. wrote the manuscript with input from all coauthors. S.K.K. supervised the study. R.J.B, R.L.W., H.P., S.K., M.S.H.F., and S.K.K. acquired the funding.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publcation date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.342378.120.
References
- Aguayo-Mazzucato C, Koh A, El Khattabi I, Li W-C, Toschi E, Jermendy A, Juhl K, Mao K, Weir GC, Sharma A, et al. 2011. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat β cells. Diabetologia 54: 583–593. 10.1007/s00125-010-2026-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen HL, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, Ferrer J, Hattersley AT, Ellard S, The International Pancreatic Agenesis Consortium. 2012. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet 44: 20–22. 10.1038/ng.1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, Spitale RC, Dai C, Gu X, Qu K, et al. 2016. Age-dependent pancreatic gene regulation reveals mechanisms governing human β cell function. Cell Metab 23: 909–920. 10.1016/j.cmet.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arda HE, Tsai J, Rosli YR, Giresi P, Bottino R, Greenleaf WJ, Chang HY, Kim SK. 2018. A chromatin basis for cell lineage and disease risk in the human pancreas. Cell Syst 7: 310–322.e4. 10.1016/j.cels.2018.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Blanchi B, Raum JC, Guo M, Kaneko T, Cordes S, Sieweke M, Stein R. 2007. Mafb is required for islet β cell maturation. Proc Natl Acad Sci 104: 3853–3858. 10.1073/pnas.0700013104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, Stein R. 2010. MafA and MafB regulate genes critical to β-cells in a unique temporal manner. Diabetes 59: 2530–2539. 10.2337/db10-0190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken TE, Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, Dalley RA, Royall JJ, Lemon T, et al. 2016. A comprehensive transcriptional map of primate brain development. Nature 535: 367–375. 10.1038/nature18637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, et al. 2016. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst 3: 346–360.e4. 10.1016/j.cels.2016.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner C, van der Meulen T, Cacéres E, Tigyi K, Donaldson CJ, Huising MO. 2014. The transcriptional landscape of mouse β cells compared to human β cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genomics 15: 620 10.1186/1471-2164-15-620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blodgett DM, Nowosielska A, Afik S, Pechhold S, Cura AJ, Kennedy NJ, Kim S, Kucukural A, Davis RJ, Kent SC, et al. 2015. Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes 64: 3172–3181. 10.2337/db15-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camunas-Soler J, Dai XQ, Hang Y, Bautista A, Lyon J, Suzuki K, Kim SK, Quake SR, MacDonald PE. 2020. Patch-seq links single-cell transcriptomes to human islet dysfunction in diabetes. Cell Metab 31: 1017–1031.e4. 10.1016/j.cmet.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyphert HA, Walker EM, Hang Y, Dhawan S, Haliyur R, Bonatakis L, Avrahami D, Brissova M, Kaestner KH, Bhushan A, et al. 2019. Examining How the MAFB transcription factor affects islet β-cell function postnatally. Diabetes 68: 337–348. 10.2337/db18-0903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, Bhushan A. 2015. DNA methylation directs functional maturation of pancreatic β cells. J Clin Invest 125: 2851–2860. 10.1172/JCI79956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210. 10.1093/nar/30.1.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, Quake SR. 2017. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell 171: 321–330.e14. 10.1016/j.cell.2017.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachiya T, Komaki S, Hasegawa Y, Ohmomo H, Tanno K, Hozawa A, Tamiya G, Yamamoto M, Ogasawara K, Nakamura M, et al. 2017. Genome-wide meta-analysis in Japanese populations identifies novel variants at the TMC6–TMC8 and SIX3–SIX2 loci associated with HbA1c. Sci Rep 7: 16147 10.1038/s41598-017-16493-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainer SJ, Bošković A, McCannell KN, Rando OJ, Fazzio TG. 2019. Profiling of pluripotency factors in single cells and early embryos. Cell 177: 1319–1329.e11. 10.1016/j.cell.2019.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Tavella S, Hanley KP, Self M, Oliver G, Grifone R, Hanley N, Ward C, Bobola N. 2010. Inactivation of Six2 in mouse identifies a novel genetic mechanism controlling development and growth of the cranial base. Dev Biol 344: 720–730. 10.1016/j.ydbio.2010.05.509 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, et al. 2016. p16Ink4a-induced senescence of pancreatic β cells enhances insulin secretion. Nat Med 22: 412–420. 10.1038/nm.4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Iacovazzo D, Flanagan SE, Walker E, Quezado R, de Sousa Barros FA, Caswell R, Johnson MB, Wakeling M, Brändle M, Guo M, et al. 2018. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc Natl Acad Sci 115: 1027–1032. 10.1073/pnas.1712262115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka K, Tomita R, Takeda J, Horikawa Y. 2012. Rat glucagon receptor mRNA is directly regulated by glucose through transactivation of the carbohydrate response element binding protein. Biochem Biophys Res Commun 417: 1107–1112. 10.1016/j.bbrc.2011.12.042 [DOI] [PubMed] [Google Scholar]
- Jeong Y, Leskow FC, El-Jaick K, Roessler E, Muenke M, Yocum A, Dubourg C, Li X, Geng X, Oliver G, et al. 2008. Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat Genet 40: 1348–1353. 10.1038/ng.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson O, Edlund T, Moss JB, Rutter WJ, Walker MD. 1987. A mutational analysis of the insulin gene transcription control region: expression in β cells is dependent on two related sequences within the enhancer. Proc Natl Acad Sci 84: 8819–8823. 10.1073/pnas.84.24.8819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Sato S, Ozaki H, Ikeda K. 2000. Six family genes—structure and function as transcription factors and their roles in development. Bioessays 22: 616–626. [DOI] [PubMed] [Google Scholar]
- Kim YJ, MAGIC consortium, Go MJ, Hu C, Hong CB, Kim YK, Lee JY, Hwang JY, Oh JH, Kim DJ, et al. 2011. Large-scale genome-wide association studies in East Asians identify new genetic loci influencing metabolic traits. Nat Genet 43: 990–995. 10.1038/ng.939 [DOI] [PubMed] [Google Scholar]
- Kim S, Whitener RL, Peiris H, Gu X, Chang CA, Lam JY, Camunas-Soler J, Park I, Bevacqua RJ, Tellez K, et al. 2020. Molecular and genetic regulation of pig pancreatic islet cell development. Development 147: dev186213 10.1242/dev.186213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Muller JY, Fan J, Kim YJR, Lee SA, Ishida E, Blaner WS, Accili D. 2016. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic β cells in diabetic mice. Nat Commun 7: 12631 10.1038/ncomms12631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Goldstein RE, Fujioka M, Paroush Z, Jaynes JB. 2001. Groucho augments the repression of multiple Even skipped target genes in establishing parasegment boundaries. Development 128: 1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. 2008. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3: 169–181. 10.1016/j.stem.2008.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantz KA, Vatamaniuk MZ, Brestelli JE, Friedman JR, Matschinsky FM, Kaestner KH. 2004. Foxa2 regulates multiple pathways of insulin secretion. J Clin Invest 114: 512–520. 10.1172/JCI21149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire K, Thorrez L, Schuit F. 2016. Disallowed and allowed gene expression: two faces of mature islet β cells. Annu Rev Nutr 36: 45–71. 10.1146/annurev-nutr-071715-050808 [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Lagutin O, Swindell E, Jamrich M, Oliver G. 2010. Neuroretina specification in mouse embryos requires Six3-mediated suppression of Wnt8b in the anterior neural plate. J Clin Investig 120: 3568–3577. 10.1172/JCI43219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Ríos J, Tessmar K, Loosli F, Wittbrodt J, Bovolenta P. 2003. Six3 and Six6 activity is modulated by members of the groucho family. Development 130: 185–195. 10.1242/dev.00185 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelbaum AD, Kredo-Russo S, Aronowitz D, Myers N, Yanowski E, Klochendler A, Swisa A, Dor Y, Hornstein E. 2019. miR-17-92 and miR-106b-25 clusters regulate β cell mitotic checkpoint and insulin secretion in mice. Diabetologia 62: 1653–1666. 10.1007/s00125-019-4916-z [DOI] [PubMed] [Google Scholar]
- Matschinsky F, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, Cohen D, Permutt MA, Tanizawa Y, Jetton TL. 1993. Glucokinase as pancreatic β cell glucose sensor and diabetes gene. J Clin Invest 92: 2092–2098. 10.1172/JCI116809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. 2008. β-Cell replication is the primary mechanism subserving the postnatal expansion of β-cell mass in humans. Diabetes 57: 1584–1594. 10.2337/db07-1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger DE, Liu C, Ziaie AS, Naji A, Zaret KS. 2014. Grg3/TLE3 and Grg1/TLE1 induce monohormonal pancreatic β-cells while repressing α-cell functions. Diabetes 63: 1804–1816. 10.2337/db13-0867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Escalada I, Bonàs-Guarch S, Cebola I, Ponsa-Cobas J, Mendieta-Esteban J, Atla G, Javierre BM, Rolando DMY, Farabella I, Morgan CC, et al. 2019. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat Genet 51: 1137–1148. 10.1038/s41588-019-0457-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mularoni L, Ramos-Rodríguez M, Pasquali L. 2017. The pancreatic islet regulome browser. Front Genet 8: 13 10.3389/fgene.2017.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraro MJ, Dharmadhikari G, Grün D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJP, et al. 2016. A single-cell transcriptome atlas of the human pancreas. Cell Syst 3: 385–394.e3. 10.1016/j.cels.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, Juang C, Li M-L, Nguyen VQ, Giacometti S, et al. 2019. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat Cell Biol 21: 263–274. 10.1038/s41556-018-0271-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien LL, Guo Q, Lee Y, Tran T, Benazet J-D, Whitney PH, Valouev A, McMahon AP. 2016. Differential regulation of mouse and human nephron progenitors by the Six family of transcriptional regulators. Development 143: 595–608. 10.1242/dev.127175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Ma W, O'Brien LL, Chung E, Guo JJ, Cheng JG, Todd Valerius M, McMahon JA, Wong WH, McMahon AP. 2012. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev Cell 23: 637–651. 10.1016/j.devcel.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KA, Kettunen J, Laakso M, Stančáková A, Laver TW, Colclough K, Johnson MB, Abramowicz M, Groop L, Miettinen PJ, et al. 2017. Heterozygous RFX6 protein truncating variants are associated with MODY with reduced penetrance. Nat Commun 8: 888 10.1038/s41467-017-00895-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris H, Park S, Louis S, Gu X, Lam JY, Asplund O, Ippolito GC, Bottino R, Groop L, Tucker H, et al. 2018. Discovering human diabetes-risk gene function with genetics and physiological assays. Nat Commun 9: 3855 10.1038/s41467-018-06249-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen TJ, Khan AM, Barton G, Butcher SA, Sun G, Rutter GA. 2010. Identification of genes selectively disallowed in the pancreatic islet. Islets 2: 89–95. 10.4161/isl.2.2.11025 [DOI] [PubMed] [Google Scholar]
- Puri S, Roy N, Russ HA, Leonhardt L, French EK, Roy R, Bengtsson H, Scott DK, Stewart AF, Hebrok M. 2018. Replication confers β cell immaturity. Nat Commun 9: 485 10.1038/s41467-018-02939-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu WL, Zhang YW, Feng Y, Li LC, Yang L, Xu CR. 2018. Deciphering pancreatic islet β cell and α cell maturation pathways and characteristic features at the single-cell level. Cell Metab 27: 702 10.1016/j.cmet.2018.01.017 [DOI] [PubMed] [Google Scholar]
- Reinert RB, Cai Q, Hong JY, Plank JL, Aamodt K, Prasad N, Aramandla R, Dai C, Levy SE, Pozzi A, et al. 2014. Vascular endothelial growth factor coordinates islet innervation via vascular scaffolding. Development 141: 1480–1491. 10.1242/dev.098657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer AE, Taylor BL, Benthuysen JR, Liu J, Thorel F, Yuan W, Jiao Y, Kaestner KH, Herrera PL, Magnuson MA, et al. 2013. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic β cell identity. PLoS Genet 9: e1003274 10.1371/journal.pgen.1003274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharp DW, Downing R, Merrell RC, Greider M. 1980. Isolating the elusive islet. Diabetes 29 Suppl 1: 19–30. 10.2337/diab.29.1.S19 [DOI] [PubMed] [Google Scholar]
- Segerstolpe Å, Palasantza A, Eliasson P, Andersson E-M, Andréasson A-C, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. 2016. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 24: 593–607. 10.1016/j.cmet.2016.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self M, Geng X, Oliver G. 2009. Six2 activity is required for the formation of the mammalian pyloric sphincter. Dev Biol 334: 409–417. 10.1016/j.ydbio.2009.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, Gloyn AL, Edghill EL, Hattersley AT, Wellauer PK, et al. 2004. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet 36: 1301–1305. 10.1038/ng1475 [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S. 2017. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6: e21856 10.7554/eLife.21856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon JB, Tang Q, Stock P, Bluestone JA, Roy S, Desai T, Hebrok M. 2018. Stem cell therapies for treating diabetes: progress and remaining challenges. Cell Stem Cell 22: 810–823. 10.1016/j.stem.2018.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spracklen CN, Shi J, Vadlamudi S, Wu Y, Zou M, Raulerson CK, Davis JP, Zeynalzadeh M, Jackson K, Yuan W, et al. 2018. Identification and functional analysis of glycemic trait loci in the China health and nutrition survey. PLoS Genet 14: e1007275 10.1371/journal.pgen.1007275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spracklen CN, Horikoshi M, Kim YJ, Lin K, Bragg F, Moon S, Suzuki K, Tam CHT, Tabara Y, Kwak S-H, et al. 2020. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 582: 240–245. 10.1038/s41586-020-2263-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh CS, Ellingsen S, Austbø L, Zhao X-F, Seo H-C, Fjose A. 2010. Autoregulatory binding sites in the zebrafish six3a promoter region define a new recognition sequence for Six3 proteins. FEBS J 277: 1761–1775. 10.1111/j.1742-4658.2010.07599.x [DOI] [PubMed] [Google Scholar]
- Svendsen B, Larsen O, Gabe MBN, Christiansen CB, Rosenkilde MM, Drucker DJ, Holst JJ. 2018. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep 25: 1127–1134.e2. 10.1016/j.celrep.2018.10.018 [DOI] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. 2005. Very slow turnover of β-cells in aged adult mice. Diabetes 54: 2557–2567. 10.2337/diabetes.54.9.2557 [DOI] [PubMed] [Google Scholar]
- Thurner M, van de Bunt M, Torres JM, Mahajan A, Nylander V, Bennett AJ, Gaulton KJ, Barrett A, Burrows C, Bell CG, et al. 2018. Integration of human pancreatic islet genomic data refines regulatory mechanisms at type 2 diabetes susceptibility loci. Elife 7: e31977 10.7554/eLife.31977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tze WJ, Tai J. 1982. Preparation of pseudoislets for morphological and functional studies. Transplantation 34: 228–231. 10.1097/00007890-198210000-00019 [DOI] [PubMed] [Google Scholar]
- Urakami T. 2019. Maturity-onset diabetes of the young (MODY): current perspectives on diagnosis and treatment. Diabetes Metab Syndr Obes 12: 1047–1056. 10.2147/DMSO.S179793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney A, Scott LJ, Welch RP, Erdos MR, Chines PS, Narisu N, Albanus RD, Orchard P, Wolford BN, Kursawe R, et al. 2017. Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proc Natl Acad Sci 114: 2301–2306. 10.1073/pnas.1621192114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazco-Cruz L, Goedegebuure MM, Maxwell KG, Augsornworawat P, Hogrebe NJ, Millman JR. 2020. SIX2 regulates human β cell differentiation from stem cells and functional maturation in vitro. Cell Rep 31: 107687 10.1016/j.celrep.2020.107687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veres A, Faust AL, Bushnell HL, Engquist EN, Kenty JHR, Harb G, Poh YC, Sintov E, Gürtler M, Pagliuca FW, et al. 2019. Charting cellular identity during human in vitro β-cell differentiation. Nature 569: 368–373. 10.1038/s41586-019-1168-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Golson ML, Schug J, Traum D, Liu C, Vivek K, Dorrell C, Naji A, Powers AC, Chang KM, et al. 2016a. Single-cell mass cytometry analysis of the human endocrine pancreas. Cell Metab 24: 616–626. 10.1016/j.cmet.2016.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Schug J, Won KJ, Liu C, Naji A, Avrahami D, Golson ML, Kaestner KH. 2016b. Single-cell transcriptomics of the human endocrine pancreas. Diabetes 65: 3028–3038. 10.2337/db16-0405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara E, Wei Z, Lin CS, Fang S, Ahmadian M, Kida Y, Tseng T, Dai Y, Yu RT, Liddle C, et al. 2016. ERRγ Is required for the metabolic maturation of therapeutically functional glucose-responsive β cells. Cell Metab 23: 622–634. 10.1016/j.cmet.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C, Mulas F, Sui Y, Guan T, Miller N, Tan Y, Liu F, Jin W, Carrano AC, Huising MO, et al. 2017. Pseudotemporal ordering of single cells reveals metabolic control of postnatal β cell proliferation. Cell Metab 25: 1160–1175.e11. 10.1016/j.cmet.2017.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Liu N, Orkin SH, Yuan G-C. 2019. CUT&RUNTools: a flexible pipeline for CUT&RUN processing and footprint analysis. Genome Biol 20: 192 10.1186/s13059-019-1802-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data discussed in this publication have been deposited in NCBI's GeneExpression Omnibus (Edgar et al. 2002) and are accessible under accession number GSE164628.