

Abstract
Mutations are the source of both genetic diversity and mutational load. However, the effects of increasing environmental temperature on plant mutation rates and relative impact on specific mutational classes (e.g., insertion/deletion [indel] vs. single nucleotide variant [SNV]) are unknown. This topic is important because of the poorly defined effects of anthropogenic global temperature rise on biological systems. Here, we show the impact of temperature increase on Arabidopsis thaliana mutation, studying whole genome profiles of mutation accumulation (MA) lineages grown for 11 successive generations at 29°C. Whereas growth of A. thaliana at standard temperature (ST; 23°C) is associated with a mutation rate of 7 × 10−9 base substitutions per site per generation, growth at stressful high temperature (HT; 29°C) is highly mutagenic, increasing the mutation rate to 12 × 10−9. SNV frequency is approximately two- to threefold higher at HT than at ST, and HT-growth causes an ∼19- to 23-fold increase in indel frequency, resulting in a disproportionate increase in indels (vs. SNVs). Most HT-induced indels are 1–2 bp in size and particularly affect homopolymeric or dinucleotide A or T stretch regions of the genome. HT-induced indels occur disproportionately in nucleosome-free regions, suggesting that much HT-induced mutational damage occurs during cell-cycle phases when genomic DNA is packaged into nucleosomes. We conclude that stressful experimental temperature increases accelerate plant mutation rates and particularly accelerate the rate of indel mutation. Increasing environmental temperatures are thus likely to have significant mutagenic consequences for plants growing in the wild and may, in particular, add detrimentally to mutational load.
Climate models predict global surface temperature rises of 0.3°C to 1.7°C (moderate scenario) or 2.6°C to 4.8°C (extreme scenario) before 2100 (subject to possible feedback effects and the impact of mitigation strategies) (IPCC 2014). Recent rises are already associated with increases in the frequencies of extreme weather events (e.g., heatwaves) (Coumou and Rahmstorf 2012). Nevertheless, the long-term consequences for biological systems of overall rising temperature and regional climatic instability remain poorly understood. One major unknown is the effect of global warming on mutation, the raw fuel of biological evolution. Mutations can arise due to faulty repair of DNA replication errors or from oxidative stress- or irradiation-induced DNA damage, and the mutagenic effects of many of these factors are likely to be increased by an increase in temperature. Because an increase in deleterious mutations can add detrimentally to the mutational load upon populations, an improved understanding of the impacts of increased temperatures on the incidence of de novo mutations is essential. Such an understanding is important for plants, because they are particularly prone to environmental temperature change. However, precise determinations of these impacts are difficult in the wild because plant populations are simultaneously subject to changes in multiple additional environmental variables that are themselves affected by temperature rise (e.g., drought, flood, wind, etc.) (Fu et al. 2019). In addition, robust detection of mutation requires a known pedigree tracing back over generations from an ancestor of known genomic DNA sequence.
A dependency of mutation rate upon temperature was first identified in Drosophila melanogaster (Muller 1928). However, with the exception of detailed studies in Escherichia coli (Chu et al. 2018) and yeast (Huang et al. 2018), systematic whole-genome sequencing (WGS) investigations of the effects of temperature on mutation rate in both prokaryotic and eukaryotic organisms have been relatively limited. The aim of this study was therefore to use WGS approaches to determine the effects of increased growth temperatures on the rates and molecular spectra of de novo mutations in laboratory-maintained mutation accumulation (MA) Arabidopsis thaliana lines of known pedigree and derived from a single genome-sequenced ancestor plant. In addition, we aimed to determine if projected global temperature increases are likely to have significant mutagenic consequences for plant populations.
Results
Growth at high temperature increases mutation rate
We first compared mutations accumulated in A. thaliana mutation accumulation lines grown at high temperature (HT; 29°C) with those accumulated in cold (CT; 16°C) or standard temperatures (ST; 23°C) (Fig. 1A,B; see Methods). Our experiment varied temperature while keeping other environmental variables constant (see Methods). A CT to ST increase accelerates growth, developmental progression, and flowering but does not appear to induce a stress response (Fig. 1A). In contrast, a HT environment causes rapid growth and elicits characteristic developmental heat stress responses, including elongated petioles, leaf hyponasty, and early flowering and seed-set (Fig. 1A). These HT-adaptive responses are thought to facilitate leaf cooling and to accelerate seed-set in suboptimal conditions (Crawford et al. 2012; Bridge et al. 2013).
Figure 1.

Stressful HT growth increases A. thaliana mutation rates. (A) Plants grown for 28 d or 58 d in cold (CT; 16°C), standard (ST; 23°C), or high temperature (HT; 29°C) conditions. HT-grown plants display characteristic high temperature developmental and stress responses. (B) Mutation rates (per genome per generation) for transitions, transversions, and indels accumulating in CT, ST (Jiang et al. 2014), and HT MA lines. (C) Overview of the 15 mutations detected at G6 in five independent CT MA lines. Deletions are 1 bp in size; SNVs are single-nucleotide variants (single-nucleotide substitutions). (D) Overview of the 52 mutations detected at G10 in nine ST MA lines from three independent lineages (Jiang et al. 2014). Deletions are 1–66 bp in size; insertion is 2 bp in size. (E) Overview of the 230 mutations (Supplemental Table 2) detected at G11 in six independent HT MA lines. Deletions are 1–22 bp in size; insertions are 1–5 bp in size. (F) SNV and indel mutation rates in ST (Jiang et al. 2014) and HT compared with those in salinity stress (Saline-ST) (Jiang et al. 2014). Error bars (B,F) indicate SEM from five (CT data), nine (ST data) (Jiang et al. 2014), nine (Saline-ST data) (Jiang et al. 2014), or six (HT data) biological replicates.
To determine the mutagenic effect of HT-growth, seeds derived from self-pollination of a single genome-sequenced A. thaliana Col-0 ancestor (Jiang et al. 2014) were used to establish parallel CT and HT MA lines, with each line being maintained by random single-seed descent for multiple successive generations. Next, we obtained Illumina WGS data from five single sixth generation (G6) CT-grown and six single 11th generation (G11) HT-grown plants (with each plant being representative of a different MA line) (see Methods for data quality controls; Supplemental Table 1). Using our previously described computational mutation-detection methods (Belfield et al. 2012, 2018; Jiang et al. 2014; see Methods for further error-rate estimates), we then identified mutations that had arisen de novo during MA line growth, with each mutation being unique to the individual G6 or G11 plant genome in which it was detected. These analyses enabled an initial comparison of mutation rates (Ossowski et al. 2010; Jiang et al. 2014; Weng et al. 2019) in CT, ST (Ossowski et al. 2010; Jiang et al. 2014), and HT. We found that, whereas mutation rates changed little in response to a low-stress ∼7°C rise from CT (16°C) to ST (23°C), a further stressful ∼6°C increase in temperature from ST (23°C) to HT (29°C) significantly increased the rate of transitions and transversions and especially increased the rate of indel mutations (Fig. 1B–E). These per generation rate comparisons are legitimate despite the differences in life span at CT, ST, and HT, because the number of DNA replications occurring (and hence, number of mutations accumulating) in A. thaliana gamete-forming cell lineages is relatively independent of life span (Watson et al. 2016).
We identified a total of 230 homozygous HT-induced G11 mutations (Fig. 1E; Supplemental Table 2; see Methods). These mutations were all SNVs, deletions, or insertions, with further analysis (comparison against a positive control WGS data set containing a known transposon transposition event) (Mirouze et al. 2009; Jiang et al. 2011; see Methods) failing to detect any HT-induced large-scale copy number variants (CNVs; e.g., duplications or transpositions of ≥1000 bp) (Supplemental Fig. 1A–E), suggesting that exposure to HT does not increase the frequency of these larger-scale events. Small-scale deletions (ranging from 1 to 22 bp) were the most abundant class of HT-induced mutation (109; 47.4%). SNVs were less frequent (92; 40.0%), and 1- to 5-bp insertions were the least frequent (29; 12.6%) (Fig. 1E; Table 1A; Supplemental Table 2). The frequencies of HT-induced mutations of each class (SNV, insertion, deletion) did not obviously differ between the six HT MA lines (Table 1A) or between each of the five A. thaliana chromosomes (Supplemental Fig. 2A,B). Further spatial clustering tests (Roberts et al. 2012) indicated that most SNVs and indels were distributed within chromosomes in a pattern that is not significantly different from the random expectation (P-values > 0.01) (see Methods). These observations suggest that HT-induced mutations do not exhibit preferential distributions or localized microclustering within the genome, although the limited number of mutations analyzed may have affected these conclusions.
Table 1.
Mutations detected in six HT MA line G11 plants

Our previous study defined the mutations arising in A. thaliana MA lineages grown in salinity-stress conditions (Jiang et al. 2014). We found that HT-induced increases in SNV and indel mutation rates were greater than those due to the previously imposed level of salinity stress (Fig. 1F). Thus, an ∼6°C increase (ST to HT) from low-stress to high-stress ambient temperature significantly increases the rates of both SNV and indel mutation, suggesting that environmental warming may be mutagenic to plant genomes in the wild.
HT growth boosts SNV frequency and maintains GC-to-AT bias
From the 92 HT-induced SNVs across six MA lines (Fig. 1E; Table 1A), we estimated a HT-induced SNV mutation rate of 11.8 × 10−9 per site per generation (Table 1B), although this may be an underestimate due to the limited number of generations (Ossowski et al. 2010; Jiang et al. 2014). This HT SNV mutation rate is approximately two- to threefold higher than previous ST SNV mutation rate estimates (4.2–6.5 × 10−9) (Ossowski et al. 2010; Jiang et al. 2014), suggesting that environmental temperature rise may significantly increase the incidence of SNV mutations.
Previous A. thaliana MA line studies have revealed a GC-to-AT SNV mutational bias, which in turn contributes to a higher than expected (by chance) transition/transversion (Ti/Tv) ratio of 2.48–2.73 in ST conditions (Fig. 2A; Ossowski et al. 2010; Jiang et al. 2014). Whereas this GC-to-AT mutational bias is maintained in HT conditions, the rates for all SNV classes increase with increasing temperature (Fig. 2A). In fact, HT growth slightly decreases the relative incidence of transitions (69.0% vs. 71.3% or 73.2%) (Jiang et al. 2014; Ossowski et al. 2010), causing a corresponding increase in transversion incidence and a depressed Ti/Tv ratio of 2.23 (Fig. 2A,B). The individual categories of mutations (AT-to-GC, GC-to-AT, AT-to-CG, AT-to-TA, GC-to-TA, and GC-to-CG) giving rise to this HT Ti/Tv ratio are not significantly different from that of ST-grown plants (6 × 2 χ2-test, P-value = 0.961; and 6 × 2 χ2-test, P-value = 0.751) (Jiang et al. 2014; Ossowski et al. 2010), indicating that although the SNV mutation rate is increased, the overall SNV mutational spectrum is little changed by growth in HT conditions.
Figure 2.
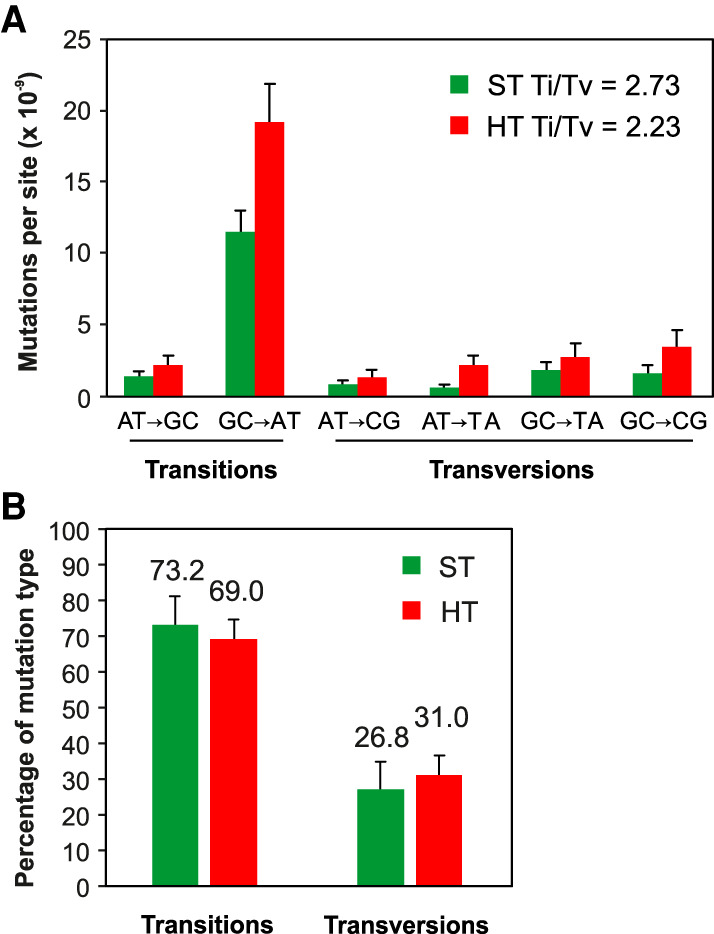
HT growth increases SNV frequency and maintains GC-to-AT bias. (A) SNV spectrum and Ti/Tv ratio in ST (Ossowski et al. 2010) and HT conditions. (B) Relative percentage of transitions versus transversions in ST (Ossowski et al. 2010) versus HT-grown MA lines. Error bars (A,B) indicate SEM from five (ST data) (Ossowski et al. 2010) or six (HT data) biological replicates.
We next used motif-detection algorithms to search for sequence context bias in HT-induced SNVs. With GC-to-AT transitions predominating (50 of 92; 54.3%), we determined if these had accumulated in preferred sequence contexts. As previously (Belfield et al. 2018), we first captured the −10-bp and +10-bp DNA sequences flanking the identified 18 HT-induced C-to-T transition sites (and those flanking the remaining 32 reverse-complemented HT-induced G-to-A transition sites) from the TAIR10 A. thaliana reference genome (Supplemental Fig. 3A). We similarly determined positional flanking nucleotide residue distributions for all C (combined with reverse-complemented G) sites in TAIR10 (>43 million sites) (Supplemental Fig. 3B). Finally, we determined flanking residue distributions for GC-to-AT transitions in ST-grown plants (38 and 47) (Jiang et al. 2014; Ossowski et al. 2010; Supplemental Fig. 3C).
Comparison of these observations revealed strong enrichment (52.8%) for an A residue at the −3 position 5′ of mutated GC-to-AT transitions (i.e., the position 3 nucleotides 5′ of each GC-to-AT transition site) in HT-grown plants (vs. the expected 31.3% in TAIR10 [P-value = 0.016, χ2 test] and vs. the observed 26.5% in ST-grown plants [P-value = 0.035, χ2 test]) (Supplemental Fig. 3A–C). An additional site, a G residue at the +8 position 3′ from mutated GC-to-AT transitions in HT-grown plants, showed no significant enrichment versus TAIR10 (30.7% vs. 18.4%, respectively, P-value = 0.076, χ2 test) but did show significant enrichment versus ST-grown plants (30.7% vs. 7.8%, respectively, P-value = 0.025, χ2 test). Thus, the most frequent HT-induced SNVs (GC-to-AT transitions) do appear to exhibit some degree of preference with respect to sequence location.
In further analysis, we found that HT growth does not detectably alter (vs. ST growth) the relative frequency (proportion of total) of SNVs in particular functional genomic sequence categories: CDS, introns, UTRs, intergenic, TE, noncoding RNAs, and pseudogenes (Fig. 3). Our previous studies showed that ST SNV distribution depends upon the function of the DNA mismatch repair (MMR) pathway and is substantially different in the absence of MMR function, suggesting that MMR preferentially protects genes from SNV mutational damage (Belfield et al. 2018). However, because the genome category-wide distribution of HT-induced SNVs is not obviously different from that of ST-grown (MMR-proficient) plants (Fig. 3), it is likely that the stress of HT growth is not impairing the function of MMR, one of the major DNA-repair pathways in eukaryotes (see Discussion). We conclude that HT growth boosts SNV frequency without detectable reduction of overall MMR bias (Belfield et al. 2018).
Figure 3.
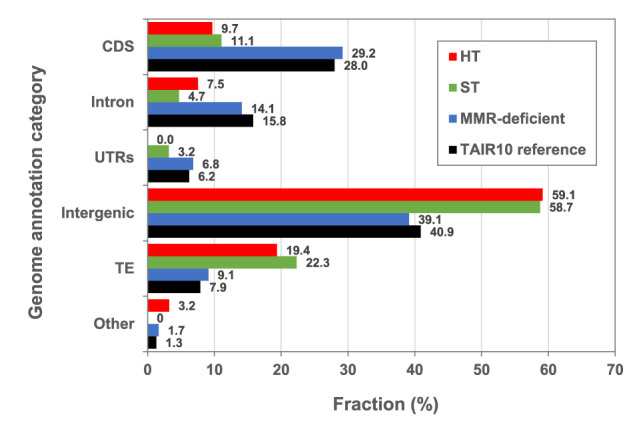
Genome-wide distribution of SNVs in HT, ST, and MMR-deficient MA lines. Black bars show the relative distribution of Arabidopsis thaliana TAIR10 reference genome annotation categories (expressed as % of total genome). (CDS) Coding DNA sequence, (UTRs) untranslated regions, (TE) transposable element, (Other) noncoding RNAs and pseudogenes. The remaining bars show relative distribution (%) of SNVs in those annotation categories in: (red) HT MA lines (N = 92); (green) ST MA lines (N = 98) (Ossowski et al. 2010) and N = 44 (Jiang et al. 2014) (averaged for each category); and (blue) MMR-deficient MA lines (N = 4048) (Belfield et al. 2018).
HT growth promotes small-scale indel mutations
We found that HT growth increases indel frequency by 19- to 26-fold (2.09 indels per genome per generation vs. 0.11–0.08 in ST) (Ossowski et al. 2010; Jiang et al. 2014; Supplemental Fig. 4). Among indels, deletions were the largest single category of HT-induced mutation (Fig. 1E). The majority of the 138 HT-induced indels impacted single bases (86; 91.9%) (Fig. 4A,B), with 1-bp deletions being the most frequent (77; 55.8%) (Fig. 4A). The second most frequent were 2-bp indels, accounting for one third of all indels (46; 33.3%) (Fig. 4A,B), with 2-bp insertions being more frequent than 1-bp insertions (19 vs. 9) (Fig. 4B). Deletions of 3 bp and larger were less frequent, with the largest indel being a single 22-bp deletion (Fig. 4A).
Figure 4.

Most HT-induced indels are of 1- to 2-bp length. Length distributions (in bp) of HT-grown MA (G11) deletions (A) and insertions (B). (C) Comparison of the frequencies of the different classes of HT-induced 1- and 2-bp indels. For example, 40 A, 32 T, 4 C, and 1 C single base deletions were detected.
The majority of HT-induced indels involved A or T, either as single bases (81 of all 86 1-bp indels; 94.2%) or as AT or TA dinucleotides (38 of all 46 2-bp indels; 82.6%) (Fig. 4C). In comparison, indels impacting C or G bases occurred relatively infrequently (Fig. 4C, Supplemental Table 2). Single C or G indels accounted for 5.8% of 1-bp indels (five of 86), and 13.0% of 2-bp indels (six of 46) included a C or a G nucleotide (three CT, one CG, one GA, and one TC). A smaller GC/AT bias was observed in indels >2 bp, with 85.7% of such indels (six of seven) being composed of a mixture of G/C and A/T nucleotides.
HT-induced indels fall predominately within homopolymeric/microsatellite stretches
We next surveyed the genome-wide distribution of the 138 HT-induced indels. First, as previously found for HT-induced SNVs, the genome-wide locations of HT-induced indels were not obviously different from the random expectation (Supplemental Fig. 2A,B). In further analyses, the majority of HT-induced indels (89; 64.5%) were found to be located in homopolymeric stretches of 4–23 bp in length (85 in A/T and four in C/G stretches) (Supplemental Table 2). The indel mutation rate was 1.55-fold higher (1.06 × 10−6 per line per generation) in homopolymeric A/T repeat regions than in homopolymeric C/G repeat regions (6.83 × 10−7 per line per generation, following normalization with respect to the numbers of homopolymeric A and T, or G and C repeat regions in the A. thaliana genome).
Next most frequent were indels within 2-bp dinucleotide repeat regions (43; 31.2%), with 38 of these located either in AT or TA repeat regions (see Supplemental Table 2). Similarly to homopolymeric site indels, the mutation rates for indels exclusively involving A or T nucleotides, either AT or TA dinucleotides, were 2.42-fold higher (1.53 × 10−5 and 1.62 × 10−5 per line per generation, respectively) than those of other identified dinucleotide repeat indels at TC, GA, or CT repeat sites (1.81 × 10−6, 2.50 × 10−6 or 8.67 × 10−6 per line per generation, respectively, following normalization with respect to the numbers of dinucleotide AT, TA, TC, GA, or CT repeat regions in the A. thaliana genome).
In addition, five indels (3.6%) were located in sequence with no particular defining characteristic (three insertions of 1–5 bp, and two deletions of 1 bp and 22 bp). Lastly, a single 3-bp deletion (TGT) was located within a 15-mer trinucleotide repeat microsatellite. Single-base C or G indels were also preferentially located within homopolymeric stretches of 6–11 bp (four of five) (Supplemental Table 2). These observations indicate that HT-grown plants are especially prone to the accumulation of indel mutations in homopolymeric repeat sequence regions and microsatellites.
We also found that the likelihood of occurrence of a HT-induced indel mutation within a homopolymeric repeat stretch relates to the length of that stretch. For example, the frequency of A/T single base indels in such stretches displays a normal (bell-shaped) distribution curve when plotted against stretch length, peaking at stretch lengths of ∼14 nucleotides, then falling with further increase in length (note that the indel frequencies shown are normalized with respect to the frequencies within the genome of stretches of particular length category, as also indicated) (Fig. 5A; Supplemental Fig. 5A,B). The biological consequences of these observations are considered further in Discussion. With most HT-induced indels being located in homopolymeric stretch/microsatellite regions, and most such regions being located in non-protein-encoding regions of the A. thaliana genome, we determined the specific genomic locations of HT-induced indels. All 138 HT-induced indels were found to be located in noncoding DNA and were especially prevalent in intergenic regions (81.9%) (Fig. 5B). We conclude that HT-growth particularly promotes the accumulation of small-scale indels in the homopolymeric repeat and microsatellite sequences characteristic of non-protein-encoding DNA.
Figure 5.

HT-induced indels cluster in homopolymeric/microsatellite stretches and cause extreme indel/SNV bias. (A) A histogram showing the frequency (left y-axis) of 1-bp A or T indels found in homopolymeric repeat regions of different lengths (x-axis: values normalized with respect to the numbers of each length category in TAIR10, numbers as indicated by the red diamond-marked line [right y-axis]) (see also Supplemental Fig. 5A,B). The black dotted line indicates a moving average trendline for indel frequency. (B) Genomic distribution of indels in HT MA lines. (CDS) Coding DNA sequence, (UTR) untranslated region, (TE) transposable element, (Other) pseudogenes and noncoding RNAs. (C) Indel/SNV ratio comparisons; data from unicellular eukaryotes (red bars), multicellular eukaryotes (green bars), and eubacteria (blue bars). Organisms were grown at relative high temperature (HT), standard temperature (ST), or alternative cold temperature (CT), as indicated. The studies shown are: Plasmodium falciparum – ST (Hamilton et al. 2017); Arabidopsis thaliana – HT (this study); A. thaliana - MMR – ST (Belfield et al. 2018); A. thaliana - FN – ST (Belfield et al. 2012); A. thaliana - Saline – ST (Jiang et al. 2014); Oryza sativa – ST (Yang et al. 2015); Escherichia coli – ST (Chu et al. 2018); E. coli – CT (28°C) (Chu et al. 2018); E. coli – CT (25°C) (Chu et al. 2018); A. thaliana A – ST (Ossowski et al. 2010); A. thaliana B – ST (Jiang et al. 2014); Saccharomyces cerevisiae – HT (Huang et al. 2018); Caenorhabditis elegans – ST (Meier et al. 2014); S. cerevisiae – ST (Liu and Zhang 2019); Homo sapiens – ST (Besenbacher et al. 2016); and Daphnia pulex – ST (Keith et al. 2016). Error bars (A,B) indicate SEM from six HT biological replicates.
HT-induced indels cause extreme genome-wide indel/SNV bias
HT growth significantly reverses the relative frequencies of indel versus SNV mutation: whereas SNVs predominate in ST, indels predominate in HT (Fig. 1F). The 138 HT-induced indels and 92 HT-induced SNVs observed correspond to an indel/SNV ratio of 1.5, significantly higher than the 0.17 or 0.16 ratios typical of ST grown plants (17 indels/99 SNVs, Fisher's exact test, P-value = 1.87 × 10−16; and seven indels/44 SNVs, Fisher's exact test, P-value = 8.82 × 10−10) (Ossowski et al. 2010: Jiang et al. 2014), or the 0.42 ratio in salinity-stressed plants (30 indels/72 SNVs, Fisher's exact test, P-value = 2.61 × 10−7) (Jiang et al. 2014). Indeed, the HT 1.5 indel/SNV ratio is higher than in all previous A. thaliana MA studies (Fig. 5C; Supplemental Table 3), even higher than the 0.7 ratio caused by fast-neutron irradiation mutagenesis (44 indels/64 SNVs, Fisher's exact test, P-value = 0.001) (Belfield et al. 2012), or the 1.2 ratio due to MMR deficiency (4663 indels/4048 SNVs, Fisher's exact test, P-value = 0.052) (Belfield et al. 2018). In almost all previously studied organisms and growth conditions, SNVs predominate over indels (a notable exception being the protozoan malarial parasite Plasmodium falciparum, which has an extremely AT-rich genome) (Hamilton et al. 2017; Katju and Bergthorsson 2019), suggesting that indel/SNV ratio is evolutionarily conserved (Fig. 5C; Supplemental Table 3). We conclude that a stressful ∼6°C increase in environmental temperature has a relatively extreme effect on the A. thaliana indel/SNV ratio, causing indels to predominate over SNVs.
HT-induced indels occur disproportionately in nucleosome-free DNA
Eukaryotic genomes are mostly packaged into nucleosomes, structural units within which ∼147 bp of DNA is wrapped around a histone octamer, with successive nucleosomes being typically connected by ∼20–40 bp of unwrapped linker DNA (Jiang and Pugh 2009). Previous analyses of the differences between the mutational spectra characteristic of the nucleosome-wrapped versus nucleosome-free genomic DNA of diverse eukaryotes suggest that nucleosome occupancy influences spontaneous mutation (Chen et al. 2012). We therefore next determined the positions of A. thaliana HT-induced and ST mutations with respect to nucleosome occupancy, performing Illumina sequencing of DNA from unamplified micrococcal nuclease (MNase-seq)-digested (nucleosome-enriched) chromatin from ST- and HT-grown A. thaliana plants (Supplemental Table 4A,B). Following read-mapping to TAIR10, we used the iNPS (Chen et al. 2014) algorithm to predict nucleosome position on the basis of read distribution across the genome. This analysis identified ST and HT nucleosome positions and coordinates, and distinguished nucleosome, edge of nucleosome, and nonnucleosome regions (as shown for an example region of Chromosome 1 in Supplemental Fig. 6).
Comparison of the genome-wide distribution of bases in nucleosome, edge of nucleosome, and nonnucleosome regions revealed little difference between ST- and HT-grown plants, suggesting relative conservancy of overall nucleosome occupancy (Supplemental Table 4A,B). However, our further analysis indicated that HT growth increases nucleosome occupancy 5′ of but not 3′ of gene transcription start sites (TSS), and that HT may therefore inhibit access of DNA-binding proteins to 5′ gene regulatory regions (Supplemental Fig. 7A,B) despite relative uniformity of nucleosome occupancy in ST- and HT-grown plants across euchromatic regions (Supplemental Fig. 8).
Next, we determined if either HT-induced (92 SNVs and 132 1- to 2-bp indels) or ST (143 SNVs and 18 1- to 2-bp indels) (Ossowski et al. 2010; Jiang et al. 2014) mutations are preferentially distributed between nucleosomal versus nonnucleosomal DNA, combining mutations located in nucleosome and edge-of-nucleosome regions into a single nucleosome-associated category. First, we overlaid the genomic positions of SNVs onto the genome-wide nucleosome occupancy location profiles generated with iNPS (Chen et al. 2014) (see Methods; Supplemental Table 2; Supplemental Fig. 6). We then found that the frequencies of nucleosome-associated HT-induced and ST SNVs (64.3% and 66.3%, respectively) were relatively similar and not significantly different from the random distribution expectation (ST exact test, P-value = 0.44; HT exact test, P-value = 0.34) (Fig. 6; Supplemental Table 4A,B; Supplemental Table 5A–D). Thus, HT-induced and ST SNVs are evenly spread between nucleosome-associated and nonnucleosomal DNA.
Figure 6.
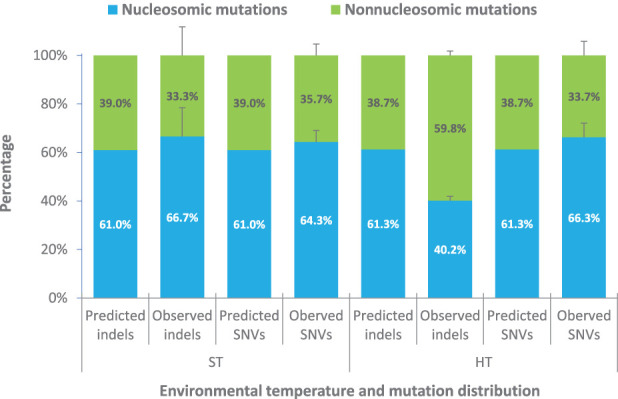
HT-induced indels are disproportionally located in nucleosome-free DNA. Nucleosome profiling of indel and SNV mutations in ST-grown (Ossowski et al. 2010; Jiang et al. 2014) and HT-grown MA lines. (Predicted) Random expectation nucleosomal-associated versus nonnucleosomal distributions, as based on the distributions of MNase-seq bases (Supplemental Table S4A,B); (Observed) actual distribution of indels and SNVs in ST (Ossowski et al. 2010; Jiang et al. 2014) and HT conditions Supplemental Table S5A–D).
However, further analysis showed that HT-induced indels are not evenly spread between nucleosome-associated and nonnucleosomal DNA. Although the frequency of nucleosome-associated ST indels (66.7%) is also not detectably different from the random distribution expectation (61.0%; exact test, P-value = 0.81) (Fig. 6; Supplemental Table 5A,B), that of nucleosome-associated HT-induced indels (40.2%) is significantly lower than expected (61.3%; exact test, P-value = 1.02 × 10−6) (Fig. 6; Supplemental Table 5C,D). This skewed distribution of HT-induced indels is unlikely to be due to sites prone to indel mutation (e.g., homopolymeric repeats) tending to be in nonnucleosomal DNA. For example, the majority of indels in both ST- and HT-grown plants were located in homopolymeric stretches (11 of 12 indels were in homopolymeric A or T stretches in ST lines; 86 of 138 HT-induced indels were in homopolymeric stretches [mainly A or T stretches]) (Ossowski et al. 2010; Jiang et al. 2014; see Supplemental Table 2; Supplemental Table 6A,B). Analysis of the nucleosome distribution profiles showed that these particular homopolymeric sequences where indels were found in ST and HT lines are more often found in nucleosomic than in nonnucleosomic regions (ST 1.23-fold, and HT 1.38-fold) (Supplemental Table 6A,B), suggesting that the homopolymeric repeat stretches analyzed tend to occur inside of rather than outside of nucleosomes.
We conclude that, whereas ST indels are evenly spread between nucleosome-associated and nonnucleosomal DNA, HT-induced indels accumulate disproportionately in nonnucleosomal DNA, suggesting the possibility that nucleosome occupancy may confer partial (relative) protection from HT-induced indel mutation (see Discussion for further consideration of this possibility), although sequence context (rather than nucleosome context) may also be important.
Discussion
Although mutation rates have long been thought to accelerate with rising temperature (Muller 1928), the likely mutagenic effects on plant populations of anthropogenic rises in global surface temperatures and resultant climatic instability remain poorly understood. In particular, the mutagenic consequences of rising temperatures and of extreme heatwaves, both increasingly prevalent global phenomena, are unknown. We therefore determined the effects of environmental temperature increase on mutation accumulation in the Col-0 strain of A. thaliana, a strain originating from temperate northern Europe, where temperatures in the 16°C–23°C range are relatively normal but 29°C relatively extreme. Our WGS analysis of Col-0 CT, ST, and HT MA lines shows that, whereas an initial low-stress ∼7°C (16°C–23°C) rise in growing temperature has little mutagenic effect, a further high-stress ∼6°C (23°C–29°C) increase significantly accelerates the rate of both SNV and small-scale indel mutation. Although the temperature increases tested in our experiments are more extreme than current 2- to 4-degree global increase projections (e.g., IPCC 2014), our observations suggest that global warming-associated increased exposure of plant populations to stressful higher-than-normal habitat range temperatures (e.g., during heatwaves) might be expected to accelerate mutation incidence in the wild.
Considering HT-induced SNVs first, whereas overall rate is significantly increased, the GC-to-AT mutational bias and molecular mutational SNV spectrum are little different from that of SNVs accumulated in ST conditions. Because SNVs are thought mostly to be the consequence of polymerase replication errors, HT-stress is therefore not detectably changing the overall nature and spectrum of those errors. HT-stress differs markedly in this respect from salinity-stress, which, although also increasing overall rate, profoundly alters the SNV spectrum (Jiang et al. 2014). These observations suggest that different stresses can have different plant mutational consequences, perhaps because they perturb cellular processes in different ways.
Next, HT growth has a strong and disproportionately promotive effect on the incidence of indels. HT growth increases indel frequency much more than SNV frequency, resulting in a relatively high indel/SNV ratio. Because a relatively low indel/SNV ratio of spontaneous mutations is evolutionarily conserved, it is likely that maintenance of that low value is selectively advantageous. Although temperature-dependent increases in indel/SNV ratio have previously been observed in E. coli (Chu et al. 2018) and in yeast (Huang et al. 2018), our observations suggest the magnitude of HT-induced increase to be much greater in A. thaliana, with potentially deleterious selective consequences. Our in-depth analysis shows the majority of HT-induced indels to occur in homopolymeric A or T or dinucleotide (microsatellite) AT repeat sequence regions. Whereas such regions are already known to be particularly prone to mutation (Golubov et al. 2010; Lujan et al. 2015), growth at HT increases that susceptibility. In addition, we show that the likelihood of a HT-induced indel occurring in any particular homopolymeric stretch is a function of the length of that stretch. As discussed previously (Belfield et al. 2018), the consequence of this phenomenon is that certain individual homopolymeric stretches have an extremely high regionally localized HT-induced mutation frequency.
Increased frequencies of DNA replication errors or of oxidative DNA damage (Cadet and Davies 2017) are both potential contributors to the increased HT mutation rate. Indeed, the disproportionate increase in small-scale indels in homopolymer or dinucleotide repeat stretches are apparently suggestive of heat-induced perturbation of DNA replication (e.g., increased polymerase slippage [Velichko et al. 2012]) or of repair functions (e.g., as similarly observed in reduced repair function MMR-deficient plants [Belfield et al. 2018]). However, the genome-wide distribution of HT-induced SNVs is dissimilar to that conferred by MMR-deficiency, suggesting that HT-induced mutation is not attributable to adverse effects on MMR function. Alternatively, as mentioned above, HT growth may be mutagenic because of increased oxidative DNA damage from heat-stress-induced increases in reactive oxygen species (ROS). We show here that nucleosome-associated DNA is partially protected from HT-induced indels, implying that a major component of HT-induced mutational damage occurs during phases of the cell cycle when DNA is wrapped within nucleosomes. Because DNA replication occurs when DNA is naked and not nucleosome-associated, these observations in turn suggest that stress-induced oxidative DNA damage is a major source of HT-induced indels and that nucleosome-occupancy provides partial protection from that damage. Perhaps ROS-induced base damage to internucleosomal DNA (sustained during the nucleosomal phase of the cell cycle) persists (Moore et al. 2017), with damaged bases then being particularly prone to slippage replication indel-inducing errors during subsequent DNA replication.
We conclude that stressful increases in environmental temperatures significantly accelerate plant mutation rates and particularly increase the incidence of small indel mutations. These changes will likely in turn increase the accumulated load of deleterious mutations on plant populations. Load accumulation may be exacerbated because HT can reduce generation time and life span, thus further accelerating mutation accumulation per year. An increased mutational load, in combination with additional increased selective pressure and reduced population diversity (Fu et al. 2019), will impact the genetic structure of populations, potentially hastening population decline and species extinctions. Accelerated mutation rates are thus a potentially important and hitherto relatively unrecognized aspect of the damaging impact of anthropogenic global climate change on natural plant populations, biodiversity, and ecosystems.
Methods
Plant materials and growth conditions
Seeds from a genome-sequenced A. thaliana Col-0 ancestor were used to initiate multiple independent high temperature (HT, 29°C) and cold temperature (CT, 16°C) mutation accumulation lines. Seeds were first sown on soil, watered, stratified at 4°C for 3 d, then transferred to controlled environment chambers (16-h light [120 μmol photons m−2 s−1]/8-h dark at constant HT or CT). MA lines were maintained by single-seed descent for 11 (G11; HT lines) or six (G6; CT lines) successive generations. For the nucleosome positioning experiment (see below); G1 plants were grown in controlled environment chambers (16-h light [120 μmol photons m−2 s−1]/8-h dark at 23°C).
DNA extraction and whole-genome sequencing.
Genomic DNA was extracted from rosette leaves of single HT and CT MA line plants using a DNeasy kit (Qiagen). This DNA was sequenced using 100-bp paired-end Illumina technology according to the manufacturer's instructions at the Beijing Genomics Institute (BGI), China (see Supplemental Table 1).
Sequence alignment, variant calling/validation, and false error-rate estimations
The computational bioinformatic mutation-detection methods used in this study are as previously described (Belfield et al. 2012, 2018; Jiang et al. 2014). Briefly, a three-step procedure enabled the robust identification of mutations arising during MA line propagation, as follows:
Step 1 (initial bioinformatic variant detection pipeline). Sequencing data set reads were aligned to the A. thaliana TAIR10 reference genome and variants subsequently called using IMR/DENOM (Gan et al. 2011). The lists of initial variants (SNVs and indels) in each sample are generated by IMR/DENOM by only considering high quality (Phred score ≥ 25) uniquely mapped reads. Between 118.1 and 118.7 million of the sites from the 119.1 Mbp reference genome passed the read coverage ≥3 and ≤100 quality requirements for each sample (Supplemental Table 1). Following initial variant calling, variants common between two or more lines (either between HT or CT MA line samples) or shared with the progenitor (Generation 0) were filtered out, as these likely represent mutational deviations from TAIR10 arising in the progenitor lineage prior to the onset of our MA line experiments. A putative unique mutation (SNV or indel) was called if 95%–100% of aligned reads in a single MA line sample differed from the progenitor reference sequence at the putative mutation position, thereby selecting only homozygous germline mutations and not somatic (heterozygous) mutations. Regions with low sequencing read coverage of <3 (that could lead to miscalling errors) or high coverage of >100 (that are associated with DNA sequences with a high degree of similarity to other sequences in the genome, such as transposable elements) were excluded from the analysis (Belfield et al. 2012).
Step 2 (visual alignment scanning using Integrative Genomics Viewer [IGV]). Each putative mutation called in Step 1 was next checked manually by visually scanning the alignment files (BAM files) generated in Step 1, using IGV (Robinson et al. 2011). In addition, putative unique indel mutations in the variant lists for each MA line generated by IMR/DENOM in Step 1, where ≥25%–95% of aligned reads at a site indicated a mutation, were checked with IGV due to the aligner gap penalties issue mentioned below (see Belfield et al. 2012 for further details). Reads with Phred scores of <25, mapping quality of ≤20 (reads with lower mapping qualities could indicate ambiguously mapped reads with high reference sequence homology and are known to produce many false positives), or reads with mates that are not mapped are ignored when visually scanning mutations in IGV. Essentially, IGV generates a visual display of multiple stacked reads from each MA line sample and from the progenitor line sample, all aligned to the reference genome (TAIR10) and highlighting nucleotide positions in reads that are variant with respect to TAIR10. For each putative mutation called, IGV enables simple visual inspection of the sequence of all sample reads covering that particular site, allowing elimination of cases where a mutation may have been erroneously called in Step 1. IGV additionally enables detection of indels missed during Step 1 because aligners such as IMR/DENOM (Gan et al. 2011) often penalize opening a gap in the alignment more heavily than allowing 2–3 mismatches toward the 3′ ends of WGS reads, which in turn can be a significant source of false-positive and false-negative variant calls. IGV-based visual analysis (Step 2) of all calls originally made in Step 1 thus enabled the elimination of erroneous calls and confirmed the identification of the 230 HT-induced mutations described in this study.
Step 3 (mutation validation via PCR/Sanger sequencing). As a quality control on the efficacy of the above combined Steps 1 and 2 in identifying genuine mutations (rather than false positives), we previously used PCR/Sanger sequencing to confirm 75 of 79 semirandomly chosen mutations, thus providing a false-positive rate estimate of 5% (Belfield et al. 2018). However, we actually think it is likely that this 5% rate is an overestimate because the four “failures” were due to PCR-amplification failure: the PCR failed to amplify the DNA region containing the putative mutation. This amplification failure may have been due to suboptimal PCR primer design, high/low GC content of the surrounding genomic region, or other causes. Nevertheless, lack of PCR product makes it impossible to confirm the presence or absence of the putative mutation, meaning that the putative mutation might still genuinely exist, despite the “failure” designation (Belfield et al. 2018).
Finally, to estimate the false-negative detection rates of our methods (Steps 1 and 2), we introduced synthetic mutations (SNVs and indels) into the reference genome, aligned sequencing reads from the A. thaliana Col-0 ancestor (Jiang et al. 2014) and all six HT MA lines individually against those modified references, and generated variant lists using IMR/DENOM (Gan et al. 2011). Briefly, we substituted 100 nucleotides in the reference with alternate bases to create 100 SNVs (i.e., 25 each of novel in silico A, T, C, and G mutations were created) in CDS, intergenic, intron, noncoding RNA, pre-tRNA, pseudogene, TE, and UTR regions across five chromosomes (see Supplemental Table 7). In addition, 1200 indels of varying sizes (100 insertions and 100 deletions of size 1 bp, 2 bp, 4 bp, 10 bp, 100 bp, and 1000 bp each) at regular intervals on Chromosome 1 (see Supplemental Tables 8, 9) and 12 tandem duplications or deletions of 2–12 bp in size (multiples of 2 bp) into microsatellite sequences present in intergenic and UTR regions across four chromosomes were created (see Supplemental Table 10). Individual modified reference genomes (synthetic SNV, indel, and microsatellite TAIR10 references) were created for each mutation category and subsequently used for alignment and variant calling by IMR/DENOM.
We found that IMR/DENOM (Gan et al. 2011) failed to detect two of 700 SNVs, thus revealing a SNV false-negative rate of 0.29% (see Supplemental Table 11). However, the indel false-negative rate was higher, at 3.30% for 1- to 100-bp indel mutations (231 of 7000 were missed) (see Supplemental Table 12A,B) and increased still further, to 7.87%, for larger indels (1000 bp) (see Supplemental Table 12A,C). Lastly, the microsatellite indel false-negative rate was found to be 26.2% (22 of 84 were missed) (see Supplemental Table 10) and was dependent on the average depth of aligned sequence coverage at these sites in particular samples.
Given that we identified 109 deletions, 92 SNVs, and 29 insertions in our HT data set, these relatively small false-negative rates are likely to have had a negligible impact on our conclusions, although they do indicate that the observed enhancement of indel incidence at HT may in actuality be even higher than our estimates suggest.
Supplemental Figure 9A shows depth of read coverage, Supplemental Figure 9B plots coverage against numbers of mutations, and Supplemental Figure 9C gives inferred mutation rates for regions with different levels of coverage. Supplemental Figure 9B shows that, although most SNVs are called in regions where read coverage is close to the peak of average coverage depth, indels are generally located in regions of lower than average coverage, thus causing the overall bias toward mutations (as a whole) to be identified at positions with lower than average coverage (Supplemental Fig. 9C). The indels likely tend to be within low coverage regions because indels within repetitive sequence (as they predominantly are) tend to reduce the frequency of succesful read alignment and hence, coverage. Nevertheless, PCR/Sanger resequencing analysis (Belfield et al. 2018) indicates that, although these indels tend to occur in regions of low coverage, they are not false positives.
We conclude that our methods for mutation detection are relatively robust and that the error-rate estimates outlined above enable relative confidence in the 230 HT-induced mutations described in this study.
Calculation of Ti/Tv ratios
HT transition and transversion SNV numbers were first normalized with respect to the GC-content bias (GC/AT content; 36/64%, respectively) of the TAIR10 reference genome. Ti/Tv ratios were then calculated as previously described (Ossowski et al. 2010; Belfield et al. 2012).
Detection of larger-scale copy-number variants
Previously described computational bioinformatic mutation-detection methods were used to identify putative CNV regions (Jiang et al. 2011, 2014; Belfield et al. 2012). Briefly, the five chromosomes of TAIR10 were segmented into consecutive 1-kb bins, after which the number of sequencing reads covering each bin (present in the BAM files generated for each sample) was summed and divided by the average depth of coverage over the whole genome for each sample (Supplemental Table 1). Next, we performed scatterplot analysis of the log2 coverage ratios of reads from HT G11 samples and from met1/+ nrpd2a (a CNV positive-control data set containing a mobilized COPIA93 transposon [At5G17125] on Chromosome 5) (Mirouze et al. 2009). Coverage ratios were normalized with respect to the number of G0 ancestor reads in corresponding 1-kb bins. Putative CNVs (log2 coverage ratio scores ≥ 1) were visually checked for authenticity using IGV (Robinson et al. 2011).
SNV flanking sequence motif detection
Using a custom Linux shell script (Belfield et al. 2018), we captured the 10-bp 5′ and 3′ sequences flanking all sites subject to HT C-to-T mutations. Sequences flanking G-to-A mutation sites were reverse-complemented to create C-to-T mutations and merged with the sequences flanking C mutation sites. These flanking sequences were captured from the TAIR10 reference forward strand. Flanking sequence motifs were created in Excel and displayed as stacked columns. Using the WebLogo 2.8.2 software (https://weblogo.berkeley.edu/logo.cgi) (Schneider and Stephens 1990; Crooks et al. 2004), we created graphical representations of these motifs. This allowed us to compare the flanking nucleotide residue distributions for HT transition sites with that for sites of transitions arising in ST conditions (Ossowski et al. 2010; Jiang et al. 2014) and with all C and all reverse-complemented G sites in TAIR10.
Analysis of small-scale indel mutations
The frequencies of 1-bp A or T indels found in different length homopolymeric A or T repeat sequence regions were calculated and normalized with respect to the frequency of those different length homopolymeric repeat regions in TAIR10 (Belfield et al. 2018).
Reference genome statistics
The proportions of TAIR10 representing CDSs, UTRs, TEs, and others—noncoding RNAs and pseudogenes (see Figs. 3, 5B)—were calculated by dividing the numbers of nucleotides annotated on Chromosomes 1–5 as CDSs, UTRs, TEs, and other regions by the total number of nucleotides in TAIR10 (119,146,348 bp).
Nucleosome preparation and MNase digestion
Between two and three rosette leaves from six to eight 4-wk-old ST- or HT-grown plants were harvested (∼1 g of leaves), frozen in liquid nitrogen, and ground into powder. Nuclei were isolated as previously described (Bernatavichute et al. 2008), with the following modifications. Plant tissue was resuspended in 10 mL of HBM buffer (25 mM Tris-HCl pH 7.6, 440 mM sucrose, 10 mM MgCl2, 10 mM β-mercaptoethanol, 2 mM spermine, 1 mM PMSF, 1 µg/mL pepstatin and EDTA-free protease inhibitor cocktail [Roche]), with the addition of 0.5% Triton X-100. Suspended nuclei were then passed through a 100-µM and then a 40-µM cell strainer (Fisherbrand). The supernatant was then spun at 3000 rpm for 5 min (SS-34, Sorvall), and the nuclei pellet resuspended in 5 mL of HBB buffer at 4°C (25 mM Tris-HCl pH 7.6, 440 µM sucrose, 10 mM MgCl2, 0.5% Triton X-100, 10 mM β-mercaptoethanol). The solution was applied to a 40%/60% Percoll (GE Healthcare) gradient in HBB floating on a bed of 2.5 M sucrose and spun at 2190 rpm for 30 min at 4°C (SS-34, Sorvall). Isolated nuclei were washed three times in HBB buffer and pelleted by spinning at 3000 rpm at 4°C for 10 min (SS-34, Sorvall).
Isolated nuclei were resuspended in 1 mL of MNase digestion buffer (16 mM Tris-HCl pH 7.6, 50 mM NaCl, 2.5 mM CaCl2), treated with 2 μL of RNase A, 20 μg/μL (Qiagen), and micrococcal nuclease (NEB) digestion performed as previously described (Chodavarapu et al. 2010). The digested DNA was then run on a 1.5% agarose gel, enabling purification of ∼150 bp mono-nucleosomic DNA with a gel purification kit (Qiagen). This DNA was used in 100-bp paired-end Illumina sequencing (BGI).
Nucleosome identification and classification
Python program iNPS (Chen et al. 2014) was used for nucleosome identification. Paired-end MNase-seq reads were first aligned using Bowtie 2 v2.3.4.1 (Langmead and Salzberg 2012) and subsequently filtered using SAMtools (Li et al. 2009) to remove reads which were not mapped in proper pairs, had unmapped mates, or had mapping quality <20, before being used for nucleosome identification. Nucleosomes reported by iNPS (Chen et al. 2014) were then filtered to remove those not reported as “main peaks” or wider than 140 bp (Lyons and Zilberman 2017). Using these accepted nucleosomes, genomic positions in TAIR10 were then classified into one of three categories: nucleosome, edge-of-nucleosome, and nonnucleosome (Supplemental Fig. 6). For this, 10 bases on either side of each accepted nucleosome were regarded as edge-of-nucleosome, and the remaining bases in the middle were designated as nucleosome. Positions outside of the accepted nucleosomes were classified as nonnucleosome.
Assessment of extent of nonrandom spatial clustering of mutations in HT MA lines
As previously described (Belfield et al. 2018), we searched for spatial clustering of mutations in the genomes of each individual HT MA line, using an approach similar to that of Roberts et al. (2012). The median number of genome-wide mutations per haploid genome was 37.5 (data from Table 1A), equivalent to 1 mutation per 3.2 Mbp of genome sequence. We identified groups of mutations for which each mutation was ≤320 kb distant from the next (this distance chosen because it reflects a 10-fold higher mutation density than the expected random genome-wide density). We next calculated the likelihood of obtaining the observed mutation distribution if all mutations were independent and random (Roberts et al. 2012). However, we found no evidence for genomic regional clustering of mutations (P-values > 0.01).
Data access
The Illumina DNA sequencing data files from this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession numbers SRP259251 (WGS for six HT MA lines) and SRP260374 (sequencing of MNase-digested DNA from plants grown under ST and HT conditions).
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank Olivier Mathieu, Jerzy Paszkowski, and Detlef Weigel for providing the met1/+ nrpd2 WGS data. We also thank three anonymous reviewers for constructive comments on initial manuscript versions of this publication, which is based on work supported by the Biological and Biotechnological Sciences Research Council (BBSRC) grants BB/N013611/1 and BB/M011224/1, as well as the Ministry of Science and Technology of the People's Republic of China 111 Project (No. B14027).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.259853.119.
References
- Belfield EJ, Gan X, Mithani A, Brown C, Jiang C, Franklin K, Alvey E, Wibowo A, Jung M, Bailey K, et al. 2012. Genome-wide analysis of mutations in mutant lineages selected following fast-neutron irradiation mutagenesis of Arabidopsis thaliana. Genome Res 22: 1306–1315. 10.1101/gr.131474.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfield EJ, Ding ZJ, Jamieson FJC, Visscher AM, Zheng SJ, Mithani A, Harberd NP. 2018. DNA mismatch repair preferentially protects genes from mutation. Genome Res 28: 66–74. 10.1101/gr.219303.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatavichute YV, Zhang X, Cokus S, Pellegrini M, Jacobsen SE. 2008. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS One 3: e3156 10.1371/journal.pone.0003156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besenbacher S, Sulem P, Helgason A, Helgason H, Kristjansson H, Jonasdottir A, Jonasdottir A, Magnusson OT, Thorsteinsdottir U, Masson G, et al. 2016. Multi-nucleotide de novo mutations in humans. PLoS Genet 12: e1006315 10.1371/journal.pgen.1006315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge LJ, Franklin KA, Homer ME. 2013. Impact of plant shoot architecture on leaf cooling: a coupled heat and mass transfer model. J Royal Soc Interface 10: 20130326 10.1098/rsif.2013.0326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J, Davies KJA. 2017. Oxidative DNA damage & repair: an introduction. Free Radic Biol Med 107: 2–12. 10.1016/j.freeradbiomed.2017.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Chen Z, Chen H, Su Z, Yang J, Lin F, Shi S, He X. 2012. Nucleosomes suppress spontaneous mutations base-specifically in eukaryotes. Science 335: 1235–1238. 10.1126/science.1217580 [DOI] [PubMed] [Google Scholar]
- Chen W, Liu Y, Zhu S, Green CD, Wei G, Han J-DJ. 2014. Improved nucleosome-positioning algorithm iNPS for accurate nucleosome positioning from sequencing data. Nat Commun 5: 4909 10.1038/ncomms5909 [DOI] [PubMed] [Google Scholar]
- Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, Hetzel JA, Kuo F, Kim J, Cokus SJ, et al. 2010. Relationship between nucleosome positioning and DNA methylation. Nature 466: 388–392. 10.1038/nature09147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XL, Zhang BW, Zhang QG, Zhu BR, Lin K, Zhang DY. 2018. Temperature responses of mutation rate and mutational spectrum in an Escherichia coli strain and the correlation with metabolic rate. BMC Evol Biol 18: 126 10.1186/s12862-018-1252-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coumou D, Rahmstorf S. 2012. A decade of weather extremes. Nat Clim Change 2: 491–496. 10.1038/nclimate1452 [DOI] [Google Scholar]
- Crawford AJ, McLachlan DH, Hetherington AM, Franklin KA. 2012. High temperature exposure increases plant cooling capacity. Curr Biol 22: R396–R397. 10.1016/j.cub.2012.03.044 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YB, Peterson GW, Horbach C, Konkin DJ, Beiles A, Nevo E. 2019. Elevated mutation and selection in wild emmer wheat in response to 28 years of global warming. Proc Natl Acad Sci 116: 20002–20008. 10.1073/pnas.1909564116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL, Lyngsoe R, Schultheiss SJ, Osborne EJ, Sreedharan VT, et al. 2011. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477: 419–423. 10.1038/nature10414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubov A, Yao Y, Maheshwari P, Bilichak A, Boyko A, Belzile F, Kovalchuk I. 2010. Microsatellite instability in Arabidopsis increases with plant development. Plant Physiol 154: 1415–1427. 10.1104/pp.110.162933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton WL, Claessens A, Otto TD, Kekre M, Fairhurst RM, Rayner JC, Kwiatkowski D. 2017. Extreme mutation bias and high AT content in Plasmodium falciparum. Nucleic Acids Res 45: 1889–1901. 10.1093/nar/gkw1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C-J, Lu M-Y, Chang Y-W, Li W-H. 2018. Experimental evolution of yeast for high-temperature tolerance. Mol Biol Evol 35: 1823–1839. 10.1093/molbev/msy077 [DOI] [PubMed] [Google Scholar]
- IPCC. 2014. Climate change 2014: Synthesis report. Contribution of working groups I, II and III to the fifth assessment report of the Intergovernmental Panel on Climate Change (Core Writing Team, ed. Pachauri RK, Meyer LA). IPCC, Geneva: https://www.ipcc.ch/report/ar5/syr/. [Google Scholar]
- Jiang C, Pugh BF. 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10: 161–172. 10.1038/nrg2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Mithani A, Gan X, Belfield EJ, Klingler JP, Zhu JK, Ragoussis J, Mott R, Harberd NP. 2011. Regenerant Arabidopsis lineages display a distinct genome-wide spectrum of mutations conferring variant phenotypes. Curr Biol 21: 1385–1390. 10.1016/j.cub.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Mithani A, Belfield EJ, Mott R, Hurst LD, Harberd NP. 2014. Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Res 24: 1821–1829. 10.1101/gr.177659.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katju V, Bergthorsson U. 2019. Old trade, new tricks: insights into the spontaneous mutation process from the partnering of classical mutation accumulation experiments with high-throughput genomic approaches. Genome Biol Evol 11: 136–165. 10.1093/gbe/evy252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith N, Tucker AE, Jackson CE, Sung W, Lucas Lledó JI, Schrider DR, Schaack S, Dudycha JL, Ackerman M, Younge AJ, et al. 2016. High mutational rates of large-scale duplication and deletion in Daphnia pulex. Genome Res 26: 60–69. 10.1101/gr.191338.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang J. 2019. Yeast spontaneous mutation rate and spectrum vary with environment. Curr Biol 29: 1584–1591.e3. 10.1016/j.cub.2019.03.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Clark AB, Kunkel TA. 2015. Differences in genome-wide repeat sequence instability conferred by proofreading and mismatch repair defects. Nucleic Acids Res 43: 4067–4074. 10.1093/nar/gkv271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DB, Zilberman D. 2017. DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. eLife 6: e30674 10.7554/eLife.30674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier B, Cooke SL, Weiss J, Bailly AP, Alexandrov LB, Marshall J, Raine K, Maddison M, Anderson E, Stratton MR, et al. 2014. C. elegans whole-genome sequencing reveals mutational signatures related to carcinogens and DNA repair deficiency. Genome Res 24: 1624–1636. 10.1101/gr.175547.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirouze M, Reinders J, Bucher E, Nishimura T, Schneeberger K, Ossowski S, Cao J, Weigel D, Paszkowski J, Mathieu O. 2009. Selective epigenetic control of retrotransposition in Arabidopsis. Nature 461: 427–430. 10.1038/nature08328 [DOI] [PubMed] [Google Scholar]
- Moore JM, Correa R, Rosenberg SM, Hastings PJ. 2017. Persistent damaged bases in DNA allow mutagenic break repair in Escherichia coli. PLoS Genet 13: e1006733 10.1371/journal.pgen.1006733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1928. The measurement of gene mutation in Drosophila, its high variability, and its dependence upon temperature. Genetics 13: 279–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas-Lledo JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327: 92–94. 10.1126/science.1180677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, et al. 2012. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell 46: 424–435. 10.1016/j.molcel.2012.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. 1990. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18: 6097–6100. 10.1093/nar/18.20.6097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velichko AK, Petrova NV, Kantidze OL, Razin SV. 2012. Dual effect of heat shock on DNA replication and genome integrity. Mol Biol Evol 23: 3450–3460. 10.1091/mbc.E11-12-1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JM, Platzer A, Kazda A, Akimcheva S, Valuchova S, Nizhynska V, Nordborg M, Riha K. 2016. Germline replications and somatic mutation accumulation are independent of vegetative life span in Arabidopsis. Proc Natl Acad Sci 113: 12226–12231. 10.1073/pnas.1609686113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng ML, Becker C, Hildebrandt J, Neumann M, Rutter MT, Shaw RG, Weigel D, Fenster CB. 2019. Fine-grained analysis of spontaneous mutation spectrum and frequency in Arabidopsis thaliana. Genetics 211: 703–714. 10.1534/genetics.118.301721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang L, Huang J, Zhang X, Yuan Y, Chen JQ, Hurst LD, Tian D. 2015. Parent–progeny sequencing indicates higher mutation rates in heterozygotes. Nature 523: 463–467. 10.1038/nature14649 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.