

Abstract
Gorham-Stout disease – also known as “disappearing bone disease” is currently considered a single entity with varying clinical manifestations. We reviewed the existent literature from the earliest historic description (Jackson in 1838) and Gorham and Stout’s original series of patients, multiple case reports and series since. After analyzing 212 reported cases, we identified 76 cases with details that recorded either a history of multifocal disease or an identifiable history of preceding trauma. From this review, we have defined two distinct Gorham-Stout entities – those characteristically associated with lymphangiomatosis [a form of GLA (generalized lymphangiomatosis) questionably distinguishable by bone biopsy and radiologic appearance] with multifocal distributed bone lesions, and those others, usually self-limited, first appearing after a traumatic event and always confined to a single bone or closely adjacent one. Multifocal disease is more likely to have chylothorax as a complication. These two Gorham-Stout entities differ in their demographic distribution, clinical history and manifestations, and they follow divergent clinical courses. The prognosis differs, and so should approaches to monitoring as well as acute and long-term treatment. Further research should seek to identify and define the differences in pathology and molecular mechanisms.
Keywords: Gorham-Stout disease (GSD), generalized lymphangiomatosis (GLA), osteolysis, trauma, disappearing bone disease, lymphangiogenesis
Gorham-Stout disease (GSD) is a rare disorder characterized by the progressive disappearance of bone that is replaced with a proliferation of blood and/or lymphatic vessels. It was first characterized clinically and histologically by Gorham and Stout in a 1955 paper reporting on 24 cases of massive osteolysis. The frequently referred to first case of Gorham-Stout disease was reported in the Boston Medical and Surgical Journal in 1838 (1). The report described a “boneless arm” that developed over 18 years in a young man following a fracture of the right humerus at the age of 18 after a serious initial fall wrestling with an “enraged cow.” He underwent surgery but before the fracture was fully healed, he had subsequent falls opening up the fracture wound twice. He was apparently otherwise healthy throughout a lengthy observation period. The original description follows:
“Not withstanding a most vigilant and untiring devotion to the injured limb, the divided extremities would not adhere; and, to the surprise of the medical attendant, the shaft of each part of the divided bone began to diminish in size, and shorten in length. By a gradual action of the absorbents, the whole of the arm bone, between the shoulder and elbow, was at length completely removed, and that too without any open ulcer, so that not a single vestige of it was left. It has now been in this state for many years, and probably will remain so for life, as there never will be a deposition of bony matter again in that place or even a cartilaginous or a condensed ligamentous substitute, which will materially change it from the present singular condition” (1).
The patient died at the age of 70 from double pneumonia without further changes to this limb or other medical conditions (2).
Was this “curious case of Mr. Brown” typical of the syndrome Gorham and Stout described in 1955 and of the link often established to a generalized lymphangiomatosis (GLA) syndrome? Or are there different forms - etiologically and pathogenetically – of what might instead be thought of as “Gorham Stout phenomenon” including one that is trauma-induced? Or is the trauma “merely incidental” as initially postulated in Gorham et al 1954 (3)? How would this different perspective alter prognostication and management?
Fewer than 1000 cases of GSD have been reported in the international literature to date. The etiopathophysiology of GSD is poorly understood, with an unpredictable course of disease. Although believed to be caused by aggressive vascular, and specifically lymphatic, proliferation or hyperactive osteoclasts, the lesions are considered benign. Massive osteolysis is known to follow a physical injury but may occur spontaneously. It has no known infectious or malignant component.
OBSERVATIONS
Primary vs. Secondary Gorham-Stout Disease
We analyzed 212 cases of GSD published in the literature, beginning with an assembled list of cases (provided courtesy of Dr. Michael Dellinger, Lymphatic Malformation Institute Scientific Director, see also reference 4) and supplemented with additional search of literature. Upon review, we identified a pattern. In cases that included multifocal, non-contiguous lesions (36 cases; 17%), there was no history of prior trauma or an inciting event. In contrast, cases of GSD traced back to a clear-cut precipitating and preceding injury (39 cases; 18.4%) resulted in lesions that remained focal, with spread limited to contiguous bones and no distant sites of disease. In the remaining cases, preceding trauma was not reported, and these cases were not included in our analysis (Fig. 1).
Fig. 1.
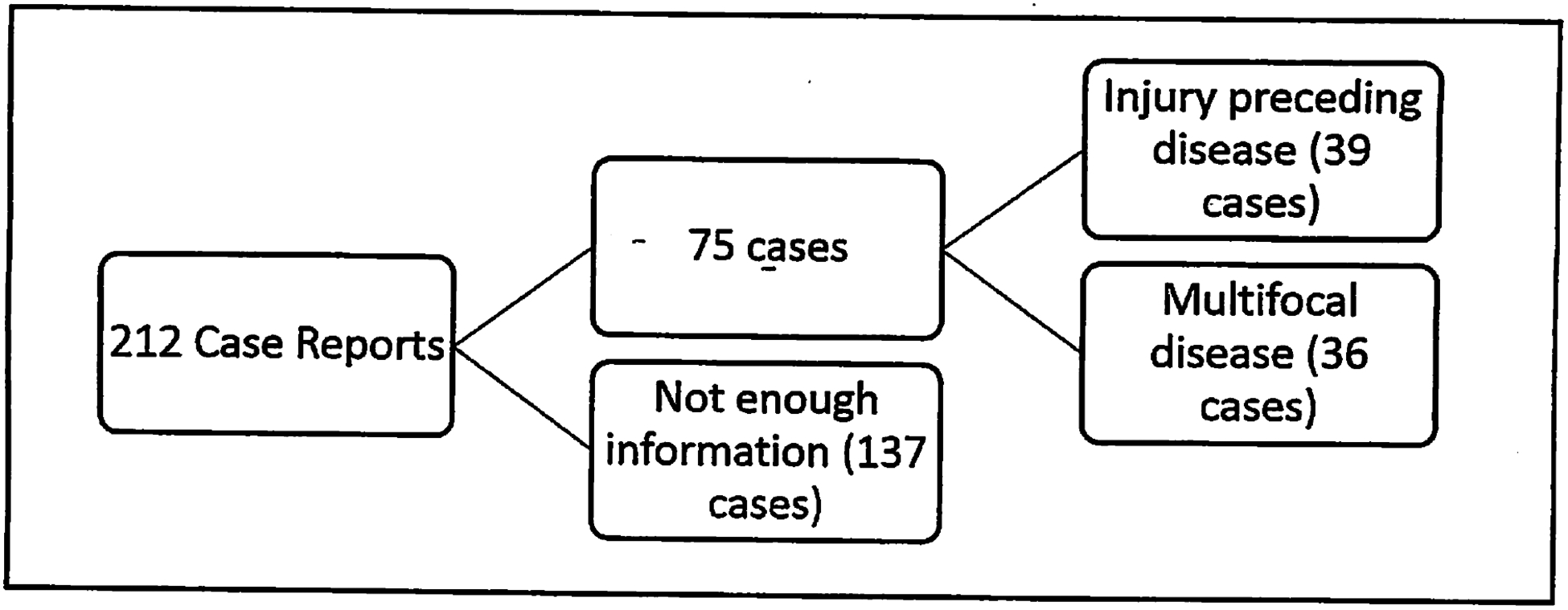
Breakdown of GSD cases analyzed.
We hypothesized that there are at least two modalities of disease. The first type will be referred to as primary GSD (Fig. 2). These cases appear to have no external cause with no preceding trauma documented and may appear as early as at birth. It is in this type that GSD with multifocal, non-contiguous bone lesions might be found. We do not know how many cases of spontaneous disease with a single focus exist, so the criteria includes only cases with multiple sites of disease involving non-contiguous bones.
Fig. 2.
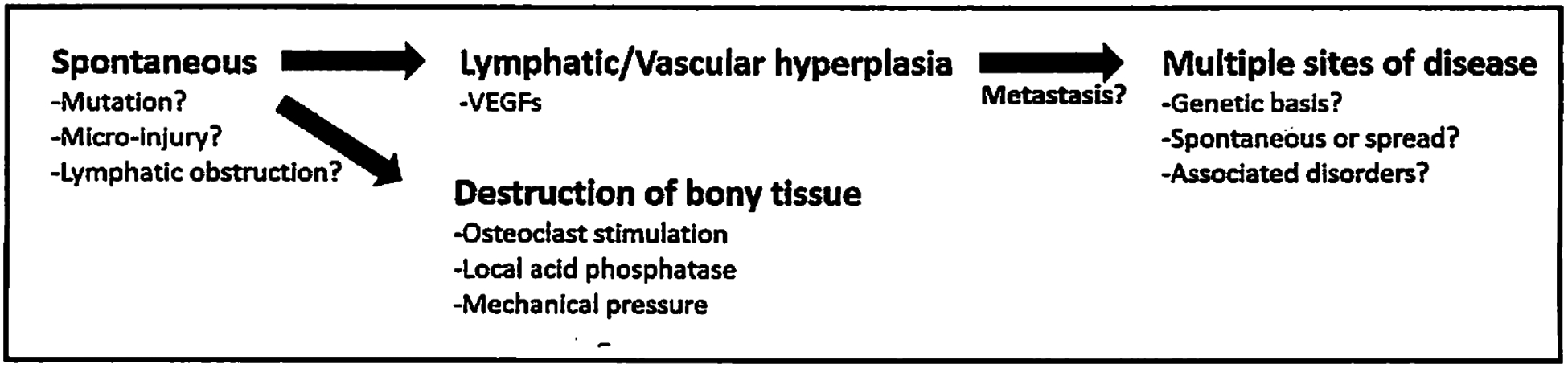
Sequence of events in primary multifocal GSD. See text for further details.
The second subtype of Gorham-Stout disease appears exclusively following significant preceding trauma (Fig. 3). Massive osteolysis following injury is well recorded. However, it has not yet been noted that the spread of disease would be limited to the immediate vicinity of the original insult and contiguous structures.
Fig. 3.

Sequence of events in secondary acquired GSD. See text for further details.
The remaining 137 cases (64.5%) are neither multifocal nor reported as preceded by trauma. It is important to note that several of the histories of trauma are noted as “minor,” without a corresponding fracture. Since minor injury (such as a fall or bruise without fracturing) may not be noticed or recalled even with a thorough interview, there may be significantly more cases of potentially trauma-induced Gorham-Stout disease.
Analysis of Cases
Age distribution (Table 1)
TABLE 1.
Age and Sex Distribution of Gorham-Stout Cases Analyzed
| Primary (Multifocal) GSD | Secondary (Acquired) GSD | |
|---|---|---|
| Age Distribution | ||
| Number of Cases (% of total) | 36 (17%) | 39 (18.4%) |
| Mean ± SD | 14.3 ± 13.4 years | 29.6 ± 19.1 years |
| Two-Tailed t-Test | p = 0.0002 | |
| Sex Distribution | ||
| Male | 16 (44.4%) | 26 (66.7%) |
| Female | 20 (55.6%) | 13 (33.3%) |
| Chi-square test* | p=0.0473 | p=0.495 |
Compared with total GSD cases analyzed
The onset of primary (multifocal) GSD occurs earlier on average than that of secondary (trauma-induced) GSD. In primary GSD, the average age at diagnosis is 14.3 years (median 9 years; standard deviation of 13.15 years) with the youngest patient diagnosed at 9 months (unpublished observations even earlier) and the oldest diagnosed at 55 years.
In contrast, those with secondary GSD tend to be older, with an average age of 29.6 years at diagnosis (median 21.5 years; standard deviation of 18.8 years), with the youngest patient diagnosed at 5 years and the oldest at 78 years. This difference between the 2 groups was significant at the p<0.0002 level by a standard two-tailed student t-test. This may suggest a genetic component where the multifocal cases present in a more severe form at an earlier age.
Sex distribution (Table 1)
There is a male predilection for disease in our review in total, which found 129 affected males (61.4%) versus 81 affected females (38.6%). These findings are consistent with the sex distribution reported by other authors (4,5).
A notable contrast results when viewing the relative male to female ratios by primary (multifocal) and secondary (acquired) GSD subtypes. In the multifocal GSD group, there were 16 males and 20 females. This ratio is unexpected given the general male preference for disease. The acquired cases included 26 males and 13 females, in a ratio consistent with the expected distribution.
The observed differences in sex distribution were analyzed with a chi-square test for significance. The real distributions in the primary (multifocal) and acquired (trauma-induced) GSD categories were compared to the expected distribution of 61.4%:38.6% male:female ratio seen in the overall distribution of the 212 cases.
We found the comparable difference in male to female distribution of the multifocal disease group to be significant, with a p-value of less than 0.05. The acquired, trauma-induced GSD pattern was not significantly different from the expected distribution (Table 1).
Presentation of Disease
Most of the secondary GSD cases presented with pain as the initial symptom. Of the multifocal GSD cases that recorded an initial sign or symptom, patients presented with pain or dyspnea (Table 2).
TABLE 2.
Presenting Symptoms in Gorham-Stout Cases Analyzed
| Presenting Symptom | Primary (Multifocal) GSD | Secondary (Acquired) GSD |
|---|---|---|
| Pain | 6 | 24 |
| Dyspnea | 6 | 6 |
| Swelling | 0 | 3 |
| Fracture | 2 | 4 |
| Not Recorded | 22 | 2 |
Distribution of Disease
The location of disease was highly variable, and cases have been recorded involving nearly every bone in the body. Upon further analysis, we found the ribs and spine to be the most common site of disease in both multifocal and acquired GSD. Other findings are summarized in Table 3.
TABLE 3.
Lesion Locations in Gorham-Stout Cases Analyzed
| Location of Lesion | Primary (Multifocal) GSD | Secondary (Acquired) GSD |
|---|---|---|
| Skull | 12 | 9 |
| Spine/Ribs | 30 | 15 |
| Scapula/Clavicle | 14 | 12 |
| Upper Limbs | 12 | 13 |
| Pelvis | 20 | 7 |
| Lower extremities (excluding pelvis) | 25 | 5 |
| Median number of bones involved | 4.5 | 2 |
Multifocal cases involved a median of 4.5 bones, with an average of 4.9 bones, while acquired cases of GSD involved a median of only 2 bones with an average of 2.45 bones.
Chylothorax
In both sets of patients, chylothorax was a common complication and an indicator for poor prognosis. However, we noted key differences between the trauma-induced GSD and multi-focal GSD groups.
When the GSD was preceded by significant trauma, chylothorax only developed in patients where disease was localized to the thoracic cage and cavity, and occurred in 11 out of 39 patients (28%). The bones involved were exclusively the clavicle, scapula, ribs, and/or the cervical or thoracic vertebrae.
For example, in 1954 Gorham et al described the case of a 16 year old boy who fractured his clavicle and later re-injured the same shoulder while playing baseball at which time imaging showed “total absence of the right clavicle except for an indefinite small fragment at the acromial end.” The following year “skeletal x-rays revealed further destruction of the right scapula. In addition, the head and neck of the right humerus, the upper three ribs on the right side and the upper three dorsal vertebrae were now involved.” He died almost 2.5 years after the initial fracture of his clavicle due to complications from chylothorax (3).
In contrast, the presence of chylothorax did not indicate the location of bone lesions in those with multifocal GSD. Most notably, chylothorax developed in a patient with lesions limited to the lower extremities (6) and with disease limited to bilateral metacarpals (7). Overall, chylothorax was significantly more likely in the multifocal disease group, occurring in 31 out of 36 patients (86%).
Chylothorax patients had worse outcomes in the trauma-induced group than in the multifocal disease group, with 63.6% of patients succumbing to their disease (7 of 11), as compared to only 19.0% of recorded outcomes of multifocal cases resulting in death (4 of 21) (Table 4).
TABLE 4.
Chylothorax Outcomes in Gorham-Stout Cases Analyzed
| Outcome | Primary (Multifocal) GSD | Secondary (Acquired) GSD |
|---|---|---|
| Death | 4 | 7 |
| Stable or Recovered | 17 | 4 |
| Outcome not recorded | 10 | 0 |
| Total | 31 | 11 |
Treatments (Table 5)
TABLE 5.
Treatments Undertaken and Outcomes in Gorham-Stout Cases Analyzed
| Primary (Multifocal) GSD | Secondary (Acquired) GSD | |||||
|---|---|---|---|---|---|---|
| Treatment | Favorable Outcome | Unfavorable Outcome | Lost to follow-up | Favorable Outcome | Unfavorable Outcome | Lost to follow-up |
| No Treatment | 0 | 0 | 1 | 5 | 2 | 1 |
| No Record of Treatment | 5 | 4 | 9 | 2 | 2 | 2 |
| Radiation only | 2 | 0 | 0 | 8 | 3 | 0 |
| Surgery (resection, curettage, graft) | 1 | 1 | 0 | 9 | 0 | 1 |
| Radiation + Surgery | 2 | 0 | 0 | 0 | 0 | 0 |
| Radiation + Bisphosphonate | 0 | 1 | 0 | 0 | 0 | 0 |
| Radiation + Interferon | 4 | 0 | 0 | 0 | 0 | 0 |
| Bisphosphonates only | 2 | 0 | 0 | 1 | 1 | 1 |
| Interferon only | 2 | 0 | 0 | 0 | 0 | 0 |
| Bisphosphonates and Interferon | 0 | 0 | 1 | 1 | 0 | 0 |
| Surgery, radiation, interferon | 1 | 0 | 0 | 0 | 0 | 0 |
| Totals | 19 | 6 | 11 | 26 | 8 | 5 |
There is no current consensus regarding therapies for GSD. Surgical, radiological, and medical interventions have all been attempted with varying degrees of success. A summary of the interventions in these cases is provided below.
Outcomes defined as favorable include cases where treatment halted osteolysis or ameliorated chylothorax and patients survived. Unfavorable outcomes include those that end with patient mortality, continued osteolysis, progressive chylous reflux, or otherwise worsening condition.
The treatments utilized in both groups of patients were varied and followed no standard practice. It is interesting to note that favorable outcomes occurred in approximately 76% of both groups. Despite significant differences in other aspects of the disease process, the treatment success rate was nearly identical.
Pathology
Fifty of the cases reviewed included pathology reports. Many echo the original description of “many anastomosing thin walled vessels,” sometimes containing visible red blood cells.
Within the pathology reports we found that the multifocal disease group included descriptions of “lymphatic vessels,” “lymphatic endothelium” or “lymphangiomatosis” in 11 of 17 histological samples. In contrast, there were only two instances of “lymphatic vessels” or “lymphangioma” described among the 41 pathology reports in the trauma-induced GSD group. Instead, many descriptions included the terms “hemangioma” or “capillary.”
It is important to note that there was no standardization in methods or stains utilized, precluding an accurate analysis. Although it is impossible to draw a true distinction between the pathology of the two groups with the given data, there is enough to warrant specific study in prospective cases. These possible differences in histology are worth investigating with stains for lymphatic-specific markers. Nonetheless, there is a suggestion of a difference in the etiopathophysiology of these two forms of GSD.
Much can be learned about Gorham-Stout disease pathogenesis from the limited existing autopsy reports. Autopsy findings from the above mentioned case of the 16 year old boy with initial clavicle fracture who died of chylothorax complications revealed “the upper part of the thoracic duct was compressed and lost in the inflamed mediastinal tissues, thus leading to its obstruction” (3). Similarly, Vigorita et al presented a case of a 35 year old man with Gorham’s disease with multicentric osteolysis with autopsy findings revealing “abnormalities of the lymphatic system including Iymphangiomatous masses, lymphangiectatic dilations (small bowel, spleen, pancreas, thymus, and bones), and absence of a portion of the thoracic duct” (8).
DISCUSSION
Gorham-Stout disease, otherwise known as massive osteolysis or vanishing bone disease, is a rare and destructive condition that involves the spontaneous local resorption of bony structures that are replaced by fibrous tissue. Since the original paper by Gorham and Stout (for whom the disease is named), there have been fewer than 1000 cases reported in the international literature.
GSD may develop at any age or stage in life, often in children or young adults. It is known to exhibit a slight male predilection. Though the disease often affects the bones of the axial skeleton, nearly every bone in the body has been recorded throughout the literature.
Although the exact mechanism of Gorham-Stout disease has remained unclear, several pathways and etiologies have been proposed. Gorham and Stout suggested that active hyperemia, changes in local pH, and mechanical forces promote bone resorption, suggesting that trauma may promote granulation tissue that invades the bone (9). Heyden and colleagues have continued with this hypothesis, observing acid phosphatase and leucine aminopeptidase near remaining bone in GSD (10). Many authors suggest that lymphatic overgrowth is the driving force behind the disease (11). More recently, several cytokines have been investigated, including IL-6 and VEGF in regard to their effect on osteoclasts and angiogenesis. Although some authors have found increased levels of circulating cytokines in GSD patients (12), these findings remain controversial due to mixed results across studies and patients. These mixed results do not even take into account our findings of the division of GSD into primary and secondary forms, which will require further investigation that could provide interesting results.
Additionally, the investigation into mutations associated with lymphatic proliferation has recently gained traction. There are now 23 mutations associated with lymphatic abnormalities (13), with a focus on VEGF (14–16) and the PI3K/AKT/mTOR (17) signaling pathways among others. Indeed, these pathways have led to targets for treatment. Bevacizumab, an anti-VEGF antibody, has been used successfully in lymphatic anomalies (18), and in one reported case of GSD. The mTOR pathway has recently become a target for treatment with sirolimus for those with lymphatic malformations (19). Sirolimus is gaining favor as an option for lymphangiomatosis and vascular malformations resistant to other therapies (20,21), and there are a small number of promising case reports of successful treatment of Gorham-Stout disease (22,23).
In light of our incomplete understanding, perhaps disappearing bone disease should no longer be described as a syndrome. Instead, as Hopman et al suggest, it “might be more appropriately defined as a phenomenon, considering the disease as a sign rather than an entity” (24). It is quite possible that several of the various theories and proposed mechanisms are correct, with multiple mechanisms leading to the characteristic bony tissue erosion in the setting of blood and/or lymphatic vascular proliferation.
Our data suggest at least two distinct modalities of disease across 75 cases. One subset appears spontaneously with multiple, distinct lesions across separate locations and at an earlier average age. The second group develops GSD after significant even repeated trauma; bony erosion remains limited to the site of injury and contiguous bones, with first onset an average of 15.3 years later than the multifocal group. Though there is a slight male predilection for disease across all cases, we found a statistically significant reversal of this trend in the primary (multifocal) disease group. Unfortunately, genetic analysis of patients in the second group is almost nonexistent and further work in this area may reveal a genetic pre-disposition to this form.
Despite a variety of treatments used and standards of care, patients in both groups experienced nearly identical outcomes, excepting cases complicated by chylothorax. Chylothorax remains an indicator for poor prognosis, and most deaths can be attributed to associated complications (25). Interestingly, we found worse outcomes for secondary (acquired) GSD with chylothorax than the corresponding multifocal cases. The importance of early imaging studies must be emphasized to localize the lymphatic dysplasia (whether primary or secondary), help monitor the elusive disease, and guide therapeutic approaches before the life-threatening complications arise (8,26). Earlier radiologic, surgical, and conventional medical interventions to control the chylous reflux are likely to improve the prognosis in both groups.
Limitations
While the data are suggestive of two etiologies of disease, we acknowledge several limitations to this study. First, many cases were excluded due to lack of information. We were unable to categorize case reports that identified a solitary lesion without a history of trauma to the site based on our criteria.
In some cases, pathologic fracture in the setting of bone disease may be difficult to separate from significant injury that then incites disease. However, not all trauma to the site of later Gorham-Stout disease involved bone fracture.
Additionally, the histopathology across cases was limited and inconsistent in both method and description of findings. Prospective study comparing both histopathology and biochemical findings (along with genetic analysis) in a standardized fashion will likely shed further light on the etiology of Gorham-Stout disease.
CONCLUSION
Gorham-Stout disease is a rare syndrome of idiopathic osteolysis. Also known as “vanishing bone disease” or “massive osteolysis,” it is associated with vascular or, specifically, lymphatic malformations in soft tissue and bone near the lesion. It is known often to affect children or young adults, sometimes after minor trauma and is frequently discovered following a fracture that may be pathologic.
In our review, we find strong indications that there are at least two types of massive osteolysis that are both classified as “Gorham-Stout disease.” Though patterns are beginning to emerge, the number of cases is low, and the data provided in the literature are inconsistent across cases.
The etiology of GSD is poorly understood despite a number of theories. A potential trauma-induced mechanism relates to wound healing inducing bone marrow stem cells with lymphangiogenic and hemangiogenic potential to be turned on uncontrollably (27,28). Without a better foundational understanding of the disease, it is difficult to precisely identify the underlying causes, and biochemical and pathophysiological differences between the primary multifocal and secondary trauma-induced subtypes. Even the semantics (“phenomenon,” “syndrome” or “disease”) of the condition remains under scrutiny. Further research ought to take place in this light.
ACKNOWLEDGMENT
Supported in part by NTH R25HL108837 (NT,LM).
Footnotes
CONFLICT OF INTEREST AND DISCLOSURE
All authors declare that no competing financial interests exist.
REFERENCES
- 1.Jackson JBS: A boneless arm. Boston Med. Surg. J 18 (1938), 368–369. [Google Scholar]
- 2.Jackson JBS: Absorption of the humerus after fracture. Boston Med. Surg. J 10 (1872), 245–247. [Google Scholar]
- 3.Gorham LW, Wright AW, Shultz HH, et al. : Disappearing bones: A Rare form of massive osteolysis. Report of two cases, one with autopsy findings. Am. J. Med 17 (1954), 674–682. [DOI] [PubMed] [Google Scholar]
- 4.Dellinger MT, Garg N,Olsen BR: Viewpoints on vessels and vanishing bones in Gorham-Stout disease. Bone 63 (2014), 47–52. [DOI] [PubMed] [Google Scholar]
- 5.Hu P, Yuan X, Hu X, et at Gorham-Stout syndrome in mainland China: A case series of 67 patients and review of the literature. J. Zhejiang Univ. Science B 14 (2014), 729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruch-Gerharz D, Gerharz C, Stege H, et al. : Cutaneous vascular malformations in disappearing bone (Gorham-Stout) disease. JAMA 289 (2003), 1479–1480. [DOI] [PubMed] [Google Scholar]
- 7.Silva S: Gorham-Stout disease affecting both hands: Stabilisation during biphosphonate treatment. Hand 6 (2011), 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vigorita VJ, Magitsky S, Bryk E: Gorham’s Disease: An autopsy report Clin. Orthop. Relat Res 451 (2006), 267–273. [DOI] [PubMed] [Google Scholar]
- 9.Gorham LW, Stout AP: Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): Its relation to hemangiomatosis. J. Bone Joint Surg 37-A (1955), 985–1004. [PubMed] [Google Scholar]
- 10.Heyden G, Kindblom LG, Nielsen JM: Disappearing bone disease: A clinical and histological study. J. Bone Joint Surg 59-A (1977), 57–61. [PubMed] [Google Scholar]
- 11.Lala S, Mulliken JB, Alomari AI, et al. : Gorham-Stout disease and generalized lymphatic anomaly – clinical, radiologic, and histologic differentiation. Skeletal Radiol. 42 (2013), 917–924. [DOI] [PubMed] [Google Scholar]
- 12.Devlin RD, Bone HG 3rd, Roodman GD: Interleukin-6: A potential mediator of the massive osteolysis in patients with Gorham-Stout disease. J. Clin. Endocrinol. Metab 81 (1996), 1893–1897. [DOI] [PubMed] [Google Scholar]
- 13.Brouillard P, Boon L,Vikkula M: Genetics of lymphatic anomalies. J. Clin. Invest 124 (2014), 898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghalamkarpour A, Holnthoner W, Saharinen P, et al. : Recessive primary congenital lymphoedema caused by a VEGFR3 mutation. J Med Genet. 46 (2009), 399–404. [DOI] [PubMed] [Google Scholar]
- 15.Mendola A, Schlögel MJ, Ghalamkarpour A, et al. : Mutations in the VEGFR3 signaling pathway explain 36% of familial lymphedema. Mol. Syndromol 4 (2013), 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dellinger MT, Hunter RJ, Bernas MJ, et al. : Chy-3 mice are VEGFc haploinsufficient and exhibit defective dermal superficial to deep lymphatic transition and dermal lymphatic hypoplasia. Dev. Dyn 236 (2007), 2346–2355. [DOI] [PubMed] [Google Scholar]
- 17.Kurek KC, Luks VL, Ayturk UM, et al. : Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am. J. Hum. Genet 90 (2012), 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang J, Lee YK, Burm JS: Treatment of tongue lymphangioma with intralesional combination injection of steroid, bleomycin and bevacizumab. Arch. Craniofac. Surg 18 (2017), 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grunewald TG, Damke L, Maschan M, et al. : First report of effective and feasible treatment of multifocal lymphangiomatosis (Gorham-Stout) with bevacizumab in a child. Ann. Oncol 21 (2010), 1733–1734. [DOI] [PubMed] [Google Scholar]
- 20.Hammill AM, Wentzel M, Gupta A, et al. : Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr. Blood Cancer 57 (2011), 1018–1024. [DOI] [PubMed] [Google Scholar]
- 21.Triana P, Do re M, Cerezo VN, et al. : Sirolimus in the treatment of vascular anomalies. Eur J. Pediatr. Surg 27 (2017), 86–90. [DOI] [PubMed] [Google Scholar]
- 22.García V, Alonso-Claudio G, Gómez-Hernández MT, et al. : AJ. Sirolimus on Gorham-Stout disease. Case report Colomb Med (Cali). 47 (2016), 213–216. [PMC free article] [PubMed] [Google Scholar]
- 23.Nozawa A, Ozeki M, Kuze B, et al. : Gorham-Stout disease of the skull base with hearing loss: Dramatic recovery and antiangiogenic therapy. Pediatr. Blood Cancer 63 (2016), 931–934. [DOI] [PubMed] [Google Scholar]
- 24.Hopman SM, Van Rijn RR, Eng C, et al. : PTEN hamartoma tumor syndrome and Gorham-Stout phenomenon. Am. J. Med. Genet A 158A (2012), 1719–1723. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Zhong DR, Zhou PR, et al. : Gorham-Stout disease: Radiological, histological, and clinical features of 12 cases and review of literature. Clin. Rheumatol 35 (2016), 813–823. [DOI] [PubMed] [Google Scholar]
- 26.Witte MH: Lymphangioleiomyomatosis and Gorham-Stout Disease: Primary or secondary disorders of the lymphatic system? Lymphology 50 (2017), 114–119. [PubMed] [Google Scholar]
- 27.Lee JY, Changwon P, Cho YP, et al. : Podoplanin-expressing cells derived from bone marrow play a crucial role in postnatal lymphatic neovascularization. Circulation 122 (2010), 1413–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buttler K, Badar M, Seiffart V, et al. : De novo hem- and lymphangiogenesis by endothelial progenitor and mesenchymal stem cells in immunocompetent mice. Cell. Mol. Life Sri 71 (2014), 1513–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]