

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is the major type of pancreatic malignancy with very poor prognosis. Despite the promising results of immune checkpoint inhibitors (ICIs) in some solid tumors, immunotherapy is less effective for PDAC due to its immunosuppressive tumor microenvironment (TME). In this report, we established an immunocompetent syngeneic PDAC model and investigated the effect of oncolytic herpes simplex virus-1 (oHSV) on the composition of TME immune cells. The oHSV treatment significantly reduced tumor burden and prolonged the survival of tumor-bearing mice. Further, by single cell RNA sequencing (scRNA-seq) and multicolor fluorescence-activated cell sorting (FACS) analysis, we demonstrated that oHSV administration downregulated tumor-associated macrophages (TAMs), especially the anti-inflammatory macrophages, and increased the percentage of tumor-infiltrating lymphocytes, including activated cytotoxic CD8+ T cells and T helper (Th)1 cells. Besides, the combination of oHSV and immune checkpoint modulators extended the lifespan of the tumor-bearing mice. Overall, our data suggested that oHSV reshapes the TME of PDAC by boosting the immune activity and leads to improved responsiveness of PDAC to immunotherapy.
Keywords: oncolytic herpes simplex virus, pancreatic ductal adenocarcinoma, tumor microenvironment, single cell RNA sequencing, cancer therapy
Graphical Abstract
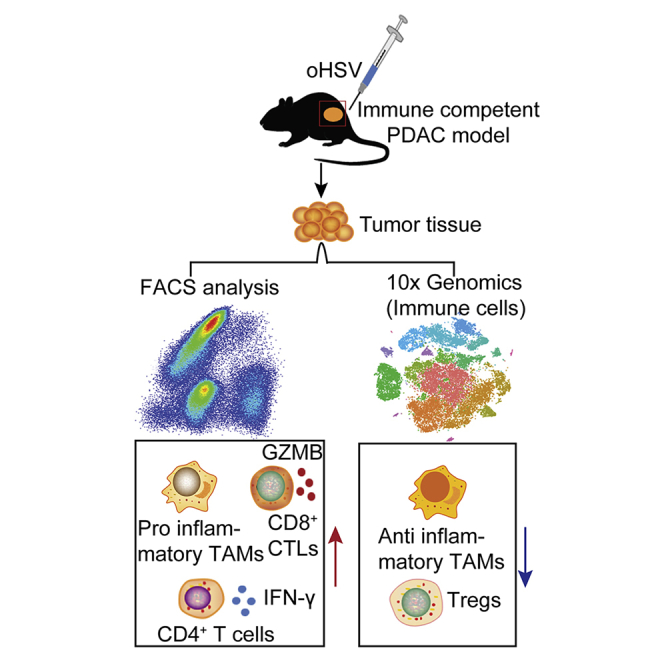
Pancreatic ductal adenocarcinoma (PDAC) is a malignant cancer, which is insensitive to immunotherapy. Zhang et al. constructed an oncolytic herpes simplex virus (oHSV) infectible murine PDAC cell line and established an immunocompetent PDAC mouse model. They found that the oHSV treatment could prolong the survival of PDAC-bearing mice by remodeling the immune microenvironment of PDAC to be less immunosuppressive, and the combination of oHSV with anti-CTLA-4/anti-OX40 antibodies extended the lifespan of the tumor-bearing mice.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most malignant cancers worldwide; once diagnosed, it is usually in the advanced stage, and the 5-year survival rate is about 9%.1 Immune checkpoint inhibitors (ICIs), such as α-programmed cell death-1 (αPD-1)/PD-L1 and α-cytotoxic T-lymphocyte-associated protein 4 (αCTLA-4) antibodies (Abs), have achieved great progress in hematopoietic malignancies, as well as a number of solid tumors.2, 3, 4, 5, 6, 7, 8, 9 However, those treatments have poor efficacy toward PDAC due to the immune-excluded tumor microenvironment (TME) that composes immunosuppressive cells, i.e., the regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and immunosuppressive cytokines.10,11 Moreover, the TME of PDAC is filled with stromal fibroblasts and pancreatic stellate cells (PSCs) that form a physical barrier for the infiltrating lymphocytes, as well as ICI therapeutics.12,13 The above factors lead to the irresponsiveness of PDAC to immunotherapy, and thus PDAC is considered an immunologically “cold” tumor. How to convert PDAC to be more susceptible to immunotherapy, namely the “hot” tumor, is the current challenge in the battle against pancreatic cancer.
Oncolytic virus is an alternative strategy to destroy the outgrowing tumor cells and thus increase the release of tumor-associated antigens (TAAs).14 Meanwhile, infection of the oncolytic virus evokes the innate immune responses of cells in TME, which in turn activates the adaptive immune responses. In this regard, many engineered viruses have been developed for oncolytic therapies.15, 16, 17, 18, 19 Herpes simplex virus 1 (HSV-1) is the most prevalent human virus and has been developed as one of the oncolytic viruses with minimum safety concern. For instance, HSV-1-derived talimogene laherparepvec (T-VEC; by Amgen, USA) has been approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) as the first oncolytic therapy for advanced melanoma, as well as many clinical trials worldwide for indications of glioma, head and neck cancer, and breast cancer.20 For the characteristic TME of PDAC, it is crucial to understand the immunological effect of oncolytic HSV (oHSV) on PDAC.
Unlike human PDAC cells, the mouse PDAC cell line lacks the receptor for HSV-1. To investigate the HSV-based oncolytic therapy for PDAC, we established a new murine pancreatic cancer cell line, Pan02_herpes virus entry mediator (HVEM), which stably expressed the HVEM and verified that tumors developed successfully in the syngeneic mice by subcutaneous (s.c.) implantation of the cells. The intratumoral (i.t.) injection of oHSV significantly inhibited tumor growth and extended survival of tumor-bearing mice. By flow cytometry and single cell RNA sequencing (scRNA-seq) techniques, we profiled the composition of the immune cells in the tumor tissue and concluded that oHSV treatment enhanced tumor-infiltrating CD8+ and CD4+ effector T cells and increased the population of proinflammatory macrophages. In contrast, the existing immunosuppressive macrophages, neutrophils, and Tregs were reduced. The transcriptome profiling also revealed that PD-1, lymphocyte-activation gene 3 (LAG-3), and T cell immunoglobulin (Ig) domain and mucin domain 3 (TIM-3) were enriched in the CD8+ T cell population, and tumor necrosis factor (TNF) receptor superfamily (TNFRSF), member 4 (OX40) and CTLA-4 were enriched in the CD4+ T cell population. Accordingly, the combination of oHSV with anti-CTLA-4 and anti-OX40 antibodies significantly prolonged the survival of PDAC mice. Our studies shed light on the remodeling of the pancreatic TME by oHSV in an immunocompetent tumor model that may lead to the development of a therapeutic strategy by combining oHSV with immune modulators for PDAC.
Results
Establishment of HSV-Infectible Murine PDAC Cells, Pan02_HVEM
First, we employed CRISPR-Cas9 system to genetically engineer the HSV-1 (F), resulting in a primary oHSV with deleted genes encoding ICP34.5 and ICP47 (Figure 1A). The construction of oHSV was verified by western blot and PCR (Figure S1).
Figure 1.

Susceptibility of PDAC Cell Lines to Oncolytic Herpes Simplex Virus (oHSV) Infection
(A) Schematic diagram of the engineered oHSV genome. By CRISPR-Cas9-based homologous recombination with two sets of sgRNAs, two copies of the ICP34.5 gene were replaced by a GFP-expressing cassette, and ICP47 was deleted. (B–F)Different cell lines PANC-1 (B), CFPAC-1 (C), MiaPaCa-2 (D), Pan02 (E), and Pan02_HVEM (F) were infected with wild-type HSV-1 (black squares) or oncolytic HSV (oHSV) (red circles) at MOI = 0.01, and the replication of viruses in those cell lines was determined by titration assays at the indicated time points. Viral titers were presented as Log pfu per mL and plotted on the vertical axis. (G) Pan02 and Pan02_HVEM cells were stained with APC-conjugated anti-HVEM antibody and then subjected to flow cytometry analysis. The histograms were shown with cell count as vertical axis and fluorescence intensity of APC as horizontal axis. The percentage of HVEM-positive cells was labeled inside. Control stands for Pan02 cells stained with APC-conjugated isotype IgG. (H) Pan02 and Pan02_HVEM cells were mock infected or infected with indicated viruses for 24 h. The expression of viral proteins was analyzed by western blot with antibodies against total HSV-1 proteins (top panel) or GAPDH as an internal loading control (bottom panel), respectively. (B–F) Data represented the mean values from three independent experiments with standard deviation. (G and H) Data were representative for three independent experiments.
Second, human PDAC cell lines were susceptible to HSV-1 and oHSV (Figures 1B–1D). However, both HSV-1 and oHSV were restricted in the C57BL/6 mice-derived PDAC cell line, Pan02 (Figure 1E), even though effective replication was seen in a colon cancer line of the C57BL/6 mice background, MC38 (Figure S2A). According to the Gene Expression Profiling Interactive Analysis 2 (GEPIA2) database, human pancreatic cancer cells highly express the HSV receptors, including HVEM, as we confirmed with human PDAC cell lines (data not shown). Thus, we established an infectible murine cell line stably expressing human HVEM, named Pan02_HVEM, as verified by fluorescence-activated cell sorting (FACS) analysis (Figure 1G). The oHSV efficiently infected and replicated in Pan02_HVEM cells, as confirmed with viral titration and viral protein expression (Figures 1F and 1H), although the viral replication efficiency was about 100-fold lower than in interferon (IFN)-incompetent Vero cells (Figure S2B).
oHSV Induced Cell Death and Cytopathology of Pan02_HVEM Cells In Vitro
The effect of oHSV infection on Pan02_HVEM cells was tested in parallel with HSV-1. Pan02_HVEM cells exhibited 49% and 25% cell death induced by wild-type HSV-1 and oHSV, respectively, whereas Pan02 cells remained intact (Figure 2A). In addition, interrupted cell proliferation appeared at 3 days postinfection (Figure 2B), and cytopathic effect (CPE) was observed in Pan02_HVEM cells but not in Pan02 cells (Figure 2C). Thus, the murine PDAC cell line, Pan02_HVEM, could be efficiently infected and lysed by oHSV in vitro.
Figure 2.

The Cytolytic Effects of oHSV
(A) Pan02 (open bar) and Pan02_HVEM (closed bar) cells were mock infected or infected with HSV-1 or oHSV at MOI = 1 for 24 h, stained with APC-conjugated Annexin V, and then subjected to flow cytometry analysis. The percentages of apoptotic cells (Annexin V+) were plotted in columns. (B) Pan02_HVEM cells were mock infected (open circles) or infected with HSV-1 (open squares) or oHSV (closed circles) at MOI = 1, and cell proliferation was monitored by a CCK8 assay. The absorbance at wavelength of 450 nm was determined and plotted against time. Bracket, differences between mock-treated and oHSV-treated groups were statistically analyzed by two-tailed Student’s t test. (C) Pan02 (top panels) and Pan02_HVEM (bottom panels) cells were mock infected or infected with indicated viruses at MOI = 1, and CPE on cells were observed at 24 hpi. Scale bar, 100 μm. The results were representative for three independent experiments. The data were expressed as the mean values with standard deviation (n = 3). Statistics analysis was performed using two-tailed Student’s t test. n.s., not significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
oHSV Inhibited Tumor Growth in the Syngeneic Mouse PDAC Model
We s.c. inoculated Pan02_HVEM cells in C57BL/6 mice (defined as day 0) and treated them with purified oHSV on days 8, 11, and 14 (Figure 3A). On day 20, the mice were euthanized, and the tumors were resected. The oHSV treatment remarkably reduced tumor sizes and weights in comparison with the control (PBS) group (Figures 3B and 3C). We followed the tumor sizes in post-treated mice and observed tumor relapse along the withdrawal of oHSV treatment (Figures 3D and S3A). Nevertheless, oHSV treatment prolonged the survival of mice, as the median survival time was 31 days for the oHSV-treated group and 23 days for the control group, respectively (Figure 3E). We further examined the viral replication in tumor tissues and other loci and found that the replicative oHSV was detected in tumor tissues on day 15 (1 day after the last treatment), and no detectable viral particles were observed on days 17 and 20 in tumor tissues (Figure S3B). No virus was detected in liver and spleen tissues (data not shown). The restrained oHSV replication and deferred enlargement of the oHSV-treated tumor implicated that oHSV alone may not be sufficient to eliminate tumor cells in such an immune-competent model in which the prompt innate immune response to oHSV may lead to the relapse of tumor.
Figure 3.

Oncolytic Effect of oHSV in the PDAC Syngeneic Mouse Model
(A) Schematic diagram of the oHSV treatment regimen. 5 × 105 Pan02_HVEM cells were implanted subcutaneously on day 0. oHSV or PBS was intratumorally injected on days 8, 11, and 14, respectively. Tumor volumes were measured every 3rd day following the first treatment. (B and C) After sacrificing the mice on day 20, the tumors were isolated (B), pictured (C, inset), and weighed (C). (D) The tumor growth curves of PBS- and oHSV-treated mice were shown until they exceeded 2,000 mm3. On day 22, for the PBS control group, n = 3; on day 29, for the oHSV-treated group, n = 4. (E) The survival of tumor-bearing mice was plotted using Kaplan-Meier analysis (n = 5). 5 × 105 Pan02_HVEM cells were implanted subcutaneously into right flanks of mice, and 5 × 105 Pan02 cells were subcutaneously inoculated into the contralateral flanks of mice. PBS or oHSV was intratumoral injected into the Pan02_HVEM tumors on the right flanks, and the growth of the Pan02_HVEM tumor (F) and the distant Pan02 tumor (G) was shown. The tumor volumes were presented as mean values + standard deviation, and tumor weights were presented as mean values ± standard deviation and analyzed by Student’s t test. Data for survival were analyzed by the log-rank test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Furthermore, we characterized the anti-tumor effects of oHSV treatment using a bilateral tumor model. Briefly, Pan02_HVEM cells were inoculated on the right flanks of mice and the HSV nonpermissive tumor cells, Pan02, on the contralateral flanks of mice. When Pan02_HVEM tumors were treated with oHSV, the outgrowth of the tumors was inhibited (Figure 3F), consistent with the single-sided inoculation model in Figure 3B. Intriguingly, Pan02 tumors in the contralateral side were also restrained in oHSV-treated mice (Figure 3G). The abscopal effect of oHSV suggested an oHSV-induced systemic immune response contributing to the anti-tumor efficacy.
oHSV Treatment Enhanced the Immune-Stimulatory Cells in TME
Wild-type HSV-1 possesses mechanisms against host immune response; for instance, ICP34.5 blocks type I IFN secretion, and ICP47 interrupts TAP1-dependent antigen presentation.21,22 Thus, the oHSV, with deletion of both ICP34.5 and ICP47, is favorable to evoke the immune responses. We analyzed the immune cells of PADC by multicolor flow cytometry, of which, the gating strategy was shown in Figure S4. Indeed, oHSV treatment increased tumor-infiltrating leukocytes (TILs) (Figures 4A and 4B), particularly the CD4+ T cells (Figure 4C). Although the proportion of CD8+ T cells in the total TILs was not significantly (n.s.) changed in oHSV treated tumors (Figure 4D), the absolute numbers increased along with TILs. Tregs were dramatically reduced, and the ratio of CD8+ T cells to Tregs increased in the oHSV treatment group compared with the control group (Figures 4E and 4F). In addition, the expression of IFN-γ in CD4+ T cells and granzyme B (GZMB) in CD8+ T cells was upregulated, respectively (Figures 4G and 4H), indicating the activation of both CD4+ and CD8+ T cells upon oHSV treatment. As noted, the expression of PD-1 in CD8+ T cells was rather reduced upon oHSV treatment (Figure 4I), implying the limited efficacy of anti-PD-1 antibody therapy for PDAC.23
Figure 4.

Effects of oHSV Treatment on Tumor-Infiltrating Immune Cells
Mice were subcutaneously implanted with 5 × 105 Pan02_HVEM cells and mock treated or treated with oHSV as described. Single cell suspension was prepared from tumor tissues on day 20 postinoculation. (A–N) Cells were then stained with fluorescence-conjugated antibodies and subjected to flow cytometry analysis for the contents of (A) CD45+ cells; (B) CD45+CD3+ cells; (C) CD45+CD4+ T cells; (D) CD45+CD8+ T cells; (E) CD4+CD25+Foxp3+Tregs; (F) the ratio of CD8+ T cells to Tregs; (G) IFN-γ+CD4+ cells; (H) GZMB+CD8+ cells; (I) PD-1+CD8+ cells; (J) macrophages (CD45+CD11b+F4/80+); (K) M1-like Mac (CD11b+F4/80+iNOS+); (L) M2-like Mac (CD11b+F4/80+CD206+); (M) M-MDSC (CD45+Ly6C+); and (N) G-MDSC (CD45+Ly6G+). The scatter graphs were plotted and presented as the percentage of an individual group of cells, as labeled on the vertical axis. Data were presented as mean values ± standard deviations, and statistics were analyzed using Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
TAMs play important roles in building the immunological circumstance in tumors. Based on their effectiveness of inflammation, macrophages are classified into two categories: proinflammatory (M1) and anti-inflammatory (M2) macrophages, respectively.24,25 Intriguingly, upon oHSV treatment, the total macrophages marked as CD11b+F4/80+/CD45+ seemed reduced by 13.48% (Figure 4J). The proinflammatory-like macrophages inducible nitric oxide synthase (iNOS)+ remained unchanged, whereas the anti-inflammatory-like macrophages (CD206+) decreased (Figures 4K and 4L). The oHSV treatment had no influence on the content of granulocytic (G)-MDSCs or monocytic (M)-MDSCs (Figures 4M and 4N). The major changes of T cells and macrophages were confirmed by immunohistochemistry of tumor samples (Figure S5). The above flow cytometry data suggested that oHSV treatment not only destroyed tumor cells directly but also shifted the TME balance toward more immunologically active.
To answer the question whether the immune balance of TME collapses eventually after oHSV therapy, we further monitored the composition of TILs as long as 32 days when all of the tumors reached 2,000 mm3. As shown in Figure S6, on day 20, the oHSV-treated group exhibited higher level of CD4+ T cells, whereas Tregs and anti-inflammatory-like macrophages were lower in the oHSV group than the control, respectively, as the data show in Figure 4. However, the contents of the oHSV treatment reduced Tregs and anti-inflammatory-like macrophages resumed on days 26 to 32, implicating a tumor relapse after the withdrawal of oHSV treatment.
Identification of PDAC-Infiltrated Immune Cells by scRNA-Seq Analysis
We next performed transcriptomic profiling by scRNA-seq with a 10× Genomics Platform to analyze immune cells in PDAC and identified 21 clusters (Figure 5A). By the characteristic markers, these clusters were identified as macrophages, T cells, natural killer (NK) cells, neutrophils, dendritic cells, and B cells (Figures 5A–5C). The oHSV treatment reduced proportions of macrophages in CD45+ cells (Figure 5D). Meanwhile, the T/NK cell populations were significantly increased by more than 3-fold in the oHSV-treated group in comparison with the control group (Figure 5D).
Figure 5.

Tumor-Infiltrating Immune Cells Delineated by Single Cell RNA Sequencing (scRNA-Seq) Analysis
(A) The viable CD45+ immune cells were isolated from tumor tissues of the PBS control mouse and oHSV-treated mouse using Microbeads, respectively. The scRNA-seq libraries of the leukocytes were prepared and sequenced. The scRNA-seq data were analyzed with Cell Ranger software. All immune cells from PBS and oHSV-treated tumors were clustered and represented as a t-distributed stochastic neighbor embedding (t-SNE) plot. Different clusters were color coded. (B) The expression of selected marker genes in total leukocytes was displayed in the t-SNE plot. Intensity of color was indicated for the level of expression. (C) Cell type-specific marker genes across all clusters were shown by bubble plot. Size of dots represented the fraction of cells expressing a particular marker, and intensity of color indicated the level of mean expression. (D) The components of tumor-infiltrating leukocytes were shown as percentage of total cells.
oHSV Treatment Recomposed the i.t. Macrophages
Macrophages were the important players in TME and contributed more than one-half of total infiltrated immune cells in the murine PDAC model. scRNA-seq analysis displayed an overall profile based on the expression of Nos2 as a marker of proinflammatory macrophages and Mrc1 as a marker of anti-inflammatory macrophages. The expression of Mrc1 decreased significantly following oHSV treatment (Figure 6A). The macrophage populations were further classified into 7 clusters (Mac_c1∼Mac_c7) (Figure 6B), and the distribution of each cluster was calculated in Figure 6C.
Figure 6.

Effect of oHSV Treatment on the Intratumoral Macrophages
(A) The expression of Nos2 and Mrc1 in tumor-associated macrophages from the PBS- and oHSV-treated group was shown by the t-SNE plot, respectively. Red, macrophages expressing Nos2; blue, macrophages expressing Mrc1. (B) Tumor-associated macrophages from PBS- and oHSV-treated tumors were clustered and displayed by the t-SNE plot, separately. Different macrophage clusters were color coded. (C) Cells in an individual cluster were plotted as percentage of CD45+ cells. (D) Heatmap displayed the normalized expression of selected genes in an individual macrophage cluster from the PBS and oHSV treatment group, respectively. (E) Pathway enrichment in macrophage clusters. (F) CD11b+ monocytes were isolated from tumor tissues of PBS (PBS-MCs)- and oHSV (oHSV-MCs)-treated mice on day 20, respectively, and the isolated cells were then intratumorally injected into Pan02_HVEM-bearing recipient mice. The tumor growth curve of the recipient mice was shown (n = 5). (G) CD8+ T cells isolated from spleens of oHSV-treated mice were stained with CFSE and cocultured with medium or with the purified monocytes from PBS-treated mice (PBS-MCs) or oHSV-treated mice (oHSV-MCs) for 4 days in the presence of anti-CD3 and anti-CD28 antibodies, and the T cell proliferation was assessed by CFSE dilution assay. ∗p < 0.05, ∗∗p < 0.01.
Cells in Mac_c1 and Mac_c7 had proinflammatory potential. Mac_c1 expressed a high level of major histocompatibility complex (MHC) class II transcripts (H2-Aa, H2-Ab1, H2-Eb1, Cd74), moderate level of Nos2, Tnf, Cd83, and Cd86, and low level of Mrc1 and Il10. The oHSV infection upregulated the expression of the proinflammatory genes (Figure 6D). The proportion of this cluster of cells was raised from 17.75% to 23.82%. Mac_c7 was similar to Mac_c1, with a relatively low expression of the above genes, and the expression of Nos2 also increased following oHSV treatment. In addition, these two clusters were marked with the upregulation of HSV-1 infection-associated pathways by Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Figure 6E). oHSV-induced elevation of Nos2, Il1, Il6, Tnf, H2-Aa, H2-Ab1, H2-Eb1, and Cd74 was seen in Mac_c2. Besides, KEGG analysis showed that Mac_c2 cells possessed the signature of the hypoxia signaling pathway (Figure 6E), which may be related to macrophage polarization.26
Mac_c3∼Mac_c5 were regarded as immunosuppressive macrophages due to the high expression of Mrc1 and Il10, which were decreased after oHSV treatment (Figure 6D). The percentage of these clusters decreased with oHSV treatment, specifically c3 from 16.65% to 4.56%, c4 from 6.43% to 1.34%, and c5 from 2.59% to 1.76% (Figure 6C). Moreover, KEGG analysis distinguished these clusters by the enrichment in the lysosome pathway and oxidative phosphorylation pathway (Figure 6E). In addition, Mac_c4 cells were rich in cell-cycle proteins and metabolic pathway (Figures 6D and 6E), indicating the active proliferating stage of macrophages.
The scRNA-seq results revealed that TAM activation states consist of a continuum of phenotypes. The classification of proinflammatory/anti-inflammatory phenotypes is simplistic to explain the heterogeneous and dynamic function of TAMs.24,25 Nevertheless, the classification can be used to indicate the potential of macrophages being more proinflammatory or anti-inflammatory.
We further investigated the effect of CD11b+ monocytes on PDAC with monocyte adoptive transfer assay. Briefly, the monocytes were isolated from PBS-treated mice (PBS-MCs) and oHSV-treated mice (oHSV-MCs), respectively, and then injected into tumor-carrying mice. The growth of tumors that received oHSV-MCs was hindered in comparison with that of PBS-MCs (Figure 6F). In addition, we assessed the effect of the above monocytes on CD8+ T cell proliferation in vitro and found that PBS-MCs markedly suppressed the proliferation of CD8+ T cells, whereas oHSV-MCs alleviated the suppressive effect of monocytes on T cells (Figure 6G).
Taken together, scRNA-seq analysis data revealed that oHSV treatment remodeled macrophage composition, especially decreasing the proportion of anti-inflammatory macrophages, and turned the immunosuppressive TME to be more proinflammatory. The adoptive cell transfer and T cell-proliferation assays indicated that the delay of tumor progression may be partly due to the change of monocytes induced by oHSV treatment.
oHSV Treatment Enhanced Tumor-Infiltrating Effector T Cells
The scRNA-seq analysis allowed further detailed profiling of the tumor-infiltrating lymphoid populations in PDAC. Twelve populations of lymphoid cells were defined as CD8+ T cells, CD4+ T cells, Tregs, NK cells, and γδ T cells (Figures 7A and 7C).
Figure 7.

Effect of oHSV Treatment on Tumor-Infiltrating Lymphoid Cells
(A) Transcription information of tumor-infiltrating lymphoid cells was extracted from total immune cells data, then merged, and clustered by the t-SNE plot. Different lymphoid cell clusters were color coded as labeled. (B) Proportions of individual clusters were presented as percentage in total lymphoid cells of PBS- or oHSV-treated groups. (C) Violin plot showed specific gene expression in distinct lymphoid cell clusters of PBS- and oHSV-treated groups. (D) Heatmap displayed the normalized expression of selected genes in lymphoid cell clusters from PBS and oHSV treatment groups, respectively. (E) Heatmap of KEGG identified enrichment of pathways in clusters of lymphoid cells.
CD8+ T cells were subdivided into 6 distinct clusters (CD8_c1∼CD8_c6) and classified into effector, memory, and exhausted T cells. CD8_c1 expressed a high level of Tcf7, Ccr7, Sell, Lef1, and Fas, indicating its central memory T (TCM) phenotype,27,28 and the percentage of which in total lymphoid cells decreased by 3-fold after oHSV treatment (Figures 7B and 7D). CD8_c2/c3 were identified as a mixture of effector/effector memory T cells based on the expression of markers, such as Cd69, Klrg1, but lack Ccr7 and Sell (Figure 7D).29 After oHSV treatment, the percentage of CD8_c3 was dramatically upregulated from 2.7% to 13.8% (Figure 7B). CD8_c2/c3 cells in the control group were characterized by no or low expression of Ifng, Gzmb, and prf1, whereas CD8_c3 of the oHSV-treated group were characterized by increased expression of Ifng, Gzmb, and Prf1 and defined as activated cytotoxic T cells (Figures 7C and 7D). Cells in CD8_c4∼CD8_c6 represented active/exhausted T cells with reduced cytotoxic activity and elevated expression of immune checkpoint receptors. CD8_c6 was the least exhausted with relatively low expression of inhibitory receptors and high proliferation capability (Figure 7D). CD8_c5 cells were at the intermediate exhaustion stage with the intermediate expression of inhibitory receptors and reduced proliferation potential, regardless of the active oxidative phosphorylation metabolic state (Figures 7D and 7E). Upon oHSV treatment, the expression of immune checkpoint genes, such as Pdcd1, Lag3, Havcr2, and Ctla4 in active/exhausted T cells, was reduced to a certain extent, suggesting that oHSV treatment may sustain the active phenotype of CD8+ T cells.
Cluster 7 (CD4_c1) and cluster 8 (CD4_c2) were CD4+Foxp3− conventional T cells (Tcon), and cluster 9 cells were CD4+Foxp3+ Tregs (Figures 7A and 7C). oHSV treatment led to a significant increase in the percentage of Tcon cells, particularly cells in CD4_c1 (Figure 7B). The gene-expression pattern of CD4_c1 represented primed T cells hallmarked by increased expression of Cd28, Icos, and Cd40lg (Figure 7D). CD4_c2 represented exhausted CD4+ helper T (Th) cells, as indicated by the expression of Pdcd1 and Lag3. Th cells consist of different subsets, such as Th1, Th2, and T follicular helper (Tfh) cell. The concomitant expression of Th1 lineage markers (interleukin [IL]-2 and IFN-γ) and Th2 lineage markers (IL-4, IL-13, and GATA3) in the control group suggests that Th1 and Th2 subsets coexisted.30,31 Although upon oHSV treatment, the expression of Th2 lineage-specific marker genes dramatically dropped, transcripts of Th1 lineage markers (TNF) and Tfh lineage markers (BCL-6 and IL-21) were elevated in CD4_c2. Thus, oHSV treatment resulted in the polarization of the CD4+ T cell toward Th1 and Tfh cells, whereas it negatively affected the expansion of pro-tumor Th2 lymphocytes. oHSV treatment caused a decrease of Tregs from 2.90% to 1.77%, and we found that Tregs expressed Cd40lg upon oHSV treatment (Figures 7B and 7D). The aberrant CD40L expression in Tregs could only be observed in severe inflammation.32 Moreover, Tbx21 expression in Tregs mediates a Th1-like response and contributes to inflammation rather than suppression phenotype.33 All of these Th1-like responses manifested the functional reprogramming of Tregs by oHSV therapy.
We also defined NK cells (NK_c1, NK_c2) by the expression of Ncr1 and Klrb1c (Figure 7C). Although the relative percentage of NK was reduced by oHSV treatment (Figure 7B), the NK cells were evidently stimulated by oHSV, as characterized by an elevated level of Gzmb and Prf1 (Figure 7D). It is likely that oHSV promoted activation of NK cells, whereas it led to the reduction of cell numbers.
Cluster 12 was identified as γδ T cells for specifically expressing Tcrg-C1 (Figure 7C). We also noted the expression of Il17a and Rorc induced by oHSV, representing the phenotype and function of innate-like IL-17-producing γδ T cells.34
The Combination Therapy of oHSV with Anti-CTLA-4 and Anti-OX40 Antibodies Prolonged the Survival of PDAC-Bearing Mice
The above scRNA-seq analysis revealed the gene-expression profile of T cells in the oHSV-treated PDAC model. We further extracted the expression patterns of the coreceptors of major T cells and analyzed the coexpression of Pdcd1, Havcr2, and/or Lag3 in CD8+ T cells (Figure 8A). Similarly, the expression of Ctla4 and Tnfrsf4 was presented in CD4+ T cells (Figure 8B). These led to a hypothetical scenario that the oHSV treatment may proactively enhance the T cell responses and thus, the efficacy of the therapeutic antibodies of the coreceptors, such as PD-1, CTLA-4, etc. We then designed a superlative therapy by combining oHSV and therapeutic antibodies (Figure 8C).
Figure 8.

Combination Therapy of oHSV and Immune Checkpoint Inhibitors or Agonist
(A) The expression of immune checkpoint genes in CD8+ T cells. P + H, cells coexpressed genes of Pdcd1 and Havcr2; P + L, cells coexpressed genes of Pdcd1 and Lag3; Triple, cells coexpressed the three genes of Pdcd1, Havcr2, and Lag3. Open bars, PBS control; closed bars, oHSV treated. (B) The expression of immune checkpoint genes in CD4+ T cells. C + T, cells coexpressed genes of Ctla4 and Tnfrsf4. Open bars, PBS control; closed bars, oHSV treated. (C) Schematic diagram of combination treatment regimen. Tumor-bearing mice were treated with oHSV or antibodies as indicated on days 8, 11, and 14. (D) The survival curve of tumor-bearing mice treated with PBS or 3 therapeutic antibodies (3Abs; anti-PD-1, anti-TIM-3, and anti-LAG-3 antibodies), or oHSV plus isotype antibody as control, or oHSV plus 3Abs as above. (E) The survival curve of tumor-bearing mice treated with PBS, or 2 antibodies (2Abs; anti-CTLA-4 plus anti-OX40 antibodies), or oHSV plus isotype antibody, or oHSV plus 2Abs as above. (D and E) PBS, Abs, and oHSV, n = 5; oHSV plus Abs, n = 6. Data for survival were analyzed by the log-rank test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
As shown in Figure 8D, the survival of mice was monitored after treatments with oHSV plus isotype antibody as a control; tri-ICIs, including anti-PD-1, anti-TIM-3, and anti-LAG-3 antibodies; and the oHSV plus the three antibodies, respectively. Compared with the control (PBS) group, all of the treatments extended the median survival days from 18 to 23 (tri-ICIs only), 26 (oHSV alone), and 28.5 (combination) days, respectively. As a result, the tri-ICI-treated group had a modest effect on the immunocompetent PDAC mouse model (survived 5 more days), whereas the combination treatment of oHSV plus tri-ICIs showed little advantage over oHSV monotherapy. Notably, oHSV treatment exhibited better efficacy than tri-ICIs in this model.
On the other hand, the combination treatment of oHSV with anti-CTLA-4 antibody plus anti-OX40 agonist antibody significantly inhibited the growth of PDAC, with the median survival time of 36 days (Figure 8E) versus 30 days by oHSV alone. Interestingly, the treatment with antibodies only, anti-CTLA-4/anti-OX40 antibodies, had merely improvement on survival compared with the PBS group (25 days versus 22 days). Thus, oHSV, in combination with anti-CTLA-4 and anti-OX40 antibodies, achieved maximal survival of PDAC-bearing mice, and may be the optimal amelioration to the oHSV therapy.
Overall, the transcriptional profiling revealed that the oHSV infection activated cytotoxic CD8+ T cells, induced the polarization of CD4+ T cells toward Th1 effector cells, and activated NK cells in the PDAC tumor model. Intriguingly, the oHSV infection led to the reprogramming of the Tregs to execute a Th1-like helper response. All of these viral-induced variations contributed an immunological-active TME that potentially enhanced the tumor response to the immunotherapy. The investigation of oHSV-induced gene-expression patterns in TILs may guide the further designing of combination therapies to improve the efficacy toward PDAC.
Discussion
Oncolytic viruses derived from HSV-1 have allured tremendous interests for cancer therapy since the characterization of ICP34.5 and ICP47.35 With the FDA approval of T-VEC (Imlygic),36 HSV-1 variants become a promising therapeutics to treat solid tumors. Here, we adopted the CRISPR-Cas9 technique to engineer the HSV-1, in which both copies of the ICP34.5 coding region and ICP47 gene were replaced or deleted (Figure 1A). The resulting oHSV was proven to induce cell death and CPE of mouse PDAC cell lines in vitro and inhibited the growth of the implanted tumor in vivo (Figures 2 and 3).
The xenografted models have been widely used to investigate the carcinogenesis and metastasis of human PDAC.37, 38, 39, 40, 41 However, oncolytic virus treatment not only lysed the tumor cells per se but also induced immune responses in the interface of tumor cells and their living-by microenvironment. In this study, we established an oHSV-infectible murine PDAC cell line, Pan02_HVEM, and the syngeneic C57BL/6 mouse model. The implantation of the Pan02_HVEM cells showed no signs of immunogenicity or tissue rejection (data not shown). Another common model for PDAC is the KPC model, a genetically engineered mouse model, in which the mutant Kras and Trp53 are expressed specifically in the pancreas.42 The KPC model is ideal for the studies of PDAC tumorigenesis, whereas our model here emphasizes the immunological aspects in TME. Besides, for the KPC model, the in situ treatment of oHSV requires an injurious surgical procedure that likely evokes wound inflammation. To this measure, the s.c. tumor model that we developed is advantageous and practical to investigate the effect of oHSV on pancreatic TME.
Successful application of oHSV has been achieved in tumors with the manipulation of the immune microenvironment. For instance, IL-12-armed oHSV, G47Δ-mIL12, significantly decreases Tregs in glioblastoma.43 Granulocyte-macrophage colony-stimulating factor (GM-CSF)-armed oHSV, OncoVEXmGM-CSF, reduces the Treg population in melanoma and B lymphoma.44,45 Apparently, these studies target Tregs, one of the major immunosuppressive players, to shift the immune balance of TME. Multiple lines of evidence revealed that the microenvironment of PDAC is immunosuppressive,46,47 so that PDAC patients are insensitive to immunotherapies, such as anti-PD-1 or anti-CTLA-4 antibodies. With the primary version of the oHSV, we analyzed the components of TILs by scRNA-seq, without interference by additional cytokines. With respect to oHSV treatment, the overall tumor-infiltrating T/NK cells were increased (Figures 5D and 7B; CD8_c3 and CD4_c1). The anti-inflammatory-like macrophages and neutrophils were reduced, whereas the proinflammatory-like macrophages were elevated (Figures 5D and 6A). In agreement with the previous reports, the relative content of Tregs decreased (Figures 4E and 7B). Thus, the oHSV treatment alone, without armed cytokines, was able to convert the immunosuppressive TME toward more immune active.
Given the importance of TAMs in regulating tumor immunity, therapeutic strategies that target macrophages have the potential to augment the effect of the oncolytic virus. oHSV significantly reduced CSF1 receptor (CSF1R)-positive monocyte subsets Mac_c3∼Mac_c5 (Figures 6C and 6D) and partially overlapped with the effect of inhibition of CSF1R, which is intensively investigated in preclinical and clinical trials.48 Besides TAM depletion, reprogramming TAMs toward an anti-tumor phenotype could be a more efficacious approach to enhance the efficacy of oHSV.
Despite the immune-stimulating effect of the oncolytic virus focused on in this study, we should also take the desmoplasia feature of the PDAC TME into account. The extracellular matrix (ECM) provides a physical barrier to inhibit penetration and dispersion of the oncolytic virus within tumor masses. A series of approaches have been attempted to partially degrade ECM proteins and improve the penetration of the oncolytic virus.49 i.t. coinjection of collagenase or hyaluronidase with an oncolytic virus in human prostate and melanoma xenograft models increased i.t. viral spread and improved virus therapeutic efficacy.50,51 A relaxin-expressing oncolytic adenovirus increased the i.t. distribution of virus by promoting the degradation of ECM.52 A mouse model that recapitulates the hallmarks of PDAC in both the immune suppression and the desmoplasia would be of necessity to understand the effect of the oncolytic virus on different components of the TME.
Finally, the transcriptome profiling of this oHSV-treated mouse model provided not only the immune variation of PDAC TME but also a guidance for the further design of oHSV derivatives. It would also lead to combination strategies incorporated with immunotherapeutics to ameliorate the efficacy and combat tumor relapse. Based on the scRNA-seq analysis, we designed the combination therapy of oHSV with ICIs/agonists. Compared with oHSV monotherapy, in which the tumor relapsed after oHSV withdrawal, the combination of oHSV and anti-CTLA-4/anti-OX40 antibodies significantly prolonged the survival of PDAC-bearing mice (Figure 8E). However, little improvement was achieved with combination of oHSV and ICIs (Figure 8D). Perhaps the oHSV-induced expression of immune checkpoint genes, including PD-1, LAG-3, and TIM-3 on CD8+ T cells, was insufficient to respond to ICIs in such a scenario. Further optimization would enhance the curative effects of oHSV and advise the designing of armed oHSV for PDAC.
In summary, we established an HSV-1-infectible murine pancreatic cancer cell line and developed a syngeneic PDAC model to explore the potential efficacy of oncolytic therapy. oHSV efficiently lysed tumor cells in vitro, reduced tumor burden, and prolonged survival in vivo. Importantly, oHSV increased the infiltration of effector T cells and downregulated immunosuppressive cells, thus turned the cold pancreatic TME into hot. The combination of oHSV and anti-CTLA-4/anti-OX40 antibodies significantly prolonged the post-treatment survival of the PDAC mice. This study may provide a rationale for the selection of immunotherapeutics in combination with oHSV for the treatment of PDAC.
Materials and Methods
Cells and Viruses
PANC-1, CFPAC-1, MiaPaCa-2, HEK293T, MC38, and Vero cells were originally obtained from ATCC (VA, USA), and cultured in DMEM, supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin/streptomycin. The murine PDAC cell line, Pan02, was purchased from Wuhan Union Hospital (Wuhan, PR China) and grew in RPMI-1640 medium containing 10% FBS and 1% penicillin/streptomycin. All cells were incubated at 37°C with 5% CO2.
HSV-1 (F strain) was prepared in the lab as previously described.53 The derived oncolytic virus oHSV with deletion of genes encoding ICP34.5 and ICP47 was obtained by CRISPR-Cas9-based homologous recombination. First, ICP34.5 was replaced with the GFP-expressing cassette. The donor plasmid containing the homology arms of ICP34.5 flanking the GFP-expressing cassette and the CRISPR plasmid containing the single guide (sg)RNAs of ICP34.5 were cotransfected into HEK293T cells, followed by infection with the wild-type HSV-1. The produced viruses were harvested and the diluted viruses then infected Vero cells. The GFP+ plaques were picked under fluorescence microscopy. The resulting ICP34.5-negative virus was confirmed by DNA sequencing and used for the next step of gene manipulation. Second, the ICP47 gene of the ICP34.5-negative virus was replaced with the red fluorescent protein (RFP)-expressing cassette by a similar approach described above. Donor DNA containing the homology arms of ICP47 flanking the RFP-expressing cassette and the CRISPR plasmid containing the sgRNAs of ICP47 were cotransfected into HEK293T cells, followed by infection with the ICP34.5-negative HSV-1, and the produced viruses were harvested for infection of Vero cells. The RFP+ plaques were picked under fluorescence microscopy. Finally, sgRNA-RFPs were applied to delete the RFP gene and made the Us11 as an early expression gene. The resulting oncolytic virus was designated as oHSV, and the deletion of both ICP34.5 and ICP47 was verified by DNA sequencing, western blot, and RT-PCR.
Virus Propagation and Purification
Vero cells were infected with indicated viruses at a multiplicity of infection (MOI) = 0.01, and cells were collected at 48 h postinfection (hpi), then resuspended in sterile milk, frozen, and thawed three times. Samples were serial diluted, and the virus titers were titrated by oncolytic plaques on Vero cells.
The viruses were further purified for in vivo injection. Vero cells were infected with indicated viruses at MOI = 0.01. The cultured medium was collected at 72 hpi, and the supernatant went through the HiScreen Capto Core 700 (17-5481-15; GE Healthcare, NJ, USA) with an ÄKTA protein purification system to obtain the purified viruses. The purified viruses were then concentrated with an ultrafiltration tube (UFC910096; Millipore, MA, USA), and virus titers were measured by oncolytic plaques on Vero cells.
We used the Vero Cell HCP ELISA Kit (F500; Cygnus, MN, USA) to test the host residual protein of the purified viruses according to the manufacturer’s protocol. Briefly, the samples, standards, and controls were pipetted into a 96-well plate and then an anti-Vero cell: horseradish peroxidase (HRP) was pipetted into each well and incubated on an orbital shaker for 2 h at room temperature. After washed four times, 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was added to each well and incubated at room temperature for another 30 min, and the reaction was stopped by adding Stop Solution into each well. The absorbance at a wavelength of 450/650 nm was determined with a microplate reader (Synergy 4; BioTek).
Establishment of Pan02_HVEM Cells
Plasmid encoding human HVEM was obtained from Sino Biological (Beijing, PR China), and the coding region was amplified and inserted in the pCDH-CMV-MCS-EF1-Puro lentivector (provided by Dr. R. Xiang, Nankai University, Tianjin, PR China) at the EcoRI/BamHI cloning sites. All constructs were verified by DNA sequencing. The lentiviral particles were prepared with HEK293T cells, as described previously.54 Basically, HEK293T cells were transfected with plasmids pCDH-CMV-HVEM-EF1-Puro together with pCMV-VSV-G, pMDLg/pRRE, and pRSV-REV. The viral particles were collected and incubated with Pan02 cells for 24 h and selected in the presence of puromycin (1.25 μg/mL) for 7 days, followed by flow cytometry sorting. The expression of HVEM was monitored by flow cytometry analysis with anti-HVEM antibody (MAB356; R&D Systems, MN, USA).
Western Blot Analysis
Western blot analysis for proteins was performed as described previously.55 Briefly, the cultured cells were collected at 24 hpi and lysed with radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10 mM PMSF, 1 mM NaF, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 1 μg/mL aprotinin) on ice for at least 30 min, and the supernatants were then boiled in 6 × loading buffer (0.3 M Tris-HCl, pH 6.8, 12% SDS (m/v), 0.6% bromophenol blue, 36% glycerol, 6% β-mercaptoethanol [v/v]). Proteins were resolved by 12% SDS-PAGE; transferred to polyvinylidene fluoride (PVDF) membranes; and detected with indicated antibodies, anti-HSV-1 antibody (B0114; Dako Denmark A/S, CA, USA), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (KM9002; Sungene, Tianjin, PR China).
CPE and Cell Death Analysis
To observe the CPE induced by viruses, cells were mock infected or infected with HSV-1 or oHSV at an MOI = 1, and images were taken under microscopy at 24 hpi. For cell death analysis, cells were mock infected or infected with indicated viruses at an MOI = 1, and cells were then collected, stained with allophycocyanin (APC)-conjugated Annexin V antibody (AO2001; Sungene, Tianjin, PR China), and subjected to flow cytometry analysis.
Cell Proliferation Assay
Cells were mock infected or infected with indicated viruses at an MOI = 1. At indicated time points, cells were incubated with Cell Counting Kit-8 (CCK8) (C6005; UE, Soochow, PR China) for 40 min at 37°C. The absorbance at wavelength of 450 nm was determined with a microplate reader (Synergy 4; BioTek).
Viral Titration Assay
Viral growth assay was performed as described.53 Briefly, cells were infected with indicated viruses at an MOI = 0.01, and the medium-containing cells were collected at indicated time points, frozen, and thawed three times. Virus titers were measured as described above by serial dilutions and titrated by oncolytic plaques on Vero cells.
To measure viral growth in vivo, tumor tissues, livers, and spleens were dissected from tumor-bearing mice at the indicated time after therapies. The tissues were minced, homogenized, frozen, and thawed three times. The supernatants were collected by centrifugation and subjected to titration assay, as previously described.
Mice and PDAC Tumor Model
All animal experiments were performed in accordance with and approved by the Institute Research Ethics Committee of Nankai University. 5-week-old male C57BL/6 mice were obtained from Vital River Laboratories (Beijing, PR China). At 6 weeks of age, suspensions of 5 × 105 viable Pan02_HVEM cells in RPMI-1640 medium were s.c. implanted in mice, and the day of tumor inoculation was set as day 0. When tumor volumes reached approximately 50 mm3, tumor-bearing mice were randomly divided into two groups, and i.t. treated with purified oHSV (1 × 107 plaque-forming units [PFU] per 100 μL per mouse) or PBS (100 μL per mouse) as control on days 8, 11, and 14 after tumor inoculation, respectively.
For the bilateral tumor model, 5 × 105 Pan02_HVEM cell suspension in RPMI-1640 medium was s.c. implanted on the right flanks of mice, and 5 × 105 Pan02 cells in RPMI-1640 medium were s.c. implanted on the contralateral flanks of mice. On day 8 post-tumor inoculation, tumors on right flanks were i.t. treated by PBS or oHSV every 3rd day for three times.
The length and width of tumors were measured every 3 days since the first treatment, and tumor volumes were calculated by formula V (mm3) = length × width2/2.
Therapeutic Administration and Survival
C57BL/6 mice were s.c. injected with 5 × 105 Pan02_HVEM cells, and tumor-bearing mice were randomly divided into groups on day 8. The mice were treated three times (days 8, 11, and 14 postinoculation) with indicated reagents, including PBS, oHSV, monoclonal antibodies against PD-1 (BE0146; Bio X Cell, NH, USA), TIM-3 (BE0115; Bio X Cell, NH, USA), LAG-3 (BE0174; Bio X Cell, NH, USA), CTLA-4 (BE0164; Bio X Cell, NH, USA), agonist antibody against OX40 (BE0031; Bio X Cell, NH, USA), and isotype control antibodies anti-IgG2a (BE0089; Bio X Cell, NH, USA) or anti-IgG1 (BE0088; Bio X Cell, NH, USA). The dose of oHSV was 1 × 107 PFUs per 100 μL per mouse, PBS was 100 μL per mouse, and each antibody was 100 μg per i.p. injection. When tumor volumes reached 2,000 mm3, mice were sacrificed following the Animal Welfare Policy. The survival of tumor-bearing mice was monitored by Kaplan-Meier analysis.56
Dissociation of Single Cells from Tumor Tissues
Tumor tissues were harvested and minced from mice on indicated days and inoculated with dissociation solution (1 mg/mL collagenase I, 0.5 mg/mL DNase, 0.05 mg/mL Dispase II dissolved in PBS) for 1 h at 37°C with gentle shaking. After passing through 70 μm cell strainers and washed with RPMI-1640/2% FBS, the sample cells were resuspended in red blood cell lysis buffer (R1010; Solarbio, Beijing, PR China), followed by RPMI-1640 supplemented with 2% FBS.
Flow Cytometry Analysis
Mice were inoculated with viable Pan02_HVEM cells on day 0, and i.t. treated with PBS or oHSV on days 8, 11, and 14. Tumor-bearing mice were sacrificed on indicated days, and single cells from tumor tissues were first stained with viability dye (423106; BioLegend, CA, USA) at 4°C for 10 min; blocked with αCD16/32 antibody (101320; BioLegend, CA, USA) at 4°C for 10 min; and then stained with antibodies against mouse CD45 (103108; BioLegend, CA, USA), CD3ε (100328; BioLegend, CA, USA), CD4 (100428; BioLegend, CA, USA), CD8 (100752; BioLegend, CA, USA), CD25 (102036; BioLegend, CA, USA), PD-1 (135228; BioLegend, CA, USA), CD11b (101224; BioLegend, CA, USA), F4/80 (123114; BioLegend, CA, USA), CD206 (141732; BioLegend, CA, USA), Ly6C (128033; BioLegend, CA, USA), Ly6G (127639; BioLegend, CA, USA), intracellular staining with Foxp3 (320014; BioLegend, CA, USA), iNOS (12-5920-82; eBioscience, MA, USA), IFN-γ (505846; BioLegend, CA, USA), and GZMB (372208; BioLegend, CA, USA) following the instruction of the True Nuclear Transcription Factor Buffer Set (424401; BioLegend, CA, USA) at 4°C for 40 min. Samples were then subjected to flow cytometry analysis, and data were analyzed using FlowJo software.
Immunohistochemistry Analysis
Mice were inoculated with Pan02_HVEM cells on day 0, and i.t. treated with PBS or oHSV on days 8, 11, and 14. Tumor tissues were isolated on day 20, fixed in 4% paraformaldehyde, and embedded in paraffin, and sections were immunostained with indicated primary antibodies against CD4 (25229; Cell Signaling Technology, MA, USA), CD8 (98941; Cell Signaling Technology, MA, USA), Foxp3 (12653; Cell Signaling Technology, MA, USA), F4/80 (70076; Cell Signaling Technology, MA, USA), iNOS (ab15323; Abcam, Cambridge, UK), and mannose receptor (ab64693; Abcam, Cambridge, UK) prior to the addition of biotinylated anti-rabbit Ig secondary antibodies.
Monocyte Adoptive Transfer
C57BL/6 mice were s.c. injected with 5 × 105 Pan02_HVEM cells on day 0, and i.t. treated with PBS or oHSV on days 8, 11, and 14. The CD11b+ monocytes were isolated from tumor tissues of PBS- and oHSV-treated mice on day 20 using CD11b Microbeads (130-126-725; Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer’s protocol, respectively. 5 × 105 purified CD11b+ monocytes were then adoptively transferred into Pan02_HVEM-bearing recipient mice (day 8 post-tumor cell inoculation) by i.t. injections. The tumor volumes were measured every 3 days.
T Cell Proliferation Assay
C57BL/6 mice were s.c. injected with 5 × 105 Pan02_HVEM cells on day 0, and i.t. treated with PBS or oHSV on days 8, 11, and 14. CD8+ T cells were enriched from spleens of oHSV-treated mice on day 20 by the CD8a+ T Cell Isolation Kit (130-104-075; Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer’s protocol. The isolated cells were stained with 6 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; 565082; BD, NJ, USA) for 10 min, and the labeled CD8+ T cells were cocultured with/without the purified monocytes (isolated from tumor tissues on day 20) at a ratio of 1:1. Cells were stimulated with well-coated anti-CD3 (102111; BioLegend, CA, USA) and anti-CD28 (100208; BioLegend, CA, USA) antibodies for 4 days, and the proliferation of CD8+ T cells was assessed by CFSE dilution assay with flow cytometry analysis.
Preparation for scRNA-Seq Library
Tumor-bearing mice treated by PBS and oHSV were killed (n = 1) on day 20 post-tumor inoculation, tumors were digested into single cells, and the viable CD45+ immune cells were sorted from single cell suspensions by the Dead Cell Removal Kit (130-090-101; Miltenyi Biotec, Bergisch Gladbach, Germany) and CD45 (TIL) Microbeads (130-110-618; Miltenyi Biotec, Bergisch Gladbach, Germany) ordinally, according to the manufacturer’s protocol. Cells were adopted only when their viabilities were more than 85%. The following experiments were manipulated by Bio Miao Biological Technology (Beijing, PR China), according to the manufacturer’s protocol (10× Genomics). Briefly, two independent single cell suspensions from PBS- and oHSV-treated tumors were loaded onto the 10× Genomics Chromium Platform to generate Gel Beads-in-Emulsion (GEM) at the concentration about 1,000 cells/μL for totally about 20,000 cells per sample. After the GEM generation, the samples were incubated at 53°C for 45 min and 85°C for 5 min in Thermal Cycle (C1000; Bio-Rad) to barcode the cDNA. GEMs were then broken, and the cDNA was purified by Dynabeads MyOne Silane Beads and amplified (98°C for 3 min, followed by 16 cycles: 98°C for 12 s, 63°C for 20 s, 72°C for 60 s, and followed by 72°C for 1 min). An Agilent Bioanalyzer 2100 was used to assess the quality of cDNA. cDNA was fragmented, end-repaired, a-tailed, then subjected to a double-sided size selection with SPRIselect beads and ligated to adaptors. cDNA was then amplified with the following parameters: 98°C for 45 s, followed by 14 cycles: 98°C for 20 s, 54°C for 30 s, 72°C for 20 s, and followed by 72°C for 1 min. The constructed libraries were subjected to double-sided size selection again, and the quality of the libraries was also assessed. Then libraries were sequenced using an Illumina NovaSeq 6000 sequencer with the paired-end 2 × 125 base pair (bp) sequencing standard.
Data Processing and Analysis
The Cell Ranger software (version [v.]3.1.0) was downloaded from the 10× Genomics website https://support.10xgenomics.com/single-cell-dna/software/release-notes/1-1#header https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest., and mouse reference dataset (mm10-3.0.0) was also downloaded from the 10× Genomics website https://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-mm10-3.0.0.tar.gz. For each individual sample’s FASTQs, the Cell Ranger count module with default parameters was used to align, filter, count barcodes, and count unique molecular identifiers (UMIs). Finally, we generated a gene-barcode matrix containing the barcoded cells and gene-expression counts for each sample.
Identification of Cell Types and Cluster Marker Genes in Seurat
Gene-barcode matrices were imported into the Seurat pipeline for quality control and downstream analysis.57 Low-quality cells (<500 genes/cells, >8,000 genes/cells, >10% mitochondrial) and suspectable populations of doublet were excluded. The data were then normalized, and most variable genes were detected by the FindVariableFeatures function. Then samples’ Seurat objects were combined together by canonical correlation analysis (CCA). Principal-component analysis (PCA) was performed on about 4,000 of the most variable genes with PCA function, and the first 50 PCA components were used for graph-based clustering (at a resolution of 0.5) to identify distinct types of cells. These cell types were projected onto the t-distributed stochastic neighbor embedding (t-SNE) analysis run using previously computed 50 PCA components. To determine lymphoid cells, cells expressing Cd3d and Ncr1 were extracted from total samples, and cell types were projected onto the t-SNE analysis run.
We used the FindAllMarkers function in the Seurat pipeline to characterize the cluster-specific marker genes, and to identify differentially expressed genes (DEGs) between two clusters, we used the FindMarkers function. The average expression of the markers within each cluster was calculated, and the heatmaps of marker gene average expression were generated by R package ComplexHeatmap.58
Pathway Enrichment Analysis
For each cluster, only the genes expressed in more than 10% of the cells with at least 0.25-fold difference and at most 0.05 adjusted p value were considered as DEGs in each cluster. We used R package clusterProfiler to perform pathway enrichment analysis for each cluster’s DEGs.59
Statistical Analysis
The quantitative data were presented as mean value ± standard deviation (SD) of at least three independent experiments. Data were analyzed using two-tailed Student’s t test.60 Animal survival was presented using Kaplan-Meier survival curves, and data for survival were analyzed by the log-rank test.56 A p value that was less than 0.05 (∗) was considered to be statistically significant (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001).
Author Contributions
Y.C. and H.Z. conceived the study. L.Z., H.Z., and Y.C. designed all experiments, analyzed all of the data, and wrote the manuscript with the input of all authors. L.Z. and R.W. performed the experiments. N.Z., H.S., Y.B., D.C., and C.Z. assisted with the viral preparation, infection, and titration. L.Z., J.Y., and L.L. performed FACS data analyses. L.Z., W.W., and H.Z. interpreted the scRNA-seq data.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors would like to thank Drs. Bin He (UIC, USA) for providing relevant information about the HSV-1 genome and critical suggestions; Xianwen Ren (Peking University, China) and Lichun Jiang (ShanghaiTech University, China) for providing relevant information about single cell RNA sequencing data analysis; and Jun Chen (Sun Yat-Sen University, China) for providing suggestions on assays for monocytes. This work was supported by the National Natural Science Foundation of China (grant numbers 81661148051 and 81672010 to Y.C. and 81872787 to H.Z.); Programme of Introducing Talents of Discipline to Universities (grant number B08011); Fundamental Research Funds for the Central Universities, Nankai University (grant number ZB19100123); Open Fund of the State Key Laboratory of Medicinal Chemical Biology for Tianjin Stomatological Hospital; National Natural Science Foundation of China (grant 31600705 to J.Y.); and Tianjin Natural Science Foundation (grant 17JCQNJC09000 to J.Y.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.10.027.
Contributor Information
Hongkai Zhang, Email: hongkai@nankai.edu.cn.
Youjia Cao, Email: caoyj@nankai.edu.cn.
Supplemental Information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Chen R., Zinzani P.L., Fanale M.A., Armand P., Johnson N.A., Brice P., Radford J., Ribrag V., Molin D., Vassilakopoulos T.P., KEYNOTE-087 Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic hodgkin lymphoma. J. Clin. Oncol. 2017;35:2125–2132. doi: 10.1200/JCO.2016.72.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McClanahan F., Riches J.C., Miller S., Day W.P., Kotsiou E., Neuberg D., Croce C.M., Capasso M., Gribben J.G. Mechanisms of PD-L1/PD-1-mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Eμ-TCL1 CLL mouse model. Blood. 2015;126:212–221. doi: 10.1182/blood-2015-02-626754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribas A., Dummer R., Puzanov I., VanderWalde A., Andtbacka R.H.I., Michielin O., Olszanski A.J., Malvehy J., Cebon J., Fernandez E. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170:1109–1119.e10. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun L., Funchain P., Song J.M., Rayman P., Tannenbaum C., Ko J., Mcnamara M., Marcela Diaz-Montero C., Gastman B. Talimogene Laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: a case series. J. Immunother. Cancer. 2018;6:36. doi: 10.1186/s40425-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmed K.A., Kim S., Arrington J., Naghavi A.O., Dilling T.J., Creelan B.C., Antonia S.J., Caudell J.J., Harrison L.B., Sahebjam S. Outcomes targeting the PD-1/PD-L1 axis in conjunction with stereotactic radiation for patients with non-small cell lung cancer brain metastases. J. Neurooncol. 2017;133:331–338. doi: 10.1007/s11060-017-2437-5. [DOI] [PubMed] [Google Scholar]
- 7.Tahara M., Muro K., Hasegawa Y., Chung H.C., Lin C.C., Keam B., Takahashi K., Cheng J.D., Bang Y.J. Pembrolizumab in Asia-Pacific patients with advanced head and neck squamous cell carcinoma: Analyses from KEYNOTE-012. Cancer Sci. 2018;109:771–776. doi: 10.1111/cas.13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muro K., Chung H.C., Shankaran V., Geva R., Catenacci D., Gupta S., Eder J.P., Golan T., Le D.T., Burtness B. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 2016;17:717–726. doi: 10.1016/S1470-2045(16)00175-3. [DOI] [PubMed] [Google Scholar]
- 9.Brahmer J.R., Tykodi S.S., Chow L.Q., Hwu W.J., Topalian S.L., Hwu P., Drake C.G., Camacho L.H., Kauh J., Odunsi K. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark C.E., Hingorani S.R., Mick R., Combs C., Tuveson D.A., Vonderheide R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 11.Padoan A., Plebani M., Basso D. Inflammation and pancreatic cancer: focus on metabolism, cytokines, and immunity. Int. J. Mol. Sci. 2019;20:676. doi: 10.3390/ijms20030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qu C., Wang Q., Meng Z., Wang P. Cancer-associated fibroblasts in pancreatic cancer: should they be deleted or reeducated? Integr. Cancer Ther. 2018;17:1016–1019. doi: 10.1177/1534735418794884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lonardo E., Frias-Aldeguer J., Hermann P.C., Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012;11:1282–1290. doi: 10.4161/cc.19679. [DOI] [PubMed] [Google Scholar]
- 14.Chiocca E.A., Rabkin S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014;2:295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Todo T., Martuza R.L., Rabkin S.D., Johnson P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang H., Rivera-Molina Y., Gomez-Manzano C., Clise-Dwyer K., Bover L., Vence L.M., Yuan Y., Lang F.F., Toniatti C., Hossain M.B., Fueyo J. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. 2017;77:3894–3907. doi: 10.1158/0008-5472.CAN-17-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirano S., Etoh T., Okunaga R., Shibata K., Ohta M., Nishizono A., Kitano S. Reovirus inhibits the peritoneal dissemination of pancreatic cancer cells in an immunocompetent animal model. Oncol. Rep. 2009;21:1381–1384. doi: 10.3892/or_00000364. [DOI] [PubMed] [Google Scholar]
- 18.Wennier S.T., Liu J., Li S., Rahman M.M., Mona M., McFadden G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol. Ther. 2012;20:759–768. doi: 10.1038/mt.2011.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z., Ravindranathan R., Kalinski P., Guo Z.S., Bartlett D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017;8:14754. doi: 10.1038/ncomms14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eissa I.R., Bustos-Villalobos I., Ichinose T., Matsumura S., Naoe Y., Miyajima N., Morimoto D., Mukoyama N., Zhiwen W., Tanaka M. The current status and future prospects of oncolytic viruses in clinical trials against melanoma, glioma, pancreatic, and breast cancers. Cancers (Basel) 2018;10:356. doi: 10.3390/cancers10100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan S., Liu X., Ma Y., Cao Y., He B. Herpes simplex virus 1 γ134.5 protein inhibits STING activation that restricts viral replication. J. Virol. 2018;92:e01015. doi: 10.1128/JVI.01015-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oldham M.L., Hite R.K., Steffen A.M., Damko E., Li Z., Walz T., Chen J. A mechanism of viral immune evasion revealed by cryo-EM analysis of the TAP transporter. Nature. 2016;529:537–540. doi: 10.1038/nature16506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansson H., Andersson R., Bauden M., Hammes S., Holdenrieder S., Ansari D. Immune checkpoint therapy for pancreatic cancer. World J. Gastroenterol. 2016;22:9457–9476. doi: 10.3748/wjg.v22.i43.9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian B.Z., Pollard J.W. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawa-Wejksza K., Kandefer-Szerszeń M. Tumor-associated macrophages as target for antitumor therapy. Arch. Immunol. Ther. Exp. (Warsz.) 2018;66:97–111. doi: 10.1007/s00005-017-0480-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Y., Zhao B., Wang X., Guan G., Xin Y., Sun Y.D., Wang J.H., Guo Y., Zang Y.J. Macrophage transcriptome modification induced by hypoxia and lactate. Exp. Ther. Med. 2019;18:4811–4819. doi: 10.3892/etm.2019.8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahnke Y.D., Brodie T.M., Sallusto F., Roederer M., Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. Eur. J. Immunol. 2013;43:2797–2809. doi: 10.1002/eji.201343751. [DOI] [PubMed] [Google Scholar]
- 28.Gattinoni L., Speiser D.E., Lichterfeld M., Bonini C. T memory stem cells in health and disease. Nat. Med. 2017;23:18–27. doi: 10.1038/nm.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reiser J., Banerjee A. Effector, memory, and dysfunctional CD8(+) T cell fates in the antitumor immune response. J. Immunol. Res. 2016;2016:8941260. doi: 10.1155/2016/8941260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gattinoni L., Ji Y., Restifo N.P. Wnt/β-catenin signaling in T-cell immunity and cancer immunotherapy. Clin. Cancer Res. 2010;16:4695–4701. doi: 10.1158/1078-0432.CCR-10-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim H.J., Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol. Res. 2014;2:91–98. doi: 10.1158/2326-6066.CIR-13-0216. [DOI] [PubMed] [Google Scholar]
- 32.Kumanogoh A., Wang X., Lee I., Watanabe C., Kamanaka M., Shi W., Yoshida K., Sato T., Habu S., Itoh M. Increased T cell autoreactivity in the absence of CD40-CD40 ligand interactions: a role of CD40 in regulatory T cell development. J. Immunol. 2001;166:353–360. doi: 10.4049/jimmunol.166.1.353. [DOI] [PubMed] [Google Scholar]
- 33.Kitz A., Dominguez-Villar M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell. Mol. Life Sci. 2017;74:4059–4075. doi: 10.1007/s00018-017-2569-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caccamo N., La Mendola C., Orlando V., Meraviglia S., Todaro M., Stassi G., Sireci G., Fournié J.J., Dieli F. Differentiation, phenotype, and function of interleukin-17-producing human Vγ9Vδ2 T cells. Blood. 2011;118:129–138. doi: 10.1182/blood-2011-01-331298. [DOI] [PubMed] [Google Scholar]
- 35.Wakimoto H., Kesari S., Farrell C.J., Curry W.T., Jr., Zaupa C., Aghi M., Kuroda T., Stemmer-Rachamimov A., Shah K., Liu T.C. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greig S.L. Talimogene Laherparepvec: first global approval. Drugs. 2016;76:147–154. doi: 10.1007/s40265-015-0522-7. [DOI] [PubMed] [Google Scholar]
- 37.Kasuya H., Nishiyama Y., Nomoto S., Goshima F., Takeda S., Watanabe I., Nomura N., Shikano T., Fujii T., Kanazumi N., Nakao A. Suitability of a US3-inactivated HSV mutant (L1BR1) as an oncolytic virus for pancreatic cancer therapy. Cancer Gene Ther. 2007;14:533–542. doi: 10.1038/sj.cgt.7701049. [DOI] [PubMed] [Google Scholar]
- 38.Yamamura K., Kasuya H., Sahin T.T., Tan G., Hotta Y., Tsurumaru N., Fukuda S., Kanda M., Kobayashi D., Tanaka C. Combination treatment of human pancreatic cancer xenograft models with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib and oncolytic herpes simplex virus HF10. Ann. Surg. Oncol. 2014;21:691–698. doi: 10.1245/s10434-013-3329-3. [DOI] [PubMed] [Google Scholar]
- 39.Gayral M., Lulka H., Hanoun N., Biollay C., Sèlves J., Vignolle-Vidoni A., Berthommé H., Trempat P., Epstein A.L., Buscail L. Targeted oncolytic herpes simplex virus type 1 eradicates experimental pancreatic tumors. Hum. Gene Ther. 2015;26:104–113. doi: 10.1089/hum.2014.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spear M.A., Sun F., Eling D.J., Gilpin E., Kipps T.J., Chiocca E.A., Bouvet M. Cytotoxicity, apoptosis, and viral replication in tumor cells treated with oncolytic ribonucleotide reductase-defective herpes simplex type 1 virus (hrR3) combined with ionizing radiation. Cancer Gene Ther. 2000;7:1051–1059. doi: 10.1038/sj.cgt.7700208. [DOI] [PubMed] [Google Scholar]
- 41.Reinblatt M., Pin R.H., Bowers W.J., Federoff H.J., Fong Y. Herpes simplex virus amplicon delivery of a hypoxia-inducible soluble vascular endothelial growth factor receptor (sFlk-1) inhibits angiogenesis and tumor growth in pancreatic adenocarcinoma. Ann. Surg. Oncol. 2005;12:1025–1036. doi: 10.1245/ASO.2005.03.081. [DOI] [PubMed] [Google Scholar]
- 42.Lee J.W., Komar C.A., Bengsch F., Graham K., Beatty G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+) ;LSL-Trp53(R172H/+) ;Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016;73:14.39.1–14.39.20. doi: 10.1002/cpph.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saha D., Martuza R.L., Rabkin S.D. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32:253–267.e5. doi: 10.1016/j.ccell.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bommareddy P.K., Aspromonte S., Zloza A., Rabkin S.D., Kaufman H.L. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci. Transl. Med. 2018;10:eauu0417. doi: 10.1126/scitranslmed.aau0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moesta A.K., Cooke K., Piasecki J., Mitchell P., Rottman J.B., Fitzgerald K., Zhan J., Yang B., Le T., Belmontes B. Local Delivery of OncoVEXmGM-CSF Generates Systemic Antitumor Immune Responses Enhanced by Cytotoxic T-Lymphocyte-Associated Protein Blockade. Clin. Cancer Res. 2017;23:6190–6202. doi: 10.1158/1078-0432.CCR-17-0681. [DOI] [PubMed] [Google Scholar]
- 46.Karamitopoulou E. Tumour microenvironment of pancreatic cancer: immune landscape is dictated by molecular and histopathological features. Br. J. Cancer. 2019;121:5–14. doi: 10.1038/s41416-019-0479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murakami T., Hiroshima Y., Matsuyama R., Homma Y., Hoffman R.M., Endo I. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019;3:130–137. doi: 10.1002/ags3.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cannarile M.A., Weisser M., Jacob W., Jegg A.M., Ries C.H., Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer. 2017;5:53. doi: 10.1186/s40425-017-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi I.K., Strauss R., Richter M., Yun C.O., Lieber A. Strategies to increase drug penetration in solid tumors. Front. Oncol. 2013;3:193. doi: 10.3389/fonc.2013.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ganesh S., Gonzalez-Edick M., Gibbons D., Van Roey M., Jooss K. Intratumoral coadministration of hyaluronidase enzyme and oncolytic adenoviruses enhances virus potency in metastatic tumor models. Clin. Cancer Res. 2008;14:3933–3941. doi: 10.1158/1078-0432.CCR-07-4732. [DOI] [PubMed] [Google Scholar]
- 51.McKee T.D., Grandi P., Mok W., Alexandrakis G., Insin N., Zimmer J.P., Bawendi M.G., Boucher Y., Breakefield X.O., Jain R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66:2509–2513. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- 52.Jung B.K., Ko H.Y., Kang H., Hong J., Ahn H.M., Na Y., Kim H., Kim J.S., Yun C.O. Relaxin-expressing oncolytic adenovirus induces remodeling of physical and immunological aspects of cold tumor to potentiate PD-1 blockade. J. Immunother. Cancer. 2020;8:e000763. doi: 10.1136/jitc-2020-000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y., Yang Y., Wu S., Pan S., Zhou C., Ma Y., Ru Y., Dong S., He B., Zhang C., Cao Y. p32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J. Biol. Chem. 2014;289:35795–35805. doi: 10.1074/jbc.M114.603845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li S.Y., Du M.J., Wan Y.J., Lan B., Liu Y.H., Yang Y., Zhang C.Z., Cao Y. The N-terminal 20-amino acid region of guanine nucleotide exchange factor Vav1 plays a distinguished role in T cell receptor-mediated calcium signaling. J. Biol. Chem. 2013;288:3777–3785. doi: 10.1074/jbc.M112.426221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wan Y.J., Yang Y., Leng Q.L., Lan B., Jia H.Y., Liu Y.H., Zhang C.Z., Cao Y. Vav1 increases Bcl-2 expression by selective activation of Rac2-Akt in leukemia T cells. Cell. Signal. 2014;26:2202–2209. doi: 10.1016/j.cellsig.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 56.Rich J.T., Neely J.G., Paniello R.C., Voelker C.C., Nussenbaum B., Wang E.W. A practical guide to understanding Kaplan-Meier curves. Otolaryngol. Head Neck Surg. 2010;143:331–336. doi: 10.1016/j.otohns.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 59.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rybarczyk B.J., Walton K.L.W., Grillo W.H. The development and implementation of an instrument to assess students’ data analysis skills in molecular biology. J. Microbiol. Biol. Educ. 2014;15:259–267. doi: 10.1128/jmbe.v15i2.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.