

Abstract
The serine/threonine protein kinase v-AKT homologs (AKTs), are implicated in typical and atypical neurodevelopment. Akt isoforms Akt1, Akt2, and Akt3 have been extensively studied outside the brain where their actions have been found to be complementary, non-overlapping and often divergent. While the neurological functions of Akt1 and Akt3 isoforms have been investigated, the role for Akt2 remains underinvestigated. Neurobehavioral, electrophysiological, morphological and biochemical assessment of Akt2 heterozygous and knockout genetic deletion in mouse, reveals a novel role for Akt2 in axonal development, dendritic patterning and cell-intrinsic and neural circuit physiology of the hippocampus and prefrontal cortex. Akt2 loss-of-function increased anxiety-like phenotypes, impaired fear conditioned learning, social behaviours and discrimination memory. Reduced sensitivity to amphetamine was observed, supporting a role for Akt2 in regulating dopaminergic tone. Biochemical analyses revealed dysregulated brain mTOR and GSK3β signaling, consistent with observed learning and memory impairments. Rescue of cognitive impairments was achieved through pharmacological enhancement of PI3K/AKT signaling and PIK3CD inhibition. Together these data highlight a novel role for Akt2 in neurodevelopment, learning and memory and show that Akt2 is a critical and non-redundant regulator of mTOR activity in brain.
Introduction
The v-AKTs are essential regulators of cell metabolism, growth, proliferation and survival (1, 2). The AKT family consists of three homologous kinases, AKT1 (PKBα), AKT2 (PKBβ) and AKT3 (PKBγ), encoded in humans by three separate genes on chromosomes 14q.32.33, 19q13.1-q13.2 and 1q44, respectively (3, 4). AKT1–3 share a high level of sequence homology, including identical phosphorylation sites at amino acid residues Serine(Ser)-473 and Threonine(Thr)-308 (5). Through activation of these sites the AKT kinases mediate their cellular functions through phosphorylation of downstream substrates, including mTOR and GSK3 (6). Although the AKTs share a high degree of sequence similarity, and exhibit overlapping functions, emerging evidence suggests physiological divergence between the isotypes (7, 8). Importantly, each of the Akt isoforms display differential tissue-specificity, with Akt1 and Akt2 being ubiquitously and highly expressed in brain, Akt2 also being expressed abundantly in skeletal muscle, heart, adipose tissue and testes, and Akt3 being restricted to the brain and testes (9–11). Consistent with non-overlapping tissue expression profiles, each Akt isoform is implicated in different human diseases (12). Akt1 has been extensively implicated in cancer (12), Akt2 with insulin resistance and diabetes mellitus (13) and Akt3 to brain growth anomalies and schizophrenia (14–16). Convergently, in rodent, targeted genetic modification results in unique physiological profiles, with Akt1 mice displaying reduced overall organismal size and disrupted placental development, Akt2 mice, mild fasting hyperinsulinemia, and Akt3 mice showing reduced brain size and corpus callosum disorganization (17–22).
Notably, it is important to recognize that all AKT isoforms are highly expressed in the human (11) and rodent brain (9,10), where Akt signaling is crucial for neuronal development and synaptic plasticity (9). In-vitro RNA interference studies show that Akt2 and Akt3, but not Akt1, are important for axonal growth and neuronal viability (23) and altered electrophysiological and neuromorphological properties are observed in Akt1 mutant mice (24, 25). Neurobehavioral characterization of Akt1 or Akt3 deficiency in mice reveals that Akt1 loss results in deficits of working memory, fear conditioning, sociability, sensorimotor gating and dopamine signalling (24–28), while Akt3 deficiency results in impairments in social cognition, sensorimotor gating and aspects of learning and memory (22). Together rodent studies of Akt1 and Akt3 support genetic association to schizophrenia (16, 26) and pharmacological targeting of AKT is posited to be a key molecular mechanism of action of existing (29) and novel preclinical antipsychotic agents (30, 31).
To date, little characterization of the neurobiological role of Akt2 has been performed, with most of the research being limited to studying its role in glucose homeostasis (9, 17–18) and studies of CNS-related phenotypes being solely limited to examination of KO animals (47, 74). Notably, with regards to neuropsychiatric phenotypes, polymorphisms in the AKT2 gene have been associated with personality traits pertaining to anxiety and depression in humans (32), and the segment of chromosome 7 in mice, which harbors Akt2, contains quantitative trait loci that predict murine anxiety-related phenotypes (33) and reduced brain AKT2 expression is observed in bipolar disorder (34). Together these studies suggest a novel and unappreciated neurological role for Akt2 that may be physiologically distinct from that of Akt1 and Akt3.
Here, we sought to comprehensively characterize the role of Akt2 in multiple behavioral domains, as well as investigation of the neuromechanistic and electrophysiological correlates of reduced Akt2 signaling in both heterozygous and homozygous Akt2 null mice. We highlight the novelty and importance of studying heterozygosity (in comparison to the null condition) for reverse translation to inform human populations and clinical findings in disorders where Akt signaling is reduced but not absent, e.g. schizophrenia, autism and intellectual disability. Through a comprehensive battery of neurobehavioral tasks, we reveal that reduced Akt2 expression negatively impacts a range of cognitive domains including learning and memory, social behaviors, anxiety-related phenotypes and dopaminergic tone. Moreover, we show that reduced Akt2 levels impair long-term potentiation (LTP) and electrophysiological properties of neurons in the cortex and hippocampus and produces a molecular signature involving mTOR and GSK3β, that does not overlap with phenotypes observed in Akt1 or Akt3 genetically modified mice. Together these studies support a unique physiological role of the Akt2 isotype in brain and identify impaired mTOR signaling as a biochemical mechanism underlying observed learning and memory impairments.
Materials / subjects and Methods
Akt2 genetically modified mice
Akt2 HET mice (strain B6.Cg-Akt2tm1.1Mbb/J, http://jaxmice.jax.org/strain/006966.html), originally donated by the laboratory of Dr. Morris J. Birnbaum (17) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were maintained on a 12:12 hours light: dark cycle in standard plastic cages with free access to food and water. A HET harem breeding trio scheme was used (2 females HET-Akt2tm1.1Mbb × 1 male HET-Akt2tm1.1Mbb) to obtain WT, HET and KO littermates. HET mice are fertile and breed properly. Pups were weaned at postnatal day 21 and housed up to 5 mice/cage with same sex littermates. All animal procedures were in accordance with and approved by the NIMH Animal Care and Use Committee and the University of Colorado Denver Institutional Animal Care and Use Committee. Sample size estimates were based on extensive prior studies in our lab (22,30,31).
Genotyping
Genotyping was performed on tail snips using standard polymerase chain reaction (PCR) (http://jaxmice.jax.org/protocolsdb/f?p=116:2:2226059766934507::NO:2:P2_MASTER_PROTOCOL_ID,P2_JRS_CODE:4745,006966). For DNA extraction, tails were digested for 2 h in lysis buffer (1 M Tris HCl pH 7.4, 0.5 M EDTA, 5 M NaCl, 10% SDS and 0.5 mg/ml Proteinase K) at 65 °C. Proteinase K was subsequently deactivated at 95 °C for 15 min, and samples were diluted to a concentration of 50–100 ng/μl. The following primers were used for genotyping: oIMR6745 TAC ACT TCA TTC TCA GTA TTG TTT TGC (mutant reverse), oIMR6746 ACC AAC CCC CTT TCA GCA CTT G (WT reverse), oIMR6747 TGC ACA ATC TGT CTT CAT GCC AC (forward) to generate a 277 bp band for the mutant gene and a 110 bp band for the endogenous gene.
Behavioral Testing
3-month-old male Akt2 HET, KO and WT littermate mice were used for behavioral analyses and subsequently euthanized at 4–5 months of age for collection of brain tissue for biochemical analyses. In preparation for behavioral testing, mice were handled for three days in alternate days of the week preceding the test and allowed to habituate for one hour before testing in a room adjacent to the behavioral testing room. Mice were divided into three cohorts and tested in the following behavioral tasks from the least to the most aversive test: Cohort 1: general health, basal locomotor activity, temporal order object recognition test, (11 WT, 12 HET, 6 KO); Cohort 2: sociability, novel object exploration, fear extinction (10 WT, 11 HET, 6 KO); Cohort 3: object location recognition, context fear, amphetamine-induced locomotor (10 WT, 10 HET, 5 KO). Investigators were blind to genotype until behavioral analyses were complete.
Amphetamine-induced locomotor activity
Mice were habituated in an open field arena (Accuscan 42×42×30, Columbus, OH, USA) for 1 h the day preceding the test. Locomotor activity was recorded by photo beam sensors installed on the edges of the arena. 24 h later the mouse was introduced into the same arena for 10 min to measure the baseline locomotor activity. Subsequently, the mouse was then removed from the arena, and randomly selected for either i.p. injection with 3 mg/kg amphetamine or vehicle (saline) and returned into the test space for an additional 75 min. Locomotor activity was measured as distance travelled in the arena; values of distance were graphed every 5 min.
Fear conditioning
The conditioned fear response is an established Pavlovian paradigm used to longitudinally study the neurological basis of learning and memory. For fear conditioning, experimental mice are exposed to a neutral conditioning stimulus (tone) and to a tone paired to an aversive unconditioned stimulus (foot shock). The conditioning chamber (Med Associates Inc, Georgia, VT,USA) was equipped with a foot shock floor, speaker and a digital video camera. Freeze Monitor software automatically scored the number of seconds the mouse spends freezing, defined as no movement except only those needed for respiration, given that the innate response to fear in mice is freezing behavior. Freezing behavior was calculated as the average time the mouse spends motionless (time frozen/allotted time × 100). Mice were tested for contextual fear memory and fear extinction learning as described in the supplementary material. Between experiments, mice were returned to their home cage for 24 h. The apparatus was cleaned with a 50% ethanol solution between experimental animals.
Recognition memory for objects, location and temporal order tasks
All three cognitive tasks consist of an acquisition or sample phase and a retention or test phase (39, 40) wherein mice are presented with small heavy glass objects of different shape and color (black cuboids and white cones) which are randomly counterbalanced between subjects during testing; between phases mice are returned to their home cage. Mice were habituated to the testing apparatus, an open field arena (Plexiglas, Accuscan 42×42×30 cm), for 1 h the day before testing. During this habituation period spontaneous locomotor activity was recorded by photo beam sensors installed on the edges of the arena. Photo beams allowed calculation of both the distance travelled and time spent in the center and borders of the arena. We quantified the overall distance travelled to evaluate baseline locomotion, and the time spent in the center as a measure of anxiety.
Temporal order recognition test, Novel object preference and Object location recognition tasks
The temporal order recognition, novel object and object location tasks were performed as described previously (31). Tasks are also described in detail in the Supplementary material.
Social function
Social behavior was tested using methods previously described for assessment of sociability and preference for social novelty (31, 41). Social approach was tested in an automated three chambered apparatus. Mice used as the novel stimulus target were C57BL/6 matched to the subject mice by sex and age.
Electrophysiology
8–10-week-old male mice were euthanized by decapitation following isoflurane anesthesia. Brains were quickly removed and sliced with a vibrating blade microtome (Leica VT1000S, Leica Systems, Wetzlar, Germany) in oxygenated (95% O2 and 5% CO2) Ice-cold Na+-free sucrose solution containing 2.5 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 0.5 mM CaCl2, 4.0 mM MgCl2, 10 mM glucose and 250 mM sucrose. 3–4 mice (8–14 brain slices)/genotype were used for the investigation of hippocampal LTP, current clamp and voltage clamp assessment of mPFC pyramidal neurons. Details are provided in the Supplementary material.
Western blotting
Proteins were extracted from fresh frozen mPFC using a lysis buffer containing 1M DDT (Dithiothreitol, Sigma, St. Louis, MO, USA), protease inhibitor cocktail and T-PER Tissue protein Extraction buffer (Thermo Scientific, Rockford, IL, USA). Tissue was homogenized with a sonicator (Ultrasonic Processor model GE50 Sonics, Newtown, CT, USA). After 30 min on ice, samples were centrifuged at 13000rpm for 3 min at 4°C, and the supernatant was collected, and protein concentration quantified using a BCA tm protein assay kit (Thermo Scientific). 25μl of protein/sample were loaded into a 15 well gel and after running, transferred onto PVDF membranes (Invitrogen Life Technology, Grand Island, NY, USA). Membranes were incubated overnight at 4°C in the appropriate primary antibody (Supplementary material) diluted in 5% milk in TBST (Tris-Buffered Saline 1% Tween 20. The following day after a series of washes, the membranes were incubated in the species appropriate HRP-conjugated secondary antibodies (Santa Cruz, CA, USA) at a 1:5000 dilution. Chemiluminescence signal was obtained using a Syngene image analyzer G:BOX (Frederick, MD, USA). The optical density of the protein bands was calculated using Image J software.
IC87114 testing in the temporal order object recognition task
To determine the preclinical potential of IC87114 to reverse recency discrimination impairments in Akt2 HET and KO mice a separate naïve cohort of mice were used at 3–5 months of age. Testing was performed as previously described (31). In brief, each genotype group was randomly split into two treatment groups, one group received a single intraperitoneal injection of 0.25% DMSO in saline, the other group received 0.1mg/kg IC87114 dissolved in 0.25% DMSO in saline. Injections were performed 30 minutes before the test phase of the temporal order recognition task as described in the Supplementary material.
Mouse ex vivo hippocampal neuronal dissociated cultures
Primary neuronal cultures were prepared from hippocampi of individual Akt2 mouse embryos on embryonic day 18 (E18) as described previously (31, 70). Briefly, hippocampi were removed and dissociated from individual embryos and resuspended in supplemented DMEM. Cells were plated at a density of 100,000 cells/well on poly-D-lysine/laminin coated glass coverslips (BD Biosciences). Culture media was changed to Neurobasal (+2% B27, +1% Pen/Strep, +1%Glutamax) 24-hours after initial plating and half of the media was changed every 7 days. Cells were transfected with a DNA plasmid containing pVenus-YFP (71) using Lipofectamine2000 (Invitrogen) per the manufacturer’s protocol apart from modifying the incubation period to one hour for optimal cell viability. Two to three days following transfection, neurons were fixed using 4% paraformaldehyde/ 4% sucrose in PBS and subsequently immunostained with an antibody against GFP (Santa Cruz Biotechnology). Images were taken on a Zeiss inverted LSM700 confocal microscope with 10 and 20 × objectives collected through a series of 5–8 z-sections max projected to create a 2D composite image for analysis in ImageJ (NIH). Measurements from at least 3 embryos/genotype were performed at DIV 3 (axon length and soma size) and DIV 10 (dendritic arborizations). Axons, somas, and dendrites were traced manually, and the sholl analysis plugin on ImageJ, with a radius step size of 5 microns, was used as an additional measure of dendritic complexity. Viability assessments were performed by co-incubating neurons at DIV 4 with Calcein-AM (CAM) and Propidium Iodide (PI) (both Invitrogen) diluted in DMEM media for 30 minutes at 37°C. Individual images of PI and CAM positive neurons were taken with a Zeiss epifluorescent inverted microscope at four quadrants of each coverslip. Images were co-localized in ImageJ (NIH) and the ratio of C-AM:PI neurons was calculated. Averages of ratios in all four quadrants of each coverslip was calculated to ensure reliability. All imaging and quantification were performed by a researcher blinded to genotype.
Data Analysis
Data are expressed as the mean ± SEM throughout. All analysis was performed using IBM SPSS Statistics 21 software (Armonk, NY, USA). T-tests, one-way or two-way ANOVA, with repeated measurement (RM ANOVA) or multivariate (MANOVA) were used when appropriate, as described in the text. Poisson regression was used to model count variables. Logistic binomial regression was used to predict categorical variables from sets of predictor variables. Post-hoc analyses were Bonferroni corrected for multiple testing. P values ≤ 0.05 were considered statistically significant.
Results
Akt2 mutant mice display anxiety-like phenotypes and hyposensitivity to amphetamine
To determine the in vivo neurological consequences of full or partial genetic deletion of Akt2, we first performed a comprehensive screen of general health and motor reflex parameters in male mice genetically engineered with heterozygous (HET) or homozygous (KO) germ-line deletion of the Akt2 gene, compared to their wild type (WT) littermates. Akt2 HET and KO mice showed no physical abnormalities of body weight, fur condition, whisker presence, reflexes (righting, eye blink, ear twitch, and whisker twitch) or body and limb tone (Table 1). Additionally, no differences were observed in open cage behaviors such as transfer freezing, digging events and grooming events. Conversely, a significantly increased number of rearing events was observed in both Akt2 HET and KO mice (Poisson regression: Wald Chi-square = 47.840, df = 2, p ≤ 0.0001; sequential Sidak post-hoc: p ≤ 0.0001 WT vs HET, p ≤ 0.0001 WT vs KO). While there were no significant differences in the wire hang performance between genotypes, significantly decreased forepaw reaching (Logistic binomial regression: Wald Chi-square = 7.798, df = 2, p = 0.02; sequential Sidak post-hoc: p ≤ 0.0001 WT vs HET, p = 0.013 WT vs KO) and increased positional passivity (42% HET, 33% KO compared to 0% of WT, one-way ANOVA: F(2,29) = 3.24, , p = 0.05) were also identified in both Akt2 HET and KO mice. These convergent findings are indicative of anxiety-like behaviors rather than a muscle weakness phenotype.
Table 1. General health profile of AKT-2 HET and KO mice.
Physical characteristics: body weight, poor coat condition, bald patches, missing whiskers, pilorerection, body tone, limb tone and physical abnormalities. Motoric abilities: trunk curl, forepaw reaching (*p ≤ 0.05, ***p ≤ 0.001 vs WT), wire hang, positional passivity (**p ≤ 0.05, *p ≤ 0. 1 vs WT). Reflexes: righting, corneal, ear twitch, reactivity to handling and petting escape. Empty cage behavior: transfer freezing, wild running, exploration, grooming time and events, rearing events (***p ≤ 0.001 vs WT) and digging events. Values represent percentage or mean ± SEM. N = 11 WT N = 12 HET, N = 6 KO.
| General Health | WT | HET | KO |
|---|---|---|---|
| Physical characteristics | |||
| Body weight (g) | 26.94±0.61 | 26.79±0.50 | 25.48±0.59 |
| Poor coat condition (%) | 0 | 0 | 0 |
| Bald Patches (%) | 0 | 0 | 0 |
| Missing whiskers (%) | 0 | 0 | 0 |
| Piloerection (%) | 0 | 0 | 0 |
| Body tone (% of good) | 100 | 100 | 100 |
| Limb tone (% of good) | 100 | 100 | 100 |
| Physical abnormalities (%) | 0 | 0 | 0 |
| Motoric abilities | |||
| Trunk curl (%) | 91 | 83 | 83 |
| Forepaw reaching (%) | 91 | 25*** | 33* |
| Wire hang (sec) | 60.0±0.0 | 60.0±0.0 | 60.0±0.0 |
| Positional passivity (%) | 0 | 42** | 33* |
| Reflexes (% of mice normal) | |||
| Righting reflex (%) | 100 | 100 | 100 |
| Corneal (%) | 100 | 100 | 100 |
| Ear twitch (%) | 100 | 100 | 100 |
| Whisker twitch (%) | 100 | 100 | 100 |
| Reactivity to handling (3-point scale) | 2.04±0.04 | 2.15±0.09 | 2.25±0.14 |
| Petting escape (%) | 36 | 46 | 25 |
| Empty cage behavior | |||
| Transfer freezing (%) | 0 | 0 | 0 |
| Wild running (%) | 0 | 0 | 0 |
| Exploration (3-point scale) | 2.36±0.15 | 2.33±0.14 | 2.33±0.21 |
| Grooming (sec) | 8.06±1.11 | 6.40±1.10 | 8.41±2.23 |
| Grooming (events) | 5.91±0.94 | 4.67±0.61 | 4.33±0.61 |
| Rearing (events) | 33.55±1.64 | 50.17±2.58*** | 54.17±2.23*** |
| Digging (events) | 10.18±2.14 | 13.83±2.30 | 13.00±2.28 |
In a separate cohort of animals we identified that heterozygous or homozygous deletion of the Akt2 gene did not impact brain weight (g) (WT=0.438±0.004, HET=0.441±0.005, KO=0.424±0.006, one-way ANOVA: F(2,45) = 2.341, p ≥ 0.05), body weight (g) (WT=26.967±0.703, HET=25.860±1.147, KO=26.667±0.804, one-way ANOVA: F(2,45) = 4.325, p ≥0.05), nor the ratio of brain:body weight (WT=0.0165±0.0052, HET=0.0173±0.00082, KO=0.0161±0.00054, one-way ANOVA: F(2,45) = 0.723, p ≥ 0.05). These observations confirm that gross basal metabolic abnormalities are not observed in the context of Akt2 deficiency (17). Next, to further examine the anxiety-like phenotype we tested locomotor performance of Akt2 HET and KO mice in the open field arena compared to WT mice. Consistently, Akt2 HET traveled significantly less distance than WT littermates during the 60 minute test (RM ANOVA: effect of genotype F(2,26) = 3.516, p = 0.045; Bonferroni post-hoc p = 0.014 WT vs HET; Figure 1A and B). Furthermore, both HET and KO mice spent significantly less time in the center of the arena than WT littermates (RM ANOVA: effect of genotype F(2,26) = 3.369, p = 0.05, Bonferroni post-hoc p = 0.04 WT vs HET, p = 0.035 WT vs KO; Figure 1C).
Fig. 1. Akt2 HET and KO mice show altered locomotor activity, response to amphetamine, associative learning of environmental context, and reassessment of a learned aversive cue.
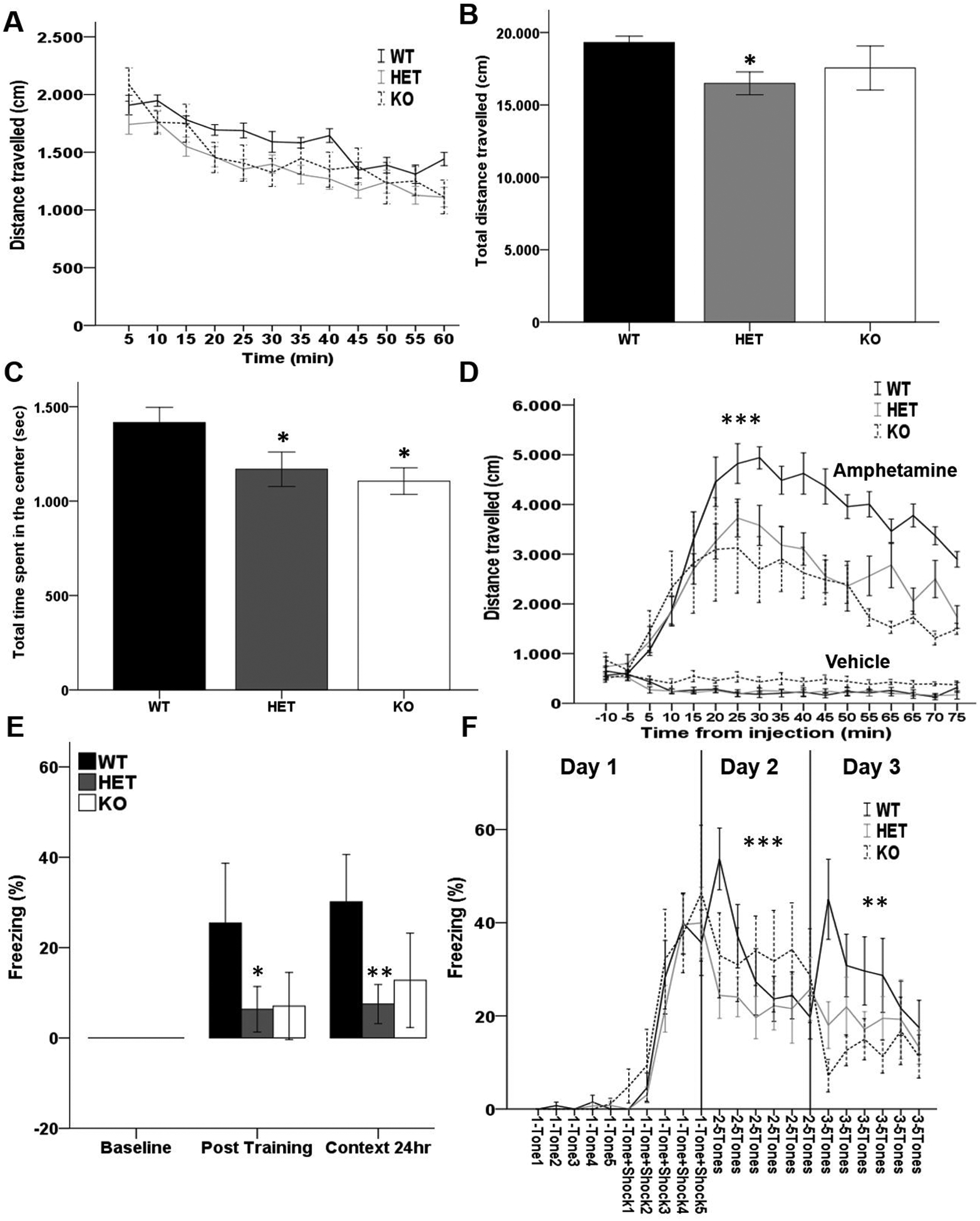
(A–B) Distance (cm) traveled during 1-hour locomotor activity (A) every 5 minutes and (B) overall traveled distance of Akt2 HET and Akt2 KO mice and their WT littermates. HET mice travel less distance than WT littermates (p = 0.014). (C) Overall time (sec) spent in center of the arena during the one-hour open field task of Akt2 HET and Akt2 KO mice and their WT littermates. HET and KO mice spend less time in the center of the arena than WT littermates (p = 0.04 and 0.035, respectively). N = 11 WT, 12 HET, 6 KO. Bars represent mean ± SEM. (D) 10 minutes locomotor activity in naïve AKT2 HET and KO mice and their WT littermates, followed by injection of amphetamine 3 mg/kg or vehicle and subsequent 75 minutes recording of locomotor activity every 5 minutes. HET and KO mice injected with amphetamine travel less distance than WT mice (p ≤ 0.0001). N = 5/genotype/treatment. (E) Contextual fear memory of Akt2 HET and Akt2 KO mice and their WT littermates. The duration of immobility (% freezing) during baseline conditions (no-stimuli), post-training and context performed after 24 hrs. % freezing is significantly lower in Akt2 mutant mice (p = 0.016) at post-training (F(2,22) = 4.912, p = 0.017; post hoc HET vs WT p = 0.025, KO vs WT p = 0.099) and at context24h (F(2,22) = 8.639, p = 0.002; post hoc HET vs WT p = 0.002, KO vs WT p = 0.057). N = 10 WT, N = 10 HET and 5 KO. (F) Fear extinction learning of Akt2 HET and Akt2 KO mice and their WT littermates. Single freezing response to 5 tones and to 5 tone+shock pairings during day 1 (conditioning phase) and to 5 tones-only during day 2 and 3 (extinction phase, 24 and 48hrs after conditioning, respectively). No extinction was observed in Akt2 HET and KO mice (day 2 tone×genotype F(10,120) = 3.434, p = 0.001; day 3 tone×genotype F(10,120) = 2.276, p = 0.018). N = 10 WT, 11 HET, 6 KO. Data represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Next, in a separate naive cohort of Akt2 WT, HET and KO mice we sought to determine if Akt2 expression impacts dopaminergic tone through sensitivity to amphetamine challenge. Intraperitoneal injection of amphetamine strongly induces locomotor activity through the inhibition of dopamine reuptake (35). Thus, we monitored locomotor response to amphetamine (3 mg/kg i.p.) in Akt2 KO and Akt2 HET mice compared to their WT littermates (Figure 1D). 3 mg/kg amphetamine injection significantly increased locomotor activity in the open field (RM ANOVA: treatment F(1,24) = 99.24, p < 0.0001). However, a significant genotype × treatment interaction was observed (genotype × treatment; F(2,24) = 32.72 p < 0.00001), whereby Akt2 HET and KO mice exhibited a significantly blunted locomotor response to amphetamine compared to WT mice (Bonferroni post-hoc WT vs HET p ≤ 0.0001, WT vs KO p = 0.001; Figure 1D). These data demonstrate that dopaminergic tone is altered in the context of reduced Akt2 expression.
AKT2 mutant mice exhibit impaired associative learning and memory
Neurons encoding fear memories are located in the lateral nucleus of the amygdala (36) and send information to the hippocampus and medial prefrontal cortex (mPFC), where extinction of fear and contextual memory are elaborated (37, 38). Akt2 WT, HET and KO mice were tested for contextual (Figure 1E) and extinction fear memory (Figure 1F) using classic Pavlovian paradigms. During contextual fear conditioning, there was a main effect of genotype (MANOVA: p = 0.016), which was significant in both the post-training (F(2,22) = 4.912, p = 0.017) and in the context condition (F(2,22) = 8.639, p = 0.002). At baseline freezing was equal to zero in all groups while at post-training a robust freezing response was observed in WT mice but a much lower extent of freezing was seen in mutant mice; post-hoc analyses showed a significant reduction of freezing in Akt2 HET mice, in Akt2 KO mice the same trend of reduction in freezing was observed (Bonferroni post-hoc: HET vs WT p = 0.025, KO vs WT p = 0.09). Re-exposure to the same context after 24 hrs, without cue, elicited a similar pattern of % freezing to that observed during the post-training with a significant reduction of freezing in Akt2 HET mice and a marginal significance in Akt2 KO mice compared to WT (Bonferroni post-hoc: HET vs WT p = 0.002, KO vs WT p = 0.057). Our data indicate that Akt2 HET and KO mice fail to associate the auditory cue and the context with the aversive event (foot shock), suggesting that Akt2 is essential for encoding of associative learning and acquisition memory.
In the fear extinction paradigm (Figure 1F) all three genotypes responded with substantial freezing to tone+shock parings during training (RM ANOVA: effect of stimulus F(9,216) = 46.387, p ≤ 0.001; tone×genotype F(18,216) = 0.336, p = 0.995) with no effect of genotype (F(2,24) = 0.355, p = 0.705). However, during the extinction phases (days 2 and 3), Akt2 HET and KO mice showed a stable % freezing without any extinction trend as indicated by a significant tone by genotype statistical interaction (day 2 RM ANOVA: within-subject effect of tone F(5,120) = 3.674, p = 0.004, tone×genotype F(10,120) = 3.434, p = 0.001; day 3 RM ANOVA: within-subject effect of tone F(5,120) = 1.761, p = 0.126, tone×genotype F(10,120) = 2.276, p = 0.018). Together, these data demonstrate that Akt2 is critical for associative learning, memory, and extinction, demonstrating that even a partial loss of Akt2 expression leads to a malfunctioning of the neural circuity between the amygdala, hippocampus and mPFC.
AKT2 mutant mice demonstrate altered PFC- and hippocampus-dependent memory tasks
To further probe the role of Akt2 signaling in the neural circuitry involving the mPFC and hippocampus we tested separate naïve cohorts of WT, HET and KO Akt2 mice in a series of three recognition memory tasks differentially associated with function of the mPFC, perirhinal cortex and hippocampus (31, 39). Recognition memory requires accurate identification of the previous occurrence of stimuli made on the basis of the relative familiarity of objects, or by integrating information concerning object location, or by using recency discrimination (31, 39–40). In the temporal order discrimination test, which requires intact functioning of the mPFC, perirhinal cortex and hippocampus, Akt2 HET and Akt2 KO mice failed to discriminate between the least recent and the most recent object (one-way ANOVA: F(2,26) = 11.667, p ≤ 0.0001; Bonferroni post-hoc p = 0.002 HET vs WT, p = 0.001 KO vs WT; Figure 2A) indicating that Akt2 is critical for recency discrimination memory and development of the mPFC, perirhinal cortex and/or hippocampus. The novel object preference task, which requires intact recruitment of the perirhinal cortex, was unimpaired in both Akt2 HET and KO mice with no effect of genotype being observed in the performance of this task (one-way ANOVA: F(2,24) = 1.095, p = 0.351; Figure 2C). Conversely, Akt2 genotype significantly impacted performance in the object location recognition test (one-way ANOVA: F(2,22) = 3.481, p = 0.049; Figure 2E), post-hoc analysis revealed that Akt2 KO (but not Akt2 HET) displayed impaired task performance dependent upon intact hippocampal functioning (Bonferroni post-hoc p = 0.049 KO vs WT). Importantly, the overall extent of exploration (object exploration time) did not differ between genotypes in any of the tasks denoting normal exploration (Figures 2B, D, F). Together, these data demonstrate that cognitive function dependent on intact PFC-hippocampal neural circuitry requires intact Akt2 signaling. Our data also support that the extent of perturbed circuit function is dependent upon the magnitude of Akt2 impairment, with HET mice displaying selective impairment in mPFC function, whereas complete knockout of Akt2 leads to impairments of both mPFC and hippocampal function.
Figure 2. Akt2 genetically modified mice display impaired recency discrimination memory and object location memory, but intact novel object memory.
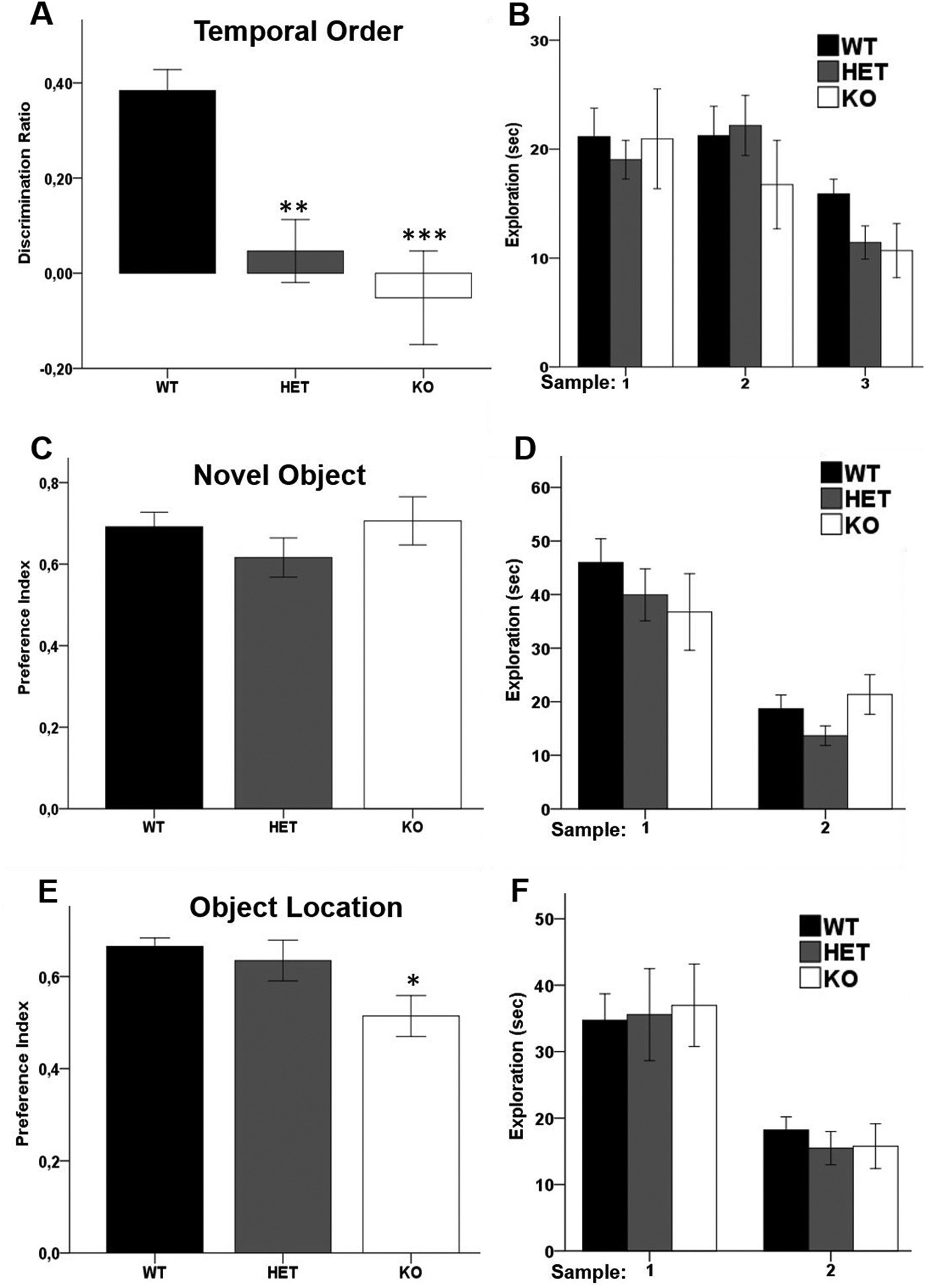
(A–B) Temporal order recognition test of Akt2 HET and KO mice and their WT littermates. (A) Recency discrimination ratio of time spent exploring the more recent object compared to a less recent. The ratio is lower in HET (p = 0.002) and KO mice (p = 0.001). (B) Overall exploration time of both objects during samples 1, 2 and 3. N = 11 WT, 12 HET, 6 KO. (C–D) Novel object recognition test of Akt2 HET and KO mice and their WT littermates. (C) Preference index for time spent exploring the novel object compared to a familiar one. (D) Overall exploration time of the objects during samples 1 and 2. N = 10 WT, 11 HET, 6 KO. (E–F) Object location recognition of Akt2 HET and KO mice and their WT littermates. (E) Preference index for the time spent exploring the misplaced object compared to object in its original location. The index is significantly lower in KO mice (p = 0.049 KO vs WT). (F) Overall exploration time of the objects during samples 1 and 2. N = 10 WT, 10 HET, 5 KO. Data represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Akt2 HET and KO mice display impaired social interaction
To assess the impact of Akt2 deficiency on social cognitive domains we tested Akt2 WT, HET and KO mice in rodent tasks of social behavior (41, 42). Using a three-chamber apparatus, murine social preference is measured as the predisposition to spend more time in the chamber containing a stranger conspecific (i.e. novel mouse 1) than the chamber containing an inanimate object (i.e. novel object). In the sociability test a significant main effect of chamber was observed for sniff time (RM ANOVA F(1,24) = 174.044, p ≤ 0.0001) and time in chambers (RM ANOVA F(1,24) = 8.919, p = 0.006). An interaction of genotype by chamber was observed for time in chamber (RM ANOVA: genotype×chamber F(2,24) = 4.359, p = 0.024), whereby Akt2 HET (p = 0.005) and WT (p = 0.003) mice, but not AKT2 KO mice, demonstrated a preference for the chamber containing the novel mouse 1 over the chamber containing the novel object (Figure 3A). These findings demonstrate that Akt2 KO mice show impaired social preference and fail to discriminate between a novel mouse and novel object. In all three genotypes, sniff time of the novel object was lower than that of novel mouse 1 (Akt2 WT p ≤ 0.0001, HET p ≤ 0.0001, and KO p = 0.006) (Figure 3B).
Figure 3. Abnormal sociability and social novelty preference in Akt2 HET and KO mice.
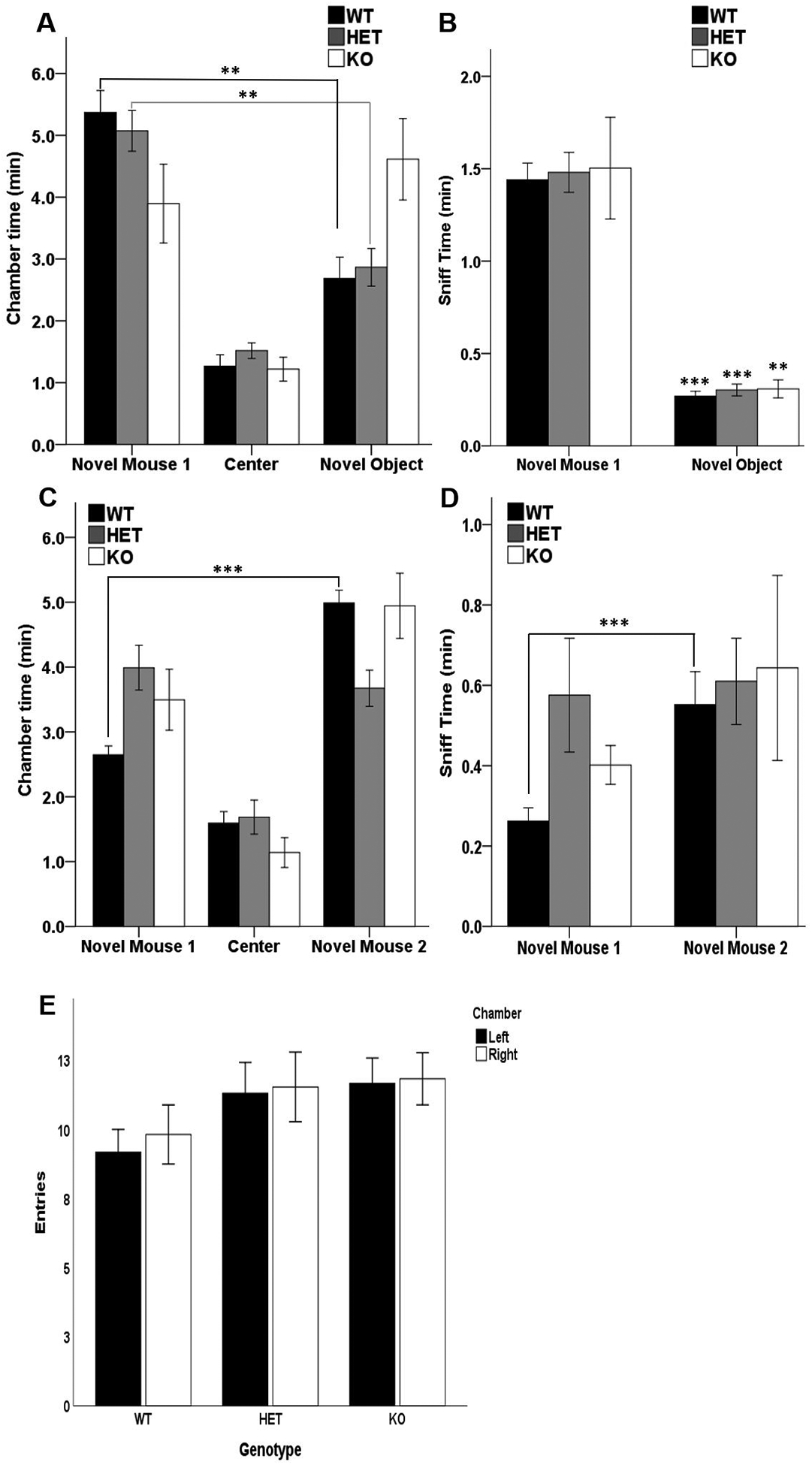
(A–B) 10 minutes social preference test of Akt2 HET and KO mice and their WT littermates. (A) Time spent in the chambers containing the novel mouse 1 or the novel object, and in the empty center chamber. WT (**p = 0.003) and HET (**p = 0.005) mice spend more time in the novel mouse 1 chamber compared to novel object chamber. (B) Time spent sniffing the novel mouse 1 and the novel object. Sniff time of the novel object is lower than that of novel mouse 1 in all genotypes (WT and HET ***p ≤ 0.0001, KO**p = 0.006). (C–D) 10 minutes social novelty preference test of Akt2 HET and KO mice and their WT littermates. (C) Time spent in the chambers containing the novel mouse 1 or the novel mouse 2, and in the empty center chamber. WT mice spend more time in the novel mouse 2 chamber compared to the novel mouse 1 chamber (***p ≤ 0.001). (D) Time spent sniffing the novel mouse 1 and the novel mouse 2. Sniff time of novel mouse 2 was higher in WT mice (**p ≤ 0.01). (E) Number of entries in left and right chambers during the initial 10-minute habituation period. N = 10 WT, 11 HET, 6 KO. Data represent mean ± SEM.
Preference for social novelty is defined as the propensity to spend more time in the chamber containing a stranger conspecific (novel mouse 2) compared to the chamber containing a familiar conspecific (novel mouse 1). A significant effect of chamber was observed on the time spent in chamber (RM ANOVA: F(1,24) = 11.794, p = 0.002). Moreover, an interaction of genotype by chamber was found (RM ANOVA: genotype×chamber F(2,24) = 6.671, p = 0.005), whereby WT (p ≤ 0.001), but not Akt2 HET or KO mice, demonstrated a preference for the chamber containing the novel mouse 2 rather than the chamber containing the novel mouse 1 (Figure 3C). These data demonstrate deficits of social novelty in both Akt2 Het and KO mice.
A significant effect of chamber was observed on sniff time in the preference for social novelty test (RM ANOVA: F(1,24) = 5.510, p = 0.027). No genotype × chamber interaction (p = 0.332) or genotype effect (p = 0.341) was observed. However, Akt2 WT mice, but not Akt2 HET and KO mice, spent more time sniffing novel mouse 2 than novel mouse 1 (p = 0.005).
Together, these results indicate a selective dose-dependent effect of Akt2 deletion on sociability and social preference, with KO mice exhibiting a pronounced deficit in sociability and social novelty, while HET mice show a selective deficit in social preference. Importantly, we did not observe any effects of genotype on entries into the left or right chambers during the habituation phase of the task (Figure 3E).
Hippocampal long-term potentiation (LTP) is impaired in AKT2 KO mice
As a neural substrate underlying the observed memory impairments dependent on hippocampal function, we investigated whether reduced Akt2 expression alters synaptic plasticity using field recordings of excitatory postsynaptic potentials (fEPSPs) and measurements of long-term potentiation (LTP). Extracellular field recording to measure LTP was conducted in the CA3-CA1 synapses of acute hippocampal slices from 8–10-week-old mice (Figure 4A and B). A main effect of genotype was observed on LTP (ANOVA, F(2,30) = 3.479, p = 0.044), whereby Akt2 KO mice displayed significantly reduced LTP compared to WT mice (Bonferroni post-hoc, p = 0.041). Specifically, LTP was only induced at 118±7% potentiation at 50–60 min in Akt2 KO mice, whereas LTP was induced at 142±11% potentiation in Akt2 HET and 153±8% in WT mice. Paired pulse facilitations (PPFs; Figure 4C) were virtually identical over a wide range of interpulse intervals in all genotypes and basal transmission in hippocampal CA3-CA1 synapses (Figure 4D) was not influenced by genotype. These data demonstrate that Akt2 KO mice exhibit impaired LTP in the absence of changes in paired pulse facilitations, indicating a postsynaptic, rather than presynaptic mechanism (43) of hippocampal circuit dysfunction.
Figure 4. Significant impairment of hippocampal LTP and decreased mPFC pyramidal neuron excitability in Akt2 KO mice.
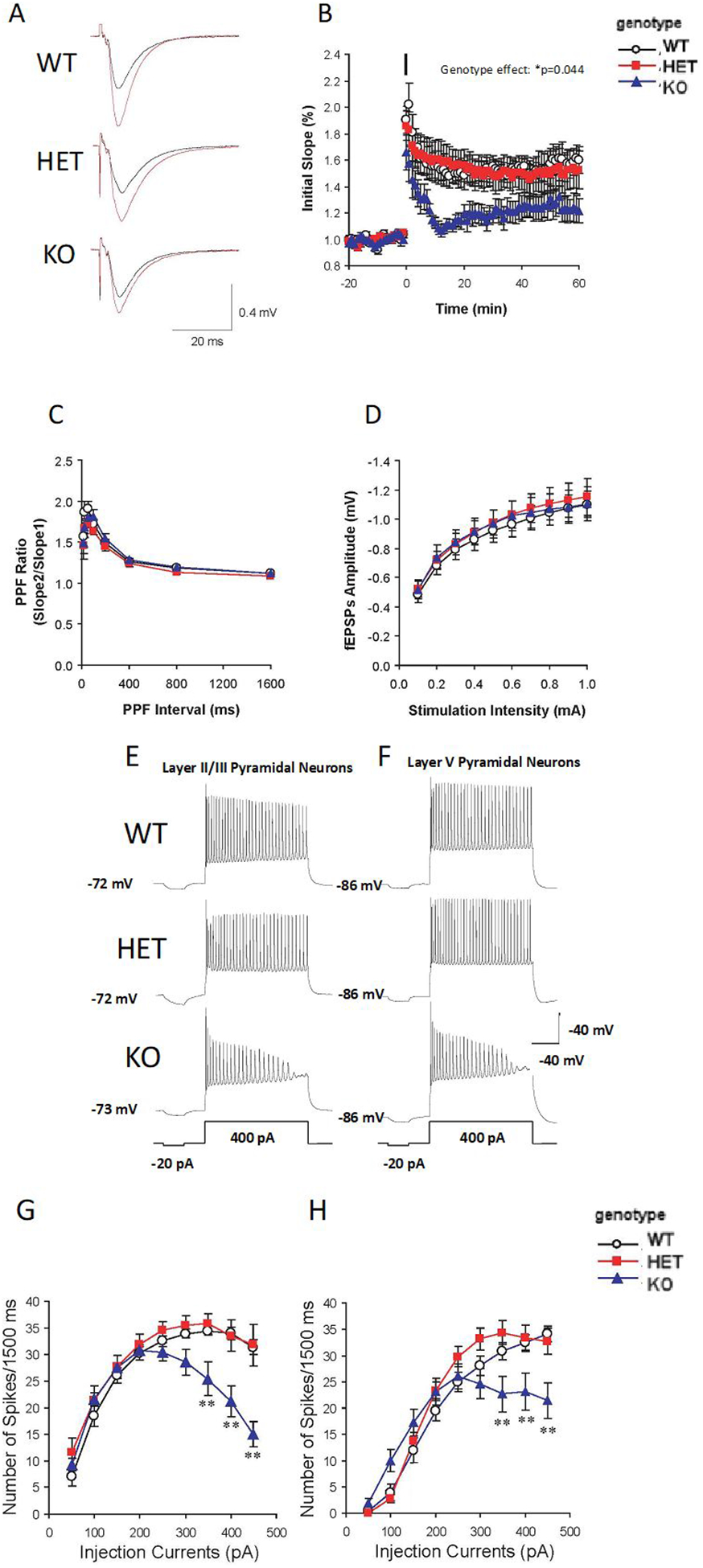
(A) Representative fEPSP recordings before (black lines) and 60 min after LTP induction (red lines) in hippocampal slices of Akt2 HET and KO mice and their WT littermates. (B) Complete time courses of fEPSP in hippocampus: the black bar represents the injection time point of 4 TBS at 100 Hz to induce LTP. fEPSP is reduced in KO mice compared to WT (KO vs WT: p = 0.044). N = 4 WT (8 slices), 4 HET (9 slices), 4 KO (8 slices). (C) PPF ratio in hippocampus. The ratios of the second and first EPSP slopes were calculated, and mean values are plotted against different inter-pulse intervals (IPI, 12.5 to 1600 ms). N = 3 WT (8 slices), 3 HET (8 slices), 3 KO (8 slices). (D) Basal synaptic transmission in hippocampus. Input-output curves were generated by plotting the postsynaptic response (initial slope of fEPSP amplitude, mV) as a function of the stimulation intensity. N = 4 WT (12 slices), 4 HET (10 slices), 4 KO (14 slices). (E) Representative traces of pyramidal neuron excitability in mPFC lamina II/III of Akt2 HET and KO and their WT littermates mice. (F) Representative traces of pyramidal neuron excitability in lamina V of mPFC of Akt2 HET and KO mice. (G) Neuronal firing frequency (spikes) of mPFC lamina II/III. Firing frequency is significantly lower in Akt2 KO mice compared mice when injection current intensities were from 350 pA to 450 pA (p value, from <0.0001 to 0.0025). N = 3 WT (7 slices, 11cells), 3 HET (6 slices, 9 cells), 3 KO (9 slices, 21 cells). (H) Neuronal firing frequency (spikes) of mPFC lamina V. Firing frequency is significantly lower in Akt2 KO mice when injection current intensities were from 350 pA to 450 pA (p value, from <0.0006 to 0.05), respectively. No difference was observed in mPFC lamina V pyramidal neuron excitability between HET and WT mice. N = 3 WT (7 slices, 12 cells), 3 HET (8 slices, 15 cells), 3 KO (9 slices, 21 cells). Data represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01.
Reduced intrinsic excitability of mPFC pyramidal neurons of laminas II/III and V in Akt2 KO mice
To study the physiological ramifications of Akt2 deficiency in the prefrontal cortex, we assayed intrinsic excitability in the form of repetitive firing patterns using whole-cell current clamp recording in pyramidal neurons of lamina II/III and V of the mPFC slices from adult mice (Figure 4E–H). A two-way ANOVA analysis revealed a significant interaction between genotype and stimulation intensity on the excitability of lamina II/III pyramidal neurons (F(16,342) = 3.792, p < 0.0001). Neuronal firing frequencies were significantly lower in Akt2 KO mice when injection current intensities ranged from 300 pA to 450 pA (p value range, from < 0.0001 to 0.0025, Figure 4G). No difference was observed in mPFC lamina II/III pyramidal neuron excitability in Akt2 HET mice (p = 0.409, Figure 4G). Next, we measured firing frequencies of mPFC lamina V pyramidal neurons. Consistently, a significant interaction between genotype and stimulation intensity on neuronal firing frequency was observed (two-way ANOVA analysis F(16,405) = 3.436, p < 0.0001). Again, neuronal firing frequencies in Akt2 KO mice were significantly lower when injection current intensities were from 350 pA to 450 pA (p value, from 0.024 to 0.001; Figure 4H), respectively. No difference was observed in mPFC lamina V pyramidal neuron in Akt2 HET mice (p = 0.343, Figure 4H). The resting membrane potential did not differ between genotypes in either lamina II/III or lamina V neurons (Figure 4 E, F). Together, these data indicate that Akt2 is critical for normal membrane potential, mediated through potential alterations in the biophysical properties of voltage gated ion channels, leading to reduced firing rates and reduced excitability of pyramidal cells within layers II/III and V of the mPFC.
Akt2 deficiency causes abnormal excitatory and inhibitory transmission in mPFC pyramidal neurons
To investigate excitatory and inhibitory pre- and post-synaptic transmission in prefrontal cortical pyramidal neurons, whole-cell voltage-clamp recording was performed in mPFC slice preparations of 8–10-week-old mice (Supplemental Figure 1). In Akt2 KO mice, lamina V pyramidal neurons exhibited significantly higher mEPSCs amplitude compared to WT (KO −7.21±0.2 pA vs. WT −5.71±0.17 pA; one-way ANOVA: F(2,45) = 5.890, p = 0.005; Bonferroni post-hoc: p = 0.006 KO vs WT, Supplemental Figure 1D) and faster mEPSCs rise time (KO 1.85±0.06 ms vs. WT 2.25±0.05 ms; one-way ANOVA: F(2,45) = 11.863, p ≤ 0.001; Bonferroni post-hoc: p ≤ 0.001 KO vs WT; Supplemental Figure 1E). No deficits in mEPSC frequency (Supplemental Figure 1C) or decay time (Supplemental Figure 1F), were observed. Together, these data demonstrate that Akt2 is critical for normative cortical lamina V pyramidal excitatory transmission and indicate altered density, conductance, or kinetics of postsynaptic AMPA-type ionotropic glutamate receptors (44, 45) rather than presynaptic release probability or excitatory synaptic number. Interestingly, whole-cell voltage-clamp recording in lamina II/III mPFC slices revealed no abnormalities of mEPSC frequency, amplitude or kinetics in Akt2 KO mice, suggesting that at least in the context of complete loss of function of Akt2, glutamatergic neurotransmission abnormalities appear laminar-specific.
In lamina V, increased frequency of miniature inhibitory postsynaptic currents (mIPSC) was observed in Akt2 KO mice (KO 7.01±0.31 Hz vs. WT 5.60±0.31; one-way ANOVA: F(2,41) = 21.637 p ≤ 0.001; Bonferroni post hoc: p = 0.001 KO vs WT; Supplemental Figure 1I). No differences were observed in mIPSC amplitude (Supplemental Figure 1J), indicating that Akt2 directly or indirectly modulates presynaptic GABAergic terminal release machinery to enhance inhibitory synaptic strength onto excitatory pyramidal neurons. Interestingly, mIPSCs event kinetics in lamina V pyramidal neurons of Akt2 KO mice also exhibited faster rise time (KO 2.82±0.06 ms vs. WT 3.42±1.92 ms; one-way ANOVA: F(2,41) = 4.147, p = 0.023; Bonferroni post-hoc: *p ≤ 0.02 KO vs WT; Supplemental Figure 1K), additionally implicating altered gating properties of postsynaptic GABAA receptor channels. Overall, these data suggest that Akt2 KO mice show altered inhibitory and excitatory synaptic transmission in pyramidal neurons within lamina V in mPFC, with complex pre- and post-synaptic mechanisms.
Interestingly, a more moderate and somewhat contrasting pattern of altered neurotransmission was observed in the context of Akt2 heterozygosity, with increased mEPSCs frequency in lamina II/III pyramidal neurons (HET 5.77±0.60 Hz vs. 2.87±0.22 Hz WT); one-way ANOVA: F(2,43) = 19.540, p ≤ 0.001; Bonferroni post-hoc: p ≤ 0.001 HET vs WT; Supplemental Figure 1C) and decreased frequency of mIPSCs in lamina V pyramidal neurons (HET 4.32±0.33 Hz vs. WT 5.60±0.31; oneway ANOVA: F(2,41) = 21.637 p ≤ 0.001; Bonferroni post hoc: p = 0.005 HET vs WT; Supplemental Figure 1I) being observed.
Pharmacological inhibition of p110δ improves recency discrimination deficits in Akt2 HET and KO mice
To determine whether modulation of AKT activity, in the context of Akt2 deficiency could rescue cognitive impairments in Akt2 HET and KO mice, we evaluated the generality of systemic effects of pharmacological inhibition of p110δ with the small molecule inhibitor, IC87114 (30,31), in a separate naïve cohort of mice. Preclinical administration of IC87114 increases cortical Akt phosphorylation and has shown antipsychotic and pro-cognitive efficacy in several rodent models of psychiatric relevance (30, 31). Here we chose to assess the efficacy of IC87114 in the temporal order recognition task as both Akt2 HET and KO animals display prominent impairments in this task, and prior work demonstrated preclinical efficacy of IC87114 to rescue such deficits in other rodent models associated with altered Akt signaling (31). Firstly, as reported in Figure 2 in a separate, replication cohort of mice, Akt2 HET and KO mice consistently showed an allele-dependent deficit in recency discrimination memory (main effect of genotype, F(5,49) = 6.524, p < 0.01). IC87114 treatment rescued deficits seen in Akt2 HET and KO mice with discrimination ratios comparable to IC87114 treated WT mice (treatment × genotype interaction F(2,49) = 3.418, p < 0.05; Figure 5A). Although discrimination ratios in treated WT mice appeared to be lower than untreated WT mice, no main effect of treatment was observed. These data suggest that cognitive deficits observed in the context of Akt2 loss-of-function are amenable to pharmacological intervention, via novel a therapy aimed at modulating PI3K/AKT signaling (30, 31).
Figure 5. Preclinical relevance of PIK3CD signaling and pharmacological inhibition in Akt2 mice and evidence that Akt2 regulates mTOR signaling.
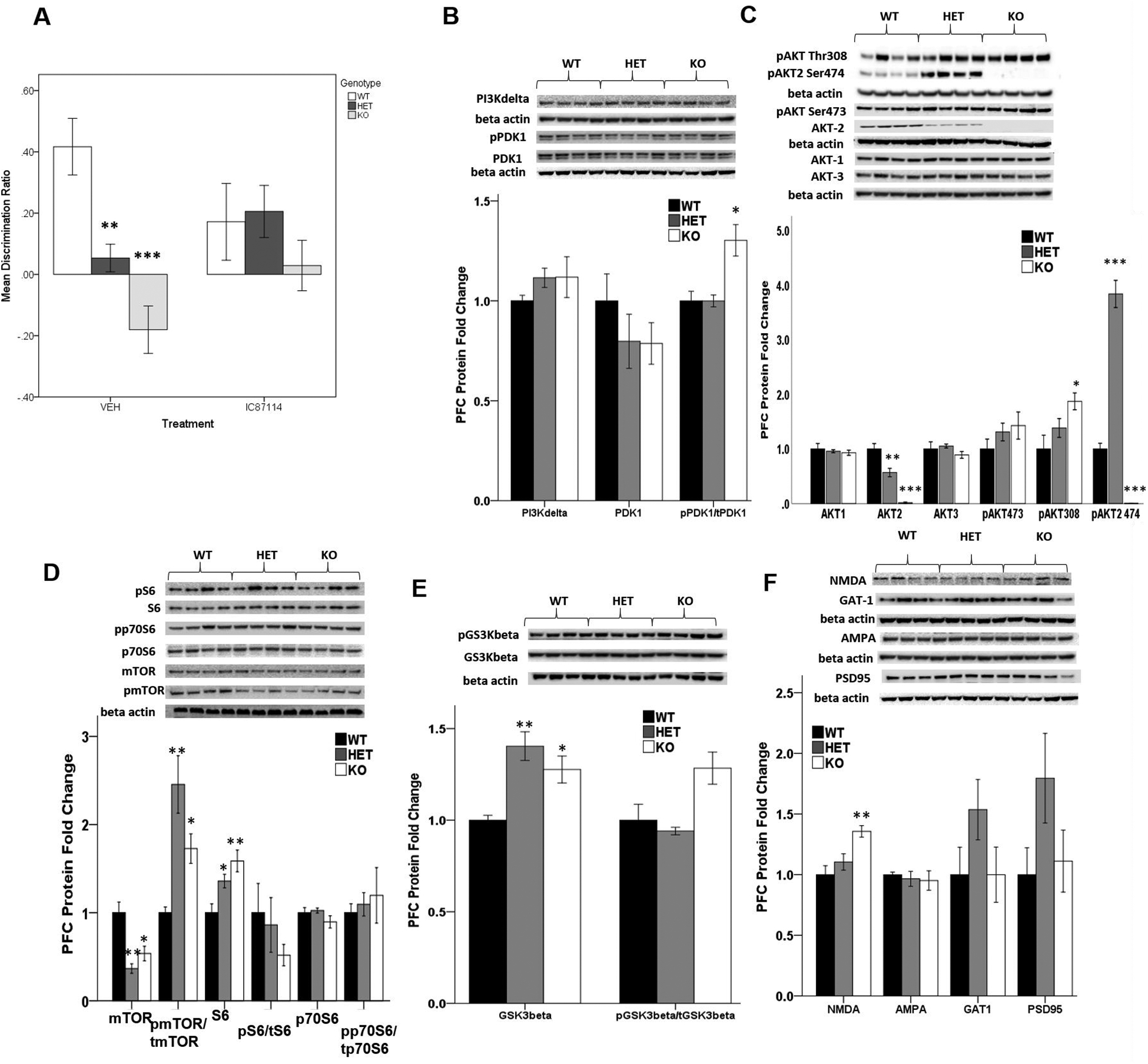
(A) Discrimination ratio in Akt2 WT, HET and KO mice in the temporal order object recognition task. Mice received a single intraperitoneal injection of 0.1mg/kg IC87114 or saline (vehicle) 30 minutes prior to the test phase. (WT/VEH n=7, WT/IC87114 n=9, HET/VEH n=13, HET/IC87114 n=14, KO/VEH n=6, KO/IC87114 n=6). (B–F) Relative expression of select signaling proteins in the AKT pathway. N = 4/genotype. Data represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.0001.
Proteomic changes in mPFC of Akt2 HET and KO mice
We next assessed a panel of select synaptic and intracellular proteins associated with PI3K/AKT/mTOR signaling in mPFC lysates of Akt2 mice. Notably, mTOR total protein was dramatically reduced in Akt2 HET (Students T-test t = 4.77; p = 0.003) and KO mice (Students T-test t = 3.15; p = 0.02; Figure 5D). Akt2 loss (KO) was also associated with increases in the activity of PDK1, as assessed by levels of phosphorylation of Ser-241, (Students T-test t = −3.28; p = 0.017; Figure 5B), implicating loss of feedback inhibition from mTORC1. Consistently, we observed significant increases in the total phospho-AKT-Thr308 in Akt2 KO mice (Students T-test t = −2.968; p = 0.025; Figure 5C). Since PDK1 is responsible for the phosphorylation of AKT at Thr308, these data demonstrate that Akt2 deficiency results in reduced mTOR levels and compensatory over-activation of remaining AKT signaling, potentially through loss of feedback inhibition to PDK1. The observation that phosphorylation levels mTOR were overall significantly increased in the context of Akt2 deficiency (WT vs HET, Students T-test t = −4.37; p = 0.005; WT vs KO, Students T-test t = 4.05; p = 0.007; Figure 5D) further supports this hypothesis. Consistently, mTOR activity is a key regulator of translation and biosynthesis of the ribosomal subunit protein S6 whose total levels were increased (WT vs HET, Students T-test t = −2.85; p = 0.029; WT vs KO; Students T-test t = 3.71; p = 0.01; Figure 5D). Activated Akt phosphorylates numerous downstream substrates, including glycogen synthase kinase-3 (GSK3β), which regulates glycogen and glucose metabolism. Notably, levels of total GSK3β were upregulated in Akt2 HET (Students T-test t = −4.88; p = 0.003) and KO mice (Students T-test t = −3.54; p = 0.012; Figure 5E).
Total protein levels of Akt1 and Akt3 were not changed in the context of Akt2 reduction, suggesting that haplo- or null- insufficiency of Akt2 does not result in compensatory expression changes in other Akt isoforms in the brain (Figure 5C). As expected, Akt2 protein was not detected in Akt2 KO mice (Students T-test t = 10.1; p < 0.0001; Figure 5C) and its level was approximately 50% in Akt2 HET mice (Students T-test t = 3.50; p = 0.013; Figure 5C). Consistently phospho-AKT2-Ser474, the specific phosphorylation site at the Serine residue for Akt2, was absent in Akt2 KO mice, however was significantly elevated in Akt2 HET mice (WT vs HET, Students T-test t = −10.548; p < 0.001 Figure 5C).
Akt2 loss-of-function impacts hippocampal neuronal morphology and dendritic anatomy
Finally, to assess the impact of genetic insufficiency of the Akt2 gene on neuronal development at the morphological level, we examined ex vivo primary hippocampal neuronal cultures from Embryonic day 18 (E18) WT, Akt2 HET and Akt2 KO embryos. At day in vitro (DIV) 3 hippocampal neuron axon length was significantly affected by Akt2 genotype (one-way ANOVA: F(2,387) = 3.946, p < 0.05), with both Akt2 HET and KO neurons having significantly shorter axons (Bonferroni post-hoc: WT vs HET, p < 0.05, WT vs KO, p < 0.01, Supplemental Figure 2A). Conversely, soma size was increased in the context of Akt2 insufficiency, this was selectively seen in neurons derived from Akt2 KO mice (one-way ANOVA: F(2,387) = p < 0.05), Bonferroni post hoc: WT vs KO, p < 0.01; Supplemental Figure 2B). Hippocampal neuron viability was unaffected by Akt2 genotype, as assessed by the percentage of calcein-AM to propidium iodide positive neurons reflective of the ratio of viable/apoptic neurons at DIV4 (Supplemental Figure 2C). Upon longer culturing conditions to assess dendritic maturation, at DIV10 hippocampal neurons derived from Akt2 HET and KO embryos had significantly altered dendritic patterning as assessed by sholl analysis. While there was the expected significant reduction in dendritic intersections as distance from soma increased (RM ANOVA: Distance F(37) = 202.191, p < 0.001), both Akt2 HET and KO neurons displayed a downwards shift in their slope resulting in a significant distance by genotype interaction (F(74) = 1.381, p = 0.017), and a main effect of genotype (F(2,238) = 4.525, p < 0.01). This simplification in dendritic maturation was observed in both heterozygous and Akt2 KO neurons (Bonferroni post hoc: WT vs KO, p < 0.01; WT vs HET, p < 0.05). This simplification was mainly evident at distances between 75–165μm from the soma (Supplemental Figure 2D). Since the structure of a neuron’s dendritic tree and axonal morphology defines how the cell receives and transmits synaptic inputs, these data provide anatomical evidence that hippocampal neuron impairment following Akt2 expression reduction may mechanistically underlie learning and memory dysfunction in Akt2 mice.
Discussion
The Akt signaling network is essential for normative brain development and alterations in the pathway are implicated in several neuropsychiatric and neurocognitive disorders, with Akt drawing attention as a potential novel target for antipsychotic, antidepressant and pro-cognitive drug development (30, 31, 46). Given that three Akt isoforms exist, all of which are highly expressed in the brain, a comprehensive understanding of the distinct physiological roles of each isoform in the context of brain development and function is necessary. While Akt1 and Akt3 have been extensively studied, little is known about the neurological functions of Akt2, with prior studies (47, 74) limited to Akt2 KO mice and examination of limited phenotypes. Together our findings demonstrate that Akt2 mice exhibit only a partial overlap in phenotypes with Akt1 and Akt3 mutant mice, providing further evidence for the non-redundant isoform-specific roles of Akt isoforms in the brain.
Consistent with a previous preliminary study (47), we demonstrate that Akt2 expression regulates anxiety-like behavioral phenotypes. We expand previous findings to demonstrate that heterozygosity of the Akt2 allele is sufficient to exert an anxiolytic phenotype, which is likely more clinically relevant to human studies of the involvement of the AKT2 locus in neurological disorders (32, 34). Anxiety-like features have not been observed in Akt3 (22), nor in Akt1 deficient mice (24, 28), suggesting that this affective domain may be selectively regulated by Akt2.
Like Akt1 deficient mice, we found that reductions in Akt2 attenuated response to amphetamine. A reciprocal regulation of AKT by dopamine type-2 receptors and serotonin has been proposed as a molecular mechanism of therapeutic efficacy of neuroleptic drugs (29). Our findings in combination with previous studies (25, 26) suggest that Akt1 and Akt2 regulate dopaminergic tone. We posit however, that Akt1 and Akt2 regulate dopaminergic neurotransmission via alternative mechanisms. Akt2 deficient mice develop hyper-insulinemia (17) and insulin growth factor (IGF) signaling in the brain has been shown to directly regulate the actions of dopamine via altering the expression of surface dopamine receptors (48). Furthermore, dopamine transporter expression is altered selectively by pharmacological Akt2 inhibition, but not by inhibition of Akt1 (49).
Heterozygous and homozygous deletion of Akt2 dramatically impaired recency discrimination memory but had no impact on novel object preference, suggestive of intact perirhinal- but impaired mPFC and hippocampal function (39). Additionally, Akt2 KO mice selectively demonstrated impaired object location recognition, an index of hippocampal dysfunction that appears to be unique to complete loss of Akt2 function. As described by rat micro-lesion studies, the combination of the three tests allows for the identification of specific brain region malfunctions (39, 40). These findings suggest that heterozygous deletion of Akt2 disrupts mPFC function, whereas complete knockout of Akt2 additionally impacts hippocampal physiology.
Interestingly, prior data show that Akt1 homozygous deletion does not affect the execution of a PFC-dependent working memory task (24). We have previously shown that Akt3 heterozygous and knockout mice also display recency discrimination and spatial location memory deficits (22), suggesting that Akt2 and Akt3 have overlapping physiological functions in these cognitive domains. In contrast, Akt3 genetic deletion mice do not display impaired associative learning and memory in Pavlovian fear-conditioning tasks (22). Given that contextual fear conditioning is commonly used to measure cortical/hippocampal communication with amygdala during emotion-associated learning (50), and that the processing of fear extinction involves the activation of the infralimbic PFC (51) these findings provide further evidence that Akt2 is critical for cortico-hippocampal circuit function.
Consistent with our neurobehavioral findings, observation that Akt2 heterozygous mice harbor a more selective prefrontal cortical deficit, whereas hippocampal function is additionally impacted in Akt2 KO; electrophysiological analyses revealed that both Akt2 KO and HET mice show alteration of inhibitory and excitatory neurotransmission in pyramidal neurons of mPFC, that serves to alter excitatory/inhibitory (E/I) balance. Additionally, Akt2 KO mice display reduced pyramidal neuron intrinsic cell excitability and hippocampal LTP. Since the pair-pulse facilitation ratio was normal, the LTP deficit is typically attributed to postsynaptic mechanisms (43). Postsynaptic currents are indicative of kinetics in AMPA or GABAA receptors, which mean the open and close speed in their receptors. Since some variation was observed between Akt2 HET and KO mice, and lack of changes in total levels of AMPA receptors were observed, this indicates that there may be a change in either 3-dimensional receptor structure or functionality. Similar to Akt2 KO mice, Akt1 KO mice showed reduced hippocampal LTP (27), whereas Akt3 KO mice display normal hippocampal LTP (52).
In addition to altered electrophysiological properties, we also found that hippocampal neuronal morphology and dendritic patterning was altered in Akt2 HET and KO mice. In keeping with a previous study (23) we report that reduced Akt2 expression leads to decreased axiogenesis, and expand on these findings to show that soma size is increased in Akt2 KO neurons, likely as a compensatory mechanism. We also found that neurons derived from Akt2 HET and KO mice display immature dendritic patterning, which may provide the cellular mechanism underlying impaired hippocampus-dependent cognitive behaviors. Impaired hippocampal and striatal neuronal dendritic complexity has also been observed in Akt1 KO mice (24,25). While it has been previously suggested that immunohistochemical localization of Akt2 is confined to astrocytes of the hippocampus (74), single cell RNA-seq transcriptomic studies in both human and mouse brain (while confirming higher expression in astrocytes), also confirm expression in neurons (75,76). The cellular dynamics and cell-type specific effects of how Akt2 reduction impacts neuronal development, therefore requires further investigation.
At the biochemical level, we demonstrate that expression of key members of the PI3K/AKT/mTOR pathway are altered in the context of Akt2 deficiency. Notably, both Akt2 HET and KO mice display dramatic (>50%) reductions in total mTOR. mTOR activity is a critical regulator of neurodevelopment and is disrupted in several neurological diseases, including autism, schizophrenia and intellectual disability (53, 54, 55). Studies of pharmacological blockade of mTOR signaling (via Rapamycin) consistently show that attenuation of mTORC1 impairs learning and memory, through inhibition of translation control, synaptic plasticity and blockade of LTP (56–59), suggesting that decreased expression of mTOR, mechanistically underlies observed learning and memory deficits in Akt2 mutant mice. In addition, the known roles of mTOR in regulating neuronal physiology and morphology (60–62) are mechanistically consistent with our observations of reduced axiogenesis and dendritic patterning. Furthermore, decreased intrinsic excitability of cortical pyramidal neurons in Akt2 mutant mice is biologically consistent with loss of mTOR function, and consistent with the converse knowledge that hyperactivation or mTOR pathogenically underlies epileptogenesis (61).
Signaling pathways are not linear but are tightly regulated by positive and negative feedback mechanisms (63). Studies of compensatory-activation of PI3K/AKT signaling in pre-clinical cancer models, mouse models and human cells have demonstrated that a prominent consequence of mTORC1 inhibition, is a dramatic increase in Akt phosphorylation at Thr308 by PDK1, due to the loss of feedback inhibition of the AKT pathway via failure of inhibition and degradation of the IRS-1 receptor (63–65). In this context our findings are consistent. Our observations of attenuated mTOR levels in the context of increased total pAKT308 and increased pmTOR demonstrate that loss of Akt2 specifically results in downregulation of mTOR protein which results in loss of normal feedback inhibition and subsequent overactivation of remaining AKT isotypes and deregulated AKT signaling. The mechanisms by which Akt2 activity influences proteostasis of mTOR require further investigation and studies of IRS signaling are currently underway. Finally, another key downstream target of AKT signaling altered in Akt2 deficient mice is GSK3β, which was increased in both genotypes. GSK3 signaling plays critical roles in neurodevelopment via regulation of glucose metabolism, insulin signaling and inflammatory mechanisms (66, 67) and is the target for the mood stabilizer lithium, the first line therapy for bipolar disorder. Lithium inhibits GSK3β activity, directly- and indirectly through modulation of AKT activity with consistent antidepressant and anxiolytic effects observed in rodent models of depression (67, 68). Our data suggest that increased GSK3β may represent a molecular mechanism underlying the anxiety and depressive (47) phenotypes observed in the context of Akt2 deficiency. Furthermore, emerging evidence suggests that impaired inhibitory regulation of GSK3β may contribute to social dysfunction (69), consistent with those observed in our model.
In summary, our convergent data demonstrate that Akt2 in the murine brain regulates learning and memory and mPFC function, in addition to depression and anxiety-like phenotypes (47). Akt2 loss-of-function produces cognitive deficits dependent upon cortico-hippocampal circuit function, which have partial overlap with previous observations in Akt1 and Akt3 genetic deletion mice and can be rescued by pharmacological modification of PI3K signaling. Moreover, we demonstrate that Akt2 is critical for physiological, morphological and biochemical properties of neurons that serve to alter the basic parameters of synaptic neurotransmission and circuit function. Together these data provide novel biological insight into the importance of Akt2 signaling in normative brain development, and in concert with future studies of Akt isoform expression in human postmortem brain, across neocortical development and aging, and in psychiatric disease, will serve to shed further light on the involvement of specific AKT isotypes in neurological syndromes such as autism, schizophrenia, and depression.
Supplementary Material
Acknowledgments:
We would like to thank Dr. Daniel Weinberger of the Lieber Institute for Brain Development and previously the NIMH Intramural Research Program, for additional resource support at the NIMH IRP. We thank Dr. Wenwei Huang, Dr. Craig Thomas and the National Center for Advancing Translational Sciences, National Institutes of Health for the synthesis of the IC87114 compound. All the work presented in this study was conducted at the NIMH Intramural Research Program, Bethesda, MD and the University of Colorado.
Funding:
This work was supported by the National Institutes of Mental Health under Award Number R01MH103716 (AJL), http://grantome.com/grant/NIH/R01-MH103716-05 and previously by funds from the National Institutes of Mental Health, Intramural Ressearch Program (AJL). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplementary information is available at MP’s website
Conflict of interests:
The authors declare that they have no conflicts of interest with the contents of this article.
References:
- 1.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275, 661–665 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. Journal of cellular and molecular medicine 9, 59–71 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones PF, Jakubowicz T, Hemmings BA. Molecular cloning of a second form of rac protein kinase. Cell regulation 2,1001–1009 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masure S, Haefner B, Wesselink JJ, Hoefnagel E, Mortier E, Verhasselt P, Tuytelaars A, Gordon R, Richardson A. Molecular cloning, expression and characterization of the human serine/threonine kinase Akt-3. European journal of biochemistry 265, 353–360 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Liao Y, Hung MC. Physiological regulation of Akt activity and stability. American journal of translational research 2, 19–42 (2010). [PMC free article] [PubMed] [Google Scholar]
- 6.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santi SA, Lee H. The Akt isoforms are present at distinct subcellular locations. American journal of physiology Cell physiology 298, C580–591 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Clark AR, Toker A. Signalling specificity in the Akt pathway in breast cancer. Biochemical Society transactions 42,1349–1355 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochemical Society transactions 35, 231–235 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Dummler B, Tschopp O, Hynx D, Yang ZZ, Dirnhofer S, Hemmings BA. Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Molecular and cellular biology 26, 8042–8051 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Pontén F. Proteomics. Tissue-based map of the human proteome. Science. 2015. January 23;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 12.Cohen MM Jr. The AKT genes and their roles in various disorders. American journal of medical genetics Part A 161A, 2931–2937 (2013). [DOI] [PubMed] [Google Scholar]
- 13.George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, Dunger DB, Barford D, Umpleby AM, Wareham NJ, Davies HA, Schafer AJ, Stoffel M, O’Rahilly S, Barroso I. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 304, 1325–1328 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boland E, Clayton-Smith J, Woo VG, McKee S, Manson FD, Medne L, Zackai E, Swanson EA, Fitzpatrick D, Millen KJ, Sherr EH, Dobyns WB, Black GC. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet 81, 292–303 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ballif BC et al. High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum Genet 131,145–156 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511,421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292,1728–1731 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, McNeish JD, Coleman KG. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. The Journal of clinical investigation 112,197–208 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, Hemmings BA. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. The Journal of biological chemistry 278, 32124–32131 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Molecular and cellular biology 25, 1869–1878 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, Michaelis T, Frahm J, Hemmings BA. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development 132, 2943–2954 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Howell KR, Floyd K, Law AJ. PKBgamma/AKT3 loss-of-function causes learning and memory deficits and deregulation of AKT/mTORC2 signaling: Relevance for schizophrenia. PloS one 12, e0175993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diez H, Garrido JJ, Wandosell F. Specific roles of Akt iso forms in apoptosis and axon growth regulation in neurons. PloS one 7, e32715 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai WS, Xu B, Westphal KG, Paterlini M, Olivier B, Pavlidis P, Karayiorgou M, Gogos JA. Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proceedings of the National Academy of Sciences of the United States of America 103, 16906–16911 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang CY, Chen YW, Wang TW, Lai WS. Akting up in the GABA hypothesis of schizophrenia: Akt1 deficiency modulates GABAergic functions and hippocampus-dependent functions. Sci Rep. 2016. September 12;6:33095. doi: 10.1038/srep33095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nature genetics 36, 131–137 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Balu DT, Carlson GC, Talbot K, Kazi H, Hill-Smith TE, Easton RM, Birnbaum MJ, Lucki I. Akt1 deficiency in schizophrenia and impairment of hippocampal plasticity and function. Hippocampus 22,230–240 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang CH, Pei JC, Luo DZ, Chen C, Chen YW, Lai WS. Investigation of gene effects and epistatic interactions between Akt1 and neuregulin 1 in the regulation of behavioral phenotypes and social functions in genetic mouse models of schizophrenia. Frontiers in behavioral neuroscience 8:455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beaulieu JM. A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. Journal of psychiatry & neuroscience : JPN 37, 7–16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Law AJ, Wang Y, Sei Y, O’Donnell P, Piantadosi P, Papaleo F, Straub RE, Huang W, Thomas CJ, Vakkalanka R, Besterman AD, Lipska BK, Hyde TM, Harrison PJ, Kleinman JE, Weinberger DR. Neuregulin 1-ErbB4-PI3K signaling in schizophrenia and phosphoinositide 3-kinase-p110delta inhibition as a potential therapeutic strategy. Proceedings of the National Academy of Sciences of the United States of America 109, 12165–12170 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papaleo F, Yang F, Paterson C, Palumbo S, Carr GV, Wang Y, Floyd K, Huang W, Thomas CJ, Chen J, Weinberger DR, Law AJ. Behavioral, Neurophysiological, and Synaptic Impairment in a Transgenic Neuregulin1 (NRG1-IV) Murine Schizophrenia Model. The Journal of neuroscience 36, 4859–4875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engeli L, Delahaye M, Borgwart S, Gallinat J, Muller D, Walter M et al. Akt2 gene is associated with anxiety and neuroticism in humans. Journal of vascular medicine and surgery 2(3), 141 (2014). [Google Scholar]
- 33.Henderson ND, Turri MG, DeFries JC, Flint J. QTL analysis of multiple behavioral measures of anxiety in mice. Behavior genetics 34, 267–293 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Thiselton DL, Vladimirov VI, Kuo PH, McClay J, Wormley B, Fanous A, O’Neill FA, Walsh D, Van den Oord EJ, Kendler KS, Riley BP. AKT1 is associated with schizophrenia across multiple symptom dimensions in the Irish study of high density schizophrenia families. Biological psychiatry 63, 449–457 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia BG, Wei Y, Moron JA, Lin RZ, Javitch JA, Galli A. Akt is essential for insulin modulation of amphetamine-induced human dopamine transporter cell-surface redistribution. Molecular pharmacology 68, 102–109 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Goosens KA, Maren S. Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learning & memory 8, 148–155 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quirk GJ, Likhtik E, Pelletier JG, Pare D. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. The Journal of neuroscience 23, 8800–8807 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kennedy PJ, Shapiro ML. Retrieving memories via internal context requires the hippocampus. The Journal of neuroscience 24, 6979–6985 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barker GR, Bird F, Alexander V, Warburton EC. Recognition memory for objects, place, and temporal order: a disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. The Journal of neuroscience 27, 2948–2957 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barker GR, Warburton EC. When is the hippocampus involved in recognition memory? The Journal of neuroscience 31, 10721–10731 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes, brain, and behavior 3, 287–302 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Paterson C, Law AJ. Transient overexposure of neuregulin 3 during early postnatal development impacts selective behaviors in adulthood. PloS one 9, e104172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. The Journal of neuroscience 14, 5325–5337 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alberto CO, Hirasawa M. AMPA receptor-mediated miniature EPSCs have heterogeneous time courses in orexin neurons. Biochemical and biophysical research communications 400, 707–712 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Queenan BN, Lee KJ, Pak DT. Wherefore art thou, homeo(stasis)? Functional diversity in homeostatic synaptic plasticity. Neural Plast 2012, 718203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol 49, 327–347 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Leibrock C, Ackermann TF, Hierlmeier M, Lang F, Borgwardt S, Lang UE. Akt2 deficiency is associated with anxiety and depressive behavior in mice. Cellular physiology and biochemistry 32, 766–777 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Williams JM, Owens WA, Turner GH, Saunders C, Dipace C, Blakely RD, France CP, Gore JC, Daws LC, Avison MJ, Galli A. Hypoinsulinemia regulates amphetamine-induced reverse transport of dopamine. PLoS biology 5, e274 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Speed NK, Matthies HJ, Kennedy JP, Vaughan RA, Javitch JA, Russo SJ, Lindsley CW, Niswender K, Galli A. Akt-dependent and isoform-specific regulation of dopamine transporter cell surface expression. ACS chemical neuroscience 1, 476–481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez RP, Biggs A, Chen G, Pine DS, Grillon C. Contextual fear conditioning in humans: cortical-hippocampal and amygdala contributions. The Journal of neuroscience 28, 6211–6219 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moustafa AA, Gilbertson MW, Orr SP, Herzallah MM, Servatius RJ, Myers CE. A model of amygdala-hippocampal-prefrontal interaction in fear conditioning and extinction in animals. Brain and cognition 81, 29–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergeron Y, Bureau G, Laurier-Laurin ME, Asselin E, Massicotte G, Cyr M. Genetic Deletion of Akt3 Induces an Endophenotype Reminiscent of Psychiatric Manifestations in Mice. Frontiers in molecular neuroscience 10, 102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gururajan A, van den Buuse M. Is the mTOR-signalling cascade disrupted in Schizophrenia? Journal of neurochemistry 129, 377–387 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Lipton JO, Sahin M. The neurology of mTOR. Neuron 84, 275–291 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lloyd BA, Hake HS, Ishiwata T, Farmer CE, Loetz EC, Fleshner M, Bland ST, Greenwood BN. Exercise increases mTOR signaling in brain regions involved in cognition and emotional behavior. Behav Brain Res 323, 56–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glover EM, Ressler KJ, Davis M. Differing effects of systemically administered rapamycin on consolidation and reconsolidation of context vs. cued fear memories. Learning & memory 17, 577–581 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 33, 67–75 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mac Callum PE, Hebert M, Adamec RE, Blundell J. Systemic inhibition of mTOR kinase via rapamycin disrupts consolidation and reconsolidation of auditory fear memory. Neurobiol Learn Mem 112,176–185 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Raab-Graham KF, Niere F. mTOR referees memory and disease through mRNA repression and competition. FEBS Lett 591, 1540–1554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 151,476–488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crino PB. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 17,734–742 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Weston MC, Chen H, Swann JW. Multiple roles for mammalian target of rapamycin signaling in both glutamatergic and GABAergic synaptic transmission. The Journal of neuroscience 32, 11441–11452 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther 13, 2477–2488 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer research 65,7052–7058 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Lane HA, Breuleux M. Optimal targeting of the mTORC1 kinase in human cancer. Curr Opin Cell Biol 21, 219–229 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. European journal of biochemistry 107, 519–527 (1980). [PubMed] [Google Scholar]
- 67.Freland L, Beaulieu JM. Inhibition of GSK3 by lithium, from single molecules to signaling networks. Frontiers in molecular neuroscience 5,14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochemical research 32,577–595 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PloS one 5, e9706 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lesuisse C, Martin LJ. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. Journal of neurobiology 51,9–23 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature Biotechnology 20,87–90 (2002). [DOI] [PubMed] [Google Scholar]
- 72.Ji Y, Yang F, Papaleo F, Wang HX, Gao WJ, Weinberger DR, Lu B. Role of dysbindin in dopamine receptor trafficking and cortical GABA function. Proceedings of the National Academy of Sciences of the United States of America 106, 19593–19598 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leavens KF, Easton RM, Shulman GI, Previs SF, Birnbaum MJ. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab 10,405–418 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Levenga J, Wong H, Milstead RA, Keller BN, LaPlante LE, Hoeffer CA. AKT isoforms have distinct hippocampal expression and roles in synaptic plasticity. Elife. 2017;6:e30640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex [published correction appears in J Neurosci. 2015 Jan 14;35(2):846–6]. J Neurosci. 2014;34(36):11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Y, Sloan SA, Clarke LE, et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 2016;89(1):37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.