

Abstract
Myelodysplastic syndrome (MDS) is an onco-hematologic disease with distinct levels of peripheral blood cytopenias, dysplasias in cell differentiation and various forms of chromosomal and cytogenomic alterations. In this study, the Chromosomal Microarray Analysis (CMA) was performed in patients with primary MDS without numerical and/or structural chromosomal alterations in karyotypes. A total of 17 patients was evaluated by GTG banding and eight patients showed no numerical and/or structural alterations. Then, the CMA was carried out and identified gains and losses CNVs and long continuous stretches of homozygosity (LCSHs). They were mapped on chromosomes 1, 2, 3, 4, 5, 6, 7, 9, 10, 12, 14, 16, 17, 18, 19, 20, 21, X, and Y. Ninety-one genes that have already been implicated in molecular pathways important for cell viability were selected and in-silico expression analyses demonstrated 28 genes differentially expressed in mesenchymal stromal cells of patients. Alterations in these genes may be related to the inactivation of suppressor genes or the activation of oncogenes contributing to the evolution and malignization of MDS. CMA provided additional information in patients without visible changes in the karyotype and our findings could contribute with additional information to improve the prognostic and personalized stratification for patients.
Subject terms: Biotechnology, Cancer, Genetics, Diseases, Medical research, Oncology
Introduction
Onco-hematologic diseases constitute a group of neoplasms with different degrees of bone marrow insufficiency, characterized by the expansion of cells with ineffective hematopoietic functions1,2. Myelodysplastic syndrome (MDS) comprises a heterogeneous subgroup of neoplasms of bone marrow clones, which present different levels of cytopenia in the peripheral blood, abnormal differentiation of the myeloid lineage, and dysplastic myeloid changes1,3. Chromosomal and/or genomic instability inherent in MDS is associated with an increased risk of unfavorable clinical evolution and the development of acute myeloid leukemia (AML), which can occur in 20 to 40% of primary MDS cases1,4.
The estimated population incidence rate of MDS is four patients per 100.000 individuals per year in the general population. With advancing age, the incidence of MDS significantly increases. For the age group ≥ 80 years, the incidence of MDS increases 12.5-fold5. The average age of onset is 60 years6 and only 10% of patients are under 50 years old5,7. Chromosomal changes are observed in approximately 50% of primary MDS and 80 to 90% of secondary neoplasms8–11. Karyotype data provides important variables for the diagnosis and prognosis of primary MDS, according to the consensus of the International Working Group for the Prognosis of MDS (IWG-PM) of the Myelodysplastic Syndrome Foundation, Inc. The MDS-Foundation proposed the International Prognostic Scoring System Revised (IPSS-R), in which cytogenetic abnormalities are used to predict the overall survival and risk of primary MDS transformation6.
The Chromosomal Microarray Analysis (CMA) based on single-nucleotide polymorphism (SNP) is a robust methodology for genomic prospection and, therefore, is useful for investigating cytogenomic rearrangements in MDS3,12. The use of CMA could improve MDS diagnosis, refine risk stratification, and provide additional genomic information that can be applied in patient handling and management13. The CMA allows for the accurate detection of copy-number variation (CNV) and long continuous stretches of homozygosity (LCSH) acquired in cell-clone neoplasms of MDS patients. This analysis is particularly important in cases where the karyotype was unable to identify chromosomal rearrangements or could not be performed on biological samples2,14–18.
Previous studies have implicated LCSHs in the genomic instability observed in patients with MDS19,20. LCSHs occur in about 50% of MDS patients with karyotypes without numerical and/or structural alterations. Therefore, genomic variations like LCSH play an important role in tumorigenesis and can alter gene expression21,22. CNVs and LCSHs demonstrate prognostic significance in MDS and AML21. Previous studies have shown that 74% of the patients evaluated by molecular or cytogenomic techniques present at least one oncogenic copy or pathogenic CNV related to the etiology and development of MDS. Changes in molecular pathways involving epigenetic regulation, chromatin modification, transcriptional regulation, signal transduction, apoptosis, RNA splicing, and cell cycle control in mesenchymal stromal cells in the medullary microenvironment may alter hematopoiesis in MDS10,23–26.
In the context previously described, the overall objective of the present study was to identify CNVs and LCSHs by applying the CMA using high-density SNP arrays in patients with primary MDS whose karyotypes presented without numerical and/or structural chromosomal alterations.
Materials and methods
Patients and biological materials
This study was conducted at the Replicon Research Center of the School of Agricultural and Biological Sciences of the Pontifical Catholic University of Goiás and in the Human Cytogenetics and Molecular Genetics Laboratory of the Secretary of Goias State for Public Health. The development of this research relied on collaboration with the South Texas Diabetes and Obesity Institute (STDOI), Department of Human Genetics, School of Medicine from the University of Texas Rio Grande Valley in the United States of America. For each of the 17 patients with MDS, the attending physician collected a total of 4 mL of bone marrow tissue. All methods were carried out in accordance with ethical and technical relevant guidelines and regulations, following both national and international consensuses. This study was approved by Human Research Ethics Committee of the Federal University of Goiás Clinics Hospital (HREC-FUG/HC), under protocol no. 1.621.064/2016. All research procedure was done in accordance with the Declaration Helsinki. All patients signed an informed consent document approved by the Human Research Ethics Committee.
Cytogenetic analysis
Karyotypes from metaphase with a 450-band resolution were obtained through cell culture in bone marrow tissue samples from patients with primary MDS. The chromosomes were banded by the GTG method (following standard procedures), modified, and optimized in accordance with previous protocols27. Short-term cultures (48 h) were established by incubating 1.0 mL of cell suspension in 5 mL RPMI1640 (Gibco Life Science, USA) supplemented with 1 mL of fetal bovine serum (Gibco Life Science, USA) and 100 μL L-glutamine (Gibco Life Science, USA) at 37 °C and in 5% CO2. To achieve metaphase, mitotic divisions were interrupted by adding 100 μL of colchicine solution (10 μg/mL) in water (Gibco Life Science, USA) to the culture system for 30 min prior to harvesting the cell suspension. Subsequently, the cells were incubated in a hypotonic solution of KCl (0.075 M) (Merck KGaA, Germany) for 30 min at 37 °C and in 5% CO2. Following hypotonization, the cells were fixed in methanol solution/glacial acetic acid (3:1), that was added by drip. The cells were mixed, centrifuged, and resuspended in fresh fixative solution. The fixation step was repeated three to four times. The fixed cells were dripped onto microscopy slides, subjected to GTG chromosome banding with the enzyme Trypsin (Invitrogen Life Technologies, USA), and stained in 4% Giemsa solution (Gibco Life Science, USA). Trypsin enzyme was diluted in phosphate-buffered saline (Invitrogen Life Technologies, USA). Twenty metaphases were analyzed per sample. The metaphase images were captured with the aid of an AxioImager 2 microscope (Carl Zeiss, Germany) connected to the blade-sweeping platform Metafer4 (MetaSystems Corporation, Germany). Chromosomal analyses were performed by the software IKAROS (Version 5.0, MetaSystems Corporation, Germany).
SNP array analysis
The platform GeneChip CytoScan HD array (Thermo Fisher Scientific, USA) was used for the identification of CNVs and LCSHs in the genome of 8/17 patients with primary MDS, who presented karyotypes without numerical or structural alterations. The genomic DNA of bone marrow tissue was extracted using the DNA GenomicPrep Mini Kit (GE Healthcare, USA) following the manufacturer’s instructions. A total of 250 ng of DNA from each sample was digested with the restriction enzyme NspI, following the manufacturer’s recommendations (Thermo Fisher Scientific, USA). Once digested, the samples were connected to the adapters, and then a universal primer was used to amplify the fragments using restriction fragment length polymorphism (RFLP) through polymerase chain reaction (PCR). PCR conditions were optimized to amplify fragments of sizes ranging from 150 to 2000 bp in length. The fragments were confirmed on 2% agarose gel in 1X TBE, subjected to a constant electric field of 10 V/cm for 1 h, and stained in an aqueous solution of ethidium bromide (5 mg/mL). The gel image was captured by the GelDoc XR + Imaging System (Bio-Rad Laboratories, USA).
The amplified products were purified with magnetic beads and quantified on the spectrophotometer NanoVue Plus (GE Healthcare, USA). Next, the amplified products were fragmented into 50- to 200-pb segments, purified, confirmed on a 4% agarose gel in TBE 1X, and separated in a constant electric field of 10 V/cm/h. The bands were revealed in an aqueous solution of ethidium bromide (5 mg/mL). The gel image was captured by the video documentation system GelDoc XR + Imaging System (Bio-Rad Laboratories, USA). DNA fragments were tagged with a terminal deoxynucleotidyl transferase and applied to the GeneChip CytoScan HD, followed by hybridization for 16–18 h at 50 ºC and 60 rpm in the GeneChip Hybridization Oven 645 (Thermo Fisher Scientific, USA). Subsequently, the chips were washed and stained in GeneChip Fluidic Station 450 (Thermo Fisher Scientific, USA) and scanned by a GeneChip Scanner 3000 7G (Thermo Fisher Scientific, USA) controlled by the Thermo Fisher Scientific GeneChip Command Console (AGCC, Version 3.2.2). The array was designed for cytogenomic diagnosis, with approximately a 2.7-million-marker copy number range, covering the entire human genome, including 743,304 SNP markers and > 1.9 million non-polymorphic markers. The CEL archives obtained by scanning each patient were compared with two databases: DGV (Database of Genomic Variants—http://projects.tcag.ca/variation) and CytoScanHD Array Database (provided by software ChAS). CNVs were also compared with the genetic diseases database DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources: https://decipher.sanger.ac.uk/application/). Additionally, 100% of the constitutional genes were covered by The International Standards for Cytogenomic Arrays Consortium (ISCA: https://www.iscaconsortium.org), including about 12,000 genes from OMIM and more than 36,000 genes from the RefSeq (Reference Sequence Database/NCBI).
In the ChAS, analysis parameters were fixed at 50 markers and a size of 100 kpb to detect gains and at 25 markers and a size of 100 kpb to detect losses. The filter was fixed to ≥ 3 Mb to detect the LCSHs. In the study, the parameters for CNV analysis followed both standard criteria based on manufacture’s recommendations and on the LCSHs analysis previously reported28,29. All analyzed chips met the quality control metrics, being MAPD and SNP-QC defined at ≤ 0.25 and ≥ 15, respectively. CNVs, LCSHs, and genes were identified, selected, and separated based on their association with molecular factors of MDS. The databases Online Mendelian Inheritance in Man (OMIM) and RefSeq were accessed by software R to support data mining, analysis, and interpretation of the cytogenomic information obtained by CMA.
Expression analysis through public databases
Expression analysis was performed on public databases of patients with primary MDS. An NCBI GEO dataset file (https://www.ncbi.nlm.nih.gov/geo/) was downloaded. The study (accession number GSE61853) was based on the analysis of gene expression in mesenchymal tissues of bone marrow stromal cells of patients with MDS and normal controls. The transcriptome was investigated with the aid of the platform Thermo Fisher Scientific Human Gene 1.0 ST Array (USA). Based on the global expression profile of bone marrow mesenchymal stem cells (BM MSCs), the top 250 genes were ranked based on P-value in the study GSE61853 where smallest P-value were the most significant. Afterwards, we compared the genes selected in the CNVs and LCSHs regions with the top 250 genes from the database. The logFC values were used to define which genes were considered to positively or negatively regulated in the individuals with MDS involved in this study.
Results
The average age of the 17 patients was 56 (26–79) years. The small sample group is a result of the reduced incidence of primary MDS in the population. Of the 17 cases, in 6/17, the results of karyotype with banding GTG demonstrated numerical or structural changes, being + 8, + 15, + 17, + 21, − X, t(4;11)(q21;q23) add(1)(q?), and polyploidy clones; in 3/17, the karyotype was not informative, given that there was no clonal expansion, so these cases were removed from subsequent analyses. In 8/17 patients, the karyotype showed no numerical and/or structural chromosomal changes, and their results were previously reported and were not included in the current analysis30. With assistance from CMA, 41 genomic variants were identified in 7/8 patients with no visible changes in karyotype, including 11/41 variations in the number of copies per gain, 2/41 regions with genomic loss, and 28/41 long continuous extensions of homozygosis, mapped on chromosomes 1, 2, 3, 4, 5, 6, 7, 9, 10, 12, 14, 16, 17, 18, 19, 20, 21, X, and Y (Figs. 1 and 2). Ninety-one genes that have already been implicated in relevant molecular pathways for cell viability were selected, including 9/91 gains and 4/91 losses, and 78/91 in regions with LCSHs.
Figure 1.
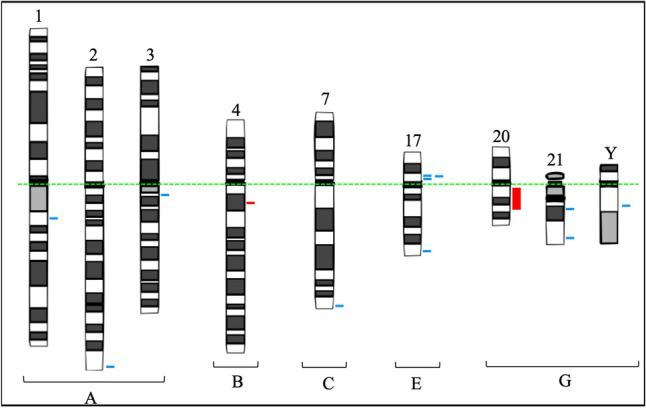
Ideograms of chromosomes following Denver’s group nomenclature summarizing the mapping of 13 unusual and clinically significant CNVs, identified by CMA in 7/8 MDS patients with karyotypes without numerical or structural chromosomal changes. Legend: Blue lines indicate CNVs with genomic gains, while red lines indicate CNVs of genomic loss. A dotted green line was added to indicate the alignment of chromosomes by centromeres.
Figure 2.
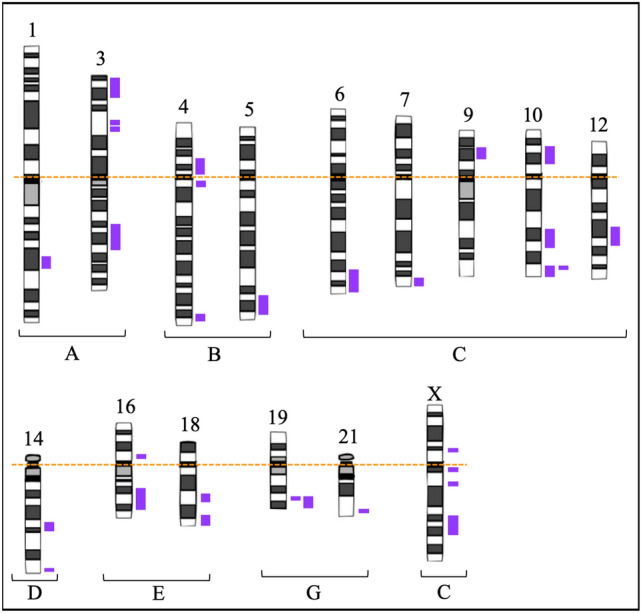
Ideogram of chromosomes following Denver’s group nomenclature summarizing the mapping of 28 unusual and clinically significant LCSHs, indicated by purple bars, identified by CMA in 7/8 patients with MDS and with karyotype without numerical and/or structural chromosomal changes. Legend: A dotted orange line was added to indicate the alignment of chromosomes by centromeres.
The cytogenetic risk of patients with karyotypes without numerical or structural alterations was rated as good, based on the International Prognostic Scoring System—Revised, while the risk of prognostic transformation of MDS ranged from very low to low. Only one patient died, while the others remained stable at the end of the study. Table 1 summarizes the results identified by the CMA and the risk estimates, according to IPSS-R, in addition to the clinical outcomes of these eight primary MDS patients.
Table 1.
Clinical features, cytogenetic and cytogenomic results, and risk estimates (according to the International Prognostic Scoring System—Revised) of a cohort of patients with myelodysplastic syndrome in addition to clinical outcomes.
| Case | Age (years) | Sex | Cytopenias | Blasts in bone marrow (%) | Karyotypic notation* | IPSS-R** cytogenetic risk | IPSS-R** score | IPSS-R** age-adjusted risk | Risk prediction | Genomic variation | Cytoband | Size (Mb) | Number of genes | Selected OMIN genes*** | Clinical outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ONCO002 | 52 | F | Thrombocytopenia | 2 | 46,XX20 | Good | 1 | 0.19 | Very low | LCSH | 3p26.1 | 16.9 | 125 | BHLHE40, LMCD1, RAD18, MTMR14, TADA3, JAGN1, RAF1 | Death |
| LCSH | 4q11 | 5.1 | 40 | *** | |||||||||||
| LCSH | 4p14 | 10.1 | 56 | KLHL5, UCHL1, TMEM33, OCIAD2 | |||||||||||
| LCSH | 10p13 | 9.1 | 47 | CUBN, HACD1, MIR511, MLLT10, BMI1, ARMC3 | |||||||||||
| LCSH | 12q21.31 | 8.7 | 41 | POC1B, BTG1 | |||||||||||
| LCSH | 16p11.2 | 4.5 | 65 | RNF40, KAT8 | |||||||||||
| Gain | 17p11.2 | 0.29 | 4 | MAP2K3 | |||||||||||
| ONCO003 | 49 | F | Leukopenia | 0 | 46,XX20 | Good | 1 | 0.05 | Very low | Gain | 1q21.3 | 0.11 | 5 | CHTOP, ILF2, NPR1, INTS3 | Stable |
| Gain | 3q12.2 | 0.1 | 2 | TFG | |||||||||||
| Loss | 4q13.2 | 0.12 | 0 | *** | |||||||||||
| LCSH | 1q31.1 | 6.88 | 16 | RGS1 | |||||||||||
| LCSH | 4q34.2 | 10.76 | 54 | KLKB1 | |||||||||||
| LCSH | 6q24.3 | 23.41 | 143 | LATS1, ULBP2, ULBP1, DYNLT1, RPS6KA2, AFDN, DLL1 | |||||||||||
| LCSH | 10q23.1 | 15.21 | 176 | SNORA12, HIF1AN | |||||||||||
| LCSH | 10q26.12 | 3.9 | 33 | C10orf88 | |||||||||||
| LCSH | 14q22.2 | 10 | 70 | BMP4, GMFB, PPM1A, PPP2R5E | |||||||||||
| LCSH | 18q22.3 | 9.21 | 38 | ZNF516 | |||||||||||
| LCSH | 19q13.32 | 13.64 | 602 | ERCC2, SIX5, SYMPK, STRN4, BBC3, BAX, GYS1, CD37, FLT3LG, BCL2L12, IL4I1, SIGLEC7, ZNF446 | |||||||||||
| LCSH | Xp11.23 | 5.38 | 108 | GATA1, PIM2, HUWE1 | |||||||||||
| LCSH | Xq13.1 | 5.65 | 41 | HDAC8 | |||||||||||
| LCSH | Xq23 | 14.29 | 85 | SEPT6 | |||||||||||
| ONCO004 | 42 | F | Thrombocytopenia | 0.5 | 46,XX20 | Good | 1 | -0.26 | Very low | Gain | 7q36.3 | 0.52 | 3 | PTPRN2 | Stable |
| ONCO005 | 57 | M | Pancytopenia | 0.5 | 46,XY20 | Good | 2.5 | 2.01 | Low | Loss | 20q11.21 | 17.1 | 224 | RALY, RAB5IF, ADA, SLC2A10 | Stable |
| Gain | 21q22.2 | 0.18 | 1 | ERG | |||||||||||
| Gain | Yq11.2 | 0.41 | 5 | *** | |||||||||||
| LCSH | 3q22.2 | 20.2 | 114 | MRAS, MME | |||||||||||
| LCSH | 9p22.1 | 9.41 | 58 | CDKN2B | |||||||||||
| LCSH | 14q32.31 | 5.45 | 72 | MOK, TRAF3, TNFAIP2, TRMT61A | |||||||||||
| LCSH | 19q13.32 | 5.48 | 244 | ERCC2, SIX5, SYMPK, STRN4, BBC3, BAX, GYS1, CD37, FLT3LG, BCL2L12, IL4I1 | |||||||||||
| LCSH | 21q22.3 | 4.9 | 120 | MX2, MX1 | |||||||||||
| ONCO012 | 73 | F | Thrombocytopenia | 0 | 46,XX20 | Good | 1 | 1.14 | Very low | LCSH | 5q33.1 | 21.1 | 96 | SPARC, HAVCR2, ITK, TLX3, NPM1 | Stable |
| LCSH | 7q36.1 | 4.9 | 72 | ZNF282 | |||||||||||
| LCSH | 10q26.12 | 10.76 | 69 | C10orf88, DHX32, DOCK1, MGMT, EBF3 | |||||||||||
| ONCO013 | 42 | F | Leukopenia | 0 | 46,XX20 | Good | 1 | -0.26 | Very low | Gain | 2q37.3 | 0.29 | 5 | *** | Stable |
| Gain | 21q21.1 | 0.27 | 4 | *** | |||||||||||
| LCSH | 16q21 | 17.91 | 202 | CBFB, NOL3, E2F4 | |||||||||||
| LCSH | Xq11.1 | 5.19 | 17 | *** | |||||||||||
| ONCO014 | 75 | F | Thrombocytopenia | 0 | 46,XX20 | Good | 1 | 1.23 | Very low | Gain | 17p11.2 | 0.13 | 1 | *** | Stable |
| Gain | 17p11.2 | 0.15 | 2 | SPECC1 | |||||||||||
| Gain | 17q25.3 | 0.15 | 1 | *** | |||||||||||
| LCSH | 18q21.1 | 8.19 | 37 | *** | |||||||||||
| ONCO15 | 60 | F | Pancytopenia | 0 | 46,XX20 | Good | 2.5 | 2.13 | Low | NAF | *** | *** | *** | *** | Stable |
F female, M male, NAF no alterations found, LCSH long continuous stretches of homozygosity.
*ISCN2016: an international system for human cytogenomic chromosome nomenclature.
**IPSS-R: risk estimates, according to the International Prognostic Scoring System—revised
***No cytogenomic rearrangements and no genes were identified using the CMA in the analyzed region.
#Estimated score according to hemoglobin levels, absolute neutrophil count, platelet count, percentage of blasts in the bone marrow, and cytogenetic risk.
The size of the genomic changes varied from 0.1 to 23.4 Mb. The LCSHs were larger when compared with the CNVs, ranging from 3.9 to 23.4 Mb. A CNV of loss identified in the cytoband 20q11.21 with 17.1 Mb was the largest genomic variant. The biggest CNV of gain was identified in the cytoband 7q36.3 with 0.52 Mb, while the lowest genomic gain was in cytoband 3q12.2 with 0.1 Mb.
Among the selected genes, 28/91 are differentially expressed by up-regulation or down-regulation in the mesenchymal stromal cells of bone marrow tissue from patients with MDS and karyotypes without numerical or structural changes. The value of logFC was used to define which of the 91 selected genes were regulated positively or negatively in the MDS patients included in this study. Table 2 shows the biological functions and expression patterns of the 28 genes at diagnosis, in addition to the logFC value.
Table 2.
Biological functions and expression patterns at diagnosis of (28/91) selected genes in MDS patients with karyotypes without numerical or structural changes.
| Selected genes | Genomic variation | Cytoband | Biological function* | logFC | Expression** |
|---|---|---|---|---|---|
|
MX1 MX2 |
LCSH LCSH |
21q22.3 21q22.3 |
Cellular antiviral response Cellular antiviral response |
2.743 1.191 |
Up-regulation Up-regulation |
| POC1B | LCSH | 12q21.31 | Centrosome | − 0.522 | Down-regulation |
| RALY | Loss | 20q11.21 | Embryonic development | 0.715 | Up-regulation |
|
SIX5 SYMPK ZNF282 ZNF446 |
LCSH LCSH LCSH LCSH |
19q13.32 19q13.32 7q36.1 19q13.32 |
Gene expression control Gene expression control Gene expression control Gene expression control |
0.589 1.115 0.392 0.373 |
Up-regulation Up-regulation Up-regulation Up-regulation |
| SLC2A10 | Loss | 20q11.21 | Glucose homeostasis | − 0.719 | Down-regulation |
|
ADA FLT3LG |
Loss LCSH |
20q11.21 19q13.32 |
Immunological response Immunological response |
0.696 0.438 |
Up-regulation Up-regulation |
| KLHL5 | LCSH | 4p14 | Immunological response | 0.235 | Up-regulation |
| UCHL1 | LCSH | 4p14 | Neuro transmission | − 1.126 | Down-regulation |
| MAP2K3 | Gain | 17p11.2 | Oncogene | 0.278 | Up-regulation |
| SNORA12 | LCSH | 10q23.1 | RNA processing | 0.774 | Up-regulation |
| HACD1 | LCSH | 10p13 | Signaling molecule | − 0.845 | Down-regulation |
| TMEM33 | LCSH | 4p14 | Transmembrane protein | − 0.572 | Down-regulation |
| TRMT61A | LCSH | 14q32.31 | tRNA stabilization | 0.593 | Up-regulation |
| GMFB | LCSH | 14q22.2 | Tumor proliferation | − 0.661 | Down-regulation |
|
KAT8 RGS1 |
LCSH LCSH |
16p11.2 1q31.1 |
Tumor proliferation Tumor proliferation |
0.681 0.414 |
Up-regulation Up-regulation |
| OCIAD2 | LCSH | 4p14 | Tumor supressor | − 0.794 | Down-regulation |
|
HIF1AN MTMR14 RNF40 SIGLEC7 |
LCSH LCSH LCSH LCSH |
10q23.1 3p26.1 16p11.2 19q13.32 |
Tumor supressor Tumor supressor Tumor supressor Tumor supressor |
0.351 0.367 0.603 0.272 |
Up-regulation Up-regulation Up-regulation Up-regulation |
|
C10orf88 RAB5IF |
LCSH Loss |
10q26.12 20q11.21 |
Identical protein binding Interacting factor protein |
− 0.535 − 0.806 |
Down-regulation Down-regulation |
*The biological functions of the genes were identified via RefSeq (NCBI Reference Sequence Database: http://www.genome.jp/dbget-bin/www_bget?ds:H01481; Kegg Disease: Myelodysplastic syndrome—Genome Net).
**For the analysis of expression through public databases of MDS patients, one GEO Dataset from NCBI (https://www.ncbi.nlm.nih.gov/geo/) was downloaded. The transcriptome was based on the study with accession number GSE61853 in which the gene expression patterns of mesenchymal bone marrow stromal cells from patients with MDS and normal controls were analyzed. The value of logFC was used to define which genes were up-regulated or down-regulated in MDS patients with karyotypes without numerical or structural changes.
Some recurrent genomic variations have been identified in patients in the present sample group. The LCSHs in cytoband 19q13.32 with sizes of 13.64 Mb and 5.48 Mb were identified in patients ONCO003 and ONCO005, respectively. In this region, the genes ERCC2, SIX5, SYMPK, STRN4, BBC3, BAX, GYS1, CD37, FLT3LG, BCL2L12, and IL4I1, which were common among these patients, were selected. That said, the genes SIX5, SYMPK, and FLT3LG were differentially expressed by up-regulation or down-regulation. The LCSHs in cytoband 10q26.12 with 3.9 Mb and 10.76 Mb were identified in the patients ONCO003 and ONCO012, respectively, and the gene C10orf88 was identified to be differentially expressed by down-regulation. CNVs of genomic gain involving cytoband 17p11.2 with different sizes were also identified. In patient ONCO002 with 0.29 Mb, the up-regulated oncogene MAP2K3 was selected, and in patient ONCO014 with 0.13 Mb and 0.15 Mb, the SPECC1 gene was selected.
Discussion
In the present study, patient karyotypes without numerical and/or structural alterations were classified according to cytogenetic risk as good, whereas risk prediction for primary MDS ranged from very low to low. These results confirmed that cytogenetic risk is an important informative variable for inferring prognosis because, according to the clinical outcomes, only one patient died, while the others remained stable at the end of the study. The CMA analysis of patients with primary MDS and no karyotype changes visible under the microscope also provided useful information for better understanding of the etiological aspects of this hematological disease3,8,10,11.
CNVs and LCSHs that are potentially associated with the etiology of MDS were identified in 87.5% (7/8) of the patients included in this study, of which 91 genes were selected that had already been implicated in molecular pathways important for cell metabolism and viability. According to the Cancer Genomics Consortium (CGC), CMA shows efficiency in detecting genomic variations in primary MDS in 10 to 80% of patient karyotypes without numerical or structural chromosomal changes13. Despite recent advances in diagnostic tools, MDS continues to show variability in its clinical course and response to treatment1,3,31,32. In this scenario, the identification of genomic variants associated with primary MDS could prove widely useful, offering additional information for the biological understanding and prognostic classification of this disease13,15,33,34.
The imbalances, characterized by gains and losses in the genome, represented 31.7% (13/41) of the genomic variations identified. Therefore, gene copy gains and losses may result in an increase or decrease/absence of any functional transcript. In this context, alterations in gene dosages play a determining role in the quantity of the expressed product and, consequently, down-regulate important molecular pathways of hematopoietic progenitor cells (HPCs), which are associated with the etiological processes of MDS35,36.
Expression analysis via a public database indicated that 30.8% (28/91) of the genes in this study located in regions with CNVs and LCSHs are up-regulated or down-regulated in the mesenchymal stromal cells of bone marrow tissue in patients with MDS, when compared with those of controls. Additionally, alterations in gene expression may be related to the inactivation of suppressor genes or the activation of oncogenes26. These altered molecular biological mechanisms may interfere with cell survival and correlate with the expansion of cells with ineffective hematopoietic functions, which may contribute to the possibility of the evolution and malignization of clinical MDS.
Of the altered genes, 32.1% (9/28) were down-regulated and 67.9% (19/28) were up-regulated. The main functional pathways in which these genes are involved have been identified; in this context, the investigation only described the biological processes that may be affected by altered gene expression. This study reports that the molecular pathways associated with gene expression control, immune response, and tumor proliferation and suppression were the most commonly deregulated. The relationship between RNA polymerase and the etiology of MDS is not well-defined26. One can infer that RNA polymerase dysregulation would alter the normal transcription of critical genes, such as tumor suppressor genes (GSTs), because RNA polymerase plays a role in modulating DNA transcription.
Identified CNVs harbor 17.9% (5/28) of genes with positive or negative regulation in the mesenchymal stromal cells of bone marrow tissue in patients with MDS. In the CNV of genomic loss in 20q, the RALY and ADA genes were found to be up-regulated and SLC2A10 and RAB5IF were down-regulated. The ADA and RALY genes are associated with immune response pathways and embryonic development, respectively, while the genes SLC2A10 and RAB5IF correlate with glucose homeostasis pathways and protein interaction factor, respectively; these genes are expressed in the mesenchymal stromal cells of normal bone marrow tissue.
According to reports previously published37,38, loss of CNVs lead to haploinsufficiency and can inactivate GSTs. In addition, the RALY and ADA genes were up-regulated in lost CNVs, suggesting compensation for protein products. Therefore, in microdeletion events in the genome, varied molecular mechanisms can occur and be associated with the pathophysiology of MDS. Thus, CNVs of genomic loss represent important markers with biological significance, providing additional information for understanding the mechanisms and molecular pathways related to the etiology and transformation potential of MDS36.
The MAP2K3 oncogene, located in the CNV with genomic gain in 17p with 0.29 Mb, shows altered expression with positive regulation. The protein encoded by this gene is a dual-specificity kinase that belongs to the family of map-kinases and is activated by mitogenic and environmental stress38. Changes in the pathways involving these kinases can deregulate molecular processes related to differentiation, cell cycle survival, and control, leading to the process of apoptosis in patients with MDS. Imbalances caused by positive gene expression, when compared with negative gene expression, may be less unfavorable for cellular biological pathways37,38. The present study confirms this statement, since about 2/3 of the genes are up-regulated and 7/8 patients with no visible changes in the karyotype exhibited stable clinical outcomes at the end of the study.
All three gain CNVs involving the 17p11.2 cytoband with different sizes, being 0.29 Mb, 0.13 Mb, and 0.15 Mb, are significant. Events involving chromosome 17p were considered important due to the proximity of the cytoband 17p13.1, which houses the GST TP53. The TP53 protein participates in the regulation and expression of several target genes, inducing cell cycle control, apoptosis signaling, cell senescence, and DNA repair39,40. Mutations in one or both alleles of TP53 can deregulate several molecular pathways involved in about 50% of human cancers and 20% of onco-hematologic malignancies, including MDS. Therefore, according to the risk prognosis and clinical results, these CNVs with a 17p gain may have a negative effect on hematological evolution and patient survival, as evidenced by the patient who died during the course of this study. The other CNVs with genomic gain in 2q, 17p, 17q, 21q and Yq, the CNV of genomic loss in 4q, and the LCSHs in 4q, 18q and Xq presented in Table 1 were selected as relevant due to the high instability inherent in the genomes of MDS patients, which is the basis for the accumulation of mutations in this disease24. Expression per positive regulation of the ADA gene, inserted in a region with genomic loss in 20q, and of the FLT3LG gene in 19q and KLHL5 in 4p, located in regions within LCSHs, may be associated with changes in interferon signaling pathways (immune response)41. These deregulated pathways in the medullary microenvironment in patients with MDS demonstrate that these factors participate in inducing the inflammatory state and immunological disorders, which alters hematopoiesis, thereby causing intramedullary apoptosis along with abnormal HPC differentiation and maturation.
LCSHs are abnormalities that represent 68.3% (28/41) of the genomic variations identified in the present study. Of the selected genes, 85.7% (78/91) were inserted in regions with LCSHs and associated with biological regulatory functions. Additionally, 82.1% (23/28) of up-regulated or down-regulated genes in mesenchymal stromal cells in the medullary microenvironment of patients with MDS were located in regions with loss of heterozygosity. The pathological potential of LCSHs emerges as a mechanism of clonal alteration in a proportion of somatic cells with uniparental disomy (UPD)19,20,42,43. In this context, recurring areas of LCSHs are strongly associated with the loss of the wild allele or an entire region, which also leads to the hypothesis of haploinsufficiency. Thus, these events indicate changes in biological processes that are important to the cells and signal a predisposition to hematological malignancies19,20. The terminal segmental LCSHS identified in 6q, 14q, 18q, 19q, and 21q presented in Fig. 2 indicates that break-induced replication (BIR) can be a dominant mechanism by which a cell duplicates a somatically acquired event, such as a mutation, microdeletion, or epigenetically suppressed region, and consequently becomes homozygous for this segment. BIR seems to be a common repair mechanism in replication, but only the reduplication of a region that contains a genetic or epigenetic alteration gives the cell a growth advantage, allowing for its dominance and clonal selection. Regions with LCSHs involving 3p, 4p, 10p, 12q, and 16p were identified only in the patient who died. The UCHL1, TMEM33, and OCIAD2 genes inserted in 4p, the HACD1 gene in 10p, and the POC1B gene in 12q, which encode neural transmission functions, transmembrane protein, tumor suppressor, signaling molecule, and centrosome, respectively, were down-regulated. Five out of nine selected and down-regulated genes were located in the LCSHs identified in the patient who died. Thus, unlike up-regulation, down-regulation events are potentially more damaging to important molecular pathways and fundamental cellular functions, as observed in this patient during the hematological evolution of the disease44.
The MX1 gene, located in an LCSH region at 21q, was the most up-regulated, being 2.74-times more expressed in bone marrow mesenchymal stromal cells in patients with MDS. This gene encodes a protein that metabolizes the guanosine triphosphate (GTP) that participates in the antiviral cellular response. The protein encoded by this MX1 gene is induced by the type I and II interferon pathways. In this study, the patients with genomic variations involving 21q were classified as having low and very low predicted risk, respectively, with stable clinical outcomes. These findings contradict previously published9 reports that changes involving chromosome 21 have been observed in more advanced cases of MDS, with a more aggressive evolution and rapid leukemic transformation1,11. On the other hand, the UCHL1 gene inserted in an LCSH region with 4p was the most down-regulated, being 1.12-times less expressed in the mesenchymal stromal cells of the medullary tissue of the MDS patient who died. The gene UCHL1 provides information for the production of the enzyme Ubiquitin Carboxyl-Terminal Esterase L1, which is involved in the cellular machinery that breaks down unnecessary substances45. In cells, damaged or excess proteins are marked with ubiquitin molecules so that the ubiquitin–proteasome system can act as a cellular quality control system.
In patients with MDS, persistent and refractory cytopenias can be induced by cytokines and associated with T-cell mediated myelosuppression46. Alongside this, changes in the medullary microenvironment inhibit the growth of HPCs through the high secretion of interferon-gamma (IFN-γ), TNF, and Interleukin-6 (IL-6)14,47. An increase in the number of stem cells could offset the phenotypic consequences of the simultaneous repression of differentiation genes, creating a pre-leukemic hematopoiesis with little or no morphological abnormalities48. As such, the association between dysplastic cell characteristics and the increase in total genomic changes is an observation that suggests a significant parallel trend. Also, the more morphological dysplasia a marrow tissue sample presents, the more extensive the underlying genomic changes can be48. Both events, CNVs and LCSHs, contributed significantly to the correlation between the identified genomic variations and the clinical and laboratory phenotypes of patients with MDS in this study, highlighting the usefulness of matrix platforms containing markers for SNPs.
In only 12.5% (1/8) of the patients, the CMA did not reveal genomic variations. It is important to note that this result without changes does not imply that there could not be some kind of deleterious mutation in the genes associated with the molecular pathways associated with the MDS phenotypes. Thus, it was not possible to suggest a genetic cause for this disease, since the CMA resolution was not comprehensive enough to identify changes in the genome. This study highlights that balanced translocations are not detectable by CMA, which is a disadvantage of this cytogenomic analysis technique49,50. The possibility of testing other more sensitive or specific genomic technologies, such as Next Generation Exome Sequencing (NGS), may be a differential approach to identify point mutations that may be associated with MDS, in which the etiology is multifactorial and extremely heterogeneous51.
The limitation of this study was the small size of the sample group. This limitation is typical of studies involving patients with primary MDS, which have reduced casuistry due to the low incidence of the disease in the population. The present findings from CNVs and LCSHs can contribute to subsequent studies of the adequate characterization of the MDS phenotype and bring additional information to improve the quality of prognostic and personalized stratification for the patient, in addition to the possibility of identifying potentially more effective therapeutic treatments in the near future13,16. The interpretation and association of CNVs that result in primary MDS is still challenging, mainly due to the unclear effects of variations in the genome that can be influenced by incomplete penetrance, variable expressiveness, and epistasis40,52,53. In this context, further investigations are needed in larger cohorts, including patients with greater heterogeneity of clinical outcomes and prognostic risks.
Conclusion
This study identified and reported CNVs and LCSHs that may be associated with primary MDS in the group of participants. Ninety-one genes involved in important molecular pathways of metabolism and cell viability were selected. From the analysis of the cytogenomic findings, when compared to information from public genomic databases, CNVs and LCSHs were related to bone marrow failure, which was characterized by the expansion of cells with ineffective hematopoietic functions. CMA is a robust and efficient cytogenomic technique for expanding the resolution of the genomic analysis of the medullary cells of primary MDS patients, allowing the identification of unusual and clinically significant genomic variations in patients without visible changes in the karyotype. Finally, 30.8% (28/91) of the selected genes showed altered expression and 2/3 of these genes showed positive regulation when compared to public data on gene expression in mesenchymal stromal cells of bone marrow tissue in primary MDS. Changes in gene expression may be related to the etiology and progression of primary MDS, with a resulting effect on the relative risk and clinical outcome of this onco-hematological disease.
Acknowledgements
The authors thank the patients for their voluntary participation. We also appreciate the possibility of carrying out this study at the Replicon Research Center of the School of Agricultural Sciences and Biological Sciences of the Pontifical Catholic University of Goiás in partnership with the Human Cytogenetics and Molecular Genetics Laboratory of the Health Department of the State of Goiás. We are grateful for the broad collaboration with the South American Institute of Diabetes and Obesity, associated with the medical school the University of Texas, Rio Grande Valley, USA. We are also particularly grateful for the encouragement of the Goiás Research Support Foundation and CAPES—The Coordination for the Improvement of Higher Education Personnel for granting scholarships. A.D.C is a research fellow PQ2 from CNPq (National Council for Scientific and Technological Development).
Author contributions
C.L.R., I.P.P., L.B.M., A.D.C. and C.C.S. designed the study. F.S.M.K. was the patients’ attending physician. C.L.R., A.D.C., C.C.S. and I.P.P. performed the experiments. C.L.R., I.P.P., M.A.A.A and A.D.C. analyzed data and performed the statistical analysis. C.L.R., I.P.P., S.S.S.P., T.E.H., C.C.S., A.M.A and A.D.C. contributed to manuscript writing. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Adès L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet. 2014;383:2239–2252. doi: 10.1016/S0140-6736(13)61901-7. [DOI] [PubMed] [Google Scholar]
- 2.Haferlach T, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prebet T, et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J. Clin. Oncol. 2011;29:3322–3327. doi: 10.1200/JCO.2011.35.8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arber DA, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leucemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 5.Neukirchen J, et al. Incidence and prevalence of myelodysplastic syndromes: Data from the Düsseldorf MDS-registry. Leuk. Res. 2011;35:1591–1596. doi: 10.1016/j.leukres.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Greenberg PL. The multifaceted nature of myelodysplastic syndromes: Clinical, molecular, and biological prognostic features. JNCCN. 2013;11(7):877–884. doi: 10.6004/jnccn.2013.0105. [DOI] [PubMed] [Google Scholar]
- 7.Lubeck DP, et al. Systematic literature review of the global incidence and prevalence of myelodysplastic syndrome and acute myeloid leukemia. Blood. 2016;128:5930. doi: 10.1182/blood.V128.22.5930.5930. [DOI] [Google Scholar]
- 8.Bejar, R. Prognostic models in myelodysplastic syndromes. Hematol. Am. Soc. Hematol. Educ. Program. 504–510 (2013). [DOI] [PubMed]
- 9.Malcovati L, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: Recommendations from the European Leukemia Net. Blood. 2013;122(17):2943–2964. doi: 10.1182/blood-2013-03-492884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Estephan F, Tiu RV. Current and novel therapeutic approaches in myelodysplastic syndromes. J. Commun. Support Oncol. 2014;12:236–249. doi: 10.12788/jcso.0057. [DOI] [PubMed] [Google Scholar]
- 11.Mohammad FZ, et al. Cytogenetic abnormalities in myelodysplastic syndromes: An overview. Int. J. Hematol. Oncol. Stem Cell Res. 2017;11(3):231–239. [PMC free article] [PubMed] [Google Scholar]
- 12.Arenillas L, et al. Single nucleotide polymorphism array karyotyping: A diagnostic and prognostic tool in myelodysplastic syndromes with unsuccessful conventional cytogenetic testing. Genes Chromosomes Cancer. 2013;52:1167–1177. doi: 10.1002/gcc.22112. [DOI] [PubMed] [Google Scholar]
- 13.Kanagal-Shamanna R, et al. Assessing copy number aberrations and copy neutral loss of heterozygosity across the genome as best practice: An evidence-based review of clinical utility from the cancer genomics consortium (CGC) working group for myelodysplastic syndrome, myelodysplastic/myeloproliferative and myeloproliferative neoplasms. Cancer Genet. 2018;228–229:197–217. doi: 10.1016/j.cancergen.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Raaijmakers MH. Myelodysplastic syndromes: Revisiting the role of the bone marrow microenvironment in disease pathogenesis. Int. J. Hematol. 2012;95(1):17–25. doi: 10.1007/s12185-011-1001-x. [DOI] [PubMed] [Google Scholar]
- 15.Greenberg PL, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–2465. doi: 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macedo LC, et al. Genetics factors associated with myelodysplastic syndromes. Blood Cells Mol. Dis. 2015;55:76–81. doi: 10.1016/j.bcmd.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Padron E, Lancet J. The molecular basis and clinical significance of genetic mutations identified in myelodysplastic syndromes. Leuk. Res. 2015;39:6–17. doi: 10.1016/j.leukres.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Diamantidis MD, Papanastasiou D. Myelodysplastic syndromes: Aiming at deciphering their secrets. Leuk. Res. 2016;51:1–2. doi: 10.1016/j.leukres.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Jerez A, et al. Topography, clinical, and genomic correlates of 5q myeloid malignancies revisited. J. Clin. Oncol. 2012;30(12):1343–1349. doi: 10.1200/JCO.2011.36.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schanz J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J. Clin. Oncol. 2012;30(8):820–829. doi: 10.1200/JCO.2011.35.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolquist KA, et al. Microarray-based comparative genomic hy- bridization of cancer targets reveals novel, recurrent genetic aberrations in the myelodysplastic syndromes. Cancer Genet. 2011;204:603–628. doi: 10.1016/j.cancergen.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad A, Iqbal MA. Significance of genome-wide analysis of copy number alterations and UPD in myelodysplastic syndromes using combined CGH-SNP arrays. Curr. Med. Chem. 2012;19:3739–3747. doi: 10.2174/092986712801661121. [DOI] [PubMed] [Google Scholar]
- 23.Liu F, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19:283–294. doi: 10.1016/j.ccr.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papaemmanuil E, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–3627. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itzykson R, Fenaux P. Epigenetics of myelodysplastic syndromes. Leukemia. 2014;28:497–506. doi: 10.1038/leu.2013.343. [DOI] [PubMed] [Google Scholar]
- 26.Kim M, et al. Increased expression of interferon signaling genes in the bone marrow microenvironment of myelodysplastic syndromes. PLoS ONE. 2015;10(3):e0120602. doi: 10.1371/journal.pone.0120602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verma RS, Babu A. Human Chromosomes: Principles and Techniques. New York: McGraw-Hill; 1995. [Google Scholar]
- 28.Kearney HM, et al. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: Consanguinity, uniparental disomy, and recessive single-gene mutations. Clin. Lab. Med. 2011;31:595–613. doi: 10.1016/j.cll.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Chaves TF, et al. Long contiguous stretches of homozygosity detected by chromosomal microarrays (CMA) in patients with neurodevelopmental disorders in the South of Brazil. BMC Med. Genomics. 2019;12:50. doi: 10.1186/s12920-019-0496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ribeiro CL, et al. High diversity of chromosomal aberrations in a Brazilian myelodysplastic syndrome cohort. Genet. Mol. Res. 2019;18(2):18322. [Google Scholar]
- 31.Akagi T, et al. Frequent genomic abnormalities in acute myeloid leukemia/myelodysplastic syndrome with normal karyotype. Haematologica. 2009;94(2):213–223. doi: 10.3324/haematol.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cordoba I, et al. The degree of neutropenia has a prognostic impact in low risk myelodysplastic syndrome. Leuk. Res. 2012;36(3):287–292. doi: 10.1016/j.leukres.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 33.Cazzola M, Malcovati L. Prognostic classification and risk assessment in myelodysplastic syndromes. Hematol. Oncol. Clin. N. Am. 2010;24(2):459–468. doi: 10.1016/j.hoc.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Giagounidis A, Haase D. Morphology, cytogenetics and classification of MDS. Best Pract. Res. Clin. Haematol. 2013;26:337–353. doi: 10.1016/j.beha.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 35.McNerney ME, et al. CUX1 is a haploinsufficient tumor suppressor gene on chromosome 7 frequently inactivated in acute myeloid leukemia. Blood. 2013;121:975–983. doi: 10.1182/blood-2012-04-426965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.- Mitelman, F., Johansson, B. & Mertens, F. Mitelman database of chromosome aberrations and gene fusions in cancer. http://cgap.nci.nih.gov/Chromosomes/Mitelman. (2016).
- 37.Bejar R, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012;30:3376–3382. doi: 10.1200/JCO.2011.40.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gomez-Segui I, et al. Novel recurrent mutations in the RAS-like GTP-binding gene RIT1 in myeloid malignancies. Leukemia. 2013;27:1943–1946. doi: 10.1038/leu.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jasek M, et al. P53 mutations in myeloid malignancies are either homozygous or hemizygous due to copy number-neutral loss of heterozygosity or deletion of 17p. Leukemia. 2010;24(1):216–219. doi: 10.1038/leu.2009.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jadersten M, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 2011;29:1971–1979. doi: 10.1200/JCO.2010.31.8576. [DOI] [PubMed] [Google Scholar]
- 41.Larrosa GM, Baer MR. FLT3 inhibitors in acute myeloid leukemia: Current status and future directions. Mol. Cancer Ther. 2017;16(6):991–1001. doi: 10.1158/1535-7163.MCT-16-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makishima H, Maciejewski JP. Pathogenesis and consequences of uniparental disomy in cancer. Clin. Cancer Res. 2011;17:3913–3923. doi: 10.1158/1078-0432.CCR-10-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Svobodova K, et al. Copy number neutral loss of heterozygosity at 17p and homozygous mutations of TP53 are associated with complex chromosomal aberrations in patients newly diagnosed with myelodysplastic syndromes. Leuk. Res. 2016;42:7–12. doi: 10.1016/j.leukres.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Hemmat M, et al. Submicroscopic deletion of 5q involving tumor suppressor genes (CTNNA1, HSPA9) and copy neutral loss of heterozygosity associated with TET2 and EZH2 mutations in a case of MDS with normal chromosome and FISH results. Mol. Cytogenet. 2014;7:35. doi: 10.1186/1755-8166-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patricia C, et al. Ubiquitin carboxyl-terminal esterase L1 (UCHL1) is associated with stem-like cancer cell functions in pediatric high-grade glioma. PLoS ONE. 2017;12(5):e0176879. doi: 10.1371/journal.pone.0176879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barrett AJ, Sloand E. Autoimmune mechanisms in the pathophysiology of myelodysplastic syndromes and their clinical relevance. Hematologica. 2009;94(4):449–451. doi: 10.3324/haematol.2009.006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng X, et al. Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. Haematologica. 2011;96:602–606. doi: 10.3324/haematol.2010.030536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeung C, et al. Impact of copy neutral loss of heterozygosity and total genome aberrations on survival in myelodysplastic syndrome. Mod. Pathol. 2018;31(4):569–580. doi: 10.1038/modpathol.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tiu RV, et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011;117(17):4552–4560. doi: 10.1182/blood-2010-07-295857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dermody J, Tolias P, Toruner GA. Chromosomal microarrays: Influential players in the diagnosis of developmental disorders. Person Med. 2012;9:167–169. doi: 10.2217/pme.11.100. [DOI] [PubMed] [Google Scholar]
- 51.Lalonde E, et al. Unexpected allelic heterogeneity and spectrum of mutations in Fowler syndrome revealed by next-generation exome sequencing. Hum. Mutat. 2010;31:918–923. doi: 10.1002/humu.21293. [DOI] [PubMed] [Google Scholar]
- 52.Pellagatti A, et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia. 2010;24:756–764. doi: 10.1038/leu.2010.31. [DOI] [PubMed] [Google Scholar]
- 53.Zanni G, et al. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc. Natl. Acad. Sci. 2012;109:14514–14519. doi: 10.1073/pnas.1207488109. [DOI] [PMC free article] [PubMed] [Google Scholar]