

Summary
MYC is a major oncogenic transcriptional driver of most human cancers that has remained intractable to direct targeting because much of MYC is intrinsically disordered. Here, we have performed a cysteine-reactive covalent ligand screen to identify compounds that could disrupt the binding of MYC to its DNA consensus sequence in vitro and also impair MYC transcriptional activity in situ in cells. We have identified a covalent ligand EN4 that targets cysteine 171 (C171) of MYC within a predicted intrinsically disordered region of the protein. We show that EN4 directly targets MYC in cells, reduces MYC and MAX thermal stability, inhibits MYC transcriptional activity, downregulates multiple MYC transcriptional targets, and impairs tumorigenesis. We also show initial structure-activity relationships of EN4 and identify compounds that show improved potency. Overall, we identify a unique ligandable site within an intrinsically disordered region of MYC that leads to inhibition of MYC transcriptional activity.
Keywords: MYC, chemoproteomics, covalent ligand, intrinsically disordered, activity-based protein profiling, cysteine, undruggable, transcription factor
eTOC Blurb
We identify a covalent ligand that functionally targets a novel ligandable site C171 within an intrinsically disordered region of MYC to reduce MYC and MAX thermal stability, inhibit MYC DNA binding, inhibit MYC transcriptional activity, downregulate MYC transcriptional targets, and impair proliferation and tumorigenesis.
Introduction
MYC is a nuclear transcription factor that regulates the expression of genes involved in cell proliferation, metabolism, and survival and is the most frequently amplified oncogene in human cancers (Kress et al., 2015; McKeown and Bradner, 2014). MYC activates its transcriptional targets through obligate heterodimerization with MAX and binding to the E-box sequences. Due to its fundamental importance to cancer pathogenesis, significant effort has been invested in trying to develop inhibitors against MYC or pathways that regulate MYC (Dang et al., 2017; McKeown and Bradner, 2014; Meyer and Penn, 2008; Whitfield et al., 2017). Strategies for MYC pathway inhibition have included targeting MYC transcription with bromodomain and extraterminal (BET) family inhibitors or CDK7/9 inhibitors, targeting MYC translation with mTORC1 or AKT/PI3K inhibitors, or targeting the heterodimeric complex between MYC and MAX. Examples of these small-molecules include bromodomain-containing 4 (BRD4) inhibitors such as JQ1 or GSK525762 that displace BRD4 binding to sites of hyperacetylated chromatin that regulate MYC transcription, and mTORC1 inhibitors such as rapamycin and everolimus that inhibit mTORC1-dependent phosphorylation of the translation initiation factor 4E binding protein 1 (4EBP1) and inhibit MYC translation (Chen et al., 2018; McKeown and Bradner, 2014). Studies using small-molecule microarrays have yielded MAX/MAX homostabilizers that sequester MAX away from MYC leading to inhibition of MYC transcriptional activity (Struntz et al., 2019). However, direct targeting of MYC itself has remained challenging and it has often been considered “undruggable” because much of MYC is intrinsically disordered and there are no obvious binding pockets or ligandable sites for pharmacological interrogation (Chen et al., 2018; Dang et al., 2017; McKeown and Bradner, 2014).
Covalent ligand screening using chemoproteomic approaches such as activity-based protein profiling (ABPP) has arisen as a powerful strategy for rapid discovery of covalent ligands, ligandable hotspots, and therapeutic modalities against proteins that are often considered intractable to standard drug discovery efforts (Backus et al., 2016; Bateman et al., 2017; Chung et al., 2019; Grossman et al., 2017; Spradlin et al., 2019; Weerapana et al., 2010). Because of the irreversible binding of covalent ligands to the target of interest, mass-spectrometry-based proteomic approaches can be used to rapidly identify the site of modification of hit ligands without structural elucidation of the protein-ligand complex—an effort that would be hindered for largely intrinsically disordered proteins like MYC (Spradlin et al., 2019; Ward et al., 2019a). In this study, we sought to identify a covalent compound directly targeting a ligandable site on MYC that would functionally impair MYC transcriptional activity.
Results
Covalent Ligand Screening to Identify MYC Inhibitors
To identify direct and covalently-acting inhibitors against c-MYC, we screened a library of 98 primarily cysteine-reactive covalent ligands consisting of acrylamides and chloroacetamides in vitro to identify compounds that would inhibit the binding of purified recombinant human MYC/MAX complex to the E-box DNA consensus sequence (Fig. 1a, Table S1). These compounds were from a collection of compounds purchased from Enamine. While human MAX does not contain any cysteine residues, we wanted to avoid potential off-chance reactivity of the covalent ligands with another nucleophilic amino acid on MAX. To maximize our chances of identifying covalent ligands that directly targeted a cysteine on MYC, we pre-incubated covalent ligands with MYC prior to the addition of MAX and then added this MYC/MAX complex to plates bearing the E-box consensus sequence. From this screen EN4 was identified as the top hit (Fig. 1a).
Figure 1. Covalent ligand screen to identify compounds that inhibit MYC activity in vitro and in situ.

(a) Screening a cysteine-reactive covalent ligand library in vitro for compounds that would inhibit MYC/MAX binding to its E-box DNA consensus sequence. DMSO vehicle or covalent ligands (50 μM) were pre-incubated with pure human full-length MYC protein for 30 min before direct addition of MAX and then addition to DNA binding plates for 1 h. Data are shown as ratio relative to DMSO vehicle treated controls set to 1. Shown in red was the top hit. (b) MYC luciferase reporter activity in HEK293T cells transfected with a MYC luciferase reporter construct encoding a Firefly luciferase reporter gene under the control of a basal promoter element, the TATA box, joined to tandem repeats of the MYC transcriptional E-box response element, alongside a constitutively expressing Renilla luciferase for normalization purposes. DMSO vehicle or cysteine-reactive covalent ligands (50 μM) were treated in HEK293T cells transfected with a MYC luciferase reporter gene for 24 h and Firefly and Renilla lucierase activity was read-out. Firefly luciferase levels were normalized to Renilla luciferase levels and then normalized to vehicle-treated controls. Data are shown as ratios relative to DMSO vehicle treated controls. Shown in red was the top hit. (c) Structure of top hit in both in vitro and in situ assays in (a) and (b) EN4. Shown in red is the cysteine-reactive acrylamide. (d) Dose-response of EN4 inhibition of MYC/MAX binding to its DNA consensus sequence. (e) Dose-response of EN4 inhibition of MYC luciferase reporter activity in cells. DMSO vehicle or EN4 was treated in HEK293T cells transfected with a MYC luciferase reporter gene for 24 h and luciferase activity was read-out. Data are shown as ratio relative to DMSO vehicle treated controls set to 1. Data shown in (a, b, d, e) are average ± sem, n=3 biologically independent samples/group. Statistical significance was calculated with unpaired two-tailed Student’s t-tests in (d, e) and is expressed as *p<0.05. This figure is related to Figure S1 and Table S1.
To further identify covalent ligands that could inhibit MYC transcriptional activity in cells in situ, we also screened these cysteine-reactive ligands in a MYC luciferase reporter assay in HEK293T cells (Fig. 1b, Table S1). This reporter assay employed a MYC-responsive Firefly luciferase reporter, encoding a luciferase reporter gene under the control of a basal promoter element, the TATA box, joined to tandem repeats of the MYC transcriptional E-box response element, alongside a constitutively expressing Renilla luciferase for normalization purposes. We were particularly interested in covalent ligands that inhibited both MYC/MAX DNA binding in vitro and MYC transcriptional activity in situ, as these compounds may be functionally inhibiting MYC activity in cells through directly binding to MYC itself. From these two screens, we identified one primary hit EN4 that bears a cysteine-reactive acrylamide warhead (Fig. 1c). EN4 showed the strongest inhibition of both MYC/MAX binding to its DNA consensus sequence in vitro as well as MYC transcriptional activity in cells (Fig. 1a-1c). EN4 inhibited MYC/MAX binding to the E-box response element DNA consensus sequence in a dose-responsive manner with a 50 % inhibitory concentration (IC50) value of 6.7 μM (Fig. 1d). We further showed that EN4 inhibited MYC/MAX DNA binding regardless of whether EN4 was pre-incubated with MYC before addition of MYC or incubated with the pre-formed MYC/MAX complex (Fig. S1a–S1b). EN4 also inhibited MYC luciferase reporter activity in a dose-responsive manner with an IC50 value of 2.8 μM (Fig. 1e). We note that EN4 is fully soluble in aqueous media at 50 μM, the maximum concentration used in this study, but this compound is not soluble at concentrations of 100 μM or higher.
Characterization of EN4 Interactions with c-MYC In Vitro
We expected that EN4 would undergo Michael addition to a cysteine on MYC (Fig. 2a). As such, we next used liquid-chromatography-tandem mass spectrometry (LC-MS/MS) to identify the single site of EN4 labeling on pure human MYC as C171 (Fig. 2b, Fig. S1c). Intriguingly, C171 belongs to a predicted intrinsically disordered region of MYC for which there is no structural information within the transactivation and transrepression N-terminal domain (NTD) far from the E-Box or MAX binding C-terminal domain (CTD) (Fig. 2c). To further confirm that EN4 was covalently binding to MYC, we synthesized 4 different alkyne-functionalized EN4 probes (EN4-alkyne-1, −2, −3, −4) (Fig. 2d, Supplemental Methods). All four of these probes showed labeling of pure human MYC protein on a denaturing gel after appending a rhodamine-azide onto probe-labeled proteins by copper-catalyzed azide-alkyne cycloaddition (CuAAC) (Fig. 2d). We also showed that EN4 displaced the labeling of EN4-alkyne-2 labeling of pure human MYC protein (Fig. 2e). Further confirming interaction of these compounds with C171, EN4-alkyne-2 labeling was not observed with pure human MYC cysteine 171-to-tyrosine mutant (C171Y) mutant protein compared to wild-type MYC (Fig. 2f). Further demonstrating that C171 on MYC is the functional target of EN4, EN4-mediated inhibition of MYC/MAX binding to its DNA consensus sequence was significantly attenuated with C171Y mutant MYC protein compared to wild-type MYC protein (Fig. 2g). Thus, our data show that EN4 directly and covalently modifies pure full-length c-MYC protein at C171 to inhibit MYC/MAX DNA binding in vitro. Interestingly, this C171 on c-MYC is not conserved in N-MYC or L-MYC and may suggest that EN4 may be selective for c-MYC over N-MYC and L-MYC (Fig. S2).
Figure 2. Characterization of EN4 binding to MYC.

(a) Structure of EN4 cysteine adduct on C171 of MYC. (b) MS/MS analysis of EN4 adduct on MYC C171. Pure human full-length MYC was incubated with EN4 (50 μM) for 30 min. MYC was then subjected to tryptic digestion and tryptic digests were analyzed by LC-MS/MS. (c) PONDR prediction of MYC protein folding and location of C171 showing that it is part of an intrinsically disordered region of MYC and a domain map of MYC. (d) 4 alkyne-functionalized EN4 probes were tested for covalent binding to MYC by gel-based ABPP methods. Pure human full length MYC protein was incubated with DMSO vehicle or each probe for 2 h at 37°C, after which rhodamine-azide was conjugated to probe-labeled proteins by CuAAC, separated by SDS/PAGE, and visualized by in-gel fluorescence. (e) Gel-based ABPP analysis of EN4 interaction with MYC. EN4 displaces EN4-alkyne-2 labeling of pure human MYC protein. DMSO vehicle or EN4 was pre-incubated with MYC for 30 min at 37°C, prior to EN4-alkyne-2 labeling (50 μM) for 2 h at 37°C. Rhodamine-azide was appended to probe-modified proteins by CuAAC, subjected to SDS/PAGE and analyzed by in-gel fluorescence. Shown also is silver staining of the gel to show equal protein loading. (f) gel-based ABPP analysis of EN4-alkyne-2 labeling of pure human wild-type (WT) and C171Y mutant MYC protein. Probe labeling with pure protein was performed with EN4-alkyne-2 (50 μM) for 2 h at 37°C. (g) MYC/MAX DNA binding with EN4 treatment. DMSO vehicle or EN4 (50 μM) were pre-incubated with pure human full-length wild-type or C171Y mutant MYC protein for 30 min before direct addition of MAX and then addition to DNA binding plates for 1 h. Gels for (d, e, f) are representative of n=3 biologically independent samples/group. Data shown in (g) show individual replicate values and average ± sem, n=5 biologically independent samples/group. Statistical significance was calculated with unpaired two-tailed Student’s t-tests in (g) and is expressed as *p<0.05 compared to respective vehicle-treated control groups and #p<0.05 compared to EN4-treated group with WT MYC protein. This figure is related to Figure S1 and Figure S2.
Characterization of EN4 Engagement of MYC in Cells
We next sought to further characterize EN4 engagement of MYC in cells. We first showed that EN4-alkyne-4 probe treatment in situ in 231MFP cells significantly enriched endogenous c-MYC compared to vehicle-treated controls after appending an azide-functionalized biotin handle by CuAAC, avidin-enrichment, and blotting for MYC (Fig. 3a). Enrichment was not observed with an unrelated protein vinculin (Fig. 3a). We note that these c-MYC enrichment studies with the EN4-alkyne probe required substantially more protein input and high-capacity avidin beads than pulldown studies that we have previously performed. This may be due to low abundance or overall instability of MYC. In this experiment, we were also not able to compete the observed c-MYC enrichment by excess EN4, since EN4 was not soluble above the 50 μM concentration of EN4-alkyne-4 needed to observe this c-MYC enrichment. Next, further reinforcing EN4 engagement of endogenous c-MYC in cells, we performed a cellular thermal shift assay (CETSA) and showed that EN4 treatment significantly reduced MYC thermal stability in 231MFP breast cancer cells (Fig. 3b) (Cimmperman et al., 2008; Martinez Molina et al., 2013). EN4 also reduced the thermal stability of the MYC heterodimer partner MAX as well (Fig. 3b). The reduced thermal stability of MYC with EN4 treatment is further confirmation that EN4 engages MYC in cells. Furthermore, our data suggest that the mechanism of EN4-mediated MYC inhibition may be through covalent targeting of MYC leading to destabilization of MYC folding. This may in-turn impair the formation of either MYC/MYC homodimer or MYC/MAX heterodimer complexes. The reduction in MAX thermal destabilization would seem to reinforce this hypothesis and would be consistent with MAX instability due to either loss of its heterodimer complex partner MYC leading to instability of free MAX and/or potentially higher levels of a less thermally stable MAX homodimer complex. These data collectively indicate that EN4 or its close analog EN4-alkyne-4 engage endogenous MYC in 231MFP cells.
Figure 3. Characterizing EN4 engagement of MYC in cells.

(a) EN4-alkyne-4 enrichment of endogenous MYC in 231MFP breast cancer cells. 231MFP cells were treated with DMSO vehicle or EN4-alkyne-4 (50 μM) for 4 h. Resulting cell lysates were subjected to CuAAC with an azide-functionalized biotin handle and EN4-alkyne-4-labeled proteins were subsequently avidin-enriched, run on SDS/PAGE, and blotted for c-MYC and loading control vinculin. Also shown is c-MYC and vinculin expression from the input protein that went into the pulldown. Gel shown is a representative image of n=3 biologically independent samples/group and fluorescence is quantified in the bar graph shown on the right. (b) Cellular thermal shift assay showing c-MYC, MAX, and loading control GAPDH protein levels in 231MFP cells treated with vehicle DMSO or EN4 (50 μM) for 2 h. Shown are representative gels and quantification of MYC and MAX protein levels, n=4 biologically independent samples/group normalized to MYC or MAX protein levels at 37o C, respectively. Data shown in (a, b) are average ± sem. Statistical significance was calculated with unpaired two-tailed Student’s t-tests and is expressed as *p<0.05 compared to vehicle-treated controls in (a) and vehicle-treated controls at each temperature in (b). This figure is related to Figure S3.
To understand the proteome-wide cysteine reactivity of EN4 in cells, we next performed isotopic tandem orthogonal proteolysis-enabled ABPP (isoTOP-ABPP) to investigate the proteome-wide cysteine-reactivity and selectivity of EN4 in 231MFP breast cancer cells using the cysteine-reactive alkyne-functionalized iodoacetamide probe (IA-alkyne). While we were not able to observe MYC itself in this isotope-ABPP experiment, likely due to its low overall abundance or potential instability, we did show that EN4 was selective across >1400 probe-modified peptides, in which we only observed 8 other off-targets of EN4 showing an isotopically light versus heavy or control versus EN4-treated probe-modified peptide ratios of greater than 3 (Fig. S3a; Table S2). Intriguingly, one of the off-targets of EN4 from isoTOP-ABPP studies was another important transcription factor JUN (Fig. S3a), which was also an off-target of previous MYC-directed probes (Berg et al., 2002). In the isoTOP-ABPP data, EN4 targets C99 of JUN. While sequence alignment analysis does not indicate that C171 is conserved between c-MYC and JUN, C99 of JUN aligns with C117 of c-MYC, suggesting potential structural features that may be in common between these proteins that give rise to common pharmacology (Fig. S2). We confirmed that JUN was an off-target of EN4 by gel-based ABPP showing competition of EN4 against labeling of pure JUN protein with the cysteine-reactive rhodamine-functionalized iodoacetamide probe (IA-rhodamine) (Fig. S3b). However, EN4 still exhibited comparable inhibitory effects upon MYC luciferase reporter activity upon JUN knockdown compared to control cells, suggesting that JUN was likely not a major driver of the EN4 inhibitory effects upon MYC transcriptional activity (Fig. S3c–S3d).
We also attempted to perform chemoproteomic pulldown studies with the EN4-alkyne-4 probes, but unfortunately, we were not able to observe c-MYC in these studies either and we were not able to perform competition studies with excess EN4 because of solubility issues encountered with beyond 50 μM. This again may be due to the relative low abundance and instability of c-MYC in cells. We performed gel-based ABPP analysis of EN4-alkyne-4 labeling in 231MFP cells and showed 5 protein bands that were competed by EN4, which was comparable to the 8 off-targets identified by isoTOP-ABPP (Fig. S3e).
Characterizing EN4 Inhibitory Activity Against MYC in Cells
Because EN4 was not completely selective for C171 of MYC, we wanted to confirm that this in situ MYC inhibitory activity was due to EN4 engagement of C171. Since knocking down the expression of endogenous MYC was not successful, likely due to its fundamental importance in cell viability, we overexpressed either wild-type (WT) or a C171Y mutant FLAG-MYC in HEK293T cells (Fig. 4a). The MYC inhibitory effect of EN4 was significantly attenuated in cells expressing C171Y FLAG-MYC compared to WT FLAG-MYC, indicating that the EN4 inhibition of MYC activity in cells was at least in-part driven through targeting C171 on MYC (Fig. 4b).
Figure 4. Characterizing EN4 inhibitory activity against MYC in cells.

(a) Protein expression of FLAG-tagged GFP, MYC wild-type (WT), or MYC C171Y mutant protein and GAPDH loading control. FLAG-GFP, FLAG-MYC WT or FLAG-MYC C171Y mutant protein were transiently overexpressed in HEK293T cells. (b) MYC luciferase reporter activity in HEK293T cells transiently expressing FLAG-MYC WT or FLAG-MYC C171Y mutant protein. DMSO vehicle or EN4 (50 μM) was treated in HEK293T cells for 24 h transiently expressing FLAG-MYC WT or FLAG-MYC C171Y protein transfected with a MYC luciferase reporter gene, and luciferase activity was read-out. Data are shown as ratio relative to DMSO vehicle treated controls set to 1. (c, d) RNA sequencing analysis of EN4-mediated transcriptional changes in 231MFP cells. 231MFP cells were treated with DMSO vehicle or EN4 (50 μM) for 24 h. Shown are log2 fold change ratios of EN4 versus DMSO vehicle-treated controls. Shown in red in (c) are direct MYC targets confirmed by CHIP-seq and in (d) are MYC-regulated gene targets that are downregulated with EN4-treatment with adjusted p-values <0.1 with fold-change <0.75 comparing EN4 to control. All RNAseq data can be found in Table S3. (e) Signature enrichment analysis of genes with an FDR < 0.1 and Fold change <= 0.75 (567 genes) using the Signature Commons platform search of the MSigDB Collection H hallmark gene set. Full analysis is shown in Table S3. (f) Protein levels of known MYC transcriptional targets CDK2 and CDC25A as well as MYC itself and loading control vinculin, assessed by Western blotting. 231MFP breast cancer cells were treated with DMSO vehicle or EN4 (50 μM) for 36 h, with a replenishment of media and with vehicle or EN4 treatment at 24 h. Shown are representative blots from n=3 biologically independent replicates/group. (g) Quantitation of Western blots from (f). (h) Cell proliferation of 231MFP breast cancer cells. Cells were treated once per day daily with DMSO vehicle or EN4 for 72 h and cell proliferation was assessed by Hoechst staining. Data are shown as percent proliferation compared to DMSO vehicle control. (i) Serum-free cell survival in MCF10A parental or MYC-expressing cells. MCF10A parental or MYC-overexpressing cells were treated with DMSO vehicle or EN4 for 48 h and cell survival as assessed by Hoechst staining. Data are shown in relation to DMSO vehicle controls for respective lines set to 1. (j) Tumor xenograft growth of 231MFP breast cancer cells in immune-deficient SCID mice. 231MFP breast cancer cells were injected subcutaneously into the flank of SCID mice and vehicle or EN4 (50 mg/kg ip) treatment was initiated after 14 days of tumor implantation. Vehicle or EN4 were treated in mice subsequently once per day and tumor volume was measured by calipers. Data shown in (a, b, f, g, h, j) are average ± sem, n=3–8 biologically independent samples/group. Statistical significance was calculated with unpaired two-tailed Student’s t-tests and is expressed as *p<0.05 compared to vehicle-treated controls and in (a, b, c, d, f, g, h, and j) and compared to respective EN4 concentrations in MCF10A cells in (i), #p<0.05 compared to EN4-treated WT group in (f). This figure is related to Figure S3 and Table S3 and Table S4.
We next investigated whether EN4 affects MYC-regulated transcriptional targets in 231MFP breast cancer cells. These highly aggressive and tumorigenic triple-negative breast cancer cells are derived from MDA-MB-231 cells that have been previously shown to be driven by MYC (Horiuchi et al., 2012; Wolfer et al., 2010; Xu et al., 2010). We performed RNA-sequencing on EN4-treated 231MFP cells to identify gene expression changes (Fig. 4c-4d). EN4 treatment significantly downregulated the expression of both direct MYC target genes previously confirmed by CHIP-Seq as well as MYC-regulated gene targets found in the Hallmark gene sets from the MSigDB Team (Zeller et al., 2003) (Fig. 4c-4d). Consistent with EN4 inhibition of the MYC pathway, Signature Commons search of EN4-mediated transcriptional changes using the MSigDB Collection H hallmark gene set showed MYC target genes as one of the most significantly enriched pathways, alongside E2F and G2M checkpoint target genes that are also related to MYC and cell proliferation pathways (Fig. 4e) (Clarke et al.; Osorio, 2015). We further demonstrated that protein levels of representative MYC-regulated target genes including CDK2 and CDC25A were significantly lowered with EN4 treatment (Fig. 4f, 4g) (Amati et al., 1998; Hydbring et al., 2017; Zörnig and Evan, 1996). Consistent with previous studies showing that MYC inhibitors downregulate MYC levels in cells either through auto-regulation or destabilization and degradation, we also showed that MYC levels were substantially reduced with EN4 treatment (Fig. 4f, 4g) (Han et al., 2019; Struntz et al., 2019). In addition, we observed the upregulation of a MYC binding partner MIZ1 or ZBTB17 with EN4 treatment (Fig. S3f) (Wolf et al., 2015).
We next investigated whether EN4 affects cell proliferation, cell survival, or tumorigenesis of 231MFP breast cancer cells. EN4 treatment significantly impaired 231MFP breast cancer cell proliferation in a dose-and time-dependent manner with >90 % inhibition of proliferation at 50 μM (Fig. 4h; Fig. S3g). Further demonstrating the dependence of observed EN4 effects on MYC, EN4 impaired the cell survival of MYC-transformed mammary epithelial MCF10A cells, but not parental MCF10A cells that are not dependent on MYC (Fig. 4i). Daily EN4 treatment initiated after establishment of a 231MFP breast tumor xenograft in immune-deficient mice also significantly attenuated tumor growth in vivo (Fig. 4j). The striking anti-tumorigenic effects of EN4 in vivo despite the relatively poor potency of EN4, may be due to the covalent nature of EN4, in which MYC would remain inhibited by EN4 until protein turnover even if EN4 compound levels may have rapidly cleared from the body. Nonetheless, future studies will be required to further ascertain pharmacokinetic and pharmacodynamic parameters of EN4.
Structure-Activity Relationship of EN4 Analogs
Finally, we sought to preliminarily define the structure-activity relationship between EN4 and MYC in the absence of any structure for the region of MYC encompassing C171. We synthesized 18 analogs and tested these analogs and the previously described EN4-derived alkyne probes in dose response studies in MYC luciferase reporter assays in HEK293T cells (Supplementary Methods). Among these analogs, we showed that EN4–2 and EN4-alkyne-4 showed the best activity with EC50s of 1.7 and 1.4 μM, respectively, compared to EN4 EC50 of 12.5 μM (Table S4). In contrast, adding the electron-withdrawing trifluoromethyl group on the acrylamide warhead (EN4–6) or changing the acrylamide warhead to a chloroacetamide group (EN4–3) led to loss of activity. Furthermore, the attachment position of the phenoxybenzene moiety is critical for activity. Five analogs (EN4–13, EN4–14, EN4–15, EN4–16, and EN4–17) with either meta or para positioning relative to the amide all showed loss of activity (Table S4; Data S1). Interestingly, we also demonstrate that the non-reactive EN4 analogs EN4–18 also inhibited MYC luciferase reporter activity, suggesting that EN4 may possess inherent affinity for the apparent site within MYC that contains C171 (Table S4). Consistent with this premise, we show that EN4–18 significantly inhibits MYC/MAX DNA binding in vitro (Fig. S4). Despite EN4 targeting an apparently intrinsically disordered region of MYC with no structural insights, we still observe some structure-activity relationships that has enabled us to improve potency of EN4 by 7–8-fold.
Discussion
Overall, we have screened a cysteine-reactive covalent ligand library against MYC and have identified a hit compound EN4 that targets a unique ligandable site C171 within MYC to reduce MYC and MAX thermal stability, inhibit MYC/MAX DNA binding in vitro, impair MYC transcriptional activity in cells, downregulate many known MYC target genes as well as MYC itself, and inhibit proliferation and tumorigenesis in breast cancer cells. Despite targeting a predicted intrinsically disordered region within MYC, preliminary medicinal chemistry efforts gave rise to structure-activity relationships and yielded improved compounds. This study adds onto the pharmacopeia of small-molecules that target the MYC pathway, which include various modalities such as BRD4 and mTORC1 inhibitors and MAX/MAX stabilizers (Chen et al., 2018; McKeown and Bradner, 2014; Struntz et al., 2019; Whitfield et al., 2017). Sequence alignment indicates that C171 of c-MYC is not conserved in N-MYC or L-MYC suggesting that EN4 may selectively target c-MYC over other MYC proteins. Additionally, other forms of MYC have been previously identified such as a cytoplasmic form of MYC, known as MYC-nick, which retains the conserved MYC box regions but lacks nuclear localization signals and the domains necessary for MAX heterodimerization and DNA binding. MYC-nick has unique cytoplasmic functions in regulating actin acetylation and cell morphology (Conacci-Sorrell et al., 2010). Since this MYC-nick retains the NTD, EN4 which targets C171 within the NTD, may be capable of also targeting this proteolytically cleaved form of MYC. Future studies are necessary to determine whether EN4 and analogs selectively target c-MYC and MYC-nick over other MYC proteins, and whether unique covalent targeting of the NTD by EN4 may open avenues to explore MYC function compared to other indirect or direct MYC inhibitors.
While EN4 showed overall selectivity on a proteome-wide scale in profiling >1500 cysteines using competitive ABPP platforms, EN4 was not fully selective for c-MYC as indicated by 8 potential off-targets identified in this study. One of the off-targets is another important transcription factor involved in cell proliferation JUN (Karin et al., 1997). While we show here that the inhibitory action of EN4 towards MYC/MAX DNA binding and MYC transcriptional reporter activity is significantly attenuated upon mutation of C171 to a tyrosine, some of the activity observed here may be due to a combination of inhibiting MYC and some of the other EN4 off-targets. Now that we have shown the feasibility of direct and functional covalent targeting of c-MYC, future medicinal chemistry and expanded covalent ligand screening efforts are necessary to further optimize the potency and selectivity of EN4 as well as to discover additional scaffolds that may target c-MYC.
Our study also highlights how intrinsically disordered regions of classically “undruggable” targets may be functionally targeted with small-molecules and underscores the utility of covalent ligand screening coupled with proteomic approaches to uncover ligands against undruggable targets like MYC. While more work needs to be done, these data along with our recent case where we discovered that nimbolide functionally targeted a predicted intrinsically disordered region within the E3 ligase RNF114 (Spradlin et al., 2019), suggest that covalent ligand discovery might be a viable approach towards drugging intrinsically disordered proteins since it allows facile mass-spectrometry-based identification of modification events and ligandable pockets that might be unique to one of several confirmations that these proteins can adopt. Most importantly, covalent ligand binders and site of covalent modification can be identified in the absence of protein structures, unlike with reversible ligands or hits arising from DNA-encoded libraries where structural biology efforts are often needed to understand mode of ligand-protein binding. While not having structural enablement for a protein such as MYC may hamper medicinal chemistry efforts, we show here that one can still generate preliminary SAR against a predicted intrinsically disordered region within MYC.
In this study, we coupled in vitro biochemical screens of covalent ligands with pure MYC/MAX DNA binding assays and cellular MYC luciferase reporter assays to identify our initial hits. Future hit finding and medicinal chemistry efforts would also be benefited by intact mass adduct screening for covalent ligand/c-MYC protein adducts by mass spectrometry-based screening platforms (Resnick et al., 2019), thermal shift assays searching for MYC/MAX protein destabilizers as employed in this study, as well as more detailed kinetic studies to investigate the reversible and irreversible binding components of EN4 and optimized inhibitors to MYC. A future optimized covalent ligand targeting C171 of c-MYC would presumably show potent MYC/MAX DNA binding inhibition, possess inherent affinity for the site that encompasses C171 of c-MYC, confer a single mass adduct on c-MYC by intact mass screening, show better proteome-wide selectivity using chemoproteomic methods, inhibit MYC transcriptional activity in cells, confer reduced thermal stability of MYC and MAX in cells, downregulate MYC target genes, and impair cancer pathogenicity in a MYC-dependent manner. Understanding these various parameters will not only aid in optimizing covalent c-MYC inhibitors, but also in future pharmacological targeting of intrinsically disordered regions.
Overall, we demonstrate the utility of covalent ligand screening and chemoproteomic platforms in discovering covalent and functional ligands that target an intrinsically disordered C171 within c-MYC. Our results also further underscore the general utility of covalent ligand screening approaches for rapidly discovering and pharmacologically interrogating unique ligandable sites within intractable disease targets.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel K. Nomura, dnomura@berkeley.edu.
Materials Availability
Plasmids, compounds generated in this study will be made available upon reasonable request.
Data and Code Availability
Data generated in this study will be made available upon reasonable request. No code was developed for this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
The 231MFP cells were obtained from Prof. Benjamin Cravatt and were generated from explanted tumor xenografts of MDA-MB-231 cells as previously described (Jessani et al., 2004). HEK293T and MCF10A cells were obtained from the American Type Culture Collection. HEK293T cells were cultured in DMEM containing 10% (v/v) fetal bovine serum (FBS) and maintained at 37 °C with 5% CO2. The 231MFP cells were cultured in L15 medium containing 10% FBS and maintained at 37 °C with 0% CO2. MCF10A cells were cultured in DMEM/ F12K medium containing 5% horse serum, 20 ng/ml epidermal growth factor, 100 ng/ml cholera toxin, 10 ng/ml insulin, and 500 ng/ml hydrocortisone, and maintained at 37C with 5% CO2. Unless otherwise specified, all cell culture materials were purchased from Gibco. 231MFP and MCF10A cells are female in origin. It is not known whether HEK293T cells are from male or female origin.
METHOD DETAILS
Covalent Ligand Library
Compounds were purchased from Enamine LLC and structures of compounds are all listed in Table S1.
MYC/MAX In Vitro DNA Binding Experiments
The protocol was adapted from the Abcam c-Myc Transcription Factor Assay kit (ab207200). 50 mM DMSO stock solutions of compound were diluted into dPBS then vortexed. 0.2 μg of c-MYC (Abcam, ab169901) was added to each Eppendorf tube then mixed gently. c-MYC was incubated with compound or DMSO control for 30 minutes at 37 °C. After incubation, 0.2 μg of MAX (Origene, TP306812) was added to each Eppendorf tube then mixed gently. Samples were added to 96-well plate containing the consensus sequence for MYC/MAX binding. Experiment proceeded following manufacturer’s protocol for binding, primary antibody, and secondary antibody incubation steps. After addition of developing and stop solution, the OD450 was read on a Molecular Devices VersaMax microplate reader. Data was normalized to DMSO control after subtracting blank wells.
MYC Luciferase Reporter Experiments
MYC luciferase reporter was purchased from Qiagen (CCS-012L) and performed according to manufacturer’s protocol. This reporter assay employed a MYC-responsive firefly luciferase reporter, encoding a luciferase reporter gene under the control of a basal promoter element, the TATA box, joined to tandem repeats of the MYC transcriptional E-box response element. The negative control construct was a non-inducible firefly luciferase under the control of the TATA box element without addition of transcriptional response elements. The positive control reporter was a construct for constitutively expressing GFP and firefly luciferase. Also included in the MYC reporter, positive and negative control is a constitutively expressing Renilla luciferase that acts as an internal control for normalization against cytotoxicity, transfection efficiency, and technical variability. In each set of assays performed, the MYC-reporter controls treated with vehicle and positive controls showed >100 x more signal than the negative control.
Prior to transfection, HEK293T cells were seeded into a 96-well plate (35,000 cells/well in 100 μL). Negative control, positive control, or Myc luciferase reporter were diluted into Opti-MEM I medium (1 μL DNA into 25 μL Opti-MEM I per each well). Lipofectamine 2000 (Invitrogen, 11668019) was diluted into Opti-MEM I medium (1 μL Lipofectamine 2000 into 25 μL Opti-MEM I per each well). DNA and diluted Lipofectamine 2000 were combined in a 1:1 ratio then incubated at room temperature for 25 minutes. 50 μL of DNA-Lipofectamine 2000 mixture was added to each well. 24 hours post-transfection, media was carefully aspirated from each well, and 50 μL of fresh media containing either DMSO or compound was added. After 24 hours of compound treatment, Dual-Glo luciferase (Promega, E2920) workup was performed according to manufacturer’s protocol. Firefly and Renilla luminescence were read on a SpectraMax i3 plate reader. Background luminescence was subtracted using a blank control then Firefly:Renilla was calculated for each individual well.
LC-MS/MS Analysis of MYC
Purified c-MYC (5 μg, Origene, 301611) in 50 μL PBS was incubated 30 min at room temperature either with DMSO vehicle or EN4 (50 μM). The DMSO control was then treated with light iodoacetamide while the EN4 treated sample was incubated with heavy iodoacetamide for 1 h each at room temperature (200 μM final concentration, Sigma-Aldrich, 721328). The samples were precipitated by additional of 12.5 μl of 100% (w/v) trichloroacetic acid and the treated and control groups were combined pairwise (10 μg total) before cooling to −80 °C for 2 h. The combined sample was then spun for at max speed for 10 min at 4 °C, supernatant was carefully removed and the sample was washed with ice-cold 0.01 M HCl/90% acetone solution. The pellet was resuspended in 4 M urea containing 0.1% Protease Max (Promega, V2071) and diluted in 40 mM ammonium bicarbonate buffer. The samples were reduced with 10 mM TCEP at 60 °C for 30 min. The sample was then diluted 50% with PBS before sequencing grade trypsin (1 μg per sample, Promega, V5111) was added for an overnight incubation at 37 °C. The next day, the sample was centrifuged at 13,200 r.p.m. for 30 min. The supernatant was transferred to a new tube and acidified to a final concentration of 5% formic acid and stored at −80 °C until mass spectrometry analysis.
Western Blotting Experiments
Antibodies to c-MYC (Cell Signaling Technology, 9402S), CDK4 (Abcam, ab199728), CCND2 (Abcam, ab207604), FLAG (Cell Signaling Technology, 14793S), and β-actin (Cell Signaling Technology, 4970S) were obtained from various commercial sources and dilutions were prepared per the recommended manufacturers’ procedures. Proteins were resolved by SDS–PAGE and transferred to nitrocellulose membranes using the iBlot system (Invitrogen). Blots were blocked with 5% Milk in Tris-buffered saline containing Tween 20 (TBST) solution for 1 h at room temperature, washed in TBST and probed with primary antibody diluted in diluent, as recommended by the manufacturer, overnight at 4 °C. Following washes with TBST, the blots were incubated in the dark with secondary antibodies purchased from Li-Cor Biosciences and used at 1:10,000 dilution in 5% Milk in TBST at room temperature for 1 h. Blots were visualized using an Odyssey Li-Cor scanner after additional washes.
Gel-ABPP Experiments
Gel-based ABPP methods were performed as previously described (Grossman et al., 2017; Spradlin et al., 2019). Recombinant pure human proteins were purchased from Origene or Abcam as indicated. Pure proteins (0.3 μg) were pre-treated with DMSO vehicle or EN4 for 30 min at 37 °C in an incubation volume of 50 μl PBS and were subsequently treated with alkyne-functionalized probes for 2 h at 37 °C. CuAAC was performed for 1 h at room temperature to append rhodamine-azide (1 μM final concentration) onto alkyne probe-labeled proteins. Samples were then diluted with 20 μl of 4x reducing Laemmli SDS sample loading buffer (Alfa Aesar) and heated at 90 °C for 5 min. The samples were separated on precast 4–20% TGX gels (Bio-Rad Laboratories, Inc.). Before scanning by ChemiDoc MP (Bio-Rad Laboratories, Inc.), gels were fixed in a solution of 10% acetic acid, 30% ethanol for 1 h.
For gel-based ABPP experiments with JUN, recombinant pure human c-JUN protein (0.1 μg per sample) was pretreated with either DMSO vehicle or EN4 at 37 °C for 1 h in 25 μl PBS, and subsequently treated with 200 nM Tetramethylrhodamine-5-Iodoacetamide Dihydroiodide (IA-rhodamine) (Setareh Biotech 6222) at room temperature for 1 h. Samples were incubated with 10 μL 4x Laemmli sample buffer, boiled at 95°C for 6 min, and separated by SDS/PAGE. Probe-labeled proteins were analyzed by in-gel rhodamine fluorescence using a ChemiDoc MP (Bio-Rad). Protein loading was assessed by Silver Staining.
Vectors for cMYC Overexpression
The pCMV6-Entry-cMYC was purchased from Origene (RC201611). The wild-type mammalian expression plasmids contained a C-terminal FLAG tag. The corresponding pCMV6-Entry-eGFP vector was constructed via Gibson Assembly with primers: GATCTGCCGCCGCGATCGCCatggtgagcaagggcgag, TCGAGCGGCCGCGTACGCGTcttgtacagctcgtccatgcc to amplify the eGFP ORF with desired overlaps, and ACGCGTACGCGGCCG, GGCGATCGCGGCGG to linearize the pCMV6-Entry backbone.
Expression of Wild-Type and C171Y Mutant MYC in HEK293T Cells with MYC Luciferase Reporter Experiments
HEK293T cells were seeded at 35,000 cells/well in a 96-well plate. Cells were transfected with the MYC luciferase reporter according to protocol described above using Lipofectamine 2000 (Invitrogen, 11668019) as a transfection reagent. Cells were also transfected with either FLAG-eGFP, FLAG-tagged wild-type c-MYC (Origene, RC201611) or C171Y cMYC mammalian expression plasmids using Lipofectamine 2000. Cells were transfected with 1 μg/well of corresponding plasmid in a final ratio of 3:1 Lipofectamine 2000:DNA. The c-MYC C171Y mutant plasmid was generated using Q5 site-directed mutagenesis kit according to the manufacturer’s instructions (New England Biolabs, E0554S). Sequencing was confirmed with Quintara Biosciences. 24 hours post-transfection, media was carefully aspirated from each well, and 50 μL of fresh media containing either DMSO or compound was added. After 24 hours of compound treatment, Dual-Glo luciferase (Promega, E2920) detection was performed according to manufacturer’s protocol. Firefly and Renilla luminescence were read on a SpectraMax i3 plate reader. Background luminescence was subtracted using a blank control then Firefly:Renilla was calculated for each individual well. Lysates were also obtained for confirmation FLAG-protein overexpression by Western blotting.
Knockdown of c-JUN by RNA interference with MYC Luciferase Reporter Experiments
RNA interference was performed by using siRNA purchased from Sigma MISSION siRNA library (SI007 siRNA Fluor Universal Negative Control#1, and PDSIRNA2D SASI_Hs01_00150279 siJUN). HEK293T cells were seeded at 35,000 cells/well in a 96-well plate. On Day 0, cells were co-transfected with 150 ng siRNA and myc luciferase reporter per well according to previous protocol using Lipofectomine 2000 (Invitrogen, 11668019). On Day 1, 24 hours post-transfection, 75 μL of fresh media containing vehicle DMSO or EN4 was added to cells in n=6 replicates. After 24 h of compound treatment, Dual-Glo luciferase (Promega, E2920) workup was performed according to manufacturer’s protocol. Firefly and Renilla luminescence were read on a SpectraMax i3 plate reader. Background luminescence was subtracted using a blank control then Firefly:Renilla was calculated for each individual well.
EN4-Alkyne-4 Labeling of Endogenous MYC
231 MFP cells were treated at 70% confluency with DMSO or 50 μM EN4-Alkyne-4 in situ for 4 h. Cells were harvested, lysed via sonication, and the resulting lysate normalized to 3.2 mg/mL per sample. Following normalization, 100 μL of each lysate sample was removed for Western blot analysis of input, and 500 μL of each lysate sample was incubated for 1 h at room temperature with 10 μL of 5 mM biotin picolyl azide (in water) (Sigma Aldrich 900912), 10 μL of 50 mM TCEP (in water), 30 μL of TBTA ligand (0.9mg/mL in DMSO:t-butanol = 1:4), and 10 μL of 50 mM Copper (II) Sulfate (12.5 mg/mL in water). Proteins were precipitated, washed 3 x with cold MeOH, resolubilized in 1 mL of 1.2% SDS/PBS (w/v), heated for 5 min at 90 °C, and centrifuged to remove any insoluble components. 1 mL of each resolubilized sample was then transferred to 15 mL conical tubes containing 5 mL PBS with 100 μL high capacity streptavidin resin (Thermo Scientific 20357) to give a final SDS concentration of 0.2%. Samples were incubated with the streptavidin beads at 4°C overnight on a rotator.
The following day the samples were warmed to room temperature and washed with 0.2% SDS, then transferred to spin columns and further washed 3 x with 50 μL PBS and 3 x with 500 μL water to remove non-probe-labeled proteins. The washed beads were resuspended in 100 μL PBS, transferred to 1.5 mL eppendorf low-adhesion tubes, combined with 30 μL Laemmli Sample Buffer (4 x) and heated to 95°C. Immunopreciptated proteins in each sample were then analyzed by Western blotting to look for enriched c-MYC (Abcam ab32072) versus non-enriched control Vinculin (Cell Signaling Technology 4650S).
IsoTOP-ABPP Chemoproteomic Experiments
IsoTOP-ABPP studies were done as previously reported(Grossman et al., 2017; Spradlin et al., 2019; Weerapana et al., 2010). Cells were lysed by probe sonication in PBS and protein concentrations were measured by BCA assay. Cells were treated for 4 h with either DMSO vehicle or EN4 (from 1,000x DMSO stock) before cell collection and lysis. Proteomes were subsequently labeled with IA-alkyne labeling (100 μM) for 1 h at room temperature. CuAAC was used by sequential addition of tris(2-carboxyethyl)phosphine (1 mM, Strem, 15–7400), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (34 μM, Sigma, 678937), copper(II) sulfate (1 mM, Sigma, 451657) and biotin-linker-azide—the linker functionalized with a tobacco etch virus (TEV) protease recognition sequence as well as an isotopically light or heavy valine for treatment of control or treated proteome, respectively. After CuAAC, proteomes were precipitated by centrifugation at 6,500g, washed in ice-cold methanol, combined in a 1:1 control:treated ratio, washed again, then denatured and resolubilized by heating in 1.2% SDS–PBS to 80 °C for 5 min. Insoluble components were precipitated by centrifugation at 6,500g and soluble proteome was diluted in 5 ml 0.2% SDS–PBS. Labeled proteins were bound to streptavidin-agarose beads (170 μl resuspended beads per sample, Thermo Fisher, 20349) while rotating overnight at 4 °C. Bead-linked proteins were enriched by washing three times each in PBS and water, then resuspended in 6 M urea/PBS, and reduced in TCEP (1 mM, Strem, 15–7400), alkylated with iodoacetamide (18 mM, Sigma), before being washed and resuspended in 2 M urea/PBS and trypsinized overnight with 0.5 μg /μL sequencing grade trypsin (Promega, V5111). Tryptic peptides were eluted off. Beads were washed three times each in PBS and water, washed in TEV buffer solution (water, TEV buffer, 100 μM dithiothreitol) and resuspended in buffer with Ac-TEV protease (Invitrogen, 12575–015) and incubated overnight. Peptides were diluted in water and acidified with formic acid (1.2 M, Fisher, A117–50) and prepared for analysis.
IsoTOP-ABPP Mass Spectrometry Analysis
Peptides from all chemoproteomic experiments were pressure-loaded onto a 250 μm inner diameter fused silica capillary tubing packed with 4 cm of Aqua C18 reverse-phase resin (Phenomenex, 04A-4299), which was previously equilibrated on an Agilent 600 series high-performance liquid chromatograph using the gradient from 100% buffer A to 100% buffer B over 10 min, followed by a 5 min wash with 100% buffer B and a 5 min wash with 100% buffer A. The samples were then attached using a MicroTee PEEK 360 μm fitting (Thermo Fisher Scientific p-888) to a 13 cm laser pulled column packed with 10 cm Aqua C18 reverse-phase resin and 3 cm of strong-cation exchange resin for isoTOP-ABPP studies. Samples were analyzed using an Q Exactive Plus mass spectrometer (Thermo Fisher Scientific) using a five-step Multidimensional Protein Identification Technology (MudPIT) program, using 0, 25, 50, 80 and 100% salt bumps of 500 mM aqueous ammonium acetate and using a gradient of 5–55% buffer B in buffer A (buffer A: 95:5 water:acetonitrile, 0.1% formic acid; buffer B 80:20 acetonitrile:water, 0.1% formic acid). Data were collected in data-dependent acquisition mode with dynamic exclusion enabled (60 s). One full mass spectrometry (MS1) scan (400–1,800 mass-to-charge ratio (m/z)) was followed by 15 MS2 scans of the nth most abundant ions. Heated capillary temperature was set to 200 °C and the nanospray voltage was set to 2.75 kV.
Data were extracted in the form of MS1 and MS2 files using Raw Extractor v.1.9.9.2 (Scripps Research Institute) and searched against the Uniprot human database using ProLuCID search methodology in IP2 v.3 (Integrated Proteomics Applications, Inc.)(Xu et al., 2015). Cysteine residues were searched with a static modification for carboxyaminomethylation (+57.02146) and up to two differential modifications for methionine oxidation and either the light or heavy TEV tags (+464.28596 or +470.29977, respectively). Peptides were required to be fully tryptic peptides and to contain the TEV modification. ProLUCID data were filtered through DTASelect to achieve a peptide false-positive rate below 5%. Only those probe-modified peptides that were evident across two out of three biological replicates were interpreted for their isotopic light to heavy ratios. For those probe-modified peptides that showed ratios greater than two, we only interpreted those targets that were present across all three biological replicates, were statistically significant and showed good quality MS1 peak shapes across all biological replicates. Light versus heavy isotopic probe-modified peptide ratios are calculated by taking the mean of the ratios of each replicate paired light versus heavy precursor abundance for all peptide-spectral matches associated with a peptide. The paired abundances were also used to calculate a paired sample t-test P value in an effort to estimate constancy in paired abundances and significance in change between treatment and control. P values were corrected using the Benjamini–Hochberg method.
Thermal Shift Assays
231MFP cells (80% confluent in 10 cm culture dishes) were treated with EN4 or vehicle (DMSO) for 120 min at 50 μM. Cells were harvested and washed once with PBS, then suspended in 1 mL of PBS containing protease inhibitor cocktail (Pierce A32955) and also maintained with 50uM compound or DMSO. The cell suspension was allocated into eight 0.2 mL PCR tubes with 100μL volume per tube, and each tube designated a temperature (37.0 °C, 38.4 °C, 40.9 °C, 45.0 °C, 49.8 °C, 53.5 °C, 56.3 °C, and 58.0 °C). Samples were heated at their respective temperatures for 3 min in a 96-well thermal cycler, then incubated at room temperature (25 °C) for 3 min. After this incubation, cells were immediately snap-lysed in liquid nitrogen (3 freeze-thaw cycles). Cell debris along with precipitated and aggregated proteins were removed from lysate by centrifuging samples at 20,000 g for 20 min at 4 °C. Samples were then boiled for 7 min at 90 °C after addition of loading buffer. Lysate samples were analyzed by Western Blot analysis. Protein intensity was quantified through Image J software.
RNA Sequencing Analysis
231MFP cells were seeded into 6 cm dishes and cells were treated with DMSO vehicle or EN4 (50 μM) for 24 h. Cells were harvested by scraping and RNA was isolated using Qiagen RNeasy mini kit with Qiagen DNase max kit to remove any DNA contamination. mRNA was enriched for using Oligo dT beads from the KAPA mRNA Capture Kit (KK8581). Subsequent library preparation steps of fragmentation, adapter ligation and cDNA synthesis were done on the enriched mRNA using the KAPA RNA HyperPrep kit (KK8540). Truncated universal stub adapters were used for ligation, and indexed primers were used during PCR amplification to complete the adapters and to enrich the libraries for adapter-ligated fragments. Samples were checked for quality on an AATI (now Agilent) Fragment Analyzer. Samples were then transferred to the Vincent J. Coates Genomics Sequencing Laboratory (GSL), another QB3-Berkeley Core Research Facility at UC Berkeley, where Illumina sequencing library molarity was measured with quantitative PCR with the Kapa Biosystems Illumina Quant qPCR Kits on a BioRad CFX Connect thermal cycler. Libraries were then pooled evenly by molarity and sequenced on an Illumina HiSeq4000 (150PE) lane. Raw sequencing data was converted into FASTQ format, sample specific files using the Illumina bcl2fastq2 software on the sequencing centers local Linux server system.
RNA-seq reads were quantified using Salmon (Patro et al., 2017) to generate Transcripts Per Million (TPM) normalized data that was then rolled into gene level summaries. Treated versus control groups were contrasted on the log transformed data after removal of low variance genes with a t-test. This was followed by Benjamini–Hochberg false discovery rate correction and log2 fold change calculation for each. We performed signature enrichment analysis of genes with an FDR < 0.1 and Fold change <= 0.75 (567 genes) using the Signature Commons platform against the MSigDB Collection H hallmark gene set.
Survival and Proliferation Experiments
Cell survival and proliferation assays were performed using Hoechst 33342 dye (Invitrogen, H3570) according to the manufacturer’s protocol and as previously described (Grossman et al., 2017; Spradlin et al., 2019). For survival assays, cells were seeded and treated with serum-free media. Cells were seeded into 96-well plates (40,000 for survival) in a volume of 150 μL and allowed to adhere overnight. Cells were treated with an additional 50 μL of media containing compound in DMSO. After the appropriate incubation period, the media was removed from each well, and 100 μL of staining solution containing 10% formalin and Hoechst 33342 dye was added to each well and incubated for 15 min in the dark at room temperature. Staining solution was then removed, and wells were washed with dPBS before imaging.
For proliferation experiments, 231MFP cells were seeded at 15,000 cells/well in a black clear-bottom 96 well plate (avoiding the outer edges of the plate) in serum-containing L-15 media. The following day, cells were treated with 150 μL media containing vehicle (DMSO) or compound (EN4) at designated concentrations. For each subsequent treatment day, media was aspirated and replaced with fresh media containing DMSO or respective treatment. Proliferation was analyzed via Hoecsht stain protocol as previously above.
Tumor Xenograft Study
C.B17 SCID female mice (6–8 weeks old) were injected subcutaneously in the flank with 231MFP cells (1 × 106 cells) suspended in serum-free media. Mice were exposed via intraperitoneal injection with either vehicle (18:1:1 PBS/ethanol/PEG40) or 50 mg/kg EN4 once per day starting 2 weeks after injection of cancer cells. Tumor size was assessed weekly by caliper measurements.
Synthetic Methods of EN4 Analogs
NMR data can be found in Data S1
Preparation of 4-(acrylamidomethyl)benzoic acid
Step 1: methyl 4-(aminomethyl)benzoate (4.03 g, 20 mmol) and TEA (5.58 ml, 40.0 mmol) were stirred in acryloyl chloride (1.625 ml, 20.00 mmol) then acryloyl chloride (1.625 ml, 20.00 mmol) was added at 0 °C. The reaction mixture was stirred for 16 hr at ambient temperature, then it was diluted with EtOAc, washed with water, brine, dried over Na2SO4 and concentrated under reduced pressure. The resulting crude product was purified by flash column chromatography on silica gel eluting (10–100% EtOAc / Heptane) to give methyl 4-(acrylamidomethyl)benzoate as a white solid (3.03 g, 66%): 1H NMR (400 MHz, Chloroform-d) δ 8.06 – 7.95 (m, 2H), 7.38 (d, J = 8.4 Hz, 2H), 6.37 (dd, J = 17.0, 1.4 Hz, 1H), 6.16 (dd, J = 17.0, 10.3 Hz, 1H), 5.72 (dd, J = 10.3, 1.4 Hz, 1H), 4.60 (d, J = 6.0 Hz, 2H), 3.93 (s, 3H); LCMS Rt 0.63 mins; m/z 220.1 (M+H)
Step 2: To a solution of methyl 4-(acrylamidomethyl)benzoate (500 mg, 2.28 mmol) in THF (5.70 ml) and water (5.70 ml) was added LiOH.H2O (383 mg, 9.12 mmol). The resulting solution was stirred at ambient temperature for 4 hr at which point HCl was added until the solution reached pH~2–3. The reaction mixture was then extracted with EtOAc (30 ml x2), the combined organic phases washed with brine, dried over Na2SO4 and concentrated to give 4-(acrylamidomethyl)benzoic acid as a white solid: LCMS Rt 0.49 mins; m/z 206.2 (M+H).
This material was used to prepare the following compounds according to general procedure A:
Procedure A
To 4-(acrylamidomethyl)benzoic acid (103 mg, 0.5 mmol), the corresponding aniline (0.500 mmol) in DMF (5 ml) was added EDC (115 mg, 0.600 mmol) and DMAP (61.1 mg, 0.500 mmol). The reaction mixture was stirred at 60 °C for 2 hr before being diluted with EtOAc (50 mL), washed with water, brine, dried over Na2SO4, filtered and the filtrate concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel eluting (20–100% EtOAc/Heptane) to give the desired product
4-(acrylamidomethyl)-N-(2-(4-(2,2,2-trifluoroethoxy)phenoxy)phenyl)benzamide
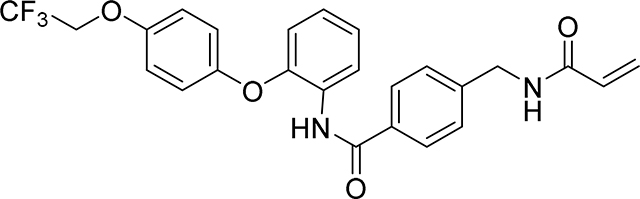
19 mg, 39 %
1H NMR (400 MHz, DMSO-d6) δ 9.75 (s, 1H), 8.66 (t, J = 6.0 Hz, 1H), 7.81 (d, J = 8.3 Hz, 2H), 7.74 (dd, J = 7.5, 2.0 Hz, 1H), 7.35 (d, J = 8.3 Hz, 2H), 7.24 – 7.12 (m, 2H), 7.08 – 7.03 (m, 2H), 7.02 – 6.96 (m, 2H), 6.89 (dd, J = 7.8, 1.7 Hz, 1H), 6.28 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.2 Hz, 1H), 5.63 (dd, J = 10.1, 2.2 Hz, 1H), 4.70 (q, J = 8.9 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H)
19F NMR (376 MHz, DMSO-d6) δ −72.59
HRMS calcd for C25H22F3N2O4(M+H)+ 471.1532, found 471.1528
4-(acrylamidomethyl)-N-(2-phenoxyphenyl)benzamide
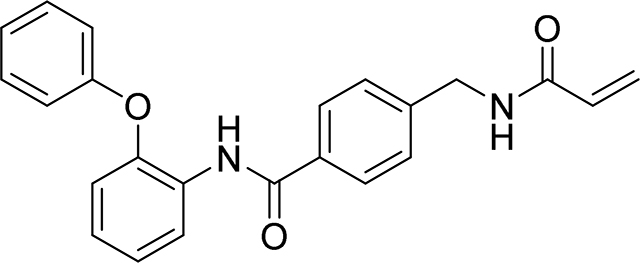
83 mg, 45%
1H NMR (400 MHz, DMSO-d6) δ 9.78 (s, 1H), 8.68 (t, J = 6.0 Hz, 1H), 7.77 – 7.69 (m, 3H), 7.33 (ddt, J = 8.4, 6.0, 3.0 Hz, 4H), 7.28 – 7.17 (m, 2H), 7.11 – 7.04 (m, 1H), 7.03 – 6.99 (m, 1H), 6.99 – 6.93 (m, 2H), 6.28 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.2 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.39 (d, J = 6.0 Hz, 2H).
13C NMR (101 MHz, DMSO-d6) δ 163.53, 163.07, 155.15, 147.69, 141.47, 131.31, 129.90, 128.22, 127.99, 126.07, 125.48, 125.10, 124.86, 124.06, 122.29, 121.51, 118.01, 116.18, 40.21
HRMS calcd for C23H21N2O3(M+H)+ 373.1552, found 373.1552
4-(acrylamidomethyl)-N-(2-(p-tolyloxy)phenyl)benzamide
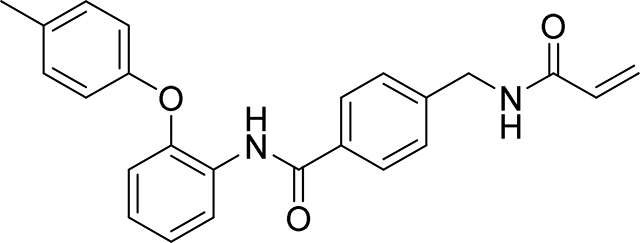
94 mg, 48%
1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.68 (t, J = 6.0 Hz, 1H), 7.78 (d, J = 8.3 Hz, 2H), 7.74 (dd, J = 7.5, 2.1 Hz, 1H), 7.34 (d, J = 8.4 Hz, 2H), 7.23 – 7.12 (m, 4H), 6.93 (dd, J = 7.7, 1.8 Hz, 1H), 6.91 – 6.86 (m, 2H), 6.28 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.2 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.40 (d, J = 6.0 Hz, 2H), 2.25 (s, 3H)
13C NMR (101 MHz, DMSO-d6) δ 165.08, 164.65, 154.35, 149.81, 143.08, 132.90, 132.28, 131.48, 130.16, 129.25, 127.67, 127.09, 126.44, 126.30, 125.63, 123.40, 118.85, 118.12, 41.80, 20.20
HRMS calcd for C24H23N2O3(M+H)+ 387.1709, found 387.1708
4-(acrylamidomethyl)-N-(2-(3-cyanophenoxy)phenyl)benzamide
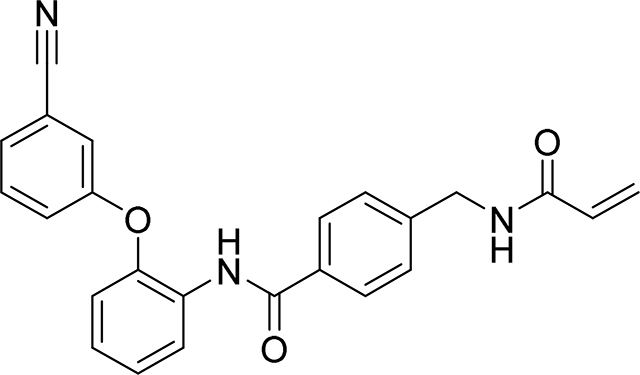
69 mg, 35%
1H NMR (400 MHz, DMSO-d6) δ 9.89 (s, 1H), 8.68 (t, J = 6.0 Hz, 1H), 7.74 – 7.66 (m, 3H), 7.55 – 7.48 (m, 2H), 7.38 – 7.24 (m, 6H), 7.17 – 7.11 (m, 1H), 6.28 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.2 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.39 (d, J = 6.0 Hz, 2H)
13C NMR (101 MHz, DMSO-d6) δ 165.23, 164.65, 157.21, 148.59, 143.14, 132.67, 131.47, 131.19, 129.83, 127.62, 127.53, 127.04, 126.68, 125.64, 124.97, 122.49, 120.55, 120.46, 118.19, 112.28, 41.79
HRMS calcd for C24H20N3O3(M+H)+ 398.1505, found 398.1503
4-(acrylamidomethyl)-N-(2-(2-methoxyphenoxy)phenyl)benzamide
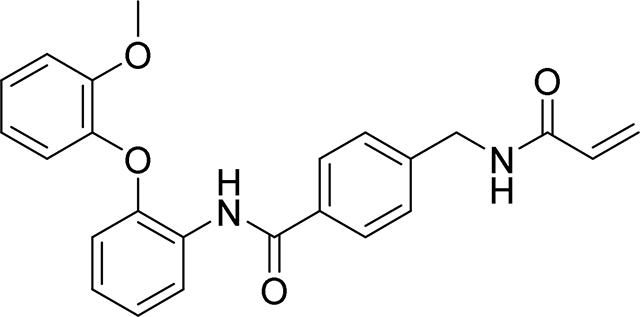
108 mg, 53%
1H NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H), 8.70 (t, J = 6.0 Hz, 1H), 7.89 (d, J = 8.3 Hz, 2H), 7.82 (dd, J = 7.3, 2.3 Hz, 1H), 7.38 (d, J = 8.3 Hz, 2H), 7.21 – 7.03 (m, 5H), 6.95 (ddd, J = 8.0, 6.7, 2.2 Hz, 1H), 6.73 – 6.68 (m, 1H), 6.29 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.2 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.42 (d, J = 6.0 Hz, 2H), 3.73 (s, 3H)
13C NMR (101 MHz, DMSO-d6) δ 162.91, 162.60, 148.98, 148.07, 142.06, 141.12, 130.90, 129.42, 126.07, 125.56, 125.16, 123.74, 123.58, 123.34, 123.25, 120.41, 119.16, 118.97, 114.32, 111.15, 53.55, 39.76
HRMS calcd for C24H23N2O4(M+H)+ 403.1658, found 403.1644
4-(acrylamidomethyl)-N-(2-(3-methoxyphenoxy)phenyl)benzamide
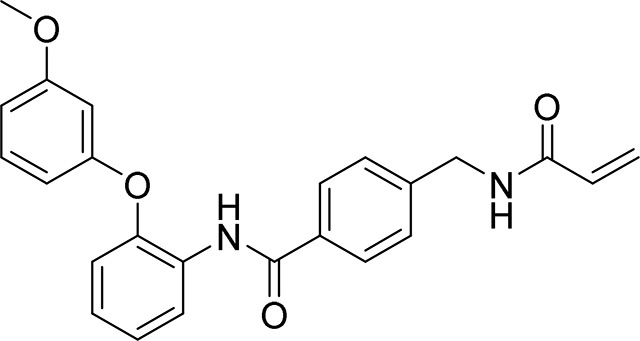
79 mg, 39%
1H NMR (400 MHz, DMSO-d6) δ 9.76 (s, 1H), 8.68 (t, J = 6.0 Hz, 1H), 7.79 – 7.72 (m, 3H), 7.32 (d, J = 8.3 Hz, 2H), 7.28 – 7.17 (m, 3H), 7.08 – 7.00 (m, 1H), 6.68 – 6.63 (m, 1H), 6.57 – 6.48 (m, 2H), 6.28 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.2 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.39 (d, J = 6.0 Hz, 2H), 3.68 (s, 3H).
13C NMR (101 MHz, DMSO-d6) δ 165.12, 164.65, 160.47, 157.93, 149.15, 143.08, 132.88, 131.48, 130.27, 129.60, 127.65, 127.06, 126.74, 126.46, 125.64, 124.00, 119.79, 109.80, 108.75, 103.82, 55.15, 41.79
HRMS calcd for C24H23N2O4(M+H)+ 403.1658, found 403.1653
4-(acrylamidomethyl)-N-(3-(o-tolyloxy)phenyl)benzamide
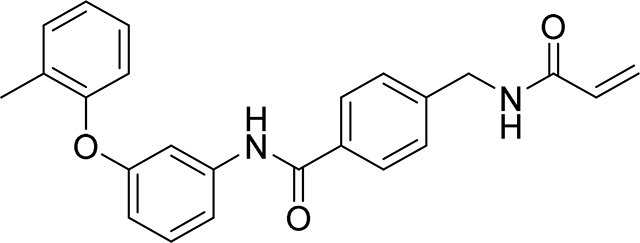
92 mg, 47%
1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 8.71 (t, J = 6.0 Hz, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.53 – 7.49 (m, 1H), 7.42 – 7.28 (m, 5H), 7.27 – 7.21 (m, 1H), 7.13 (td, J = 7.4, 1.0 Hz, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.72 – 6.61 (m, 1H), 6.29 (dd, J = 17.1, 10.1 Hz, 1H), 6.14 (dd, J = 17.1, 2.2 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.42 (d, J = 6.0 Hz, 2H), 2.18 (s, 3H)
13C NMR (101 MHz, DMSO-d6) δ 164.01, 163.32, 156.26, 152.30, 141.80, 139.35, 131.97, 130.17, 130.14, 128.48, 128.01, 126.43, 126.18, 125.76, 124.30, 123.02, 118.58, 112.86, 110.77, 107.08, 40.49, 14.48
HRMS calcd for C24H23N2O3(M+H)+ 387.1709, found 387.1705
4-(acrylamidomethyl)-N-(4-(p-tolyloxy)phenyl)benzamide
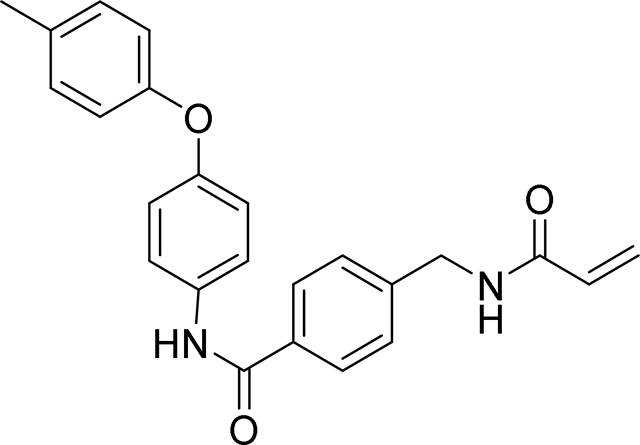
93 mg, 48%
1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 8.72 (t, J = 6.0 Hz, 1H), 7.92 (d, J = 8.3 Hz, 2H), 7.79 – 7.72 (m, 2H), 7.40 (d, J = 8.3 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 7.04 – 6.95 (m, 2H), 6.93 – 6.87 (m, 2H), 6.30 (dd, J = 17.1, 10.1 Hz, 1H), 6.14 (dd, J = 17.1, 2.2 Hz, 1H), 5.65 (dd, J = 10.1, 2.2 Hz, 1H), 4.43 (d, J = 6.0 Hz, 2H), 2.28 (s, 3H)
13C NMR (101 MHz, DMSO-d6) δ 165.04, 164.67, 154.80, 152.69, 143.02, 134.63, 133.42, 132.18, 131.50, 130.33, 127.71, 127.14, 125.64, 121.98, 118.66, 118.23, 41.85, 20.21
HRMS calcd for C24H23N2O3(M+H)+ 387.1709, found 387.1702
4-(acrylamidomethyl)-N-(4-(2-methoxyphenoxy)phenyl)benzamide
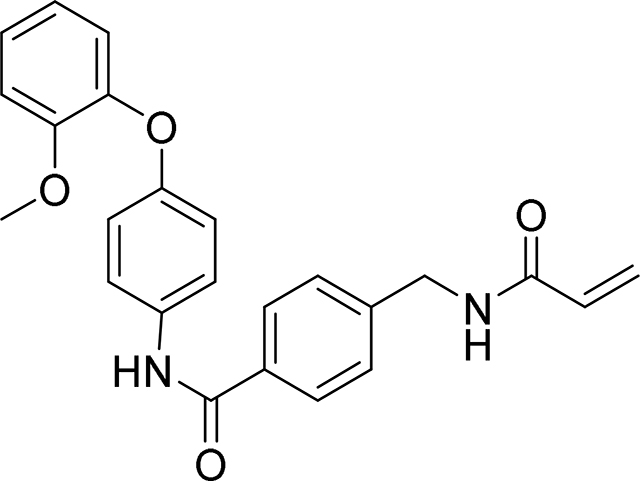
99 mg, 48%
1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.71 (t, J = 6.0 Hz, 1H), 7.91 (d, J = 8.3 Hz, 2H), 7.71 – 7.65 (m, 2H), 7.39 (d, J = 8.3 Hz, 2H), 7.22 – 7.14 (m, 2H), 7.05 – 6.92 (m, 2H), 6.89 – 6.81 (m, 2H), 6.30 (dd, J = 17.1, 10.1 Hz, 1H), 6.14 (dd, J = 17.1, 2.2 Hz, 1H), 5.65 (dd, J = 10.1, 2.2 Hz, 1H), 4.43 (d, J = 6.0 Hz, 2H), 3.75 (s, 3H)
13C NMR (101 MHz, DMSO-d6) δ 162.79, 162.51, 151.51, 149.01, 141.89, 140.81, 131.55, 131.29, 129.35, 125.53, 124.97, 123.49, 123.07, 119.82, 118.93, 118.88, 114.23, 111.16, 53.40, 39.69
HRMS calcd for C24H23N2O4(M+H)+ 403.1658, found 403.1648
4-(acrylamidomethyl)-N-(4-(3-methoxyphenoxy)phenyl)benzamide
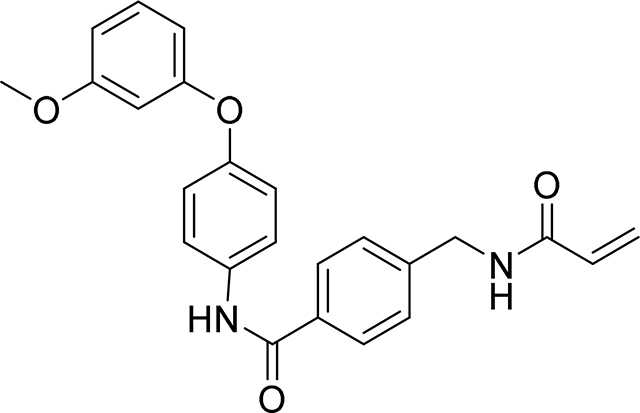
121 mg, 58%
1H NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 8.72 (t, J = 6.0 Hz, 1H), 7.96 – 7.89 (m, 2H), 7.83 – 7.76 (m, 2H), 7.45 – 7.37 (m, 2H), 7.27 (t, J = 8.2 Hz, 1H), 7.07 – 7.00 (m, 2H), 6.69 (ddd, J = 8.5, 2.4, 0.9 Hz, 1H), 6.56 (t, J = 2.3 Hz, 1H), 6.52 (ddd, J = 8.2, 2.3, 0.9 Hz, 1H), 6.30 (dd, J = 17.1, 10.1 Hz, 1H), 6.14 (dd, J = 17.1, 2.2 Hz, 1H), 5.65 (dd, J = 10.2, 2.2 Hz, 1H), 4.43 (d, J = 5.9 Hz, 2H), 3.73 (s, 3H)
13C NMR (101 MHz, DMSO-d6) δ 164.44, 164.01, 159.98, 157.86, 151.13, 142.40, 134.46, 132.75, 130.84, 129.78, 127.07, 126.49, 124.99, 121.29, 118.75, 109.12, 108.00, 103.26, 54.55, 41.19
HRMS calcd for C24H23N2O4(M+H)+ 403.1658, found 403.1656
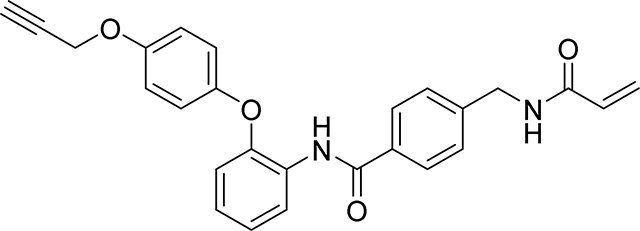
Preparation of 4-(acrylamidomethyl)-N-(2-(4-(prop-2-yn-1-yloxy)phenoxy)phenyl)benzamide
Step 1: To a suspension of K2CO3 (10.04 g, 72.7 mmol) in DMAc (45.4 ml) was added hydroquinone (2 g, 18.16 mmol) and 3-bromoprop-1-yne (80% in toluene w/w) (2.023 ml, 18.16 mmol). The resulting dark solution was stirred at 40 °C for 4 hr when the reaction was partitioned between EtOAc and water, washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude residue was purified by flash chromatography on silica gel eluting with (0–50% EtOAc / Heptanes) to give the dsired product 4-(prop-2-yn-1-yloxy)phenol as a pale solid (533 mg, 20%); 1H NMR (400 MHz, DMSO-d6) δ 9.01 (s, 1H), 6.83 – 6.76 (m, 2H), 6.71 – 6.64 (m, 2H), 4.65 (d, J = 2.4 Hz, 2H), 3.52 (t, J = 2.4 Hz, 1H); LCMS Rt 0.64 mins; m/z 149.0 (M+H)+
Step 2: A mixture of 4-(prop-2-yn-1-yloxy)phenol (548 mg, 3.70 mmol), 1-fluoro-2-nitrobenzene (390 μl, 3.70 mmol), K2CO3 (767 mg, 5.55 mmol) and DMAc (3.70 ml) was stirred at 160° C for 2 hr under an inert atmosphere. The reaction was diluted with EtOAc, washed with water and saturated brine, and dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give a residue which was purified by flash chromatography on silica gel eluting with (0–20% EtOAc / Heptanes) to give 1-nitro-2-(4-(prop-2-yn-1-yloxy)phenoxy)benzene as a pale oil (857 mg, 86%); 1H NMR (400 MHz, Chloroform-d) δ 7.95 (dd, J = 8.2, 1.6 Hz, 1H), 7.49 (ddd, J = 8.8, 7.5, 1.7 Hz, 1H), 7.22 – 7.14 (m, 1H), 7.09 – 7.00 (m, 4H), 6.96 (dd, J = 8.4, 1.0 Hz, 1H), 4.72 (d, J = 2.4 Hz, 2H), 2.57 (t, J = 2.4 Hz, 1H)
Step 3: To a solution of 1-nitro-2-(4-(prop-2-yn-1-yloxy)phenoxy)benzene (857 mg, 3.18 mmol) in EtOH (18.100 ml) and water (1.80 ml) was added iron powder (978 mg, 17.51 mmol) and ammonium chloride (119 mg, 2.228 mmol). The resulting mixture was heated at reflux at 95 °C for 2 hr when the reaction mixture was passed through filtered through Celite™ rinsing the pad with MeOH and the filtrate evaporated in vacuo. The residue was partitioned between EtOAc / sat NaHCO3 solution and the combined organic layers then dried over anhydrous magnesium sulphate, filtered and the filtrate evaporated to dryness to afford a light brown oil. The crude 2-(4-(prop-2-yn-1-yloxy)phenoxy)aniline was used to next step without further purification: LCMS Rt 0.94 mins; m/z 240.2 M+H
Step 4: To 4-(acrylamidomethyl)benzoic acid (103 mg, 0.5 mmol), 2-(4-(prop-2-yn-1-yloxy)phenoxy)aniline (120 mg, 0.500 mmol) in DMF (5.00 ml) was added EDC (115 mg, 0.600 mmol) and DMAP (61.1 mg, 0.500 mmol). The mixture was stirred at 60 °C for 2 hr before it was diluted with EtOAc (50 mL), the combined organics then washed with water, brine, dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica gel eluting with (20–100% EtOAc/Heptane) to give a white soild which was then subjected to HPLC (C18 OBD 30 × 50mm; MeCN / H20 w 0.1%formic acid; 35–65% MeCN gradient) to give a 4-(acrylamidomethyl)-N-(2-(4-(prop-2-yn-1-yloxy)phenoxy)phenyl)benzamide as white solid (71 mg, 34%)
1H NMR (400 MHz, DMSO-d6) δ 9.76 (s, 1H), 8.70 (t, J = 6.0 Hz, 1H), 7.82 (d, J = 8.3 Hz, 2H), 7.74 (dd, J = 7.7, 1.9 Hz, 1H), 7.35 (d, J = 8.3 Hz, 2H), 7.22 – 7.11 (m, 2H), 6.98 (s, 4H), 6.87 (dd, J = 7.9, 1.7 Hz, 1H), 6.29 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.2 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.75 (d, J = 2.4 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H), 3.56 (t, J = 2.4 Hz, 1H)
13C NMR (101 MHz, DMSO-d6) δ 165.02, 164.57, 153.15, 150.33, 150.27, 143.00, 132.85, 131.39, 128.77, 127.60, 127.04, 126.21, 126.17, 125.56, 122.96, 119.71, 117.97, 115.91, 79.17, 78.12, 55.65, 41.73
HRMS calcd for C26H23N2O4(M+H)+ 427.1658, found 427.1652
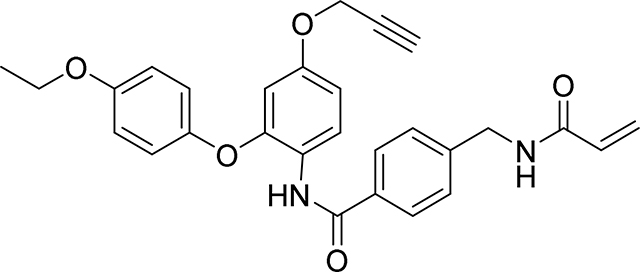
Preparation of 4-(acrylamidomethyl)-N-(2-(4-ethoxyphenoxy)-4-(prop-2-yn-1-yloxy)phenyl) benzamide
Step 1: To a solution of 2,4-difluoro-1-nitrobenzene (1.992 ml, 18.17 mmol) in DMF (91 ml) was added propargyl alcohol (1.586 ml, 27.2 mmol) and K2CO3 (7.53 g, 54.5 mmol). The solution was stirred at 40 °C for 18 hr when the reaction mixture was diluted with EtOAc, washed with water, brine, dried over Na2SO4, filtered and the filtrate concentrated in vacuo. The residue was purified by flash chromatography on silica gel (0–20% EtOAc/Heptane) to give the undesired regioisomer (1.1g, 30%) and the desired product 2-fluoro-1-nitro-4-(prop-2-yn-1-yloxy)benzene (545 mg, 16%): 1H NMR (400 MHz, Chloroform-d) δ 8.24 – 8.07 (m, 1H), 6.93 – 6.78 (m, 2H), 4.81 (d, J = 2.4 Hz, 2H), 2.64 (t, J = 2.4 Hz, 1H)
Step 2: A mixture of 4-ethoxyphenol (370 mg, 2.67 mmol), 2-fluoro-1-nitro-4-(prop-2-yn-1-yloxy)benzene (522 mg, 2.67 mmol), K2CO3 (555 mg, 4.01 mmol) in DMA (2.68 ml) was stirred at 160° C for 2 hr under an inert atmosphere. The reaction mixture was then diluted with EtOAc, washed with water and saturated brine, and the combined organics dried over anhydrous sodium sulfate and filtered. The filtrate was evaporated under reduced pressure to yield a crude product which was then purified by flash chromatography on silica gel eluting (0–20% EtOAc / Heptanes) to give 2-(4-ethoxyphenoxy)-1-nitro-4-(prop-2-yn-1-yloxy)benzene as a light pale solid (720 mg, 86%): 1H NMR (400 MHz, Chloroform-d) δ 8.08 (d, J = 9.2 Hz, 1H), 7.08 – 7.01 (m, 2H), 6.97 – 6.90 (m, 2H), 6.70 (dd, J = 9.2, 2.6 Hz, 1H), 6.45 (d, J = 2.5 Hz, 1H), 4.66 (d, J = 2.3 Hz, 2H), 4.06 (q, J = 7.0 Hz, 2H), 2.55 (t, J = 2.3 Hz, 1H), 1.45 (t, J = 7.0 Hz, 3H); LCMS Rt 1.15 mins; m/z 314.1 (M+H)
Step 3: To a solution of 2-(4-ethoxyphenoxy)-1-nitro-4-(prop-2-yn-1-yloxy)benzene (710 mg, 2.27 mmol) in EtOH (12.900 ml) and water (1.29 ml) was added iron powder (696 mg, 12.46 mmol) and ammonium chloride (85 mg, 1.586 mmol). The resulting mixture was heated at reflux at 90 °C for 18 hr. The reaction mixture was cooled to ambient temperature, filtered through Celite™ rinsing the pad with MeOH and the filtrate evaporated in vacuo. The residue was partitioned between EtOAc / sat NaHCO3 solution and the combined organic layers then dried over anhydrous magnesium sulphate, filtered and the filtrate evaporated to dryness to afford a light brown oil. The crude product was purified by flash chromatography on silica gel eluting (0–50% EtOAc / Heptanes) to give 2-(4-ethoxyphenoxy)-4-(prop-2-yn-1-yloxy)aniline as a light brown oil (477 mg, 74%): 1H NMR (400 MHz, Chloroform-d) δ 6.99 – 6.94 (m, 2H), 6.90 – 6.85 (m, 2H), 6.78 (d, J = 8.6 Hz, 1H), 6.61 (dd, J = 8.6, 2.7 Hz, 1H), 6.47 (d, J = 2.7 Hz, 1H), 4.56 (d, J = 2.4 Hz, 2H), 4.03 (q, J = 7.0 Hz, 2H), 3.83 (br s, 2H), 2.49 (t, J = 2.4 Hz, 1H), 1.44 (t, J = 7.0 Hz, 3H); LCMS Rt 0.89 mins; m/z 284.1 (M+H).
Step 4: 4-(acrylamidomethyl)benzoic acid (103 mg, 0.5 mmol) and 2-(4-ethoxyphenoxy)-4-(prop-2-yn-1-yloxy)aniline (142 mg, 0.500 mmol) were dissolved in DMF (5 ml) to give a clear solution, then EDC (115 mg, 0.600 mmol) and DMAP (61.1 mg, 0.500 mmol) were added. The mixture was stirred at 60 °C for 2 hr before the mixture was diluted with EtOAc (50 mL), washed with water, brine, and the combined organics dried over Na2SO4, filtered and concentrated. The crude product was purified by flash chromatography on silica gel eluting (20–100% EtOAc/Heptane) to give 4-(acrylamidomethyl)-N-(2-(4-ethoxyphenoxy)-4-(prop-2-yn-1-yloxy)phenyl) benzamide as a white soild (153 mg, 63%): 1H NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H), 8.69 (t, J = 6.0 Hz, 1H), 7.81 (d, J = 8.2 Hz, 2H), 7.53 (d, J = 8.8 Hz, 1H), 7.34 (d, J = 8.3 Hz, 2H), 6.99 – 6.85 (m, 4H), 6.77 (dd, J = 8.8, 2.8 Hz, 1H), 6.45 (d, J = 2.7 Hz, 1H), 6.29 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.2 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.74 (d, J = 2.3 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H), 3.97 (q, J = 7.0 Hz, 2H), 3.58 (t, J = 2.3 Hz, 1H), 1.30 (t, J = 7.0 Hz, 3H)
13C NMR (101 MHz, DMSO-d6) δ 164.43, 163.94, 154.79, 154.00, 151.29, 148.66, 142.23, 132.25, 130.78, 127.19, 126.94, 126.35, 124.92, 121.46, 119.24, 114.66, 108.10, 104.48, 78.27, 77.75, 62.60, 54.97, 41.10, 13.93
HRMS calcd for C28H27N2O5(M+H)+ 471.1920, found 471.1915
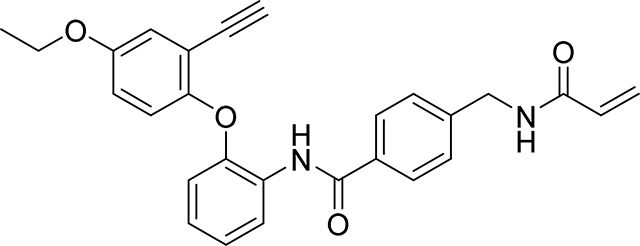
Preparation of 4-(acrylamidomethyl)-N-(2-(4-ethoxy-2-ethynylphenoxy)phenyl)benzamide
Step1: A mixture of 5-ethoxy-2-hydroxybenzaldehyde (405 mg, 2.44 mmol), 1-fluoro-2-nitrobenzene (257 μl, 2.44 mmol), K2CO3 (505 mg, 3.66 mmol) in DMA (2.44 ml) was stirred at 160° C for 2 hr under an inert atmosphere. The reaction mixture was cooled to room temperature, diluted with water and extracted with ethyl acetate (3 × 25 ml). The combined organic phases were washed with water, brine, and dried over anhydrous sodium sulfate, filtered and the filtrate evaporated under reduced pressure. The residue obtained was purified by flash column chromatography on silica gel eluting (0–30% EtOAc/Heptane) to give the desired compound 5-ethoxy-2-(2-nitrophenoxy)benzaldehyde as a yellow solid (690 mg, 80%): 1H NMR (400 MHz, Chloroform-d) δ 10.39 (s, 1H), 8.02 (dd, J = 8.2, 1.6 Hz, 1H), 7.55 (ddd, J = 8.4, 7.5, 1.7 Hz, 1H), 7.44 (d, J = 3.2 Hz, 1H), 7.30 – 7.23 (m, 1H), 7.17 (dd, J = 9.0, 3.2 Hz, 1H), 7.04 – 6.93 (m, 2H), 4.11 (q, J = 7.0 Hz, 2H), 1.46 (t, J = 7.0 Hz, 3H); LCMS Rt 1.08 mins; m/z 288.0 (M+H)
Step 2: To a solution of 5-ethoxy-2-(2-nitrophenoxy)benzaldehyde (690 mg, 2.402 mmol) in MeOH (2.40 ml) was added K2CO3 (1095 mg, 7.93 mmol) and dimethyl (1-diazo-2-oxopropyl)phosphonate (923 mg, 4.80 mmol). The reaction was stirred at ambient temperature for 18 hr before the reaction was diluted with EtOAc, washed with water, brine, dried over Na2SO4 and concentrated to give a solid 4-ethoxy-2-ethynyl-1-(2-nitrophenoxy)benzene (680 mg, 100%): 1H NMR (400 MHz, Chloroform-d) δ 7.96 (dd, J = 8.1, 1.6 Hz, 1H), 7.47 (ddd, J = 8.9, 7.5, 1.6 Hz, 1H), 7.19 – 7.12 (m, 1H), 7.09 – 7.02 (m, 2H), 6.94 (dd, J = 9.0, 3.0 Hz, 1H), 6.88 – 6.82 (m, 1H), 4.05 (q, J = 7.0 Hz, 2H), 3.13 (s, 1H), 1.45 (t, J = 7.0 Hz, 3H); LCMS Rt 1.15 mins; m/z 284.1 (M+H)
Step 3: To a solution of 4-ethoxy-2-ethynyl-1-(2-nitrophenoxy)benzene (680 mg, 2.4 mmol) in EtOH (14.5 ml) and water (1.45 ml) was added iron (0.737 g, 13.20 mmol) and ammonium chloride (0.090 g, 1.680 mmol). The resulting mixture was heated at reflux at 90 °C for 2 hr before it was cooled, the crude reaction mixture was then filtered through Celite™ and rinsing the pad with MeOH. The combined organic solvent was removed in vacuo and the resulting residue dissolved in EtOAc, washed with sat. NaHCO3 solution, the combined organic phases washed with brine, dried over Na2SO4 and concentrated to give a pale solid 2-(4-ethoxy-2-ethynylphenoxy)aniline in quantitative yield {LCMS: Rt 1.05 mins; 254.2 (M+H)}. The crude product was used without further purification.
Step 4: To 4-(acrylamidomethyl)benzoic acid (103 mg, 0.5 mmol), 2-(4-ethoxy-2-ethynylphenoxy)aniline (127 mg, 0.500 mmol) was added DMF (5 ml) giving a clear solution, then EDC (115 mg, 0.600 mmol) and DMAP (61.1 mg, 0.500 mmol) were added. The mixture was stirred at 60 °C for 2 hr when LC-MS indicated all the acid was consumed. The reaction mixture was diluted with EtOAc (50 mL), washed with water, brine, dried over Na2SO4 and concentrated. Purification by flash silica gel chromatography eluting with (20–100% EtOAc / Heptane) gave 4-(acrylamidomethyl)-N-(2-(4-ethoxy-2-ethynylphenoxy)phenyl)benzamide as a white soild (75 mg, 33%): 1H NMR (400 MHz, DMSO-d6) δ 9.69 (s, 1H), 8.70 (t, J = 6.0 Hz, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.80 (dd, J = 7.4, 2.1 Hz, 1H), 7.38 (d, J = 8.3 Hz, 2H), 7.19 – 7.09 (m, 2H), 7.05 (t, J = 1.7 Hz, 1H), 6.99 (d, J = 1.7 Hz, 2H), 6.83 – 6.69 (m, 1H), 6.29 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.2 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.42 (d, J = 6.0 Hz, 2H), 4.26 (s, 1H), 4.01 (q, J = 7.0 Hz, 2H), 1.30 (t, J = 7.0 Hz, 3H)
13C NMR (101 MHz, DMSO-d6) δ 160.24, 159.65, 150.16, 145.17, 141.45, 136.95, 128.93, 125.12, 123.83, 122.87, 122.37, 122.13, 118.76, 118.39, 115.80, 115.34, 113.70, 112.04, 110.82, 110.33, 77.10, 73.42, 58.87, 37.98, 9.54
HRMS calcd for C27H25N2O4(M+H)+ 441.1814, found 441.1805
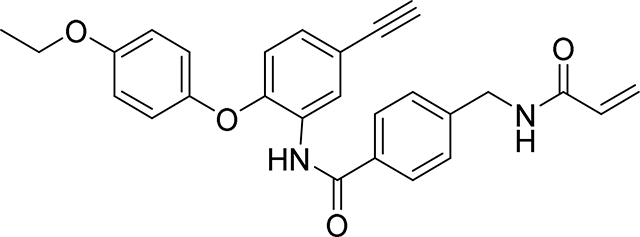
Preparation of 4-(acrylamidomethyl)-N-(2-(4-ethoxyphenoxy)-5-ethynylphenyl)benzamide
Step 1: A mixture of 4-ethoxyphenol (418 mg, 3.03 mmol), 4-ethynyl-1-fluoro-2-nitrobenzene (500 mg, 3.03 mmol), K2CO3 (628 mg, 4.54 mmol) and DMA (3.02 ml) was stirred at 160° C. for 2 hours under N2 atmosphere. The reaction mixture was cooled to room temperature then diluted with water, and extracted with ethyl acetate (x3). The combined organic layers were washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered and the filtrate evaporated in vacuo. The residue obtained was purified by flash column chromatography on silica gel (0–50% EtOAc / Heptane) to give 1-(4-ethoxyphenoxy)-4-ethynyl-2-nitrobenzene as a yellow solid (703 mg, 82%): 1H NMR (400 MHz, Chloroform-d) δ 8.06 (d, J = 2.0 Hz, 1H), 7.54 (dd, J = 8.7, 2.1 Hz, 1H), 7.06 – 7.02 (m, 2H), 6.97 – 6.91 (m, 2H), 6.86 (d, J = 8.7 Hz, 1H), 4.06 (q, J = 7.0 Hz, 2H), 3.13 (s, 1H), 1.45 (t, J = 7.0 Hz, 3H); LCMS m/z 283.0
Step 2: To a solution of 1-(4-ethoxyphenoxy)-4-ethynyl-2-nitrobenzene (703 mg, 2.482 mmol) in EtOH (Volume: 14.100 mL, Ratio: 10) and Water (Volume: 1.410 mL, Ratio: 1) was added iron powder (762 mg, 13.65 mmol) and ammonium chloride (93 mg, 1.737 mmol). The resulting mixture was heated at reflux at 90 °C for 18 hr. The reaction mixture was filtered through Celite™ and the pad rinsed with MeOH. The filtrate was reduced in vacuo and the residue then partitioned between EtOAc/sat NaHCO3 solution. The combined organic layers dried over anhydrous magnesium sulphate and evaporated to dryness, to afford 2-(4-ethoxyphenoxy)-5-ethynylaniline as a light brown oil which was used in the next step without further purification (609 mg, 89%): LCMS Rt 1.09 mins; m/z 254.2
Step 3: To 4-(acrylamidomethyl)benzoic acid (103 mg, 0.5 mmol), 2-(4-ethoxyphenoxy)-5-ethynylaniline (127 mg, 0.500 mmol) and DMF (5 ml) was added EDC (115 mg, 0.600 mmol) and DMAP (61.1 mg, 0.500 mmol). The reaction mixture was stirred at 60 °C for 2 hr. The reaction mixture was diluted with EtOAc (50 mL), washed with water, brine, dried over Na2SO4, filtered and concentrated to a crude product. Purification by flash silica gel chromatography eluting with (20–100% EtOAc / Heptane) gave the desired product 4-(acrylamidomethyl)-N-(2-(4-ethoxyphenoxy)-5-ethynylphenyl)benzamide as a white solid (77 mg, 33%):
1H NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 8.70 (t, J = 6.0 Hz, 1H), 7.90 – 7.83 (m, 3H), 7.37 (d, J = 8.3 Hz, 2H), 7.27 (dd, J = 8.5, 2.1 Hz, 1H), 7.05 – 6.97 (m, 2H), 6.99 – 6.90 (m, 2H), 6.77 (d, J = 8.5 Hz, 1H), 6.29 (dd, J = 17.1, 10.1 Hz, 1H), 6.13 (dd, J = 17.1, 2.3 Hz, 1H), 5.64 (dd, J = 10.1, 2.2 Hz, 1H), 4.41 (d, J = 6.0 Hz, 2H), 4.15 (s, 1H), 3.99 (q, J = 6.9 Hz, 2H), 1.31 (t, J = 6.9 Hz, 3H)
13C NMR (101 MHz, DMSO-d6) δ 164.10, 163.53, 154.05, 150.27, 147.49, 142.19, 131.56, 130.35, 128.50, 127.79, 127.33, 126.65, 126.03, 124.51, 119.66, 115.99, 114.49, 114.39, 81.77, 78.86, 62.22, 40.69, 13.50
HRMS calcd for C27H25N2O4(M+H)+ 441.1814, found 441.1809
Preparation of N-(2-(4-ethoxyphenoxy)phenyl)-4-(propionamidomethyl)benzamide
Step 1: To a solution of 4-(((tert-butoxycarbonyl)amino)methyl)benzoic acid (13.06 g, 52.0 mmol) in toluene (200 ml) under inert atmosphere, was added anhydrous DMF (0.5 ml), pyridine (25.2 ml, 312 mmol) and oxalyl chloride (8.64 ml, 99 mmol), and the resulting mixture was stirred for 5 hr, with the formation of a precipitate being observed. This precipitate was filtered off and washed with toluene (x2). The combined filtrates were dried over Na2SO4 and evaporated under reduced pressure, to obtain the corresponding tert-butyl (4-(chlorocarbonyl)benzyl)carbamate as a light pale solid. The crude product was used to next step without further purification.
Step 2: To tert-butyl (4-(chlorocarbonyl)benzyl)carbamate (1471 mg, 2.181 mmol), 2-(4-ethoxyphenoxy)aniline (500 mg, 2.181 mmol) in DCM (10.9 ml) was added TEA (0.912 mL, 6.54 mmol) and DMAP (133 mg, 1.090 mmol). The reaction mixture was stirred for 16 hr before being diluted with DCM, washed by sat. NaHCO3 solution, brine, dried over Na2SO4, filtered and concentrated under reduced pressure to give a crude product. The crude product was purified by flash column chromatography on silica gel eluting (0–40% EtOAc / Heptane) to give tert-butyl (4-((2-(4-ethoxyphenoxy)phenyl)carbamoyl)benzyl) carbamate as a white solid (690 mg, 69%): 1H NMR (400 MHz, Chloroform-d) δ 8.65 – 8.57 (m, 2H), 7.84 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 8.1 Hz, 2H), 7.14 (t, J = 7.8 Hz, 1H), 7.07 – 6.99 (m, 3H), 6.96 – 6.90 (m, 2H), 6.84 – 6.78 (m, 1H), 4.95 (s, 1H), 4.39 (d, J = 4.3 Hz, 2H), 4.05 (q, J = 7.0 Hz, 2H), 1.48 (s, 9H), 1.45 (t, J = 7.0 Hz, 3H); LCMS Rt 1.20 mins; m/z 463.2 (M+H)
Step 3: To a solution of tert-butyl (4-((2-(4-ethoxyphenoxy)phenyl)carbamoyl)benzyl) carbamate (690 mg, 1.492 mmol) in MeOH (7.46 ml) was added 4N HCl in 1,4-dioxane (7.46 ml, 29.8 mmol). The resulting solution was stirred at ambient temperature or 18 hr before the solvent was removed under reduced pressure to give a white solid, 4-(aminomethyl)-N-(2-(4-ethoxyphenoxy)phenyl)benzamide hydrochloride. The crude product was used to next step without furtheer purification: LCMS Rt 0.81 mins; m/z 363.2 (M+H).
Step 4: To crude 4-(aminomethyl)-N-(2-(4-ethoxyphenoxy)phenyl)benzamide hydrochloride (120 mg, 0.3 mmol) in DCM (Volume: 1.5 ml) at 0 °C was added TEA (84 μl, 0.600 mmol) followed by propionyl chloride (26.2 μl, 0.300 mmol). The reaction mixture was stirred for 16 hr at ambient temperature before it was diluted with EtOAc, washed with water, brine, dried over Na2SO4, filtered and concentrated under reduced pressure to give the crude product. The crude product was purified by flash column chromatography on silica gel eluting (30–100% EtOAc/Heptane) to give N-(2-(4-ethoxyphenoxy)phenyl)-4-(propionamidomethyl)benzamide as a white solid (103 mg, 82%): 1H NMR (400 MHz, Chloroform-d) δ 8.61 (dd, J = 8.3, 1.4 Hz, 2H), 7.83 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.19 – 7.10 (m, 1H), 7.08 – 6.99 (m, 3H), 6.96 – 6.89 (m, 2H), 6.81 (dd, J = 8.1, 1.3 Hz, 1H), 5.85 (s, 1H), 4.52 (d, J = 5.9 Hz, 2H), 4.05 (q, J = 7.0 Hz, 2H), 2.30 (q, J = 7.6 Hz, 2H), 1.45 (t, J = 7.0 Hz, 3H), 1.22 (t, J = 7.6 Hz, 3H).
13C NMR (101 MHz, Chloroform-d) δ 173.74, 165.02, 155.75, 149.18, 147.10, 142.59, 134.20, 129.14, 128.06, 127.52, 124.09, 123.37, 120.54, 116.23, 115.66, 63.96, 43.13, 29.70, 14.89, 9.86
HRMS calcd for C25H27N2O4(M+H)+ 419.1971, found 419.1973
The following compounds were prepared using a general procedure B (as used to prepare N-(2-(4-ethoxyphenoxy)phenyl)-4-(propionamidomethyl)benzamide: step 4) from tert-butyl (4-(chlorocarbonyl)benzyl)carbamate and the necessary aniline with subsequent deprotection.
Procedure B
To the corresponding crude 4-(aminomethyl)-benzamide hydrochloride (0.3 mmol) in DCM (1.5 ml) at 0 °C was added TEA (0.60 mmol) followed by either crotonyl chloride, acyrloyl chloride or chloro acetyl chloride (0.300 mmol). The reaction mixture was stirred for 16 hr at ambient temperature before it was diluted with EtOAc, washed with water, brine, dried over Na2SO4, filtered and concentrated under reduced pressure to give the crude product. The crude product was then purified by flash column chromatography on silica gel eluting (20–100% EtOAc / Heptane) to give the desired product
4-((2-chloroacetamido)methyl)-N-(2-(4-ethoxyphenoxy)phenyl)benzamide
73 mg, 56%
1H NMR (400 MHz, Chloroform-d) δ 8.61 (dd, J = 8.2, 1.3 Hz, 2H), 7.87 (d, J = 8.2 Hz, 2H), 7.42 (d, J = 8.1 Hz, 2H), 7.19 – 7.11 (m, 1H), 7.07 – 7.00 (m, 3H), 6.97 (s, 1H), 6.95 – 6.89 (m, 2H), 6.81 (dd, J = 8.2, 1.3 Hz, 1H), 4.59 (d, J = 6.0 Hz, 2H), 4.15 (s, 2H), 4.05 (q, J = 7.0 Hz, 2H), 1.45 (t, J = 7.0 Hz, 3H).
13C NMR (101 MHz, Chloroform-d) δ 166.02, 164.90, 155.77, 149.15, 147.12, 141.34, 134.55, 129.09, 128.05, 127.66, 124.13, 123.37, 120.57, 120.54, 116.20, 115.66, 63.96, 43.39, 42.62, 14.89
HRMS calcd for C24H24ClN2O4(M+H)+ 439.1425, found 439.1421
(E)-4-(but-2-enamidomethyl)-N-(2-(4-ethoxyphenoxy)phenyl)benzamide
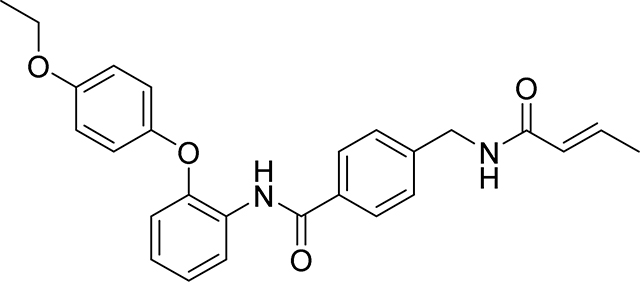
66 mg, 46%
1H NMR 1H NMR (400 MHz, Chloroform-d) δ 8.65 – 8.54 (m, 2H), 7.83 (d, J = 8.2 Hz, 2H), 7.41 (d, J = 8.3 Hz, 2H), 7.15 (td, J = 7.8, 1.4 Hz, 1H), 7.07 – 6.99 (m, 3H), 6.95 – 6.90 (m, 3H), 6.81 (dd, J = 8.2, 1.4 Hz, 1H), 5.85 (dd, J = 15.2, 1.5 Hz, 1H), 5.77 (s, 1H), 4.59 (d, J = 6.0 Hz, 2H), 4.05 (q, J = 7.0 Hz, 2H), 1.90 (dd, J = 7.0, 1.7 Hz, 3H), 1.45 (t, J = 7.0 Hz, 3H).
13C NMR 101 MHz, Chloroform-d) δ 165.91, 165.05, 155.73, 149.19, 147.08, 142.56, 140.99, 134.17, 129.16, 128.05, 127.50, 124.54, 124.08, 123.38, 120.53, 120.51, 116.26, 115.66, 63.96, 43.10, 17.84, 14.89
HRMS calcd for C26H27N2O4(M+H)+ 431.1971, found 431.1968
4-(acrylamidomethyl)-N-(2-(4-(trifluoromethoxy)phenoxy)phenyl)benzamide
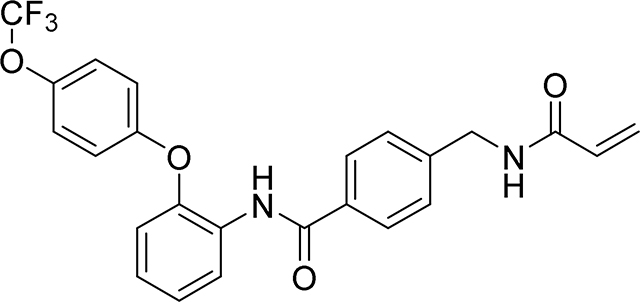
86 mg, 57%
1H NMR (400 MHz, Chloroform-d) δ 8.61 (dd, J = 8.2, 1.5 Hz, 1H), 8.40 (s, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.41 (s, 1H), 7.39 (s, 1H), 7.23 (t, J = 7.7 Hz, 3H), 7.15 – 7.05 (m, 3H), 6.93 (dd, J = 8.1, 1.3 Hz, 1H), 6.37 (dd, J = 17.0, 1.3 Hz, 1H), 6.15 (dd, J = 17.0, 10.3 Hz, 1H), 5.98 (s, 1H), 5.72 (dd, J = 10.3, 1.3 Hz, 1H), 4.59 (d, J = 6.0 Hz, 2H)
13C NMR NMR (101 MHz, Chloroform-d) δ 165.51, 165.05, 154.84, 145.50, 145.07 (d, J = 2.1 Hz), 142.41, 134.04, 130.31, 129.87, 128.13, 127.47, 127.44, 124.78, 124.47, 122.98, 121.17, 120.46 (q, J = 257.1 Hz), 119.51, 117.99, 43.20
19F NMR (376 MHz, Chloroform-d) δ −58.26
HRMS calcd for C24H20F3N2O4(M+H)+ 457.1375, found 457.1378
4-(acrylamidomethyl)-N-(2-(4-methoxyphenoxy)phenyl)benzamide
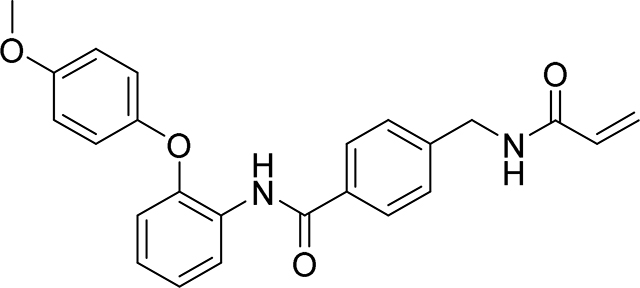
105 mg, 73%
1H NMR (400 MHz, Chloroform-d) δ 8.66 – 8.56 (m, 2H), 7.84 (d, J = 8.2 Hz, 2H), 7.42 (d, J = 8.2 Hz, 2H), 7.19 – 7.10 (m, 1H), 7.08 – 7.00 (m, 3H), 6.97 – 6.89 (m, 2H), 6.81 (dd, J = 8.2, 1.2 Hz, 1H), 6.38 (dd, J = 16.9, 1.3 Hz, 1H), 6.16 (dd, J = 16.9, 10.3 Hz, 1H), 5.96 (s, 1H), 5.73 (dd, J = 10.3, 1.3 Hz, 1H), 4.61 (d, J = 6.0 Hz, 2H), 3.84 (s, 3H).
13C NMR (101 MHz, Chloroform-d) δ 165.49, 165.02, 156.37, 149.30, 147.09, 142.22, 134.27, 130.36, 129.13, 128.13, 127.54, 127.38, 124.12, 123.41, 120.56, 116.24, 115.07, 55.72, 43.23
HRMS calcd for C24H23N2O4(M+H)+ 403.1658, found 403.1651.
4-(acrylamidomethyl)-N-(2-(4-ethoxyphenoxy)-5-fluorophenyl)benzamide
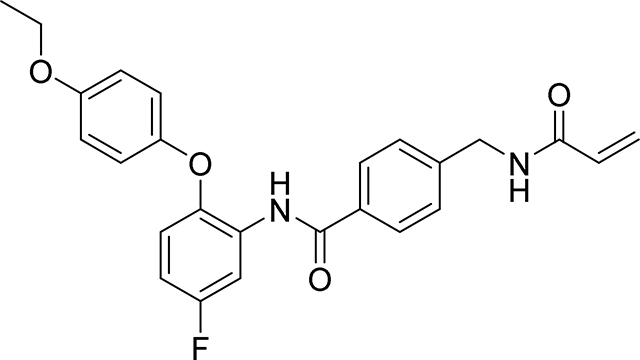
116 mg, 77%
1H NMR (400 MHz, Chloroform-d) δ 8.57 (s, 1H), 8.45 (dd, J = 10.6, 2.9 Hz, 1H), 7.80 (d, J = 8.3 Hz, 2H), 7.41 (d, J = 8.3 Hz, 2H), 7.01 – 6.95 (m, 2H), 6.93 – 6.88 (m, 2H), 6.80 (dd, J = 9.0, 5.2 Hz, 1H), 6.73 (ddd, J = 9.0, 7.7, 3.0 Hz, 1H), 6.37 (dd, J = 17.0, 1.3 Hz, 1H), 6.15 (dd, J = 17.0, 10.3 Hz, 1H), 5.95 (s, 1H), 5.72 (dd, J = 10.3, 1.4 Hz, 1H), 4.60 (d, J = 6.0 Hz, 2H), 4.04 (q, J = 7.0 Hz, 2H), 1.44 (t, J = 7.0 Hz, 3H)
13C NMR (101 MHz, Chloroform-d) δ 165.50, 164.98, 158.42 (d, J = 240.3 Hz), 155.69, 149.56, 142.68 (d, J = 2.8 Hz), 142.53, 133.80, 130.31, 130.20, 128.17, 127.54, 127.45, 119.88, 117.47 (d, J = 9.5 Hz), 115.72, 110.07 (d, J = 23.7 Hz), 107.95 (d, J = 29.9 Hz), 63.98, 43.20, 14.87
19F NMR (376 MHz, Chloroform-d) δ −117.09
HRMS calcd for C25H24FN2O4 (M+H)+ 435.1720, found 435.1721
4-(acrylamidomethyl)-N-(2-(4-ethoxyphenoxy)-5-(trifluoromethyl)phenyl)benzamide
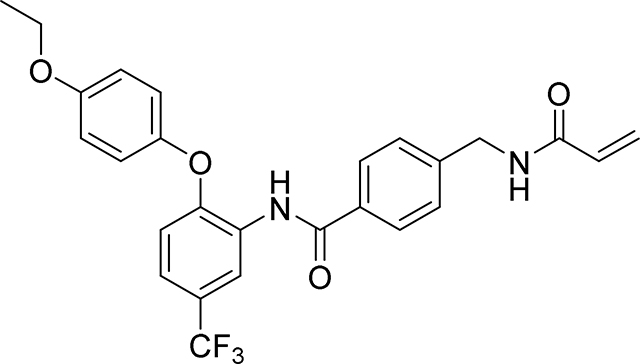
125 mg, 76%
1H NMR (400 MHz, Chloroform-d) δ 8.98 (d, J = 2.1 Hz, 1H), 8.69 (s, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.45 (d, J = 8.3 Hz, 2H), 7.31 – 7.23 (m, 1H), 7.08 – 7.02 (m, 2H), 6.99 – 6.93 (m, 2H), 6.81 (d, J = 8.5 Hz, 1H), 6.37 (dd, J = 17.0, 1.3 Hz, 1H), 6.16 (dd, J = 17.0, 10.3 Hz, 1H), 5.95 (s, 1H), 5.73 (dd, J = 10.3, 1.3 Hz, 1H), 4.62 (d, J = 6.0 Hz, 2H), 4.07 (q, J = 7.0 Hz, 2H), 1.46 (t, J = 7.0 Hz, 3H)
13C NMR (101 MHz, Chloroform-d) δ 165.53, 165.13, 156.49, 149.86, 147.78, 142.64, 133.74, 130.30, 128.83, 128.20, 127.58, 127.45, 125.00 (q, J = 32.7 Hz), 124.04 (q, J = 271.8 Hz), 121.44, 121.02 (q, J = 3.7 Hz), 117.57 (q, J = 3.8 Hz), 115.85, 114.75, 63.99, 43.21, 14.85
19F NMR (376 MHz, Chloroform-d) δ −61.84
HRMS calcd for C26H24F3N2O4 (M+H)+ 485.1688, found 485.1684
Preparation of (E)-N-(2-(4-ethoxyphenoxy)phenyl)-4-((4,4,4-trifluorobut-2-enamido)methyl) benzamide
Step 1: (E)-4,4,4-trifluorobut-2-enoic acid (56.0 mg, 0.40 mmol), crude 4-(aminomethyl)-N-(2-(4-ethoxyphenoxy)phenyl)benzamide hydrochloride (160 mg, 0.40 mmol) and DIPEA (210 μl, 1.20 mmol) were stirred in THF (2 ml) at 0 °C before EDC (115 mg, 0.600 mmol) was added. The reaction mixture was stirred for 16 hr at ambient temperature before the mixture was diluted with EtOAc, washed with water, brine, dried over Na2SO4, filtered and concentrated under reduced pressure to give the crude product. The crude product was purified by flash column chromatography on silica gel eluting (0–80% EtOAc/heptane) to give (E)-N-(2-(4-ethoxyphenoxy)phenyl)-4-((4,4,4-trifluorobut-2-enamido)methyl) benzamide as a white solid (59 mg, 30%): 1H NMR (400 MHz, Chloroform-d) δ 8.61 – 8.54 (m, 2H), 7.80 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 7.18 – 7.11 (m, 1H), 7.07 – 6.97 (m, 3H), 6.95 – 6.78 (m, 4H), 6.56 (dd, J = 15.4, 1.9 Hz, 1H), 6.38 (s, 1H), 4.61 (d, J = 5.9 Hz, 2H), 4.04 (q, J = 7.0 Hz, 2H), 1.44 (t, J = 7.0 Hz, 3H)
13C NMR (101 MHz, Chloroform-d) δ 165.05, 162.54, 155.79, 149.10, 147.17, 141.32, 134.47, 130.31 (q, J = 5.8 Hz), 129.19 (q, J = 35.2 Hz), 128.94, 128.17, 127.59, 124.29, 123.36, 122.41 (q, J = 270.0 Hz), 120.55, 116.23, 115.68, 63.97, 43.58, 14.88
19F NMR (376 MHz, Chloroform-d) δ −65.07
HRMS calcd for C26H24F3N2O4 (M+H)+ 485.1688, found 485.1690
QUANTIFICATION AND STATISTICAL ANALYSIS
For covalent ligand screening and dose-response studies of covalent ligands in DNA binding and MYC transcriptional assays, EN4-alkyne MYC enrichment studies, thermal shift assays, proliferation and survival assays, tumor xenograft studies, and quantitation of Western blots, we quantified the data as described in the Methods section and statistical analysis was performed using a Student’s unpaired two-tailed t-test.
For isoTOP-ABPP data analysis, data were extracted in the form of MS1 and MS2 files using Raw Extractor v.1.9.9.2 (Scripps Research Institute) and searched against the Uniprot human database using ProLuCID search methodology in IP2 v.3 (Integrated Proteomics Applications, Inc.)(Xu et al., 2015). Cysteine residues were searched with a static modification for carboxyaminomethylation (+57.02146) and up to two differential modifications for methionine oxidation and either the light or heavy TEV tags (+464.28596 or +470.29977, respectively). Peptides were required to be fully tryptic peptides and to contain the TEV modification. ProLUCID data were filtered through DTASelect to achieve a peptide false-positive rate below 5%. Only those probe-modified peptides that were evident across two out of three biological replicates were interpreted for their isotopic light to heavy ratios. For those probe-modified peptides that showed ratios greater than two, we only interpreted those targets that were present across all three biological replicates, were statistically significant and showed good quality MS1 peak shapes across all biological replicates. Light versus heavy isotopic probe-modified peptide ratios are calculated by taking the mean of the ratios of each replicate paired light versus heavy precursor abundance for all peptide-spectral matches associated with a peptide. The paired abundances were also used to calculate a paired sample t-test P value in an effort to estimate constancy in paired abundances and significance in change between treatment and control. P values were corrected using the Benjamini–Hochberg method.
For quantification and statistical analysis of the RNA sequencing data, we performed Contrast analysis to perform t-tests to generate p-values, Benjamini-Hochberg false discovery rates (FDRs) and log2 fold change for each gene comparing treated versus control groups. We performed signature enrichment analysis of genes with an FDR < 0.1 and Fold change <= 0.75 (567 genes) using the MSigDB Collection H hallmark gene set.
Supplementary Material
Table S1. Related to Figure 1. Covalent ligand structures and screening data for data shown in Figure 1a, 1b.
Table S2. Related to Figure 2 and Figure S3. IsoTOP-ABPP analysis of EN4 in 231MFP breast cancer cells shown in Figure S3.
Table S3. Related to Figure 4. RNA sequencing analysis of EN4-mediated transcriptional changes in 231MFP cells. 231MFP cells were treated with DMSO vehicle or EN4 (50 μM) for 24 h, n=3 biological replicates/group. Tab 1 shows total RNA sequencing data, quantitation, and statistical analysis. Contrast analysis produced t-test p-values, Benjamini-Hochberg false-discovery rates (FDRs) and log 2 fold changes for each gene with treated versus DMSO vehicle-treated controls. Tab 2 shows signature enrichment. For the signature enrichment, genes with an FDR < 0.1 and Fold change <= 0.75 (567 genes) were selected and signature search was performed using the MSigDB Collection H hallmark gene set: https://www.gseamsigdb.org/gsea/msigdb/collections.jsp#H.
Significance.
MYC is a major oncogenic transcriptional driver of most human cancers that has remained intractable to direct targeting because much of MYC is intrinsically disordered. We have screened a cysteine-reactive covalent ligand library against MYC and have identified a hit compound EN4 that targets a unique ligandable site C171 within MYC to reduce MYC and MAX thermal stability, inhibit MYC/MAX DNA binding in vitro, impair MYC transcriptional activity in cells, and inhibit proliferation and tumorigenesis in breast cancer cells. Preliminary medicinal chemistry efforts also gave rise to structure-activity relationships and yielded improved compounds. Our study also highlights how intrinsically disordered regions of classically “undruggable” targets may be functionally targeted with small-molecules and underscores the utility of covalent ligand screening coupled with chemoproteomic approaches to uncover ligands against undruggable targets like MYC.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rb pAb to MAX | Abcam | ab101271 |
| Rb mAb to c-Myc | Abcam | ab32072 |
| Rb mAB to DYKDDDDK Tag (D6W5B) | Cell Signaling Technology | Cat#14793S |
| Vinculin Rabbit Ab | Cell Signaling Technology | Cat#4650S |
| Rb mAb to c-Jun (60A8) | Cell Signaling Technology | Cat#9165S |
| Rb CDC25A Ab | Cell Signaling Technology | Cat#3652S |
| Rb mAb to CDK2 (78B2) | Cell Signaling Technology | Cat#2546S |
| Rb Anti-MAX | Sigma-Aldrich | HPA003474-100uL |
| GAPDH Ms McAb | Proteintech | Cat#60004-I-Ig |
| IRDye 680RD Goat anti-Mouse | LI-COR | 926-60870 |
| IRDye 800CW Goat anti-Rabbit | LI-COR | 926-32211 |
| B-Actin Rabbit mAb | Cell Signaling Technology | Cat#3700 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemically Competent E. coli | Invitrogen | Cat#C7373-03 |
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| c-MYC human recombinant protein | Origene | Sku#TP301611 |
| MAX human recombinant protein | Origene | AR09449PU-N |
| c-Jun human recombinant protein | Origene | AR09900PU-S |
| Invitrogen Hoechst 33342 dye | Fisher Scientific | Cat#H3570 |
| Dimethyl Sulfoxide | Fisher Chemical | D128-500 (CAS 67-68-5) |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Sigma-Aldrich | D8537-500mL |
| c-MYC human recombinant protein | Abcam | Ab169901 |
| MAX human recombinant protein | Origene | Sku#TP306812 |
| Opti-MEM Reduced Serum Media | ThermoFisher Scientific | Cat#31985062 |
| Lipofectamine 2000 Transfection Reagent | ThermoFisher Scientific | Cat#11668027 |
| Iodoacetamide 98% | ACROS Organics | Cat# AC122270050 |
| Iodoacetamide-13C2, 2-d2 | Sigma-Aldrich | Cat#721328 |
| Urea | Fisher Chemical | U15-500 (CAS 57-13-6) |
| ProteaseMAX Surfactant, lyophilized | Promega | REF#V2072 |
| Ammonium Bicarbonate | Fisher Chemical | A643-500 (CAS 1066-33-7) |
| TCEP 99% | Strem Chemicals | Cat#15-7400 (CAS 51805-45-9) |
| Formic Acid, 99.0+%, Optima LC/MS Grade | Fisher Chemical | A117-50 (CAS 64-18-6) |
| Sequencing Grade Modified Trypsin, Porcine | Promega | V511A |
| Covalent Ligand Library | Enamine | N/A |
| TAMRA-PEG4-Azide | Click Chemistry Tools, Inc. | AZ109-5 |
| Laemmli SDS sample buffer, reducing (4x) | Alfa Aesar | J60015 |
| Dharmafect I | Dharmacon | T-2001-02 |
| Anti-DYKDDDDK G1 Affinity Resin | GenScript | Cat#L00432 |
| 3X (DYKDDDDK) Peptide | APExBIO | A6001 |
| Tris(benzyltriazole methylamine) (TBTA) | Sigma-Aldrich | 678937 (CAS 510758-28-8) |
| Biotin-TEV-azide | Weerapana et. al., 2010 | N/A |
| Copper (II) Sulfate | Sigma-Aldrich | 451657 |
| AcTEV Protease | Invitrogen | 12575-015 |
| N-Hex-5-ynyl-2-iodo-acetamide (IAyne) | Chess Gmbh | 3187 |
| Spectra Multicolor Broad Range Protein Ladder | Thermo Scientific | Prod#26634 |
| Pierce Protease inhibitor Mini Tablets, EDTA-free | Thermo Scientific | Prod#A32955 |
| 5-TMRIA [Tetramethylrhodamine-5-Iodoacetamide Dihydroiodide] | Setareh Biotech | 6222 |
| High Capacity Streptavidin Agarose Resin | Thermo Scientific | Ref#20357 |
| Streptavidin Agarose Resin | Thermo Scientific | Ref#20349 |
| Critical Commercial Assays | ||
| Abcam c-Myc Transcription Factor Assay Kit | Abcam | Ab207200 |
| MYC Luciferase Reporter | Qiagen | CCS-012L |
| Q5 site-directed mutagenesis kit | New England Biolabs | E0554S |
| Dual-Glo Luciferase | Promega | E2920 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| 231MFP | (Jessani et al., 2004) | N/A |
| HEK293T/17 | ATCC | CRL-11268 |
| MCF10A | ATCC | CRL-10317 |
| Experimental Models: Organisms/Strains | ||
| Mouse model-- C.B-Igh-1b/IcrTac-Prkdcscid female mice | Taconic | CB17SC-F |
| Oligonucleotides | ||
| Primer: MYC C171Y forward ACT CCA CCT CCA GCT TGT ACC T | This paper | N/A |
| Primer: MYC C171Y reverse AGA CGC TGT GGC CGC | This paper | N/A |
| SASI_HS01_0015-0279/JUN (siJUN) 5’-CCUUCUAUGACGAUGCCCU[dt][dt]-3’ (sense) 5’- AGGGCAUCGUCAUAGAAGG-3’ (antisense) | Sigma-Aldrich | PDSIRNA2D SASI_HS01_0015-0279/ |
| siRNA Fluorescent Universal Negative Control#1, 6-FAM | Sigma-Aldrich | SIC007 |
| Recombinant DNA | ||
| Plasmid: pCMV6-Entry-cMYC (tag: c-term Myc-DDK) | Origene | RC201611 |
| Plasmid: pCMV6-Entry-cMYC (C171Y mutant, tag: c-term Myc-DDK) | This study | N/A |
| Plasmid: pCMV6-Entry-eGFP (tag: c-term Myc-DDK) | (Ward et al., 2019) | N/A |
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| IP2 proteomics pipeline 5.0.1 | Integrated Proteomics Applications | N/A |
| Other | ||
Highlights.
We have identified a covalent ligand EN4 that targets C171 of MYC.
EN4 functionally and covalently targets an intrinsically disordered region in MYC.
EN4 directly targets MYC in cells and reduces MYC and MAX thermal stability.
EN4 inhibits MYC activity, downregulates MYC targets, and impairs tumorigenesis.
Acknowledgements
We thank the members of the Nomura Research Group and Novartis Institutes for BioMedical Research for critical reading of the manuscript. We thank Matthew D. Shirley and Joshua M. Korn for development and support of the PISCES package for quantitation and QC of mRNA-seq data. This work was supported by Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies (NB-CPACT) for all listed authors. This study was also funded by the Mark Foundation for Cancer and Chordoma Foundation ASPIRE Award and the National Institutes of Health (R01CA240981). LB was also supported by a National Science Foundation Graduate Research Fellowship.
Declaration of Interests
J.A.T., J.M.K., G.L., and M.S. are employees of Novartis Institutes for BioMedical Research. This study was funded by the Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies. D.K.N. is a co-founder, share-holder, and adviser of Frontier Medicines. We also have a patent application filed based on this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amati B, Alevizopoulos K, and Vlach J (1998). Myc and the cell cycle. Front. Biosci. J. Virtual Libr 3, d250–268. [DOI] [PubMed] [Google Scholar]
- Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman LA, Nguyen TB, Roberts AM, Miyamoto DK, Ku W-M, Huffman TR, Petri Y, Heslin MJ, Contreras CM, Skibola CF, et al. (2017). Chemoproteomics-enabled covalent ligand screen reveals a cysteine hotspot in reticulon 4 that impairs ER morphology and cancer pathogenicity. Chem. Commun. Camb. Engl 53, 7234–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, and Vogt PK (2002). Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci 99, 3830–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Liu H, and Qing G (2018). Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY-S, Shin HR, Berdan CA, Ford B, Ward CC, Olzmann JA, Zoncu R, and Nomura DK (2019). Covalent targeting of the vacuolar H + -ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmperman P, Baranauskiene L, Jachimoviciūte S, Jachno J, Torresan J, Michailoviene V, Matuliene J, Sereikaite J, Bumelis V, and Matulis D (2008). A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J 95, 3222–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DJB, Lachmann A, Evangelista JE, and Ma’avan A Signature Commons Signature Search.
- Conacci-Sorrell M, Ngouenet C, and Eisenman RN (2010). Myc-nick: a cytoplasmic cleavage product of Myc that promotes alpha-tubulin acetylation and cell differentiation. Cell 142, 480–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Reddy EP, Shokat KM, and Soucek L (2017). Drugging the “undruggable” cancer targets. Nat. Rev. Cancer 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman EA, Ward CC, Spradlin JN, Bateman LA, Huffman TR, Miyamoto DK, Kleinman JI, and Nomura DK (2017). Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-cancer Natural Products. Cell Chem. Biol 24, 1368–1376.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, Sagar V, Luan Y, Chalmers ZR, Unno K, et al. (2019). Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 36, 483–497.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, Creasman KJ, Bazarov AV, Smyth JW, Davis SE, et al. (2012). MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med 209, 679–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hydbring P, Castell A, and Larsson L-G (2017). MYC Modulation around the CDK2/p27/SKP2 Axis. Genes 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessani N, Humphrey M, McDonald WH, Niessen S, Masuda K, Gangadharan B, Yates JR, Mueller BM, and Cravatt BF (2004). Carcinoma and stromal enzyme activity profiles associated with breast tumor growth in vivo. Proc. Natl. Acad. Sci. U. S. A 101, 13756–13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Liu Z g, and Zandi, E. (1997). AP-1 function and regulation. Curr. Opin. Cell Biol 9, 240–246. [DOI] [PubMed] [Google Scholar]
- Kress TR, Sabò A, and Amati B (2015). MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593. [DOI] [PubMed] [Google Scholar]
- Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, and Nordlund P (2013). Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87. [DOI] [PubMed] [Google Scholar]
- McKeown MR, and Bradner JE (2014). Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect. Med 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, and Penn LZ (2008). Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990. [DOI] [PubMed] [Google Scholar]
- Osorio J (2015). Cell cycle: repurposing MYC and E2F in the absence of RB. Nat. Rev. Mol. Cell Biol 16, 516–517. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon: fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick E, Bradley A, Gan J, Douangamath A, Krojer T, Sethi R, Geurink PP, Aimon A, Amitai G, Bellini D, et al. (2019). Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc 141, 8951–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L, To M, Proudfoot A, Ornelas E, Woldegiorgis M, et al. (2019). Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol 15, 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struntz NB, Chen A, Deutzmann A, Wilson RM, Stefan E, Evans HL, Ramirez MA, Liang T, Caballero F, Wildschut MHE, et al. (2019). Stabilization of the Max Homodimer with a Small Molecule Attenuates Myc-Driven Transcription. Cell Chem. Biol 26, 711–723.e14. [DOI] [PubMed] [Google Scholar]
- Ward CC, Kleinman JI, Brittain SM, Lee PS, Chung CYS, Kim K, Petri Y, Thomas JR, Tallarico JA, McKenna JM, et al. (2019a). Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol 14, 2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CC, Kleinman JI, Brittain SM, Lee PS, Chung CYS, Kim K, Petri Y, Thomas JR, Tallarico JA, McKenna JM, et al. (2019b). Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol 14, 2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, and Cravatt BF (2010). Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield JR, Beaulieu M-E, and Soucek L (2017). Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E, Lin CY, Eilers M, and Levens DL (2015). Taming of the beast: shaping Myc-dependent amplification. Trends Cell Biol. 25, 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfer A, Wittner BS, Irimia D, Flavin RJ, Lupien M, Gunawardane RN, Meyer CA, Lightcap ES, Tamayo P, Mesirov JP, et al. (2010). MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. U. S. A 107, 3698–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chen Y, and Olopade OI (2010). MYC and Breast Cancer. Genes Cancer 1, 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015). ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller KI, Jegga AG, Aronow BJ, O’Donnell KA, and Dang CV (2003). An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 4, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zörnig M, and Evan GI (1996). Cell cycle: On target with Myc. Curr. Biol 6, 1553–1556. [DOI] [PubMed] [Google Scholar]
- Reflecting on 25 years with MYC | Nature Reviews Cancer.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 1. Covalent ligand structures and screening data for data shown in Figure 1a, 1b.
Table S2. Related to Figure 2 and Figure S3. IsoTOP-ABPP analysis of EN4 in 231MFP breast cancer cells shown in Figure S3.
Table S3. Related to Figure 4. RNA sequencing analysis of EN4-mediated transcriptional changes in 231MFP cells. 231MFP cells were treated with DMSO vehicle or EN4 (50 μM) for 24 h, n=3 biological replicates/group. Tab 1 shows total RNA sequencing data, quantitation, and statistical analysis. Contrast analysis produced t-test p-values, Benjamini-Hochberg false-discovery rates (FDRs) and log 2 fold changes for each gene with treated versus DMSO vehicle-treated controls. Tab 2 shows signature enrichment. For the signature enrichment, genes with an FDR < 0.1 and Fold change <= 0.75 (567 genes) were selected and signature search was performed using the MSigDB Collection H hallmark gene set: https://www.gseamsigdb.org/gsea/msigdb/collections.jsp#H.
Data Availability Statement
Data generated in this study will be made available upon reasonable request. No code was developed for this study.