

Abstract
A better understanding of the human central nervous system (CNS) pharmacokinetics is critical to the selection of the right drug and refinement of dosing regimen for more effective treatment of primary and metastatic brain cancer. Using the physiologically‐based pharmacokinetic (PBPK) modeling approach, we systematically compared the CNS pharmacokinetics of three cyclin D‐cyclin dependent kinase 4 and 6 (CDK4/6) inhibitors (ribociclib, palbociclib, and abemaciclib) in patients with cancer. A PBPK model platform was developed and verified for predicting plasma and CNS pharmacokinetics. Target engagement ratio (TER), defined as the ratio of the average steady‐state unbound drug brain concentration to the in vitro half‐maximal inhibitory concentration (IC50) for CDK4/6 inhibition, was used as a crude predictor of efficacy. As compared with ribociclib and palbociclib, abemaciclib penetrated into the human brain to a larger extent, but at a slower rate, and was retained in the brain longer. Following the standard dosing regimens, the predicted CDK4/6 TERs were 26/5.2 for abemaciclib, 2.4/0.62 for ribociclib, and 0.36/0.27 for palbociclib. Simulations suggested that abemaciclib achieved comparable TERs following twice daily or daily dosing; ribociclib may sufficiently inhibit both CDK4 and CDK6 at the maximum tolerated dose; whereas, palbociclib achieved TERs < 0.5 even at a dose 50% higher than the standard dose. In conclusion, the PBPK modeling, supported by available preclinical and clinical evidence, suggests that abemaciclib is the best CDK4/6 inhibitor for brain cancer treatment, whereas palbociclib is not recommended. The model refined dosing regimen is 300 mg daily on a 4‐weeks‐on schedule for abemaciclib, and 900 mg daily on a 3‐weeks‐on/1‐week‐off schedule for ribociclib.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Cyclin D‐cyclin dependent kinase 4 and 6 (CDK4/6) inhibitors (ribociclib, palbociclib, and abemaciclib) are under clinical development for treating primary and metastatic brain cancer. Sufficient drug penetration across the human blood‐brain barrier is the prerequisite for sustained in vivo target inhibition and efficacious treatment of brain cancer. Selection of the drug with favorable central nervous system (CNS) pharmacokinetics and design of optimal dosing regimen will maximize the success of the development and treatment. However, the human CNS pharmacokinetics of the CDK4/6 inhibitors remain understudied and poorly understood.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Using the physiologically‐based pharmacokinetic (PBPK) modeling approach, we systematically compared the human CNS pharmacokinetics of the three CDK4/6 inhibitors.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The PBPK modeling, supported by available preclinical and clinical evidence, suggests that abemaciclib is the best CDK4/6 inhibitor for brain cancer treatment, whereas palbociclib is not recommended. The model refined dosing regimen is 300 mg daily on a 4‐weeks‐on schedule for abemaciclib, and 900 mg daily on a 3‐weeks‐on/1‐week‐off schedule for ribociclib. Further prospective clinical trials are needed to confirm this.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Our study composes a framework for the application of mechanism‐based PBPK modeling to guide selection of the right drug and refinement of dosing regimen for more effective treatment of brain cancer.
Despite significant advances in understanding cancer biology, the mortality rates for primary and metastatic brain cancer have remained steady for more than 30 years. For patients with newly diagnosed glioblastoma (the most common and aggressive malignant primary brain cancer), the median overall survival is 14.6 months with the current standard of care. 1 , 2 For patients with metastatic brain cancer, occurring in 10–35% of adult patients with non‐brain primary cancer, 3 the median overall survival is 7.2 months. 4 There is an unmet need for the development of more effective drug therapies for both primary and metastatic brain cancer.
Loss of cell cycle regulation leading to uncontrolled cellular proliferation is a hallmark of cancer. The cyclin D‐cyclin dependent kinase 4 and 6 (CDK4/6)‐retinoblastoma (Rb) pathway is a key regulator of cell cycle, where CDK4/6 interacts with cyclin D to phosphorylates the tumor suppressor protein Rb and subsequently release the transcription factor E2F, thus promoting the G1 to S phase cell cycle progression. The cyclin D‐CDK4/6‐Rb pathway is often dysregulated in virtually all human cancers via both genetic and epigenetic mechanisms, resulting in increased kinase activity. 5 , 6 , 7 Excessive CDK4/6 activity may suppress senescence and directly contribute to both initiation and maintenance of the cell transformed state. 8 , 9 Inhibition of the cyclin D‐kinase activity has emerged as a novel cancer therapeutic strategy. Three orally bioavailable, selective, small molecule CDK4/6 inhibitors, including ribociclib, palbociclib, and abemaciclib, have been approved by the US Food and Drug Administration (FDA) as monotherapy or combination with hormone therapy for treating hormone receptor‐positive, HER2‐negative metastatic breast cancers. 10 In light of the central role of the cyclin D‐CDK4/6‐Rb pathway in controlling cellular proliferation and its frequent dysregulation in glioblastoma and cancers with a high incidence of brain metastases, CDK4/6 inhibitors are currently under active clinical investigation in patients with glioblastoma 11 , 12 and brain metastases of breast cancer, non‐small cell lung cancer, or melanoma (ClinicalTrials.gov Identifier: NCT02308020).
Although sharing a similar mechanism of action, ribociclib, palbociclib, and abemaciclib are distinct with respect to physicochemical properties, selectivity and potency for CDK4/6 inhibition, pharmacokinetics, and clinical toxicity profiles. 13 , 14 In addition, preclinical studies and preliminary clinical data suggest that the three CDK4/6 inhibitors plausibly penetrate into the human central nervous system (CNS) to a different extent. 12 , 14 , 15 , 16 Sufficient drug penetration into brain tumors, particularly into the invasive, infiltrating tumor cells behind an intact blood‐brain barrier (BBB), is the prerequisite for consistent in vivo target inhibition and efficacious treatment of primary brain cancer or brain metastasis. 17 Selection of the CDK4/6 inhibitor(s) with favorable CNS pharmacokinetics and design of optimal dosing regimen will maximize the success in the development of these novel agents for brain cancer treatment. However, as many other new or existing anticancer drugs, the CNS pharmacokinetics of the three CDK4/6 inhibitors in patients with cancer remain understudied and poorly understood largely due to the difficulty in accessing human brain tissues and the lack of in vitro or animal models that reliably predict human BBB permeability. Therefore, it is imperative that innovative approaches are developed to more reliably predict drug disposition in the human CNS. One such an approach is the in vitro‐in vivo extrapolation, physiologically‐based pharmacokinetic (IVIVE‐PBPK) modeling that enables prediction of pharmacokinetic processes in the system and individual organs by integrating human physiological parameters (e.g., enzyme and transporter protein abundances in organs) with drug‐specific parameters (e.g., physicochemical properties, in vitro metabolism, and transporter kinetics). 18
In this study, we developed and verified a whole‐body PBPK model integrated with a four‐compartment permeability‐limited brain model platform that accounted for the general anatomic structure of the human CNS and key processes governing the CNS pharmacokinetics of a drug. This mechanism‐based model platform allowed prediction of the plasma and CNS pharmacokinetics of CDK4/6 inhibitors following various dosing regimens. Our study provided critical mechanistic and quantitative information to guide selection of the right drug and refinement of dosing regimen for the rational development and optimal use of CDK4/6 inhibitors for brain cancer treatment.
METHODS
In vitro studies
In vitro studies were performed to determine drug‐specific parameters for ribociclib, palbociclib, and abemaciclib, including in vitro metabolic intrinsic clearance by human liver and intestinal microsomes, fraction unbound in patient plasma and brain tumor tissues, apparent transcellular passive permeability, and ABCB1/ABCG2‐mediated intrinsic efflux clearance. Details for in vitro experiments are provided in Supplementary Material s.
PBPK modeling and simulation
PBPK model development
A whole‐body PBPK model integrated with a 4Brain model (Figure 1 ) was developed for predicting the system and CNS pharmacokinetics of each CDK4/6 inhibitor, using the Simcyp Simulator V18 (Simcyp Ltd., Sheffield, UK). System‐specific parameters were derived from the existing Simcyp virtual cancer patient population, unless stated otherwise. Drug‐specific parameters are summarized in Table 1 . The model development is described in detail in Supplementary Material s.
Figure 1.
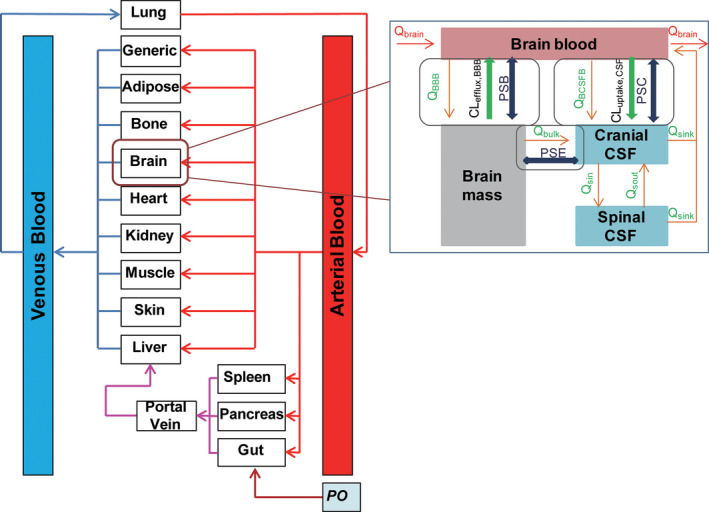
Model structure of the whole‐body physiologically‐based pharmacokinetic model integrated with a 4‐compartment permeability‐limited brain (4Brain) model, which has 4 compartments representing the brain blood, brain mass, cranial cerebrospinal fluid (CSF), and spinal CSF. The 4Brain model assumes: (1) drug transport across the blood‐brain barrier (BBB) is governed by bidirectional passive permeability (PSB) and ABCB1‐mediated and ABCG2‐mediated active efflux clearance (CLefflux,BBB); (2) drug transport across the blood‐CSF barrier was controlled by bidirectional PSB and ABCB1‐mediated active influx clearance (CLuptake,CSF); (3) drug transport between the brain mass and cranial CSF is diffusive transport at a bi‐directional clearance (PSB); (4) fluid balance is maintained by the circulation of CSF between spinal and cranial compartments and reabsorbed into the brain blood; (5) the cerebral blood flow rate (Q Brain) links the 4Brain model to whole‐body model; (6) only unbound and unionized drug can passively pass through all barriers, while transporters act upon unbound drug (including both unionized and ionized species); (7) metabolism in brain mass is negligible; and (8) all compartments are well‐stirred with defined volumes. Flow rates are described by the CSF secretion rate (Q BCSFB), bulk flow rate from brain mass to cranial CSF (Q bulk), CSF flow rate out of cranial and spinal compartments (Q sink), CSF shuttle flow rate between cranial and spinal compartments (Q Sin and Q Sout), and water transfer rate from the brain blood to brain mass (Q BBB).
Table 1.
Drug‐specific parameters in the whole‐body‐4Brain PBPK models
| Ribociclib | Palbociclib | Abemaciclib | Comments/reference | |
|---|---|---|---|---|
| Physicochemical | ||||
| MW, g/mol | 434.6 | 447.5 | 506.6 | PubChem |
| LogP | 2.38 | 2.77 | 4.25 | ChemAxon |
| PKa1, strongest acidic | 11.59 | 11.34 | 10.27 | ChemAxon |
| PKa2, strongest basic | 8.87 | 8.86 | 7.94 | ChemAxon |
| B/P | 1.5 | 1.9 | 1.2 | Investigator brochure |
| f u,p | 0.125 | 0.126 | 0.06 | Experimental determined |
| Absorption | ||||
| Fa | 0.9 | 0.5 | 0.6 | Assigned |
| Ka | 0.8 | 0.367 | 0.197 | Observed |
| Lag time, hours | 0.5 | 0.658 | 0.5 | Observed |
| fugut | 0.0061 | 0.007 | 0.004 | Simcyp predicted |
| Q gut, L/h | 9.61 | 14.48 | 11.08 | Simcyp predicted |
| Distribution | ||||
| V ss, L/kg | 12.19 | 16.31 | 7.86 | Predicted by Method 3, K p scalar of 1 |
| Elimination | ||||
| HLMs: | ||||
| Vmax, pmol/min/mg | 313.7 | 518.5 | 2003 | Experimental determined from HLM |
| K m, µM | 13.4 | 78.1 | 126.2 | Experimental determined from HLM |
| CLint, µL/min/mg | 23.4 | 6.64 | 15.9 | Experimental determined from HLM |
| fuinc | 0.35 | 0.2 | 0.2 | Experimental determined from HLM |
| HIMs: | ||||
| Vmax, pmol/min/mg | 104.8 | 280.7 | 494.3 | Experimental determined from HIM |
| K m, µM | 28.8 | 70.4 | 115.1 | Experimental determined from HIM |
| CLint, µL/min/mg | 3.64 | 3.99 | 4.29 | Experimental determined from HIM |
| fumic | 0.35 | 0.2 | 0.2 | Experimental determined |
| CLR, L/h | 7.5 | 10.5 | 1.5 | Assigned |
| 4Brain model | ||||
| BBB | ||||
| PSB, L/h | 135 | 307 | 25 | Estimated by Eq. 1 a |
| fu,br | 0.044 | 0.015 | 0.006 | Experimental determined from patient brain tumor tissue |
| CLABCB1,vitro, µL/min/mg | 21770 | 26160 | 800 | Experimental determined Eq. 2 b |
| CLABCG2,vitro, µL/min/mg | 0 | 8580 | 90 | Experimental determined Eq. 2 b |
| ABCB1 RAF at BBB | 87.3 | 87.3 | 87.3 | Determined based on ABCB1 abundance in normal human brain microvessels by Eq. 3 c |
| ABCB1 RAF at BBB | 5.88 | 5.88 | 5.88 | Determined based on ABCB1 abundance in human glioblastoma microvessels by Eq. 3 c |
| ABCG2 RAF at BBB | 125.3 | 125.3 | 125.3 | Determined based on ABCG2 abundance in normal human brain microvessels by Eq. 3 c |
| ABCG2 RAF at BBB | 55.47 | 55.47 | 55.47 | Determined based on ABCG2 abundance in human glioblastoma microvessels by Eq. 3 c |
| Blood‐CSF barrier | ||||
| PSC, L/h | 67.5 | 150 | 13 | Assumed to be half of PSB |
| fucsf | 1 | 1 | 1 | Assigned given low CSF protein concentration |
| CLABCB1,vitro, µL/min/mg | 21770 | 26160 | 800 | Experimental determined Eq. 2 b |
| ABCB1 RAF at BCCSF | 8.7 | 8.7 | 8.7 | Assigned and validated by observed ribociclib CSF data |
| Brain‐cranial CSF barrier | ||||
| PSE, L/h | 300 | 300 | 300 | Assigned assuming no barrier function |
BBB, blood‐brain barrier; B/P, blood‐to‐plasma partition ratio; CLABCB1,vitro and CLABCB1,vitro, ABCB1‐mediated and ABCG2‐mediated in vitro efflux clearance, respectively; CLint, in vitro intrinsic metabolic clearance; CLR, renal clearance; f u,br, fraction unbound drug in brain tissue; f u,p, fraction of unbound drug in plasma; fucsf, fraction unbound in cerebrospinal fluid; fugut, fraction of unbound drug in enterocytes; fumic, fraction of unbound in microsomal incubation; K m, substrate concentration at which half of Vmax is achieved; logP, logarithm of the neutral species octanol‐to‐buffer partition ratio; MW, molecule weight; PKa, acid dissociation constant; PBPK, physiologically‐based pharmacokinetic; PSB, passive permeability‐surface area product at the BBB; PSC, passive permeability‐surface area product at the blood‐cranial CSF barrier; PSE, passive permeability‐surface area product at the brain‐cranial CSF barrier; Q gut, gut blood flow; RAF, in vivo‐in vitro relative activity factor; Vmax, maximum metabolic rate; V ss, volume of distribution at steady‐state.
(Eq. 1), where P app,A→B is the apparent permeability determined from MDCKII cell monolayer; SA is the human brain microvasculature surface area (mean, 20 m2); and λ is unionization efficiency.
(Eq. 2), where CLefflux,vitro (µL/min/mg) is the in vitro efflux transporter‐mediated intrinsic clearance; NER is the net efflux ratio determined from MDCKII‐ABCB1or MDCKII–ABCG2; P app,A‐B is the apparent passive permeability determined from MDCKII; SA is the filter surface area (0.143 cm2) in a 96‐well transwell; λ is unionization efficiency; and Procell is the cell membrane protein amount in a 96‐well transwell.
(Eq. 3), where RAF is the relative activity factor; BMvPGB is the milligrams of brain microvessels per gram brain; BW is the average human brain weight; abundance in vivo or in vitro represents the ABCB1/ABCG2 transporter protein expression level in human brain microvessels or in MDCKII‐ABCB1 and –ABCG2 cells, respectively.
Model verification
The total and unbound drug concentration—time profiles of each CDK4/6 inhibitor in the plasma, brain, and CSF were predicted in a Simcyp virtual cancer population by simulations of 10 trials with 10 subjects in each trial. The dosing regimens for simulations were matched to the FDA‐approved standard dose regimens or that used in the published clinical trials. The developed PBPK models were verified by comparing the model‐predicted plasma and CNS pharmacokinetics with the observed clinical plasma and CNS (if available) data.
Simulations
To assist the design of optimal dosing regimens, simulations of 10 trials with 10 patients with cancer in each trial were performed to predict the plasma and CNS pharmacokinetics of each CDK4/6 inhibitor following the FDA‐approved standard and modified dosing regimens. The standard dosing regimen for ribociclib is 600 mg q.d. oral dosing on a 3‐weeks‐on/1‐week‐off schedule; for palbociclib is 125 mg q.d. oral dosing on a 3‐weeks‐on/1‐week‐off schedule; and for abemaciclib is 150 mg b.i.d. oral dosing on a 4‐weeks‐on continuous schedule. Modified dosing evaluated were 900 mg q.d. (maximum tolerated dose) for ribociclib, 187.5 mg q.d. (50% increase of the standard dose) for palbociclib, and 300 mg q.d. for abemaciclib.
RESULTS
Drug‐specific parameters
In vitro metabolic kinetics
The overall metabolic kinetics of ribociclib, palbociclib, and abemaciclib in human liver and intestinal microsomes were well‐described by Michaelis–Menten kinetics (Figure S1 ). The estimated in vitro metabolic kinetic parameters, including the maximum metabolic velocity (Vmax), substrate concentration at which 50% of Vmax is obtained (K m), and in vitro intrinsic metabolic clearance (CLint) estimated as Vmax/K m, are summarized in Table 1 and were used for predicting the whole‐organ liver and intestine metabolic clearance in the PBPK models.
Binding to patient plasma proteins and brain tumor tissues
In the plasma of patients with cancer, ribociclib and palbociclib were bound to plasma proteins to a similar extent, with the mean fraction unbound in plasma (f u,p) 0.125 and 0.126, respectively, whereas abemaciclib had higher plasma binding (mean f u,p, 0.06). Similarly, abemaciclib showed the highest binding to human brain tumor tissues with the mean fraction unbound in brain tissue (f u,br) 0.006, followed by palbociclib (mean f u,br, 0.015) and ribociclib (mean f u,br, 0.044).
Passive transcellular permeability
The three CDK4/6 inhibitors exhibited similar apical‐to‐basolateral apparent transcellular passive permeability (P app,A‐B) across MDCKII cell monolayers at pH 7.4 in both apical and basolateral chambers (Table 2 ). Based on pH partition theory (Henderson–Hasselbalch equation), the unionization efficiency (λ) at pH 7.4 for ribociclib (PKa, 8.87), palbociclib (PKa, 8.86), and abemaciclib (PKa, 7.94) was estimated to be 0.033, 0.034, and 0.224, respectively. Because only unbound and unionized drugs pass through biological membranes, the intrinsic passive transcellular permeability (P app,A‐B/λ) for unionized drugs were estimated (Table 2 ), and were then scaled to the human brain microvasculature surface area to predict the BBB passive permeability clearance (PSB) of the unbound and unionized drug in the PBPK model (Table 1 ).
Table 2.
Transcellular permeability and efflux ratios of the three CDK4/6 inhibitors, determined from the parental MDCKII, MDCKII‐ABCB1, and MDCKII‐ABCG2 cell monolayers
| Ribociclib | Palbociclib | Abemaciclib | |
|---|---|---|---|
| Parental MDCKII | |||
| Apparent permeability (P app,A‐B) (×10−6 cm/s) a | 7.17 ± 1.85 | 9.38 ± 1.16 | 9.09 ± 2.21 |
| Net efflux ratio b | 1.02 ± 0.21 | 1.55 ± 0.17 | 1.03 ± 0.30 |
| Unionization efficiency (λ) c | 0.033 | 0.034 | 0.224 |
| Intrinsic permeability (P app,A‐B/λ) d | 218.6 | 280.0 | 40.6 |
| MDCKII‐ABCB1 | |||
| Apparent permeability (P app,A‐B) (×10−6 cm/s) a | 1.77 ± 0.82 | 5.56 ± 1.10 | 5.65 ± 0.86 |
| Net efflux ratio b | 10.5 ± 3.30 | 5.92 ± 1.04 | 2.84 ± 0.49 |
| MDCKII‐ABCG2 | |||
| Apparent permeability P app,A‐B (×10−6 cm/s) a | 5.90 ± 0.91 | 7.73 ± 0.25 | 7.98 ± 0.42 |
| Net efflux ratio b | 1.02 ± 0.20 | 2.64 ± 1.05 | 1.21 ± 0.11 |
Data are expressed as the mean ± SD from at least three independent experiments (with duplicates in each experiment).
Bidirectional permeability experiments were performed at pH 7.4 in both apical and basolateral chambers.
Net efflux ratio was the efflux ratio in the absence of an ABCB1 inhibitor (elacridar) or ABCG2 inhibitor (Ko145) divided by the efflux ratio in the presence of the inhibitor.
Unionization efficiency (λ) is the ratio of unionized form to total drug (the sum of unionized and ionized forms), where the unionized‐to‐ionized ratio is calculated based on Henderson‐Hasselbalch equation: .
Intrinsic permeability is the transcellular permeability of the unbound and unionized drug, estimated as the mean P app,A‐B normalized by λ.
The three CDK4/6 inhibitors are weak base drugs. Similar to AZD1775 (a weak base drug), 19 the three CDK4/6 inhibitors exhibited pH‐dependent apparent permeability across the MDCKII, MDCKII‐ABCB1, and MDCKII‐ABCG2 cell monolayers (Figure 2 ). As the pH decreased (7.4, 7.0, 6.5, and 6.0) in the basolateral chamber (mimicking brain/brain tumor interstitium) whereas remaining the same (7.4) in the apical chamber (mimicking blood circulation), the apparent permeability increased in the apical‐to‐basolateral direction while decreasing in the basolateral‐to‐apical direction, overall resulting in reduced efflux ratios (Figure 2 ). This observation was in accordance with the pH partition theory. At a relatively acidic basolateral pH, the ionization of a weak base drug was increased, and, as a result, the basolateral‐to‐apical permeability decreased and apical‐to‐basolateral permeability increased, thus leading to the trap of ionized drugs in the basolateral compartment.
Figure 2.
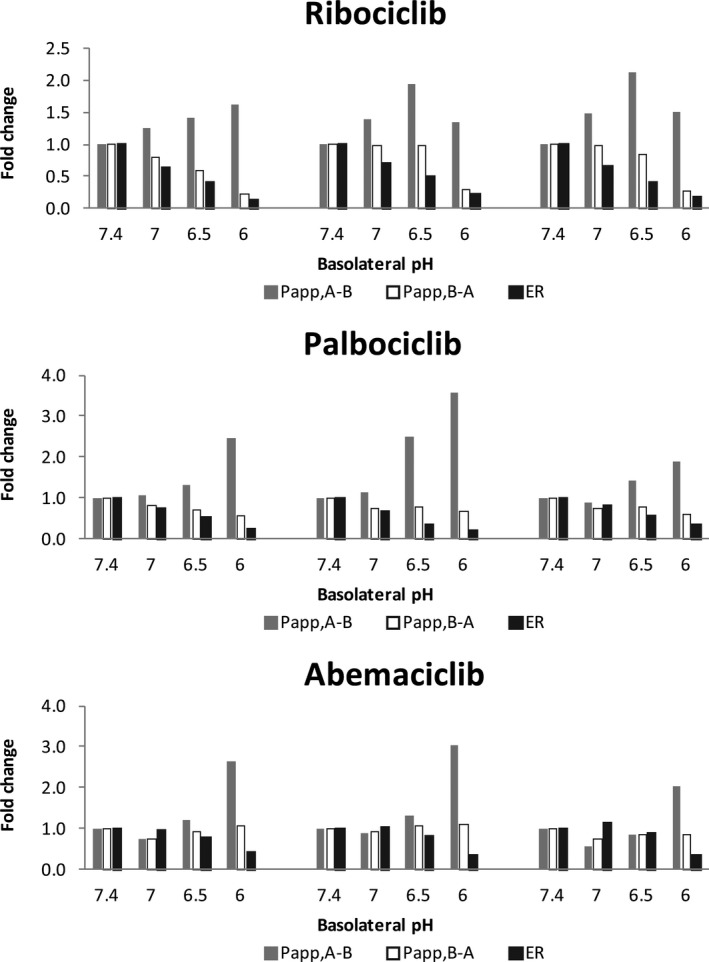
The pH‐dependent apparent permeability of the three cyclin D‐cyclin dependent kinase 4 and 6 inhibitors across the MDCKII, MDCKII‐ABCB1, and MDCKII‐ABCG2 cell monolayers. Bi‐directional permeability experiments were performed at a fixed apical pH (7.4) and varying basolateral pH (7.4, 7.0, 6.5, and 6.0). The apparent permeability in the apical‐to‐basolateral (P app,A‐B) and basolateral‐to‐apical (P app,B‐A) directions and efflux ratio (ER) are expressed as the mean fold change relative to those determined in the control (at pH 7.4 in both apical and basolateral chambers).
ABCB1‐mediated and ABCG2‐mediated intrinsic efflux clearance
MDCKII cells with stable expression of ABCB1 or ABCG2 were used to determine the interactions of CDK4/6 inhibitors with ABCB1 and ABCG2, two major efflux transporters that are expressed at the human BBB. The positive controls, loperamide (a typical ABCB1 substrate) and gefitinib (a typical ABCG2 substrate) exhibited a mean net efflux ratio of 6.11 and 8.86 on MDCKII‐ABCB1 and MDCKII‐ABCG2 cell monolayers, respectively, confirming the functional expression of ABCB1 and ABCG2 in these cellular models. All three CDK4/6 inhibitors were ABCB1 substrates, whereas ABCB1 efflux efficiency appeared the highest for ribociclib (net efflux ratio 10.5), followed by palbociclib (net efflux ratio 5.92) and abemaciclib (net efflux ratio 2.84; Table 2 ). Palbociclib was a substrate of ABCG2 (net efflux ratio 2.64), but ribociclib and abemaciclib were not good substrates for ABCG2 (net efflux ratio 1.02 and 1.21, respectively; Table 2 ). Based on the intrinsic transcellular passive permeability and net efflux ratio, in vitro ABCB1‐mediated intrinsic efflux clearance was determined to be 4,354, 5,232, and 160 µL/min/mg for ribociclib, palbociclib, and abemaciclib, respectively, and the respective in vitro ABCG2 intrinsic efflux clearance was 0, 1,716, and 18 µL/min/mg (Table 1 ). These values were then scaled by IVIVE strategy to predict in vivo ABCB1‐mediated and ABCG2‐mediated efflux clearance at the BBB.
In vivo‐in vitro relative activity factor of ABCB1 and ABCG2
Quantitative proteomics analyses of 30 human normal brain and 47 glioblastoma samples suggest that the median protein abundances of ABCB1 and ABCG2 are 3.38 and 6.21 pmol/mg in human normal brain microvessels, respectively; and their respective levels are reduced to 0.14 and 1.69 pmol/mg in human glioblastoma microvessels. 20 In the cell membranes of MDCKII‐ABCB1 and MDCKII‐ABCG2 cell lines, ABCB1 and ABCG2 protein abundances are 10.3 and 13.2 pmol/mg, respectively. 20 The mean recovery of microvessels isolated from human normal brain and glioblastoma is 0.19 and 0.31 mg per gram tissue, respectively. 20 Based on these data and given an average brain weight of 1,400 g, the relative activity factor (RAF) for ABCB1 and ABCG2 was determined to be 87.3 and 125.1 mg at the intact human BBB, respectively, whereas being 5.88 and 55.5 mg at the BBB in glioblastoma.
ABCB1 has been located at the apical (cerebrospinal fluid (CSF) facing) membrane of the blood‐cranial CSF barrier, and therefore ABCB1‐mediated active influx from the blood to cranial CSF was considered at the blood‐cranial CSF barrier. However, ABCB1 protein abundance data at this barrier is not available, and thus the RAF for ABCB1‐mediated active influx clearance at the blood‐CSF barrier was assigned and verified by the observed clinical CSF data of ribociclib.
PBPK modeling and simulations
Model verification for prediction of plasma pharmacokinetics
Following the dosing regimen (900 mg q.d. for 5 days) as used in the clinical trial, the PBPK model well‐predicted ribociclib mean plasma concentration time profiles and interindividual variabilities, as demonstrated by > 98% of observed plasma concentration data falling within the 5th and 95th percentiles of the simulated population mean profile (Figure 3 a). The model‐simulated population mean maximum plasma concentration (Cmax) and apparent oral clearance (CL/F) of ribociclib were 1.03‐fold and 0.90‐fold of the respective observed parameters (i.e., predicted vs. observed Cmax, 3.87 vs. 3.75 µmol/L; CL/F, 55.5 vs. 61.6 L/h). 12
Figure 3.
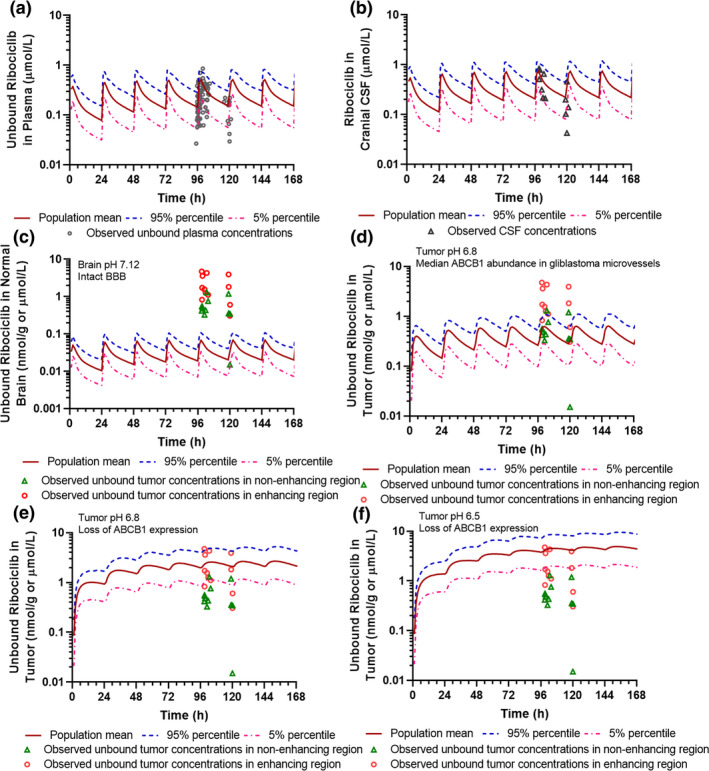
Physiologically‐based pharmacokinetic model‐simulated and clinically observed plasma and central nervous system (CNS) pharmacokinetics of ribociclib. (a) Simulated and observed unbound ribociclib plasma concentration‐time profiles. (b) Simulated and observed ribociclib cerebrospinal fluid concentration‐time profiles. (c) Simulated unbound ribociclib brain concentration‐time profiles in the human normal brain (with brain pH 7.12 and ABCB1 abundance 3.38 pmol/mg at the BBB). (d) Simulated unbound ribociclib concentration‐time profiles in brain tumors with pH 6.8 and ABCB1 abundance 0.14 pmol/mg at the BBB. (e) Simulated unbound ribociclib concentration‐time profiles in brain tumors with pH 6.8 and loss of ABCB1 expression at the BBB. (f) Simulated unbound ribociclib concentration‐time profiles in brain tumors with pH 6.5 and loss of ABCB1 expression at the BBB. Simulations of 10 trials with 10 subjects in each were performed in the Simcyp virtual cancer patient population following 5‐day ribociclib treatment at a daily oral dose of 900 mg. Observed clinical plasma and CNS pharmacokinetic data were obtained from 12 glioblastoma patients treated with ribociclib (900 mg q.d. for 5 days).
Similarly, the developed PBPK models well‐predicted the plasma pharmacokinetics of palbociclib and abemaciclib. The simulated population mean plasma pharmacokinetic parameters were within 0.90–1.15‐fold of the respective parameters estimated by population pharmacokinetic modeling analysis of observed data in patients with cancer. 21 , 22 Specifically, the predicted vs. observed palbociclib CL/F was 59.4 vs. 63.1 L/hour, elimination half‐life (T 1/2) was 30 vs. 29 hours, apparent volume of distribution (V/F) was 2,283 vs. 2,342 L, and oral bioavailability (F) was 0.47 vs. 0.46. The predicted vs. observed abemaciclib CL/F was 35.9 vs. 36.0 L/hour, T 1/2 was 22 vs. 20 hours, V/F was 1,041 vs. 1,050 L, and F was 0.52 vs. 0.45.
Model verification for prediction of CNS pharmacokinetics
Given the availability of ribociclib CNS pharmacokinetic data (i.e., CSF and tumor concentrations) observed in patients with glioblastoma, 12 the 4Brain model was first developed and verified for the prediction of CNS pharmacokinetics of ribociclib. The simulated ribociclib CSF concentrations were in line with observed CSF data in patients with glioblastoma, with all except for one observed data falling within the 5th and 95th percentiles of the simulated population mean profile (Figure 3 b). The model‐predicted population mean CSF‐to‐unbound plasma ratio (CSF Kp,uu, 1.6) was 0.9‐fold of the observed median CSF K p,uu (1.8) in 12 patients with glioblastoma. 12
The physical and biochemical barriers of the BBB in glioblastomas are often disrupted, as indicated by leaky tight junctions and loss or significant reduction in protein expression of major BBB efflux transporters. 20 It has been reported that the median protein abundances of ABCB1 and ABCG2 in glioblastoma microvessels are reduced to 3.7% and 35% of the normal levels, respectively. 20 In addition, electrode measurements of pH in human brain tumors are as low as 5.9 with a mean around 6.8; whereas, normal human brain maintains a pH at ~ 7.1–7.2. 23 , 24 , 25 As demonstrated by the in vitro pH‐dependent apparent permeability data (Figure 2 ), the relative acidic tumor environment would be favorable to the tumor penetration and trap of weak base drugs, regardless whether they are the substrates of ABCB1/ABCG2. The regional pH difference in the normal brain and brain tumors may contribute to the heterogeneous brain/tumor distribution of CDK4/6 inhibitors. Therefore, to predict ribociclib concentrations in the human normal brain as well as in contrast non‐enhancing and enhancing tumor regions of glioblastoma, different ABCB1/ABCG2 protein abundances at the BBB and varying brain pH were applied in the simulations following the dosing regimen used in the clinical trial (900 mg q.d. for 5 days; Figure 3 c‐f). In the normal brain with an intact BBB (i.e., brain pH 7.12, ABCB1 abundance 3.38 pmol/mg), the simulated population mean steady‐state average concentration (C ss,ave) of unbound ribociclib was 0.036 µmol/L and unbound brain‐to‐plasma ratio (K p,uu) was 0.14 (Figure 3 c). In brain tumors with increasing BBB disruption, specifically in brain tumors with (i) pH 6.8 and ABCB1 abundance 0.14 pmol/mg (Figure 3 d), (ii) pH 6.8 and loss of ABCB1 expression (Figure 3 e), and (iii) pH 6.5 and loss of ABCB1 expression (Figure 3 f), the simulated population mean C ss,ave of unbound ribociclib was 0.453, 2.608, 5.096 µmol/L, respectively, and the respective K p,uu was 1.84, 10.6, and 20.8. Notably, the predicted ribociclib tumor concentrations and extent of tumor penetration (K p,uu) were in accordance with the observed data in non‐enhancing and enhancing tumor regions in patients with glioblastoma, showing the observed median (range) unbound ribociclib tumor concentrations of 0.458 (0.015–1.295) µmol/L and 1.644 (0.307–4.743) µmol/L, respectively, and the observed K p,uu of 1.8 (0.7–11.3) and 10.1 (2.4–37.4), respectively. 12 Collectively, these data suggested that the developed 4Brain model well‐predicted the CNS pharmacokinetics of ribociclib as observed in patients with glioblastoma. Therefore, the system‐specific parameters for the 4Brain model (e.g., the transporter RAFs at the BBB and blood‐CSF barrier) were verified, and could be confidently applied to predict the CNS pharmacokinetics of palbociclib and abemaciclib (for which no or limited clinical CNS data were available).
The model‐simulated steady‐state concentrations of unbound abemaciclib and palbociclib in the human normal brain and CSF are summarized in Table 3 . For abemaciclib, limited CSF data were obtained from 10 patients following varying dosing regimens (i.e., daily or twice daily oral dosing at 50–275 mg for 15 days). 14 A direct comparison between the observed and predicted abemaciclib CSF data was not feasible because of varying dosing regimens and varying sampling times in the reported clinical study. Nevertheless, the observed abemaciclib CSF concentrations (range, 0.004–0.030 µmol/L) generally overlapped with the predicted data (population mean C ss,ave, 0.038 µmol/L; 5th and 95th percentiles, 0.013–0.091 µmol/L) following the standard dosing regimen (150 mg b.i.d. for 28 days). In addition, the predicted mean CSF K p,uu was 1.10‐fold of the observed data (predicted vs. observed mean, 0.77 vs. 0.70).
Table 3.
PBPK model‐predicted unbound drug exposure in the plasma, normal brain, cranial CSF, and spinal CSF at the steady‐state following the standard or modified dosing regimens a
| Ribociclib | Palbociclib | Abemaciclib | ||||
|---|---|---|---|---|---|---|
| Standard (600 mg q.d., 21 days) | Alternative (900 mg q.d., 21 days) | Standard (125 mg q.d., 21 days) | Alternative (187 mg q.d., 21 days) | Standard (150 mg b.i.d., 28 days) | Alternative (300 mg q.d., 28 days) | |
| Plasma | ||||||
| Tss,max, hours | 1.32 | 1.32 | 3.84 | 3.84 | 2.91 | 3.84 |
| Css,max, µmol/L | 0.298 | 0.448 | 0.038 | 0.057 | 0.055 | 0.062 |
| Css,min, µmol/L | 0.092 | 0.139 | 0.022 | 0.033 | 0.045 | 0.040 |
| Css,ave, µmol/L | 0.176 | 0.264 | 0.029 | 0.044 | 0.050 | 0.050 |
| AUC24h, µmol/L*h | 4.291 | 6.440 | 0.779 | 1.167 | 1.683 | 1.326 |
| Brain | ||||||
| Tss,max, hours | 3.84 | 3.84 | 3.84 | 3.84 | 6.27 | 10.56 |
| Css,max, µmol/L | 0.039 | 0.059 | 0.005 | 0.007 | 0.052 | 0.053 |
| Css,min, µmol/L | 0.014 | 0.020 | 0.003 | 0.004 | 0.051 | 0.050 |
| Css,ave, µmol/L | 0.024 | 0.036 | 0.004 | 0.005 | 0.052 | 0.052 |
| AUC24h, µmol/L*h | 0.624 | 0.935 | 0.096 | 0.143 | 1.746 | 1.391 |
| Brain K p,uu b | 0.14 | 0.14 | 0.12 | 0.12 | 1.04 | 1.05 |
| TER for CDK4 c | 2.4 | 3.6 | 0.36 | 0.45 | 26 | 26 |
| TER for CDK6 c | 0.62 | 0.92 | 0.27 | 0.33 | 5.2 | 5.2 |
| Cranial CSF | ||||||
| Tss,max, hours | 1.32 | 1.32 | 3.84 | 3.84 | 6.27 | 10.56 |
| Css,max, µmol/L | 0.475 | 0.713 | 0.068 | 0.102 | 0.039 | 0.040 |
| Css,min, µmol/L | 0.149 | 0.223 | 0.038 | 0.058 | 0.038 | 0.036 |
| Css,ave, µmol/L | 0.281 | 0.421 | 0.052 | 0.078 | 0.038 | 0.038 |
| AUC24h, µmol/L*h | 6.890 | 10.33 | 1.385 | 2.078 | 1.311 | 1.045 |
| CSF K p,uu d | 1.60 | 1.60 | 1.78 | 1.78 | 0.78 | 0.79 |
| Spinal CSF | ||||||
| Tss,max, hours | 3.84 | 3.84 | 6.36 | 6.36 | 6.27 | 10.56 |
| Css,max, µmol/L | 0.404 | 0.606 | 0.063 | 0.094 | 0.039 | 0.039 |
| Css,min, µmol/L | 0.159 | 0.238 | 0.040 | 0.060 | 0.038 | 0.037 |
| Css,ave, µmol/L | 0.263 | 0.394 | 0.051 | 0.076 | 0.038 | 0.038 |
| AUC24h, µmol/L*h | 6.742 | 10.11 | 1.379 | 2.069 | 1.293 | 1.030 |
| CSF K p,uu d | 1.57 | 1.57 | 1.77 | 1.77 | 0.77 | 0.78 |
AUC24h, area under the concentration time curve during 24 hours at the steady‐state; CDK, cyclin D‐cyclin dependent kinase; CSF, cerebrospinal fluid; Css,ave, average steady‐state concentration; Css,max, maximum steady‐state concentration; Css,min, trough steady‐state concentration; PBPK, physiologically‐based pharmacokinetic; TER, target engagement ratio.
Simulations of 10 trials with 10 subjects in each trial were performed in the Simcyp cancer patient population. Data are presented as the population mean values.
Brain K p,uu is estimated as the AUC24h ratio of unbound brain to unbound plasma at the steady‐state.
TER is calculated as the ratio of the average steady‐state unbound brain concentrations to the in vitro IC50 for inhibiting CDK4/6 enzymes.
CSF K p,uu is estimated as the AUC24h ratio of CSF to unbound plasma at the steady‐state.
For palbociclib, no clinical CNS pharmacokinetic data was available. Preclinical studies showed that the brain penetration of palbociclib (K p,uu, 0.01 in rats and mice) was ~ 10‐fold less than that of abemaciclib (K p,uu, 0.11 in rats and 0.17 in mice). 15 Despite the species difference in the absolute K p,uu values between humans and rodents, the model‐predicted 10‐fold less brain penetration of palbociclib than abemaciclib in humans (K p,uu, 0.12 vs. 1.04) was consistent with preclinical observation. The brain penetration of ABCB1 substrates is expected to be larger in humans than rodents because ABCB1 protein abundance is significantly lower at the human BBB than rodent BBB and also because of species difference in the CSF dynamics and brain physiology. 20 , 26 For example, the brain penetration (K p,uu) of ABCB1 substrate drugs (including verapamil, GR205171, altanserin, and AZD1775) has been reported to be several to over 10‐fold greater in humans than in rodents. 19 , 27 , 28
Comparison of the CNS pharmacokinetics among the three CDK4/6 inhibitors
Ribocliclib and palbociclib are similar with respect to the chemical structure, physicochemical properties, binding to plasma proteins and brain tissues, as well as intrinsic passive transcellular permeability and intrinsic active efflux clearance; whereas abemaciclib is significantly dissimilar than either one (Table 1 and Table 2 ). Therefore, it was not surprising that the CNS pharmacokinetics of abemaciclib was different from the other two CDK4/6 inhibitors, which was characterized by a slower brain penetration but to a larger extent (Figure 4 a‐c). It is known that the rate of drug brain penetration is largely driven by the drug binding to brain tissue and passive or active efflux clearance at the BBB. Specifically, higher brain tissue binding and lower passive or active efflux clearance at the BBB lead to slower brain penetration. 19 Hence, the slower brain penetration of abemaciclib was expected given its significantly higher brain tissue binding and lower passive and active efflux clearance at the BBB as compared with ribociclib and palbociclib.
Figure 4.
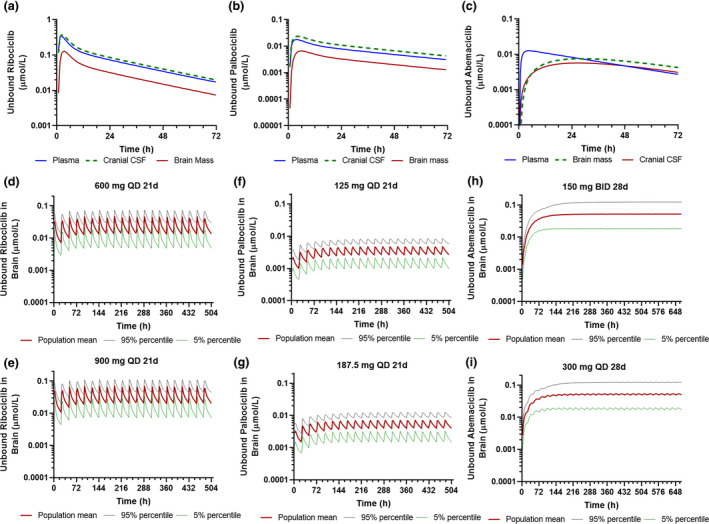
Physiologically‐based pharmacokinetic model‐simulated plasma and central nervous system pharmacokinetics of the three cyclin D‐cyclin dependent kinase 4 and 6 inhibitors following varying dosing regimens. (a) Simulated population mean plasma, CSF, and brain concentration time profiles of unbound ribociclib following a single oral dose (600 mg). (b) Simulated population mean plasma, CSF, and brain concentration time profiles of unbound palbociclib following a single oral dose (125 mg). (c) Simulated population mean plasma, CSF, and brain concentration time profiles of unbound abemaciclib following a single oral dose (150 mg). (d, e) Simulated unbound ribociclib brain concentration time profiles following the standard dosing regimen (600 mg q.d. for 3‐weeks) and modified dosing regimen (900 mg q.d. for 3 weeks). (f, g) Simulated unbound palbociclib brain concentration time profiles following the standard dosing regimen (125 mg q.d. for 3‐weeks) and modified dosing regimen (187 mg q.d. for 3 weeks). (h, i) Simulated unbound abemaciclib brain concentration time profiles following the standard dosing regimen (150 mg b.i.d. for 4‐weeks) and modified dosing regimen (300 mg q.d. for 4 weeks). Simulations of 10 trials with 10 subjects in each trial were performed in the Simcyp virtual cancer patient population.
Despite slow brain penetration, abemaciclib penetrated into the brain to a larger extent than the other two CDK4/6 inhibitors. The PBPK models predicted that ribociclib and palbociclib shared a similar brain K p,uu (~ 0.14 vs. 0.12), whereas abemaciclib had a K p,uu of ~ 1.0 (Table 3 ). It is known that the K p,uu (the extent of brain penetration) is determined by the relative contribution of passive clearance and transporter‐mediated active clearance at the BBB. 29 If the BBB transport of a drug is dominated by passive permeability, the K p,uu would approach 1; whereas when a drug is actively transported by efflux or uptake transporters at the BBB, the K p,uu would be smaller or larger than 1.0. Although the three CDK4/6 inhibitors were ABCB1 substrates, abemaciclib showed ~ 30‐fold lower ABCB1‐mediated intrinsic efflux clearance as compared with ribociclib and palbociclib (Table 1 ). In addition, although palbociclib and abemaciclib were identified as the substrates for mouse abcg2, 15 our study suggested that palbociclib was a substrate of human ABCG2, but ribociclib and abemaciclib were not, at least not good substrates for human ABCG2 (Table 2 ). ABCB1 and ABCG2 are two predominant efflux transporters at the human BBB, acting synergistically to restrict the brain penetration of dual ABCB1/ABCG2 substrates. 30 , 31 , 32 Palbociclib was a dual substrate for ABCB1 and ABCG2, whereas ribociclib and abemaciclib appeared to be transported mainly by ABCB1. Collectively, the IVIVE predicted that the overall efflux transporter‐mediated in vivo efflux clearance at the human BBB was 1.0 L/hour for abemaciclib, 22.8 L/hour for ribociclib, and 40.3 L/hour for palbociclib, which accounted for 0.2‐fold, 6.4‐fold, and 4.9‐fold of their respective BBB passive clearance at a normal brain pH. These data suggested that the BBB transport of abemaciclib was dominated by passive permeability, whereas the BBB transport of ribociclib and palbociclib was dominated by active efflux.
Also noted was that the CSF pharmacokinetics of abemaciclib was different from the other two CDK4/6 inhibitors (Figure 4 a‐c). The predicted mean CSF‐to‐unbound plasma ratio of abemaciclib (CSF K p,uu, 0.8) was close to its brain K p,uu (1.0), indicating that abemaciclib CSF concentration could be used as a surrogate for unbound brain concentration. By contrary, the predicted CSF K p,uu of ribociclib (~ 1.6) and palbociclib (~ 1.8) was > 10‐fold of their respective brain K p,uu, consistent with the predicted ~ 10‐fold higher CSF concentrations than their respective unbound brain concentrations (Table 3 ). Drug penetration from the circulation blood to CSF is controlled by the blood‐CSF barrier, which is formed by the choroid plexus epithelial cells and the arachnoid membrane. 33 ABCB1 expression has been identified on the apical, CSF‐facing side of the blood‐CSF barrier, suggesting that ABCB1 facilitates the transport of substrates into the CSF. 34 , 35 Therefore, the dominant ABCB1‐mediated active CSF uptake of ribociclib and palbociclib at the blood‐CSF barrier resulted in CSF K p,uu > 1.0. By contrary, ABCB1 played a minor role in the BBB or CSF penetration of abemaciclib because of the insignificant ABCB1 intrinsic clearance of abemaciclib. The PBPK model predictions supported the notion that CSF concentrations may be useful as a reflection of unbound drug brain concentrations for drugs (e.g., abemaciclib) that pass the BBB and blood‐CSF barrier mainly by passive transcellular permeability, whereas for drugs (e.g., ribociclib and palbociclib) that undergo dominant active transport at the BBB and blood‐CSF barrier, the relationship between unbound drug brain and CSF concentrations may not be obvious. 36
Refining dosing regimens
Target engagement ratio, defined as the ratio of the average steady‐state unbound drug brain concentration to the in vitro IC50 for CDK4/6 inhibition, was used as a crude predictor of efficacy. 15 Theoretically, a target engagement ratio ≥ 1 would lead to 50% or greater inhibition of CDK4/6. Abemaciclib is the most potent CDK4/6 inhibitor (with the in vitro IC50 of 2 and 10 nM for inhibition of CDK4 and CDK6, respectively) as compared with palbociclib (IC50, 11 and 15 nM) and ribociclib (IC50, 10 and 39 nM). 13 Simulations following the standard dosing regimens suggested that the predicted population mean brain C ss,ave of unbound ribociclib, palbociclib, and abemaciclib were 0.024, 0.004, and 0.052 µmol/L, respectively, and correspondingly the CDK4/6 target engagement ratios were 2.4/0.62 for ribociclib, 0.36/0.27 for palbociclib, and 26/5.2 for abemaciclib (Table 3 ). Based on the criterion of target engagement ratio ≥ 1 required for potential clinical efficacy, abemaciclib achieved adequate inhibition for CDK4 and CDK6 in the human brain at the standard dosing regimen (150 mg b.i.d.). However, ribociclib may sufficiently inhibit CDK4, but not CDK6 in the brain at the standard dosing regimen (600 mg q.d.). Palbociclib did not achieve adequate brain concentrations to inhibit CDK4 or CDK6 following the standard dosing regimen (125 mg q.d.).
Additional simulations were performed to refine dosing regimens (Figure 4 ). Following 21‐day treatment of ribociclib at the maximum tolerated dose (900 mg q.d.), the simulated population mean brain Css,ave of unbound ribociclib was 0.036 µmol/L and the corresponding CDK4/6 target engagement ratios were 3.6/0.92 (Table 3 ), indicating that the treatment of ribociclib at the maximum tolerated dose may sufficiently inhibit both CDK4 and CDK6. However, palbociclib did not achieve sufficient brain concentration for CDK4/6 inhibition (target engagement ratios < 0.5) even at a dose 50% higher than the standard dose (Table 3 ). For abemaciclib, simulations suggested that 150 mg b.i.d. and 300 mg q.d. dosing resulted in comparable unbound brain exposure in terms of the maximum, trough, or average steady‐state brain concentrations of unbound abemaciclib (Table 3 ).
DISCUSSION
Despite extensive efforts, progress in developing effective therapies for primary and metastatic brain cancer has been disappointing. Lack of efficacy may stem from genetically heterogeneous features of the tumor, emergence of intrinsic resistance mechanisms, and, particularly, poor penetration of potentially effective therapeutic agents across the human BBB. 37 , 38 , 39 The BBB is a dynamic interface separating the brain parenchyma from the circulatory system, which protects the CNS from potentially harmful chemicals while regulating transport of essential molecules and maintaining the homeostasis of the CNS. 39 The structure and function of the BBB in brain tumors is often disrupted and heterogeneous. 40 , 41 , 42 For example, glioblastoma consists of different regions presenting various degrees of BBB integrity varying from completely compromised in bulky tumor areas, slightly “leaky” in invasive peripheral regions, and to completely intact in infiltrating tumor regions. 17 , 20 , 40 , 41 , 42 In spite of enhanced drug penetration into bulky tumor areas, insufficient penetration of potentially effective therapeutic agents into invasive, infiltrative tumor regions or micro‐metastases behind an intact BBB, which are often unresectable and give rise to recurrent disease, is a significant barrier to long‐term, efficacious treatments. 17 Therefore, both primary and metastatic brain cancer should be treated as a disease of the whole brain. A potentially clinical successful therapeutic agent must penetrate across the intact human BBB to achieve adequate pharmacologically active concentrations in the brain. When selecting drug candidates and designing dosing regimens for brain cancer treatment, one needs to consider the drug brain penetration, system pharmacokinetics, and potency for target inhibition.
The K p,uu, as the measure of the extent of drug brain penetration, is a key parameter considered in CNS drug discovery and development. Nevertheless, it should be emphasized that brain exposure to unbound drug, which drives pharmacological activity and efficacy, is determined by both the K p,uu and systemic (or plasma) drug exposure. PBPK model simulations, supported by in vitro cellular permeability and transporter kinetic data, suggest that the transport of abemaciclib at the human BBB is dominated by passive permeability resulting in a K p,uu of ~ 1.0, whereas the transport of ribociclib (K p,uu 0.14) and palbociclib (K p,uu 0.12) is dominated by ABCB1/ABCG2‐medidated active efflux. Notably, although ribociclib and palbociclib share similar K p,uu, unbound ribociclib brain concentration is greater than five‐fold higher than palbociclib following the standard dosing regimens (Table 3 ). Likewise, although the extent of brain penetration of abemaciclib is approximately sevenfold larger than ribociclib (K p,uu, 1.0 vs. 0.14), the brain exposure to two drugs only differs by about twofold following the standard dosing regimens (Table 3 ). The relatively higher unbound ribociclib brain exposure is largely driven by its higher unbound plasma exposure. For example, the population mean plasma C ss,ave of unbound ribociclib is ~ 6.‐fold and 3.5‐fold of that for palbociclib and abemaciclib, respectively, following the standard dosing regimens (Table 3 ). Hence, in spite of a relatively low K p,uu, ribociclib has the potential to achieve adequate target inhibition in the brain at the maximum tolerate dose. These data underscore the importance for consideration of not only the extend of brain penetration (K p,uu) but also drug plasma exposure achievable at a safe or tolerable amount when selecting candidate drugs for brain cancer treatment.
Another critical consideration is the drug potency for target inhibition. In light of the notion that adequate target inhibition is the prerequisite for clinical efficacy, the target engagement ratio, which takes into account both unbound drug brain concentration and the potency for target inhibition, appears to be the best criteria for selection of brain cancer therapeutic agents. It has been demonstrated that the adequate inhibition of cyclin D‐CDK4/6‐Rb pathway (as indicated by a decrease in pRb > 60% in skin) is significantly associated with the clinical efficacy (stable disease or response) of abemaciclib in patients with HR‐positive breast cancer. 14 In our study, a target engagement ratio ≥ 1 achieved in the normal brain is used as the threshold to ensure CDK4/6 inhibition in brain tumors especially in microscopic tumor regions behind an intact BBB. Abemaciclib is the most potent CDK4/6 inhibitor as compared with palbociclib and ribociclib. 13 In addition, abemaciclib exhibits excellent brain penetration (K p,uu, 1.0) and reasonably good systemic exposure. These together lead to abemaciclib target engagement ratio of 26 (for CDK4) and 5.2 (for CDK6) following the standard dosing regimen. The potential clinical efficacy of abemaciclib for brain cancer treatment has been demonstrated in a phase I/II clinical trial in which 3 patients with glioblastoma achieved stable disease on single‐agent abemaciclib treatment, and 2 of them continued the treatment without progression for nearly 2 years. 14 By contrary, because of the low brain exposure and modest potency of palbociclib, the CDK4/6 target engagement ratio is < 0.5 even at a dose 50% higher than the standard dose. On the other hand, due to the relatively low potency for CDK4/6 inhibition, ribociclib may sufficiently inhibit CDK4 (target engagement ratio, 2.4) but not CDK6 (target engagement ratio, 0.62) although it achieves similar brain exposure as abemaciclib following the standard dosing regimens. These data collectively suggest that abemaciclib is the best CDK4/6 inhibitor for brain cancer treatment, whereas palbociclib is not recommended to use. Ribociclib has the potential to sufficiently inhibit both CDK4 and CDK6 at the maximum tolerated dose.
Different dosing strategies have been used for the three CDK4/6 inhibitors in the treatment of breast cancer, given their different system pharmacokinetics and clinical toxicities. Due to significant myelotoxicity, ribociclib and palbociclib require an intermittent dosing schedule (3‐weeks‐on/1‐week‐off) to allow bone marrow recovery. By contrast, abemaciclib has a safety profile allowing continuous dosing schedule (4‐week‐on), which provides important opportunities for combining abemaciclib with a variety of targeted or cytotoxic agents. Previous clinical study suggests that the administration of abemaciclib at 200 mg or 150 mg b.i.d. can achieve sustained pharmacodynamic effects in proliferating keratinocytes and tumors in patients with non‐CNS cancers, including breast cancer, non‐small cell lung cancer, and melanoma. 14 Hence, 200 mg and 150 mg b.i.d. was recommended as the starting dose for single‐agent and combination treatment, respectively. However, because of the reasonable plasma elimination half‐life of abemaciclib (observed vs. PBPK‐model predicted population mean T 1/2, 22 vs. 20 hours) and a broad dose range associated with clinical efficacy, a once‐daily dosing schedule may also achieve sustained target inhibition in the human brain. PBPK simulations suggest that 150 mg b.i.d. and 300 mg q.d. dosing result in comparable plasma or brain exposure to unbound abemaciclib in terms of the maximum, trough, or average steady‐state concentrations (Table 3 and Figure 4 ). Notably, similar to slow‐released formulations, the slow BBB penetration, and sustained brain retention of abemaciclib result in a small fluctuation between the peak and trough steady‐state brain concentrations following either twice‐daily or once‐daily dosing (Figure 4 ). Thus, 300 mg q.d. dosing would be expected to produce a similar, if not better, safety profile and clinical efficacy as 150 mg b.i.d. dosing. Considering the treatment convenience and likely better safety profile for once‐daily dosing, it would be reasonable to initiate abemaciclib for brain cancer treatment at 300 mg q.d. on a 4‐weeks‐on continuous schedule. Further prospective clinical trials are needed to confirm the refined dosing regimen.
Funding
This study was funded by the United States National Institute of Health (NIH) Cancer Center Support Grant P30 CA022453.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
J.L. wrote the manuscript. J.L. designed the research. J.L., J.J., J.W., X.B., and N.S. performed the research. J.L. analyzed the data.
Supporting information
Figure S1
References
- 1. Batich, K.A. & Sampson, J.H. Standard of care and future pharmacological treatment options for malignant glioma: an urgent need for screening and identification of novel tumor‐specific antigens. Expert Opin. Pharmacother. 15, 2047–2061 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ohgaki, H. & Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 109, 93–108 (2005). [DOI] [PubMed] [Google Scholar]
- 3. Gavrilovic, I.T. & Posner, J.B. Brain metastases: epidemiology and pathophysiology. J. Neuro‐Oncol. 75, 5–14 (2005). [DOI] [PubMed] [Google Scholar]
- 4. Sperduto, P.W. et al Summary report on the graded prognostic assessment: an accurate and facile diagnosis‐specific tool to estimate survival for patients with brain metastases. J. Clin. Oncol. 30, 419–425 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hall, M. & Peters, G. Genetic alterations of cyclins, cyclin‐dependent kinases, and CDK inhibitors in human cancer. Adv. Cancer Res. 68, 67–108 (1996). [DOI] [PubMed] [Google Scholar]
- 6. Sherr, C.J. Cancer cell cycles. Science 274, 1672–1677 (1996). [DOI] [PubMed] [Google Scholar]
- 7. Shapiro, G.I. Cyclin‐dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 24, 1770–1783 (2006). [DOI] [PubMed] [Google Scholar]
- 8. Yu, Q. et al Requirement for CDK4 kinase function in breast cancer. Cancer Cell 9, 23–32 (2006). [DOI] [PubMed] [Google Scholar]
- 9. Choi, Y.J. et al The requirement for cyclin D function in tumor maintenance. Cancer Cell 22, 438–451 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shah, M. , Nunes, M.R. & Stearns, V. CDK4/6 inhibitors: game changers in the management of hormone receptor‐positive advanced breast cancer? Oncology (Williston Park) 32, 216–222 (2018). [PMC free article] [PubMed] [Google Scholar]
- 11. Geoerger, B. et al A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin. Cancer Res. 23, 2433–2441 (2017). [DOI] [PubMed] [Google Scholar]
- 12. Tien, A.C. et al A phase 0 trial of ribociclib in recurrent glioblastoma patients incorporating a tumor pharmacodynamic‐ and pharmacokinetic‐guided expansion cohort. Clin. Cancer Res. 25, 5777–5786 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klein, M.E. , Kovatcheva, M. , Davis, L.E. , Tap, W.D. & Koff, A. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell 34, 9–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patnaik, A. et al Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non‐small cell lung cancer, and other solid tumors. Cancer Discov. 6, 740–753 (2016). [DOI] [PubMed] [Google Scholar]
- 15. Raub, T.J. et al Brain exposure of two selective dual CDK4 and CDK6 inhibitors and the antitumor activity of CDK4 and CDK6 inhibition in combination with temozolomide in an intracranial glioblastoma xenograft. Drug Metab. Dispos. 43, 1360–1371 (2015). [DOI] [PubMed] [Google Scholar]
- 16. Parrish, K.E. , Pokorny, J. , Mittapalli, R.K. , Bakken, K. , Sarkaria, J.N. & Elmquist, W.F. Efflux transporters at the blood‐brain barrier limit delivery and efficacy of cyclin‐dependent kinase 4/6 inhibitor palbociclib (PD‐0332991) in an orthotopic brain tumor model. J. Pharmacol. Exp. Ther. 355, 264–271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarkaria, J.N. et al Is the blood‐brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro. Oncol. 20, 184–191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones, H. & Rowland‐Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst. Pharmacol. 2, e63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li, J. et al Quantitative and mechanistic understanding of AZD1775 penetration across human blood‐brain barrier in glioblastoma patients using an IVIVE‐PBPK modeling approach. Clin. Cancer Res. 23, 7454–7466 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bao, X. et al Protein expression and functional relevance of efflux and uptake drug transporters at the blood‐brain barrier of human brain and glioblastoma. Clin. Pharmacol. Ther. 107, 1116–1127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tate, S.C. , Sykes, A.K. , Kulanthaivel, P. , Chan, E.M. , Turner, P.K. & Cronier, D.M. A population pharmacokinetic and pharmacodynamic analysis of abemaciclib in a phase I clinical trial in cancer patients. Clin. Pharmacokinet. 57, 335–344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Luca, A. et al Pharmacokinetic drug evaluation of palbociclib for the treatment of breast cancer. Expert Opin. Drug Metab. Toxicol. 14, 891–900 (2018). [DOI] [PubMed] [Google Scholar]
- 23. Honasoge, A. & Sontheimer, H. Involvement of tumor acidification in brain cancer pathophysiology. Front. Physiol. 4, 316 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang, X. , Lin, Y. & Gillies, R.J. Tumor pH and its measurement. J. Nucl. Med. 51, 1167–1170 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Casey, J.R. , Grinstein, S. & Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 11, 50–61 (2010). [DOI] [PubMed] [Google Scholar]
- 26. de Lange, E.C. Utility of CSF in translational neuroscience. J. Pharmacokinet. Pharmacodyn. 40, 315–326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Syvanen, S. et al Species differences in blood‐brain barrier transport of three positron emission tomography radioligands with emphasis on P‐glycoprotein transport. Drug Metabol. Dispos. 37, 635–643 (2009). [DOI] [PubMed] [Google Scholar]
- 28. Pokorny, J.L. et al The efficacy of the Wee1 inhibitor MK‐1775 combined with temozolomide is limited by heterogeneous distribution across the blood‐brain barrier in glioblastoma. Clin. Cancer Res. 21, 1916–1924 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hammarlund‐Udenaes, M. , Friden, M. , Syvanen, S. & Gupta, A. On the rate and extent of drug delivery to the brain. Pharm. Res. 25, 1737–1750 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bauer, M. et al Pilot PET study to assess the functional interplay between ABCB1 and ABCG2 at the human blood‐brain barrier. Clin. Pharmacol. Ther. 100, 131–141 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Polli, J.W. et al An unexpected synergist role of P‐glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N‐{3‐chloro‐4‐[(3‐fluorobenzyl)oxy]phenyl}‐6‐[5‐({[2‐(methylsulfonyl)ethyl]amino }methyl)‐2‐furyl]‐4‐quinazolinamine; GW572016). Drug Metab. Dispos. 37, 439–442 (2009). [DOI] [PubMed] [Google Scholar]
- 32. Zamek‐Gliszczynski, M.J. , Kalvass, J.C. , Pollack, G.M. & Brouwer, K.L. Relationship between drug/metabolite exposure and impairment of excretory transport function. Drug Metabol. Dispos. 37, 386–390 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Redzic, Z. Molecular biology of the blood‐brain and the blood‐cerebrospinal fluid barriers: similarities and differences. Fluids Barriers CNS 8, 3 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Daood, M. , Tsai, C. , Ahdab‐Barmada, M. & Watchko, J.F. ABC transporter (P‐gp/ABCB1, MRP1/ABCC1, BCRP/ABCG2) expression in the developing human CNS. Neuropediatrics 39, 211–218 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rao, V.V. et al Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance‐associated protein contribute to the blood‐cerebrospinal‐fluid drug‐permeability barrier. Proc. Natl. Acad. Sci. USA 96, 3900–3905 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Lange, E.C. & Danhof, M. Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target concentrations in the clinical setting: implications of the barriers between blood and brain. Clin. Pharmacokinet. 41, 691–703 (2002). [DOI] [PubMed] [Google Scholar]
- 37. Miller, J.J. & Wen, P.Y. Emerging targeted therapies for glioma. Expert Opin. Emerg. Drugs 21, 441–452 (2016). [DOI] [PubMed] [Google Scholar]
- 38. Henderson, J.T. & Piquette‐Miller, M. Blood‐brain barrier: an impediment to neuropharmaceuticals. Clin. Pharmacol. Ther. 97, 308–313 (2015). [DOI] [PubMed] [Google Scholar]
- 39. Bhowmik, A. , Khan, R. & Ghosh, M.K. Blood brain barrier: a challenge for effectual therapy of brain tumors. Biomed. Res. Int. 2015, 1–20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Watkins, S. , Robel, S. , Kimbrough, I.F. , Robert, S.M. , Ellis‐Davies, G. & Sontheimer, H. Disruption of astrocyte‐vascular coupling and the blood‐brain barrier by invading glioma cells. Nat. Commun. 5, 4196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lockman, P.R. et al Heterogeneous blood‐tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. 16, 5664–5678 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gerstner, E.R. & Fine, R.L. Increased permeability of the blood‐brain barrier to chemotherapy in metastatic brain tumors: establishing a treatment paradigm. J. Clin. Oncol. 25, 2306–2312 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1