

Abstract
Since the publication of the first draft of the human genome, there has been an explosion of new technologies with increasing power to interrogate the totality of biological molecules (e.g. DNA, RNA, proteins, metabolites) and their modifications (e.g. DNA methylation, histone modifications). These technologies, collectively called omics, have been widely applied in the last two decades to study biological systems to gain deeper insight into mechanisms driving the physiology and pathophysiology of human health and disease. Due to its complex, multifactorial nature, food allergy is especially well-suited to be investigated using omics approaches. In this rostrum, we review how omic technologies have been applied to explore diverse aspects of food allergy, including adaptive and innate immune processes in food allergic responses, the role of the microbiome in food allergy risk, metabolic changes in the gut and blood associated with food allergy, as well as the identification of biomarkers and potential therapeutic targets for the condition. We discuss the strengths and limitations of the studies performed thus far and the need to adopt systems biology approaches that integrate data from multiple omics to fully leverage the potential of these technologies to advance food allergy research and care.
Keywords: Omics, Food allergy
INTRODUCTION
Food allergy (FA), the aberrant immune response from exposure to certain foods, is a group of IgE-dependent and non-IgE dependent conditions1 whose complexity and heterogeneity make both research on their biological mechanisms and the identification of potential therapeutic targets challenging. Currently, avoidance of the culprit food, injection of epinephrine in times of severe reaction, and desensitization2 are the only tools available, with none of these options curative1. There is strong evidence that the prevalence of FA has been increasing over the last 2–3 decades, with some estimates pointing at a prevalence as high at 10% in some populations3. Deepening our understanding of the molecular and cellular mechanisms driving FA and parsing the biological heterogeneity hidden under its clinical umbrella is paramount to improve the prevention, diagnosis and clinical management of FA.
While reductionist approaches have contributed to our understanding of the biological alterations underlying FA and other atopic conditions, they may be insufficient to provide a picture of disease mechanisms detailed and comprehensive enough to optimize the design of safe and effective intervention strategies4,5. A more holistic approach where the biology of FA is investigated comprehensively and at several levels may help identify solutions to the challenge4,5,6. Increasingly available high-throughput omic technologies enable system-wide characterization of biological systems at multiple levels6. The capacity to profile whole genomes, transcriptomes, proteomes, metabolomes and microbiomes, and to integrate data across several levels brings great promise to the field of FA (Figure 1).
Figure 1:

The integration of data generated via high-throughput omic technologies that describe different layers of biological complexity will enable the identification of the next generation of biomarkers, therapeutic targets, and molecular endpoints for clinical trials in food allergy. To that aim, tools from systems biology, such as network modelling, will be invaluable to finding solutions for the challenges posed by the integration of heterogeneous biological data. GC, gas chromatography; LC, liquid chromatography; MS, mass spectrometry; PBMC, peripheral blood mononuclear cell; SNP, single nucleotide polymorphism; WES, whole exome sequencing; WGS, whole genome sequencing.
The potential of omics technologies to unravel hidden mechanisms and shed light on disease heterogeneity in FA is recognized6,7. While their use in the field so far has been somewhat limited, their application has already started to provide insight into some of the challenges in FA.
OMICS AND THE ETIOLOGY OF FOOD ALLERGY
Genomics
While there is no consensus on the heritability of FA and food sensitization, with values ranging between 0.15 and 0.828–10), there is compelling evidence for the involvement of genetic factors in FA, as we review in more detail in a complementary article11. Since the completion of the Human Genome Project and the development of technological platforms that allow for the interrogation of millions of SNPs across the genome, genome-wide association studies (GWAS) have become the main tool for investigating the genetic architecture of complex diseases. Several studies have already been conducted investigating genetic variation in FA (Figure 1).
The first GWAS of well-defined FA was performed in the United States with 2759 participants and included several FA subtypes (peanut, milk and egg allergy)12. This GWAS identified two loci for peanut allergy in the HLA-DR/-DQ region of the major histocompatibility complex (MHC) and suggested that differential DNA methylation partially mediates the association between corresponding SNPs and peanut allergy12. The involvement of the MHC region in peanut allergy was later replicated in an additional small Australian study that found a significant association with a variant located in the HLA-DRB1 gene13. However, the importance of the MHC region in the genetic architecture of peanut allergy had already been shown by previous candidate gene studies14,15.
Later GWAS studies performed on larger samples revealed several loci apart from the MHC region that contribute to the risk of developing FA in general16 and peanut allergy in particular17. A study of 497 FA patients and 2387 controls identified four genome-wide significance loci (the SERPINB gene cluster, a cytokine gene cluster in chromosome 5, the filaggrin gene, and the C11orf30/LRRC32 locus) in addition to the MHC region16. The authors found that while the MHC locus was specific to peanut allergy, the other four loci increased risk for any FA. A subsequent GWAS of peanut allergy in 850 cases and 926 controls was accompanied by a meta-analysis of seven studies addressing two phenotypes (FA and peanut allergy)17. This study identified a variant near integrin α6 (ITGA6) and another variant near the C11orf30 locus that showed genome-wide significant association with peanut allergy and FA, respectively, as well as several loci with suggestive evidence for association, many containing genes involved in epigenetic regulation of gene expression. Overall, these results point to an important role for the HLA genes of the MHC in peanut allergy, specifically, as well as the involvement of immune regulation and epithelial barrier function pathways and epigenetic mechanisms in the pathogenesis of FA.
Despite the potential of GWAS to unravel the genetic architecture of FA, the degree to which they have been able to explain the heritability of FA has been limited; for example, the five genome-wide significant loci identified by Marenholtz et al. account for less than 50% of heritability estimated from whole-genome data16. This suggests that a substantial number of variants that contribute to disease risk remain to be identified, likely due to limited power in the FA GWASs to date. One way to circumvent this is using genetic knowledge generated for other atopic diseases for which larger GWAS studies have been performed to guide genetic discovery in FA. An example of this is a study in which 19 and 7 genome-wide significant susceptibility loci for atopic dermatitis and eosinophilic esophagitis, respectively, were selectively targeted, of which 14 were found to be associated with FA18. Additionally, most of the identified GWAS loci have had no follow up to hone in on true causal variants and genes that account for these associations. To obtain a comprehensive understanding of the genetic architecture of FA, it will be necessary to perform coordinated multi-center studies with phenotype harmonization to reach the sample sizes needed to identify common variants with mild effect sizes on disease risk (the majority of variants) complemented by targeted investigations of rarer source of genetic variance such as copy number variation and point mutations.
Microbiome
The realization of the crucial role played by the human microbiome (i.e. the aggregate of ecological communities of commensal, symbiotic, and pathogenic microorganisms that inhabit the human body), in human health and disease has led to an explosion of studies exploring how changes in these microbial communities contribute to or prevent disease development19–22. In part, this explosion has been possible thanks to the increasing deployment of 16S rRNA and shotgun metagenomic sequencing, two high-throughput technologies that enable comprehensive profiling of microbial communities (Figure 1)6,23 There is growing evidence supporting an important role of the gut microbiome in the development and progression of FA23–30.
One of the approaches used to shed light on the potential role of microbiota in FA has been to identify alterations in gut microbial composition in subjects with FA. A study of 141 infants with and without egg allergy enrolled in the Consortium for Food Allergy Research (CoFAR) Observational Study31 revealed different abundances of bacteria from the Lachnospiraceae family of the Clostridia class, and the Streptococcaceae and Leuconostocaceae families of the Bacilli class (all belonging to the Firmicutes phylum) in infants with egg allergy compared to non-food allergic controls28 (Table 1, Figure 2). The authors also found increased microbial (alpha) diversity in egg allergic infants. In another CoFAR-based study25, the researchers focused on identifying gut microbiota features associated with milk allergy resolution in children. They identified 226 infants with milk allergy, collected fecal samples for 16S rRNA sequencing, and followed participants via clinical and immunological assessments until the age of 8 years. Their results revealed that infant gut microbiome composition at ages 3 to 6 months was significantly associated with later milk allergy resolution, observing increased abundance of Clostridia and Firmicutes in infants whose allergy subsequently resolved. Their results suggest that bacteria from the Firmicutes phylum, generally, and, more specifically, of the Clostridia class, as potential probiotic candidates to treat milk allergy (Table 1, Figure 2)25. Another study from the Vitamin D Antenatal Asthma Reduction Trial cohort explored the gut microbiome of early infancy (3–6 months) and its association with sensitization to several foods at age 3 years. Among the 225 children, 87 had food sensitization and 14 had FA by age 3 years27. There were no differences in microbial diversity between cases and controls, but three genera (Haemophilus, Dialister and Clostridium), were found to be underrepresented in sensitized subjects, and another three (Citrobacter, Oscillospira and Lactococcus), were underrepresented in the 14 children with FA (Table 1, Figure 2)27. These results further confirmed the important role of bacteria from the Firmicutes phylum (to which the Dialister, Clostridium, Oscillospira, Lactoccocus and Dorea genera belong) in FA and sensitization and suggested that Proteobacteria (Heamophilus and Citrobacter) as also being involved.
Table1:
Summary of the main findings of the studies included in this review except for the genomic studies, which we have reviewed in a complementary article11.
| Microbiome/gut metabolomics | Transcriptomics | Epigenomics (methylomics) | Multi-omics |
|---|---|---|---|
|
Food
allergy/sensitization |
Acute response to
peanut |
Differential DNA methylation in
FA |
Transcriptomics/metabolomics response
to profilin |
|
Lachnospiraceae
↑28 Streptococcaceae ↑28 Ruminococcaceae ↑28 Citrobacter ↓27 Oscillospira ↓27 Lactococcus ↓27 Haemophilus ↓27 Dialister ↓27 Clostridium↓27 Leuconostoc ↓28 De novo sphingolipid synthesis ↓30 |
Acute-phase inflammatory
response ↑42 Positive regulation of NK cell chemotaxis ↑42 Key drivers: LTB4R, PADI4, IL1R2, PP1R3D, KLHL2, ECHDC342 Macrophages ↑42 Neutrophils ↑42 CD4+ Tcells (naïve) ↓42 |
MAPK signalling pathway genes53 Butirosin and neomycin biosynthesis54 Starch & sucrose metabolism54 Fructose & mannose metabolism54 Th1-Th2 balance genes54 |
Platelet function genes
↓65 Protein synthesis genes ↓65 Histone modification genes ↓65 Fatty acid metabolism genes ↓65 Carbohydrates, pyruvate ↓65 Lactate ↑65 Carnitine ↓ (with severity)65 |
|
Allergy resolution/tolerance development | |||
|
IgE clonal response in peanut allergy |
Th2 cytokine methylation
↑55 Th1 cytokine methylation ↓55 FOXP3 methylation ↓56,57 |
||
| Convergence of IgE gene
rearrangements43 IgEs cross-reactivity for Ara h2 – Ara h343 |
Transcriptomics/epigenomics TCR activation in FA |
||
| E2F/MYC TF network genes
↓66 Diff methylation metabolic genes66 Diff methylation inflammatory genes66 Diff methylation TCR cascade genes66 | |||
|
Allergy resolution | |||
|
Firmicutes ↑25 Clostridia ↑25 Fatty acid metabolism 25 |
T cell clonal expansion signature |
Biomarkers of FA |
|
| CD154, CD69, TNFRSA, GATA3
↑44 CCR7, LEF1, SELL ↓44 Th2 signature enrichment44 |
96 CpG signature: 79.2% accuracy66 | ||
|
FA and biological age | |||
| DNA methylation age
↑62 DNA meth. age acceleration ↑62 |
Transcriptomics/epigenomics of reaction severity |
||
|
Therapeutic potential |
Reactive vs hyporeactive T cell response |
||
|
Clostridiales (several
genera)33 Bacteroidales (several genera)33 Subdoligranulum variabile33 |
Macrophages ↑45 Th2 and Th17 cell genes ↑45 Treg and immune regulation genes ↓45 |
Neutrophil immunity involvement67 NFKBIA and ARG1 are hub genes67 PHACTR1/ZNF121 expression mediate CpG associations67 |
|
Figure 2:
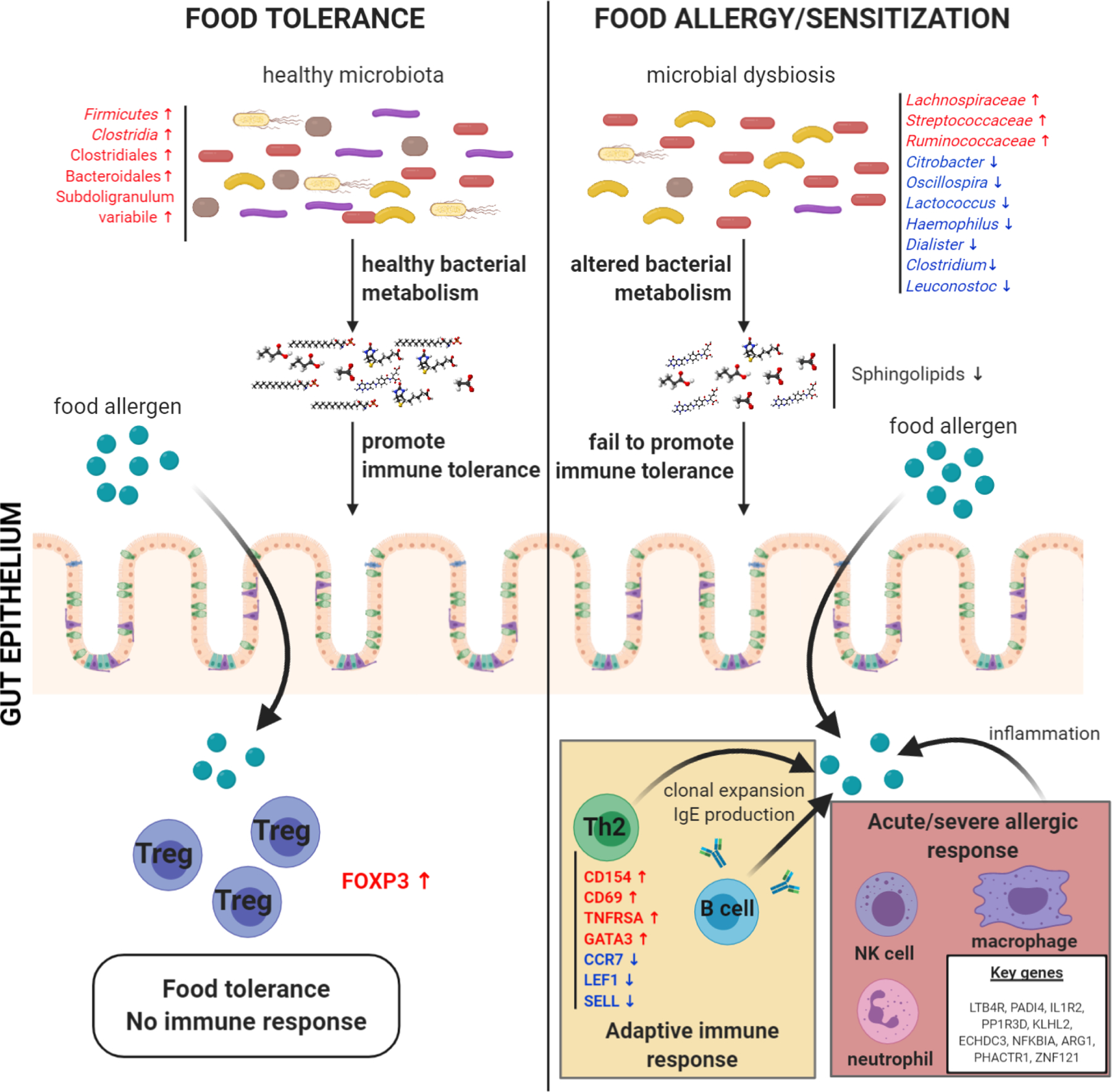
Visual representation of a selection of gut microbiota and metabolomic changes, immune mechanisms, and gene expression alterations involved in food allergy/sensitization, as well as the potential relationships between them, as revealed by the studies reviewed in this rostrum.
Microbiome analysis of animal models, especially of germ-free mice, has also yielded important insights into the role of the microbiota and dysbiosis in FA32–34. Fecal microbiota transplantation (FMT) from healthy infants has been shown to protect sensitized germ-free mice from milk allergy32,34. In one study, mice receiving FMT from healthy controls plus milk formula demonstrated protection from milk allergy, while mice receiving FMT from milk allergic infants plus hypoallergenic (i.e. hydrolyzed casein formula) demonstrated symptoms consistent with allergic reaction after sensitization and challenge to β-lactoglobulin34. The abundance of a Clostridiales species was also found to be associated with regulatory gene expression in the ileum34. In another study, FMT from food allergic infants to germ-free Il1raF709 mice did not prevent the development of a strong allergic reaction following sensitization with chicken egg ovalbumin, while FMT from healthy controls did result in a significant reduction of the reaction33. 16S rRNA sequencing of fecal samples collected from 56 allergic and 98 healthy control children starting at 1–15 months of age and, then, every 4–6 months up to 30 months of age revealed that, while FA subjects did not show a difference in overall microbial diversity, the relative abundance of 77 operational taxonomic units (OTUs) was different compared to controls33. These results enabled the identification of various Clostridiales and Bacteroidales species affected by the dysbiosis that, when used as combination therapy, suppressed FA in mice (Table 1, Figure 2)33. The use of Subdoligranulum variabile as monotherapy had similar results (Table 1, Figure 2)33. The results of these studies suggest therapeutic potential of microbiome manipulation for FA.
One of the most important roles of the gut microbiota is to perform metabolic functions that may be helpful for the host, including, for example, the fermentation of carbohydrates into short-chain fatty acids35, the synthesis of vitamins like biotin and folate, and the absorption of dietary minerals36. Thus, it is reasonable to hypothesize that at least part of the effects of gut microbiota on food sensitization and allergies is mediated by those metabolic functions. Within the study of milk allergy resolution among 226 CoFAR subjects25, investigators explored this through predicted metagenome function analysis that revealed significant differences in microbial fatty acid metabolism in children whose milk allergy resolved versus persisted. The role of microbiota and metabolites was also investigated in another study30 where microbiota composition was profiled by 16S rRNA sequencing and metabolomic profiles were generated via liquid chromatography tandem mass spectrometry of stool samples collected at 3–6 months of age from 12 children with FA, 32 with food sensitization, and 37 healthy controls; FA and sensitization at age 3 years were determined based on parental responses to a questionnaire and serum-levels of specific IgE antibodies. The results from this study revealed a significant association between FA or sensitization and a group of highly correlated metabolites involved in de novo sphingolipid synthesis (sphingolipid module) (Table 1, Figure 2), as well as 6 OTUs of the Bacteroides genus positively associated to that group of metabolites30.
The results of all these studies reveal an important role for the gut microbiome and microbial metabolism in FA, sensitization, and tolerance, raising the potential of microbial interventions for these conditions, which have been reviewed elsewhere23,29,37,38.
OMICS AND THE MOLECULAR MECHANISMS OF FOOD ALLERGY
Gene expression and regulation
The investigation of gene expression and its regulation genome-wide is a powerful tool to elucidate the molecular and cellular mechanisms acting in complex diseases6. There is ample evidence that many of the common genetic variants found to increase risk for any given complex disease exert their effects through changes in gene expression39,40, often by modulating regulatory factors such as transcription factor binding, DNA methylation, and histone acetylation. Thus, the comprehensive characterization of the transcriptome (the group of RNA transcripts generated by genes across the genome), the epigenome (the compendium of factors that regulate genome-wide gene expression without changes to the genetic code), and the regulatory networks that govern the behavior of biological systems at the RNA level is paramount to understand the mechanisms that drive complex disease. As it has happened with other molecular features of biological systems, the completion of the sequence of the human genome has enabled the development of high-throughput technologies with which both the transcriptome and the epigenome can be characterized in a systematic and quantitative way genome-wide. The investigation of the transcriptome and its alterations across cell-types, tissues, organisms, and phenotypic states was initially performed by oligonucleotide microarrays, a technology based on the binding of fluorescently labeled fragments of cDNA generated from the RNA under study to complementary oligonucleotide probes. However, this technology is hampered by several problems, including the limitation of only capturing the expression of well-characterized genes, high levels of background noise, and signal non-specificity that limit its power to provide an unbiased and comprehensive picture of the transcriptome. The development of RNA-sequencing41 has enabled the field to overcome many of the problems imposed by microarray technology when studying transcriptomes. Sequencing-based technology is free of the constraints of pre-specified probes and has enabled the comprehensive and in-depth characterization of transcriptomes.
Transcriptomics
Transcriptomics has also entered the research arena in FA, and while the number of studies performed thus far has been limited, it has already revealed deep insights into the molecular, cellular, and immunological mechanisms driving FA. RNA sequencing (RNA-seq) technologies, spanning bulk and single-cell RNA-seq, have enabled the exploration of mechanistic aspects previously inaccessible (Figure 1). An in vivo transcriptomic study of longitudinal peripheral blood samples from 40 peanut allergic children collected before, during and after double-blind, placebo-controlled peanut oral challenges enabled a fresh perspective on molecular mechanisms underlying acute peanut allergic reactions.42 In addition to identifying peripheral blood gene signatures that characterized allergic reaction to peanut but not placebo, the investigators built weighted gene coexpression networks to identify broader constructs for the signature, identifying acute-phase response and pro-inflammatory processes as significant biological processes. From transcriptome data deconvolution, they also observed an increase of neutrophils and macrophages, and a decrease of naïve CD4+ T cells over the course of the acute peanut allergy reactions (Table 1, Figure 2)42. Importantly, the investigators then built probabilistic causal networks and performed key driver analyses to identify six master regulator genes (LTB4R, PADI4, IL1R2, PP1R3D, KLHL2 and ECHDC3) predicted to causally modulate acute allergic reactions to peanut42 (Table 1, Figure 2)42.
Subsequently, other investigators applied single-cell RNAseq to characterize the clonal architecture of IgE B cells from a pool of B cells isolated from the peripheral blood of 6 peanut allergic subjects43. Apart from performing differential expression and splicing analyses, they used the RNA-seq reads to assemble heavy and light antibody IgE chains and define clonotypes based on V and J genes combinations and CDR3 length. They observed a surprising convergence of immunoglobulin gene rearrangement of IgE antibodies across different individuals (Table 1)43. Affinity assays identified several IgE antibodies with cross-reactivity for Ara h 2 and Ara h 3, a capacity the authors conclude was gained through the acquisition of mutations in VH and VL43 (Table 1). The ability of single-cell RNA-seq data to characterize the clonal architecture of adaptive immune responses in FA has also been studied in T cells by recovering TCR sequences from 3′ single-cell RNAseq reads44. The results of single-cell RNA-seq of CD154+ T cells sorted from peripheral blood mononuclear cells (PBMCs) of FA subjects and incubated with peanut showed a weaker clonal expansion than in immunized mice and allowed the identification of signatures shared by the expanded T cell clones. Clonally expanded T cells showed increased expression of CD154, CD69, and TNFRSFA, as well as of GATA3, a Th2 cell transcription factor, while non-expanded T cells expressed central memory genes (CCR7, LEF1 and SELL) (Table 1, Figure 2)44. A signature-enrichment analysis showed a positive enrichment of Th2 signatures in expanded T cells, indicating a shift towards the Th2 phenotype among the T cells undergoing clonal expansion in response to peanut allergen (Table 1, Figure 2)44.
Further evidence of the importance of the Th2 phenotype in FA has come from another transcriptomic study exploring the biological mechanisms of clinical sensitivity to peanut allergens in peanut allergic subjects45. As there is huge variability in clinical sensitivity to peanut across peanut allergic patients, and current testing has very limited power to predict the level of clinical sensitivity, these investigators sought to identify the characteristics of peanut-specific CD4+ T-cell responses that are associated with clinical sensitivity. They recruited 62 subjects enrolled in a peanut oral immunotherapy trial, 41 of whom showed a strong reaction during food challenge (reactive patients) and 21 who tolerated the highest dose (hyporeactive patients), and isolated PBMCs from them that were then either stimulated with a peanut extract or left unstimulated. Analysis of the RNA-seq data generated from the CD154+ CD4+ T cells isolated from the PBMC cultures revealed that the transcriptomic response of the reactive individuals was much stronger than that of the hyporeactive ones (1585 and 608 differentially expressed genes (DEGs), respectively) and yielded 31 DEGs between reactive and hyporeactive patients45. Th2 and Th17 cell genes were found to be upregulated in reactive patients, whereas hyporeactive individuals showed a significantly higher expression of Treg and immune regulation genes45, suggesting that the balance between Th2 and Th17-mediated pro-inflammatory activity and immune response regulatory activity as one of the mechanisms behind the variation in clinical sensitivity to peanut allergens (Table 1).
Epigenomics
While direct study of the transcriptome and its alterations in FA provides invaluable insights into disease mechanism and has the potential to identify new biomarkers and therapeutic targets, factors that regulate gene activity are also of interest. The epigenome, the set of genome-wide modifications to DNA and histones that regulate gene expression, plays a role in mediating gene-environment interactions46,47. Epigenomics may help us understand how genetic and environmental forces contribute to the pathogenesis of complex disorders, including FA6. The two most widely studied epigenetic mechanisms are DNA methylation (the addition of methyl groups to cytosine bases), and histone modifications (a wide range of chemical modifications to the residues of the histone proteins). While hypermethylation of DNA is usually associated with downregulation of expression of a nearby gene48, different histone modifications can have either activation or repression effects on gene expression49,50. Microarray technology has been widely used to study both DNA methylation and histone modifications. However, as with genomics and transcriptomics, sequencing-based methods have enabled a more comprehensive study of the epigenome as well as the investigation of epigenetic mechanisms that could not be explored using microarray technology. Of the next-generation epigenomic technologies that have been developed, ChIP-seq (the chromatin immuno-precipitation coupled with a high-throughput sequencing to investigate histone modifications)51 and FAISE-seq (the sequencing method to explore open chromatin areas of the genome), and 3C-seq (a technology that allows the characterization of the spatial organization of the chromatin), are among the most widely used approaches (Figure 1). While the initial high cost of the bisulfite sequencing technology limited its use for genome-wide epigenome profiling, recent advances have reduced costs in addition to overcoming the technology’s initial limitations, such as limited capacity to distinguish methylation and hydroxymethylation52.
Several studies have been performed examining the role of epigenomic variation in FA. An early epigenome-wide association study (EWAS) sought to identify DNA methylation changes in CD4+ T cells associated with FA53. The comparison of ~450,000 CpG loci across the genome in DNA from peripheral blood CD4+ T cells isolated at birth and at 12 months of age from 12 children with IgE-mediated FA and 12 non-allergic controls revealed 170 differentially methylated probes (DMP) at 12 months of age and 136 DMPs at birth, 96 of which were present before the development of clinical symptoms53. Pathway enrichment analysis of the differential methylation signatures revealed enrichment for genes involved in the MAP kinase (MAPK) signaling pathway, leading the authors to conclude that the aberrant methylation-mediated dysregulation of the MAPK pathway in early CD4+ T-cell development contributes to the pathogenesis of FA in early childhood53(Table 1). Two years later, another group performed the first EWAS of cow’s milk allergy (CMA) in US children54. Genome-wide profiling of DNA methylation using microarrays on whole blood from 106 children with CMA and 76 nonatopic controls identified 568 differentially hypomethylated and 7 hypermethylated probes54. Pathway enrichment analysis of the 576 DMPs revealed genes involved in butirosin and neomycin biosynthesis, starch & sucrose metabolism, and fructose and mannose metabolism54 (Table 1). Close inspection of DMPs annotated to Th1 and Th2 genes confirmed an association between methylation of genes involved in Th1 and Th2 pathways and CMA54. The relevance of methylation-mediated regulation of Th1 and Th2 genes in CMA had already been observed in a previous study investigating DNA methylation profiles of genes for Th1 (IL-10, IFN-γ) and Th2 (IL-4, IL-5) cytokines, where, not only did the methylation profiles for the four cytokines distinguish between CMA and control individuals, but the children who had outgrown the allergy showed significantly higher methylation levels for the Th2 cytokines and significantly lower methylation levels for Th1 cytokines55 (Table 1). All these results suggest that the regulation of Th1 and Th2 responses via methylation/demethylation of corresponding genes is a mechanism in FA.
Epigenomics has also been used to explore the mechanisms behind the development of tolerance to food allergens. A genome-wide DNA methylation study performed of peripheral blood from 43 peanut allergy subjects enrolled in an oral immunotherapy (OIT) trial, 23 of whom were undergoing active OIT and 20 of whom were receiving standard of care, reported hypomethylation of forkhead box protein 3 (FOXP3) CpG sites in antigen-induced regulatory T cells associated with the development of immune tolerance to peanut56 (Table 1, Figure 2). The importance of FOXP3 methylation in FA was further examined in a separate study where significantly decreased demethylation of the Treg-specific demethylated region of Foxp3 in mononuclear cells from children with active IgE-mediated CMA was observed compared to children that had outgrown the allergy and healthy controls57.
Among the most interesting applications of epigenomics is to use DNA methylation-based molecular clocks as measures of biological age58. There is an increasing body of evidence suggesting that epigenetic clocks metrics (including DNA methylation age and age acceleration) are associated with several adult diseases including physical and cognitive decline59, Parkinson’s disease60, and lung cancer61. As little is known about biological aging in allergy and asthma, one group of investigators aimed to explore the relationship between DNA methylation age (DNAmAge) and age-acceleration with asthma and several allergies, including FA62. They calculated DNAmAge and age-acceleration using genome-wide DNA methylation data from cord blood and peripheral blood from 408 children at birth, early-childhood, and mid-childhood. They found that both DNA methylation age and DNA methylation aging rate were associated with risk of developing food allergen sensitization (OR = 1.21, P = 0.006 and OR = 1.29, P < 0.001, respectively), indicating that biological aging is accelerated in the peripheral blood of subjects with FA62 (Table 1).
Finally, the epigenome has also been studied in the search of biomarkers of FA. Given that currently used diagnostic tests can predict sensitization but not clinical reactivity to food allergens, one study aimed at finding epigenetic biomarkers of clinical reactivity to food allergens63. The authors investigated the genome-wide DNA methylation profiles on PBMCs obtained from 71 infants aged 11–15 months who underwent either a peanut or an egg food challenge and were classified into three groups: sensitized infants who did and did not show an allergic reaction (N = 29 in both groups) and non-allergic controls (N = 13). Using a supervised learning approach, a DNA methylation signature across 96 CpG sites was identified that could predict FA status in an independent replication cohort with 79.2% accuracy63.
THE POTENTIAL OF MULTIOMICS INTEGRATION
Studies of the genome, microbiome, transcriptome, epigenome and metabolome have already yielded results that have advanced our understanding of FA. However, these individual molecular layers to do not act in isolation, so it is unlikely that we will achieve an understanding of the biological complexity driving FA unless we integrate the information from all these levels by characterizing interactions between them64. A deep, detailed, mechanistic understanding of the dynamic molecular processes involved in FA will only come from a holistic, integrative, multiscale approach; that is, a systems biology approach6. The main objective of systems biology is to develop comprehensive models of biological systems with a focus on describing not only the molecular components of those systems, but also the interactions between them that govern the behavior of the system (Figure 1).
There has been limited work done thus far on integrating multiple omics data to inform on FA, but a few recent studies have started to pave the way by combining transcriptomics with metabolomics65 and epigenomics66,67. A recent study on blood transcriptomic and metabolomic changes in response to a profilin oral challenge65 showed a decrease of carbohydrates and pyruvate and an increase of lactate in plasma, as well as a gradual decrease in carnitine levels along the gradient of the severity of reaction to profilin (Table 1). They also showed an underexpression of genes involved in platelet function, protein synthesis, histone modification, and fatty acid metabolism in PBMCs (Table 1). Another study characterized the transcriptomic and epigenomic responses to TCR activation in CD4+ T-cells from patients with egg allergy and healthy controls66, finding that the poorer lymphoproliferative response of CD4+ T-cells observed in the children who did not outgrow their egg allergy before age 4 was associated with a reduced expression of genes involved in the E2F and MYC transcription factor networks and with a remodeling of DNA methylation at metabolic and inflammatory genes. The investigators also found that early resolution vs. no resolution of egg allergy was associated to DNA methylation changes in T cell activation genes located upstream of the TCR transduction cascade.
The potential of the multi-omic approach to deeply explain biological mechanisms driving complex disease cannot be fully reached without examining interactions between molecular forces. This applies not only to multi-omic studies but also to investigating molecular entities within an –ome, as each of these interact with one other in specific manners to drive the behavior of the system. The comprehensive modeling of the molecular interactions inside and across omic levels that drive the behavior of complex biological systems requires the use of mathematical tools tailored to the study of complex system architecture and behavior. Ever since the seminal paper by Barabasi & Albert proposing their free-scale model of real world networks and showing its pervasiveness across a wide variety of systems from genetic networks to the internet68, there has been a revival of research in graph theory and its applied branch, network science, that has reached fields as diverse as sociology, transport engineering, and medicine. Due to their capacity to model complex systems as sets of elements and the interactions between them, networks have enormous potential to solve the challenges of data integration in systems biology69,70. Network medicine, the application of network modeling to biomedical problems, is an emerging research area that, coupled with the increasingly large biomedical data generation enabled by high-throughput omic technologies, has the potential to dramatically increase our power to understand the biological bases of health and disease and design radically better diagnostic, prognostic and therapeutic strategies71.
A good example of the application of network modeling to multi-omics data to investigate FA can be found in a recent study aimed at shedding light on the biological mechanisms driving the severity of allergic reactions to peanut67. The authors generated whole-blood transcriptome and peripheral blood CD4+ T cell epigenome data from children with peanut allergy undergoing double-blind placebo-controlled peanut challenges and applied linear mixed models, weighted gene coexpression network analysis (WGCNA)72, multi-omics network integration analysis (xMWAS)73, and causal mediation to identify genes, CpG loci, and their interactions that mediate reaction severity. Their results demonstrated the involvement of neutrophil immunity in mediating severe allergic reactions and highlighted NFKBIA and ARG1 as hubs in the gene-CpG island interaction network (Table 1, Figure 2). Causal mediation analysis showed that the expression of PHACTR1 and ZNF121 mediates the association between severe allergic reactions and methylation levels in the corresponding CpG sites (Table 1, Figure 2). The comprehensive, integrative approach used in this study enabled the identification of key immune processes, genes, and methylation sites driving severe allergic reactions to peanut, and, thus, suggested potential biomarkers and therapeutic targets for peanut allergy. Multi-omic, integrative, network-based modeling strategies such as those used in this study set the way forward for FA omics research and reinforce the notion that a systems biology approach holds huge potential to lead us to the next generation of biomarkers and therapy for FA.
CONCLUDING REMARKS
The application of omic technologies to the study of FA has already generated invaluable new insights into this group of diseases. The 16S rRNA and shotgun metagenomic sequencing technologies have allowed the comprehensive study of the gut microbiome to reveal early and late dysbiosis associated with FA and sensitization, with alterations of bacteria from the Firmicutes and Proteobacteria phyla as one of the main hallmarks27,28. We have also learned that the composition of gut microbiota in early childhood is associated to FA resolution25 and that changes in the bacterial community are reflected in alterations of bacterial metabolism25,30. Further, these studies have allowed the identification of several bacterial genera and species (in the Clostridiales and Bacteroidales orders) with therapeutic potential33.
Omics have also enabled the exploration of risk factors and biological mechanisms in the host. GWAS have revealed the involvement of the major histocompatibility complex (MHC) genes, emphasizing the importance of the adaptive immune system among the mechanisms driving allergic responses to food, as well as other loci like the filaggrin gene11,12,13,16,17. The mechanisms through which the adaptive immune system contributes to the pathogenesis of FA have been explored in, among others, transcriptomic studies, which, thanks to the recent development of the RNA-sequencing technology, have been able to characterize the clonal IgE B cell and T cell responses in FA from sequencing data43,44 and reveal the importance of the Th1/Th2 balance in FA44,45. A balance that epigenomic studies exploring genome-wide DNA methylation patterns have also revealed to fulfill a central role54,55. Epigenomic studies have also enabled the observation of the accelerated biological aging that happens in patients with FA62 and the identification of methylation signatures as biomarkers of disease63. The central role played by the Th1/Th2 balance has also been revealed by epigenomic studies exploring genome-wide DNA methylation patterns.34,36
Despite all the new knowledge gained from the application of omics to FA research, the complexity of the biological systems we are studying (the microbiome, the immune system, the epithelial barrier of the gut etc.) will most likely require a systems biology approach integrating data from several omes into comprehensive models so we can gain an understanding detailed and deep enough to develop efficacious diagnostic, prognostic, preventive, and therapeutic tools in the future. While there have been a few attempts at combining data from two omic technologies (like transcriptomics and microbiomics or transcriptomic and epigenomics)34,66 and even some work on using network modelling to integrate multi-omics data67, the field of multi-omics data integration in FA is still in its infancy and it needs to be further developed as the high-throughput technologies themselves and, especially, the mathematical tools that will allow data integration, also develop. We are still at the beginning of the journey of FA omics and multi-omics, but the path ahead looks promising.
Acknowledgments
Funding: Drs. Kanchan and Mathias receive funding from the Immune Tolerance Network and the National Institutes of Health (NIH) UM1AI109565. Dr. Bunyavanich receives funding from NIH grants R01 AI147028, U19 AI136053, and R01AI118833.
Abbreviations
- CMA
cow’s milk allergy
- CoFAR
Consortium for Food Allergy Research
- DEG
differentially expressed gene
- DMP
differentially methylated probe
- DNAmAge
DNA methylation age
- FA
food allergy
- FMT
fecal microbiota transplantation
- GWAS
genome-wide association study
- MHC
major histocompatibility complex
- OUT
operational taxonomic unit
- PBMC
peripheral blood mononuclear cell
- TCR
T cell receptor
GLOSSARY
- Complex diseases
conditions caused by a large number of genetic and environmental factors and the interactions between them. Many common diseases, such as asthma, diabetes, epilepsy, hypertension, major depression, Alzheimer’s and food allergy, are complex diseases
- Dysbiosis
refers to the imbalance of one or various microbial communities. In humans, dysbiosis of the gastrointestinal tract has been reported to be associated to inflammatory bowel disease, obesity, cancer, colitis, and food allergy
- Epigenome
the compendium of DNA modifications, such as DNA methylation and histone modification, that regulate gene expression without altering the DNA sequence. The study of the epigenome is well suited to explore mechanisms of gene expression regulation and of gene-environment interactions
- Gene regulatory network
is a group of molecular regulators and the interactions among them and with other molecules that govern gene expression of a cell at the RNA and protein levels
- Genetic architecture
the compendium of genetic variation that is responsible for the heritable variability of a phenotypic trait. Among others, common single nucleotide polymorphisms, rare point mutations, and rare copy number variants are sources of genetic variation that have been implicated in the heritability of complex diseases
- Genome
the complete set of genetic material of an organism that includes protein coding and non-coding regions and, in eukaryotes, the DNA contained in organelles. The human genome is comprised of ~ 3.1 billion base pairs organized in 23 pairs of chromosomes
- Metabolome
the complete set of small molecular chemicals found within a biological system such as within a cell, an organ, a tissue, or an organism
- Microbiome
the complete set of ecological communities of symbiotic and pathogenic microorganisms found in and on a multicellular organism. In humans, the composition of the microbiome varies markedly between different tissues like the skin, mouth, nose, digestive tract, and vagina. In food allergy, the most widely studied bacterial community has been the gut microbiome
- Network
Also called a graph, it is a mathematical construct that models a system as a set of entities (represented as nodes) and the relationships between them (represented as edges). Graphs are essential modelling tools in systems biology and are extensively used to model protein-protein interactions and gene regulatory networks, for example
- Systems biology
An interdisciplinary field of biology aimed at the computational and mathematical modeling of biological systems that uses a holistic approach taking into account not only the molecular agents of the system, but the interactions between them as well. Coupling high-throughput generation of data on the molecular components with bioinformatic and computational techniques, the objective of the system biology approach is to generate comprehensive, dynamic and multiscale models of biological systems that will enable an accurate prediction of the phenotypic landscape from the molecular information
- Transcriptome
the set of all RNA transcripts in a cell or group of cells. While the set of distinct transcripts that comprise the human transcriptome is not fully known, Gencode currently contains > 200,000 human transcript entries
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Sicherer SH, Warren CM, Dant C, Gupta RS, Nadeau KC. Food Allergy from Infancy Through Adulthood. J Allergy Clin Immunol Pract. 2020; [DOI] [PMC free article] [PubMed]
- 2.Vickery BP, Vereda A, Casale TB, Beyer K, Toit G Du, Hourihane JO, et al. AR101 oral immunotherapy for peanut allergy. N Engl J Med. 2018; [DOI] [PubMed]
- 3.Lopes JP, Sicherer S. Food allergy: epidemiology, pathogenesis, diagnosis, prevention, and treatment. Current Opinion in Immunology. 2020. [DOI] [PubMed]
- 4.Vogt H, Hofmann B, Getz L. The new holism: P4 systems medicine and the medicalization of health and life itself. Med Heal Care Philos. 2016; [DOI] [PMC free article] [PubMed]
- 5.Manzoni C, Kia DA, Vandrovcova J, Hardy J, Wood NW, Lewis PA, et al. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief Bioinform. 2018; [DOI] [PMC free article] [PubMed]
- 6.Bunyavanich S, Schadt EE. Systems biology of asthma and allergic diseases: A multiscale approach. J Allergy Clin Immunol. 2015; [DOI] [PMC free article] [PubMed]
- 7.Fiocchi A, Wang J. -omic sciences: new horizons in food allergy. Curr Opin Allergy Clin Immunol. 2015;15:234–6. [DOI] [PubMed] [Google Scholar]
- 8.Sicherer SH, Furlong TJ, Maes HH, Desnick RJ, Sampson HA, Gelb BD. Genetics of peanut allergy: A twin study. J Allergy Clin Immunol. 2000; [DOI] [PubMed]
- 9.Liu X, Zhang S, Tsai HJ, Hong X, Wang B, Fang Y, et al. Genetic and environmental contributions to allergen sensitization in a Chinese twin study. Clin Exp Allergy. 2009; [DOI] [PMC free article] [PubMed]
- 10.Tsai HJ, Kumar R, Pongracic J, Liu X, Story R, Yu Y, et al. Familial aggregation of food allergy and sensitization to food allergens: A family-based study. Clin Exp Allergy. 2009; [DOI] [PMC free article] [PubMed]
- 11.Kanchan K, Clay S, Irizar H, Bunyavanich S, Mathias RA. Current Insights Into The Genetics Of Food Allergy. Journal of Allergy and Clinical Immunology. 2020, accepted 10/30/2020 [DOI] [PMC free article] [PubMed]
- 12.Hong X, Hao K, Ladd-Acosta C, Hansen KD, Tsai H-J, Liu X, et al. Genome-wide association study identifies peanut allergy-specific loci and evidence of epigenetic mediation in US children. Nat Commun. 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martino DJ, Ashley S, Koplin J, Ellis J, Saffery R, Dharmage SC, et al. Genomewide association study of peanut allergy reproduces association with amino acid polymorphisms in HLA-DRB1. Clin Exp ALLERGY. 2017;47:217–23. [DOI] [PubMed] [Google Scholar]
- 14.Howell WM, Turner SJ, Hourihane JOB, Dean TP, Warner JO. HLA class II DRB1, DQB1 and DPB1 genotypic associations with peanut allergy: Evidence from a family-based and case-control study. Clin Exp Allergy. 1998; [DOI] [PubMed]
- 15.Madore AM, Vaillancourt VT, Asai Y, Alizadehfar R, Ben-Shoshan M, Michel DL, et al. HLA-DQB1*02 and DQB1*06:03P are associated with peanut allergy. Eur J Hum Genet. 2013; [DOI] [PMC free article] [PubMed]
- 16.Marenholz I, Grosche S, Kalb B, Rueschendorf F, Bluemchen K, Schlags R, et al. Genome-wide association study identifies the SERPINB gene cluster as asusceptibility locus for food allergy. Nat Commun. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asai Y, Eslami A, van Ginkel CD, Akhabir L, Wan M, Ellis G, et al. Genome-wide association study and meta-analysis in multiple populations identifies new loci for peanut allergy and establishes C11orf30/EMSY as a genetic risk factor for food allergy. J Allergy Clin Immunol. 2018;141:991–1001. [DOI] [PubMed] [Google Scholar]
- 18.Hirota T, Nakayama T, Tamari M, Sato S, Yanagida N, Ebisawa M, et al. Association study of childhood food allergy with genome-wide association studies–discovered loci of atopic dermatitis and eosinophilic esophagitis. J Allergy Clin Immunol. 2017; [DOI] [PubMed]
- 19.Marchesi JR, Adams DH, Fava F, Hermes GDA, Hirschfield GM, Hold G, et al. The gut microbiota and host health: A new clinical frontier. Gut. 2016; [DOI] [PMC free article] [PubMed]
- 20.Boulangé CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Medicine. 2016. [DOI] [PMC free article] [PubMed]
- 21.Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016. [DOI] [PubMed]
- 22.Lynch SV, Pedersen O The human intestinal microbiome in health and disease. New England Journal of Medicine. 2016. [DOI] [PubMed]
- 23.Bunyavanich S, Berin MC. Food allergy and the microbiome: Current understandings and future directions. J Allergy Clin Immunol. 2019;144:1468–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rivas MN, Burton OT, Wise P, Zhang Y, Hobson SA, Lloret MG, et al. A microbiota signature associated with experimental food allergy promotes allergic sensitization and anaphylaxis. J Allergy Clin Immunol. 2013;131:201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bunyavanich S, Shen N, Grishin A, Wood R, Burks W, Dawson P, et al. Early-life gut microbiome composition and milk allergy resolution. J Allergy Clin Immunol. 2016;138:1122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang YJ, Marsland BJ, Bunyavanich S, O’Mahony L, Leung DYM, Muraro A, et al. The microbiome in allergic disease: Current understanding and future opportunities-2017 PRACTALL document of the American Academy of Allergy, Asthma & Immunology and the European Academy of Allergy and Clinical Immunology. J Allergy Clin Immunol. 2017;139:1099–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savage JH, Lee-Sarwar KA, Sordillo J, Bunyavanich S, Zhou Y, O’Connor G, et al. A prospective microbiome-wide association study of food sensitizationand food allergy in early childhood. Allergy. 2018;73:145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fazlollahi M, Chun Y, Grishin A, Wood RA, Burks AW, Dawson P, et al. Early-life gut microbiome and egg allergy. Allergy. 2018;73:1515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho H, Bunyavanich S. Role of the Microbiome in Food Allergy. Curr Allergy Asthma Rep. 2018;18. [DOI] [PubMed] [Google Scholar]
- 30.Lee-Sarwar K, Kelly RS, Lasky-Su J, Moody DB, Mola AR, Cheng TY, et al. Intestinal microbial-derived sphingolipids are inversely associated with childhood food allergy. J Allergy Clin Immunol. 2018; [DOI] [PMC free article] [PubMed]
- 31.Sampson HA, Berin MC, Plaut M, Sicherer SH, Jones S, Burks AW, et al. The Consortium for Food Allergy Research (CoFAR): The first generation. J Allergy Clin Immunol. 2019; [DOI] [PMC free article] [PubMed]
- 32.Rodriguez B, Prioult G, Hacini-Rachinel F, Moine D, Bruttin A, Ngom-Bru C, et al. Infant gut microbiota is protective against cow’s milk allergy in mice despite immature ileal T-cell response. FEMS Microbiol Ecol. 2012; [DOI] [PubMed]
- 33.Abdel-Gadir A, Stephen-Victor E, Gerber GK, Noval Rivas M, Wang S, Harb H, et al. Microbiota therapy acts via a regulatory T cell MyD88/RORγt pathway to suppress food allergy. Nat Med. 2019; [DOI] [PMC free article] [PubMed]
- 34.Feehley T, Plunkett CH, Bao R, Hong SMC, Culleen E, Belda-Ferre P, et al. Healthy infants harbor intestinal bacteria that protect against food allergy. Nat Med. 2019;25:448+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M, Wichienchot S, He X, Fu X, Huang Q, Zhang B. In vitro colonic fermentation of dietary fibers: Fermentation rate, short-chain fatty acid production and changes in microbiota. Trends in Food Science and Technology. 2019.
- 36.Steinert RE, Lee YK, Sybesma W. Vitamins for the Gut Microbiome. Trends in Molecular Medicine. 2020. [DOI] [PubMed]
- 37.Zhao W, Ho H, Bunyavanich S. The gut microbiome in food allergy. Ann ALLERGY ASTHMA Immunol. 2019;122:276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho H, Bunyavanich S. Microbial Adjuncts for Food Allergen Immunotherapy. Current Allergy and Asthma Reports. 2019. [DOI] [PubMed]
- 39.Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun. 2018; [DOI] [PMC free article] [PubMed]
- 40.Gamazon ER, Segrè AV, Van De Bunt M, Wen X, Xi HS, Hormozdiari F, et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet. 2018; [DOI] [PMC free article] [PubMed]
- 41.Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009. [DOI] [PMC free article] [PubMed]
- 42.Watson CT, Cohain AT, Griffin RS, Chun Y, Grishin A, Hacyznska H, et al. Integrative transcriptomic analysis reveals key drivers of acute peanut allergic reactions. Nat Commun. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Croote D, Darmanis S, Nadeau KC, Quake SR. High-affinity allergen-specific human antibodies cloned from single IgE B cell transcriptomes. Science (80-). 2018;362:1306+. [DOI] [PubMed] [Google Scholar]
- 44.Tu AA, Gierahn TM, Monian B, Morgan DM, Mehta NK, Ruiter B, et al. TCR sequencing paired with massively parallel 3 ` RNA-seq reveals clonotypic T cell signatures. Nat Immunol. 2019;20:1692+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruiter B, Smith NP, Monian B, Tu AA, Fleming E, Virkud YV, et al. Expansion of the CD4(+) effector T-cell repertoire characterizes peanut-allergic patients with heightened clinical sensitivity. J Allergy Clin Immunol. 2020;145:270–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leenen FAD, Muller CP, Turner JD. DNA methylation: conducting the orchestra from exposure to phenotype? Clinical Epigenetics. 2016. [DOI] [PMC free article] [PubMed]
- 47.Ladd-Acosta C, Fallin MD. The role of epigenetics in genetic and environmental epidemiology. Epigenomics. 2016. [DOI] [PubMed]
- 48.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011; [DOI] [PMC free article] [PubMed]
- 49.Lawrence M, Daujat S, Schneider R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends in Genetics. 2016. [DOI] [PubMed]
- 50.Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nature Reviews Molecular Cell Biology. 2017. [DOI] [PMC free article] [PubMed]
- 51.Ku CS, Naidoo N, Wu M, Soong R. Studying the epigenome using next generation sequencing. Journal of Medical Genetics. 2011. [DOI] [PubMed]
- 52.Barros-Silva D, Marques CJ, Henrique R, Jerónimo C. Profiling DNA methylation based on next-generation sequencing approaches: New insights and clinical applications. Genes. 2018. [DOI] [PMC free article] [PubMed]
- 53.Martino D, Joo JE, Sexton-Oates A, Dang T, Allen K, Saffery R, et al. Epigenome-wide association study reveals longitudinally stable DNA methylation differences in CD4+T cells from children with IgE-mediated food allergy. EPIGENETICS. 2014;9:998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hong X, Ladd-Acosta C, Hao K, Sherwood Benand Ji H, Keet CA, Kumar R, et al. Epigenome-wide association study links site-specific DNA methylationchanges with cow’s milk allergy. J Allergy Clin Immunol. 2016;138:908+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Canani RB, Paparo L, Nocerino R, Cosenza L, Pezzella V, Di Costanzo M, et al. Differences in dna methylation profile of th1 and th2 cytokine genes are associated with tolerance acquisition in children with ige-mediated cow’s milk allergy. Clin Epigenetics. 2015; [DOI] [PMC free article] [PubMed]
- 56.Syed A, Garcia MA, Lyu SC, Bucayu R, Kohli A, Ishida S, et al. Peanut oral immunotherapy results in increased antigen-induced regulatory T-cell function and hypomethylation of forkhead box protein 3 (FOXP3). J Allergy Clin Immunol. 2014; [DOI] [PMC free article] [PubMed]
- 57.Paparo L, Nocerino R, Cosenza L, Aitoro R, D’Argenio V, Del Monaco V, et al. Epigenetic features of FoxP3 in children with cow’s milk allergy. Clin Epigenetics. 2016; [DOI] [PMC free article] [PubMed]
- 58.Horvath S DNA methylation age of human tissues and cell types. Genome Biol. 2013; [DOI] [PMC free article] [PubMed]
- 59.Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol. 2015; [DOI] [PMC free article] [PubMed]
- 60.Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY). 2015; [DOI] [PMC free article] [PubMed]
- 61.Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, Horvath S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging (Albany NY). 2015; [DOI] [PMC free article] [PubMed]
- 62.Peng C, Cardenas A, Rifas-Shiman SL, Hivert M-F, Gold DR, Platts-Mills TA, et al. Epigenetic age acceleration is associated with allergy and asthma in children in Project Viva. J Allergy Clin Immunol. 2019;143:2263+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martino D, Dang T, Sexton-Oates A, Prescott S, Tang MLK, Dharmage S, et al. Blood DNA methylation biomarkers predict clinical reactivity in food-sensitized infants. J Allergy Clin Immunol. 2015;135:1319–U345. [DOI] [PubMed] [Google Scholar]
- 64.Sun YV, Hu YJ. Integrative Analysis of Multi-omics Data for Discovery and Functional Studies of Complex Human Diseases. Adv Genet. 2016; [DOI] [PMC free article] [PubMed]
- 65.Obeso D, Mera-Berriatua L, Rodriguez-Coira J, Rosace D, Fernandez P, Adoracion Martin-Antoniano I, et al. Multi-omics analysis points to altered platelet functions in severe food-associated respiratory allergy. Allergy. 2018;73:2137–49. [DOI] [PubMed] [Google Scholar]
- 66.Martino D, Neeland M, Dang T, Cobb J, Ellis J, Barnett A, et al. Epigenetic dysregulation of naive CD4+T-cell activation genes in childhood food allergy. Nat Commun. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Do AN, Watson CT, Cohain AT, Griffin RS, Grishin A, Wood RA, et al. Dual transcriptomic and epigenomic study of reaction severity in peanut-allergic children. J Allergy Clin Immunol. 2020;145:1219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barabási AL, Albert R. Emergence of scaling in random networks. Science (80-). 1999; [DOI] [PubMed]
- 69.Li Y, Wu FX, Ngom A. A review on machine learning principles for multi-view biological data integration. Briefings in Bioinformatics. 2018. [DOI] [PubMed]
- 70.Lee B, Zhang S, Poleksic A, Xie L. Heterogeneous Multi-Layered Network Model for Omics Data Integration and Analysis. Frontiers in Genetics. 2020. [DOI] [PMC free article] [PubMed]
- 71.Sonawane AR, Weiss ST, Glass K, Sharma A. Network medicine in the age of biomedical big data. Frontiers in Genetics. 2019. [DOI] [PMC free article] [PubMed]
- 72.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008; [DOI] [PMC free article] [PubMed]
- 73.Uppal K, Ma C, Go YM, Jones DP. XMWAS: A data-driven integration and differential network analysis tool. Bioinformatics. 2018; [DOI] [PMC free article] [PubMed]