

Abstract
Chronic hepatitis B viral infection is a significant health problem world-wide, and currently available antiviral agents suppress HBV infections, but rarely cure this disease. It is presumed that antiviral agents that target the viral nuclear reservoir of transcriptionally active cccDNA may eliminate HBV infection. Through a series of chemical optimization, we identified a new series glyoxamide derivatives affecting HBV nucleocapsid formation and cccDNA maintenance at low nanomolar levels. Among all the compounds synthesized, GLP-26 displays a major effect on HBV DNA, HBeAg secretion and cccDNA amplification. In addition, GLP-26 shows a promising pre-clinical profile and long-term effect on viral loads in a humanized mouse model.
Keywords: Capsid, hepatitis B virus, antiviral, cccDNA
Graphical Abstract
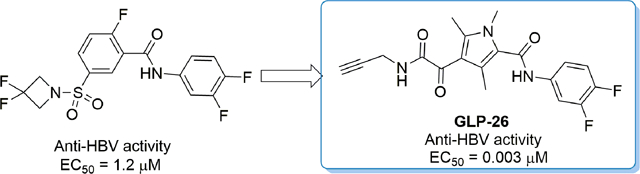
1. Introduction
Hepatitis B virus (HBV) remains a major global health concern. Currently, it is estimated that 257 million people worldwide are suffering from HBV, and are at high risk for development of more life-threatening diseases such as liver failure, cirrhosis or hepatocellular carcinoma (HCC).1 Despite the widespread availability of vaccines, they are not effective for those already infected. The current standard of care (SOC) for treating chronic HBV patients relies on pegylated interferon alpha in combination with a nucleoside analog (Entecavir, tenofovir disoproxil fumarate or alafenamide, lamivudine, adefovir dipivoxil or telbivudine). Although these therapies can suppress HBV replication and prevent disease progression, they are not a cure since they do not affect the nuclear pool of covalently closed circular DNA (cccDNA), the viral minichromosome accumulating in the nucleus of hepatocytes. Since, cccDNA has been shown to play a central role in the establishment of HBV infection and persistence, cessation of treatment leads most of the time to a virus rebound.
Capsid assembly modulators (CAMs) have recently been proven to be effective agents for disrupting HBV capsid assembly and thus affecting several steps of HBV replication and impairing the cccDNA stability. Over the years, several classes of CAMs including heteroaryldihydropyrimidines (HAPs), phenylpropenamides (PPAs), sulfamoylpyrroloamides (SPAs) and sulfamoylbenzamides (SBAs) have been studied.2 Among these classes, several compounds, including JNJ 56136379 (1) (Phase II),3 ABI-H0731 (2) (Phase II),4 ABI-H2158 (Phase I),5 RG7907 (3) (Phase I), QL-007 (Phase I) and EDP-514 (Phase I) are being evaluated in humans (Figure 1).6
Figure 1:
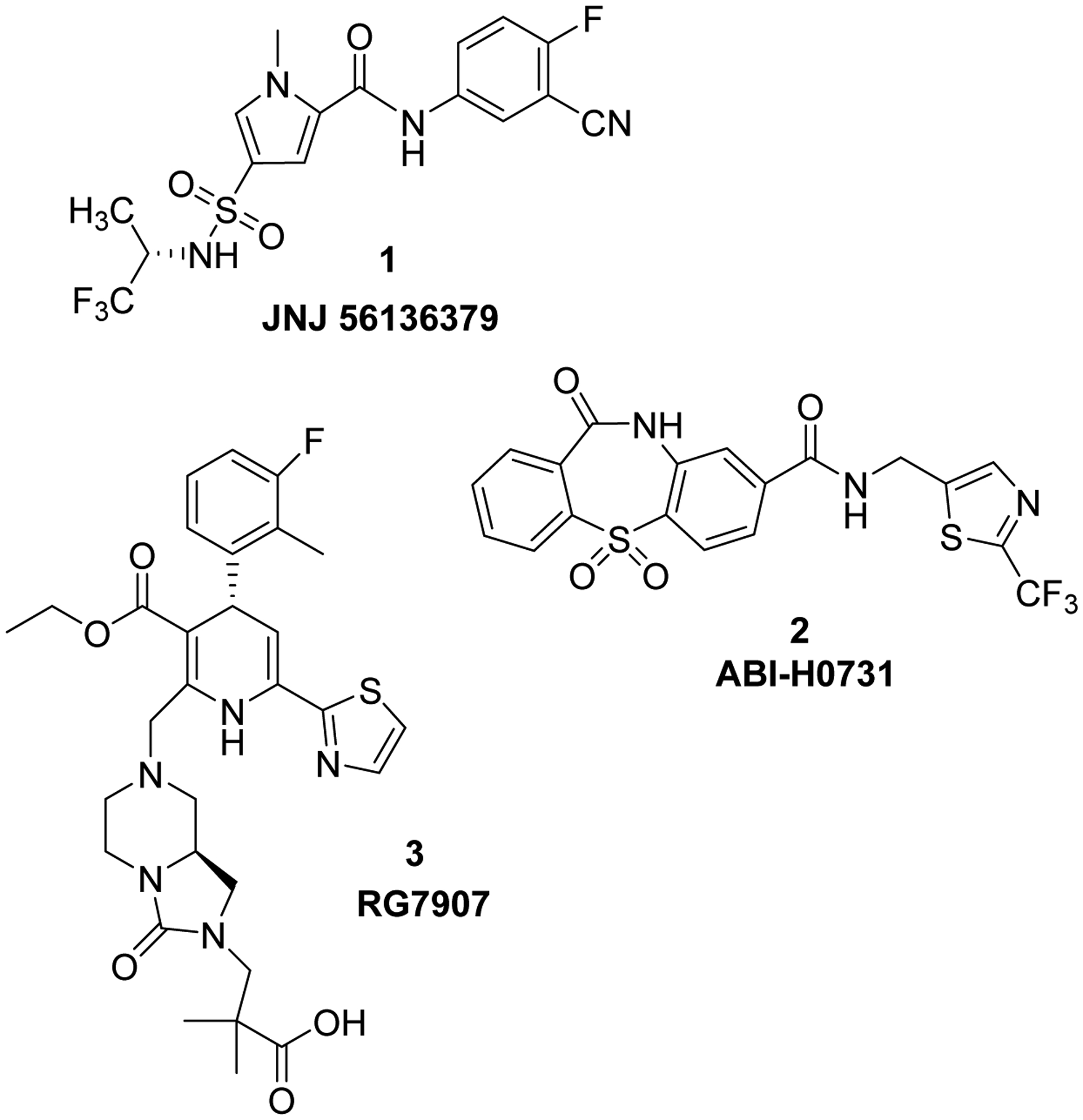
Structures of HBV capsid assembly modulators 1, 2 and 3
Our group recently identified GLP-26, a highly potent, non-toxic, glyoxamoylpyrroloxamide (GLP) derivative that can alter HBV nucleocapsid assembly, prevents transport of capsid to the nucleus and inhibits viral DNA replication.7,8 We also demonstrated that GLP-26 inhibits HBeAg secretion, and reduces cccDNA amplification.
More importantly, in a humanized mouse model, treatment with GLP-26 in combination with entecavir led to a sustained antiviral response up to 12 weeks after treatment cessation. Herein, we wish to report how GLP-26 was discovered through chemical optimization of compound 4, a sulfamoyl derivative affecting capsid assembly and displaying anti-HBV activity in the low micromolar range (Figure 2). More specifically, we will report how the replacement of the phenyl sulfamoyl moiety in compound (4) by a diketopyrole group (Part A) and the introduction of variously functionalized amine on Part B greatly improved the potency of our lead compound and ultimately led our group to the discovery of GLP-26 and additional bioactive molecules that could be used as a treatment for HBV.7,8
Figure 2.
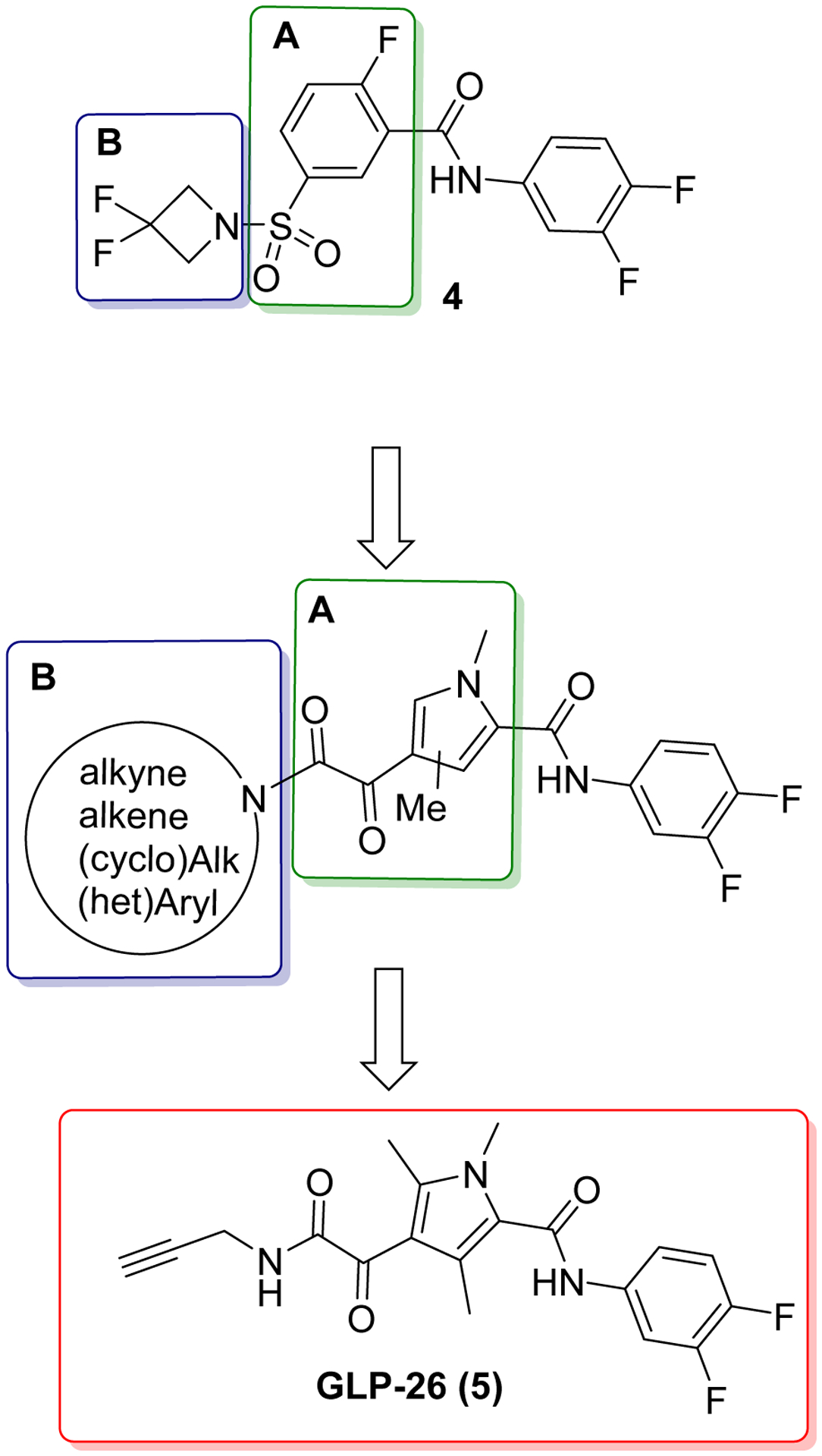
Chemical optimization of lead compound 4 leading to the discovery of GLP-26.
2. Results and discussion
2.1. Chemistry
Preliminary in silico evaluation of the diketopyrole scaffold was performed. The conformation of the phenyl sulfamoyl derivative 4 was derived from the co-crystal structure of the HBV capsid Y132A complexed to a sulfamoyl benzamide inhibitor (PDBID 5T2P).9 The diketopyrole scaffold (Fig 3: Compound in purple) was flexibly aligned to the phenyl sulfamoyl derivative (Fig 3: Compound in green) using the BIOVIA Discovery Studio suite.10 The overall structural similarity between the two small molecules was 0.88 supporting that the diketopyrole moiety is a suitable isostere for the benzyl-sulfonamide scaffold.
Figure 3.
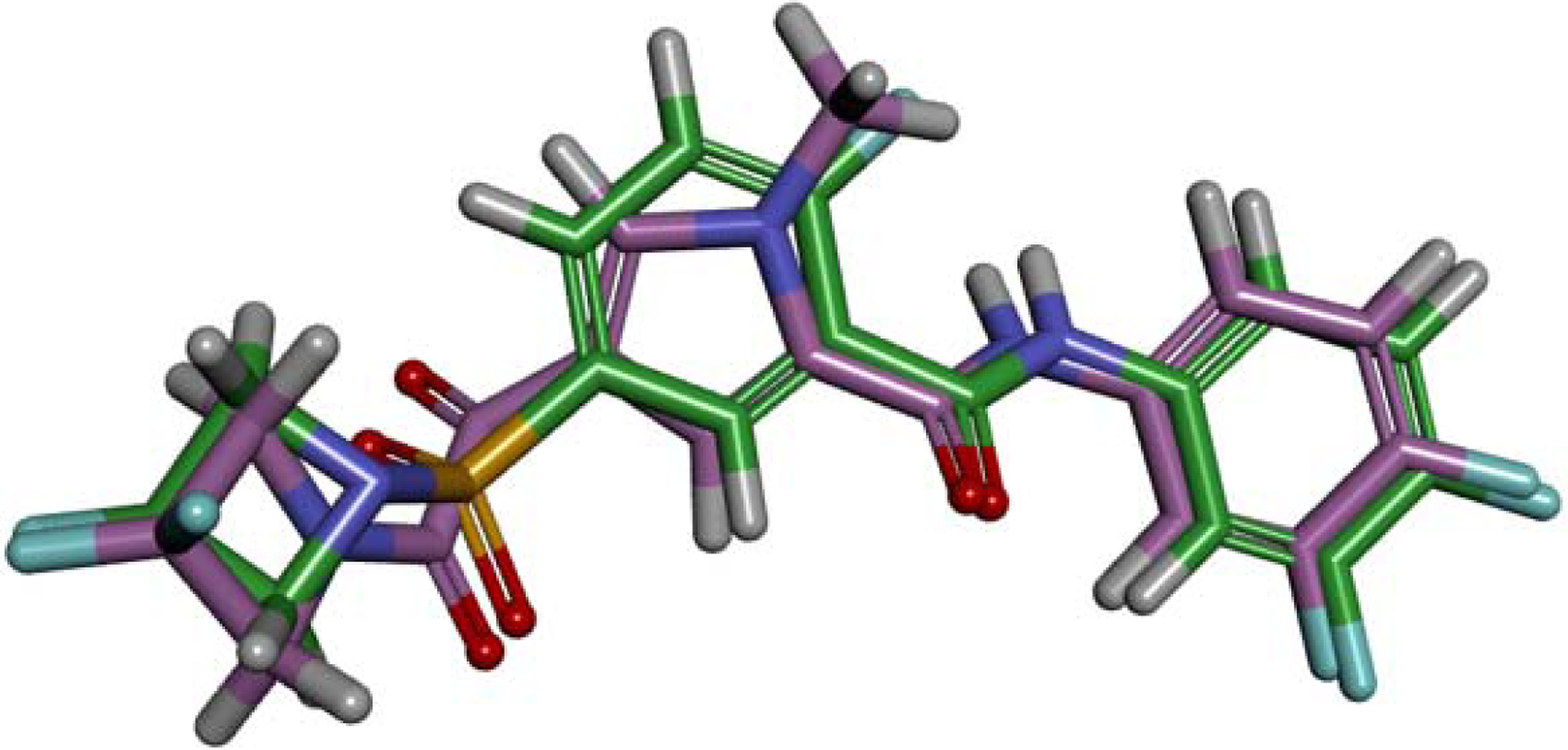
Alignment of a diketopyrole derivative (in purple) and its phenyl sulfamoyl analog (in green) using the BIOVIA Discovery Studio suite.
Initial diketopyrole derivatives 10a-i were prepared according to the chemistry described in Scheme 1. Commercially available 1-methyl-1H-pyrrole-2-carboxylic acid 6 was coupled with 3,4-difluoroaniline in presence of HATU and DIPEA to give amide 7 which was then reacted with ethyl chlorooxoacetate in presence of AlCl3 to afford compound 8. Key acid intermediate 9 was obtained by treatment of 8 under basic condition and final coupling with a variety of amines, in presence of CDI gave access to the desired glyoxamide derivatives 10a-i.
Scheme 1.
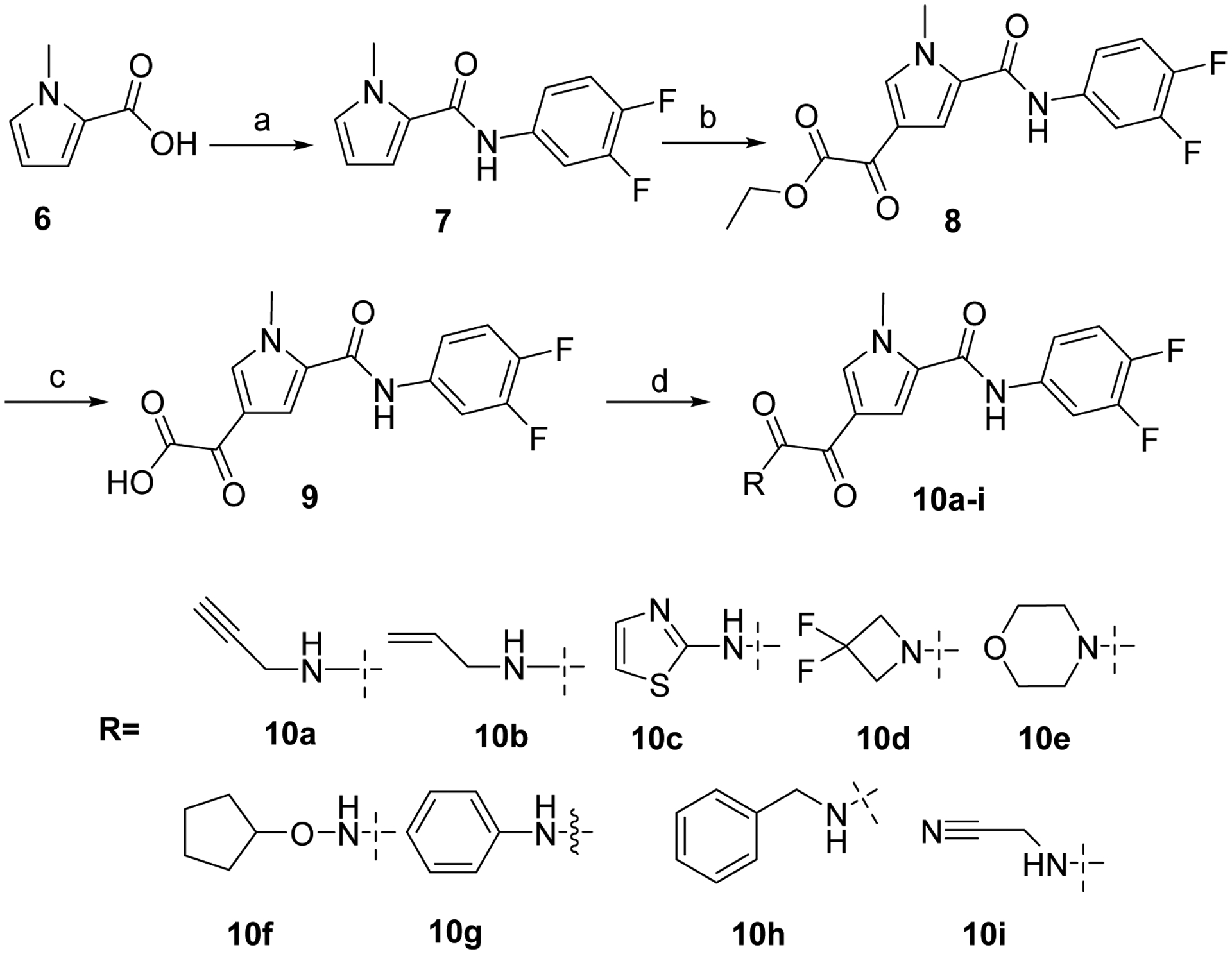
Reagents and conditions: a) HATU, 3,4-difluoroaniline, DIPEA, DMF, 65 °C, overnight; b) ClC(O)COOEt, AlCl3, CH2Cl2, 0 °C to rt, overnight; c) i) NaOH 5%, MeOH, rt, 15 min; ii) HCl 1N; d) CDI, amine, DMF, CH2Cl2, rt, 2 h.
In a similar manner, trimethyl pyrrole derivatives 17a-f were prepared from 11 by first, methylation, using MeI in presence of KOH, and then saponification of the intermediate 12. Carboxylic acid 13 was then coupled with 3,4-difluoroaniline before being reacted with ethyl chlorooxoacetate in presence of AlCl3 to afford compound 15. Final saponification of 15 followed by coupling of the resulting acid 16 with various amines in presence of CDI afforded targeted compounds 17a-f. (Scheme 2)
Scheme 2.
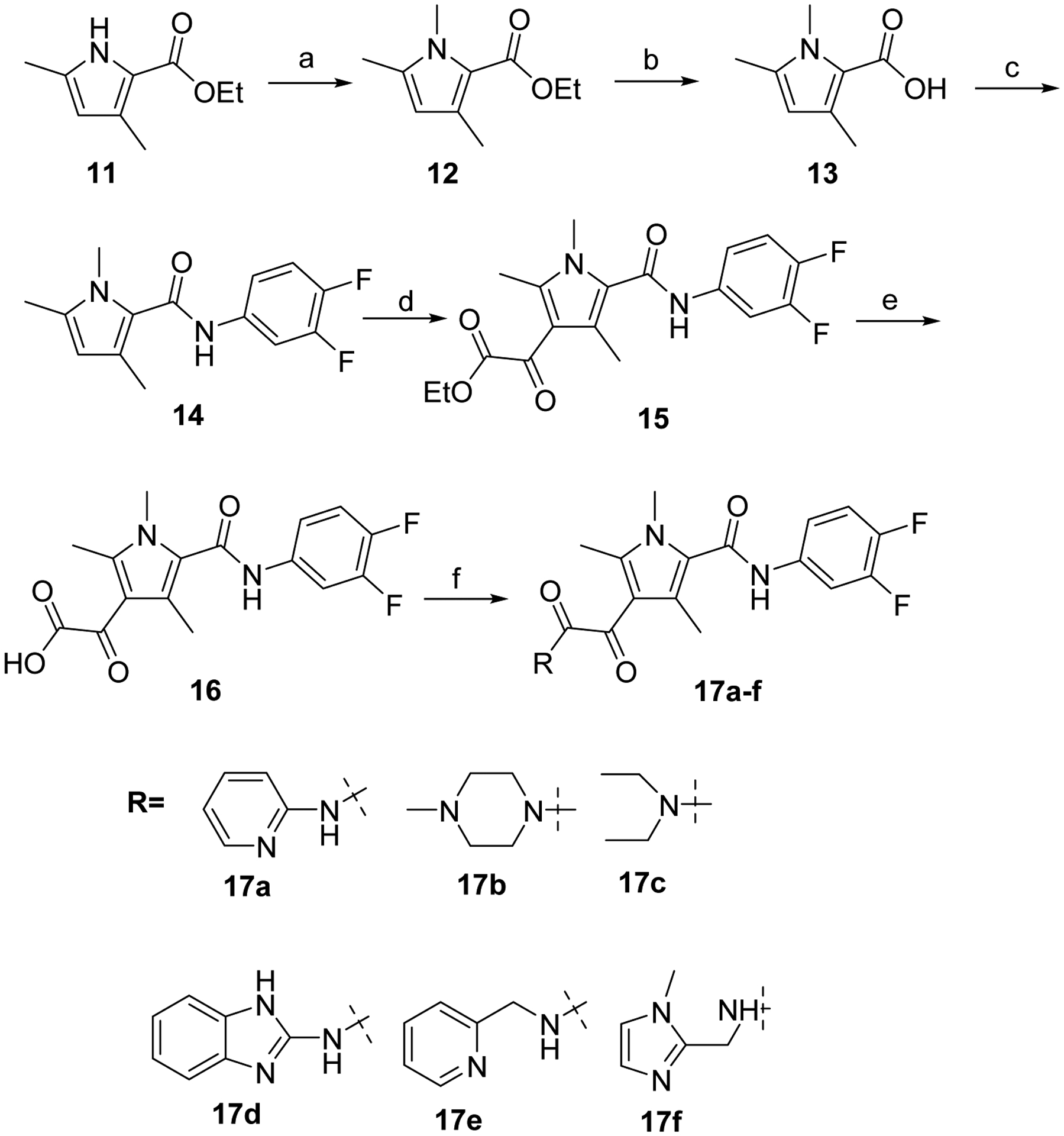
Reagents and conditions: a) MeI, KOH, DMSO; b) NaOH, EtOH, 100 °C, 6 h; c) 3,4-difluoroaniline, HATU, DIPEA, DMF, 60 °C; d) ClC(O)COOEt, AlCl3, DCM, 0 °C to rt, 16 h; e) NaOH, EtOH, rt, 1 h; f) Amine, CDI, DMF, DCM, rt, 1 h.
Derivatives 5 and 22 were prepared from trimethyl pyrrole ester 12 by first reaction with ethyl chlorooxoacetate in presence of AlCl3 followed by saponification to form key intermediate 19. Coupling of 19 with either propargyl amine or thiazol-2-amine in presence of CDI afforded compounds 20a-b respectively. Finally, treatment under basic condition and coupling of carboxylic acids 21a-b with difluoroaniline using HATU gave the desired compounds 5 and 22 (Scheme 3).
Scheme 3.
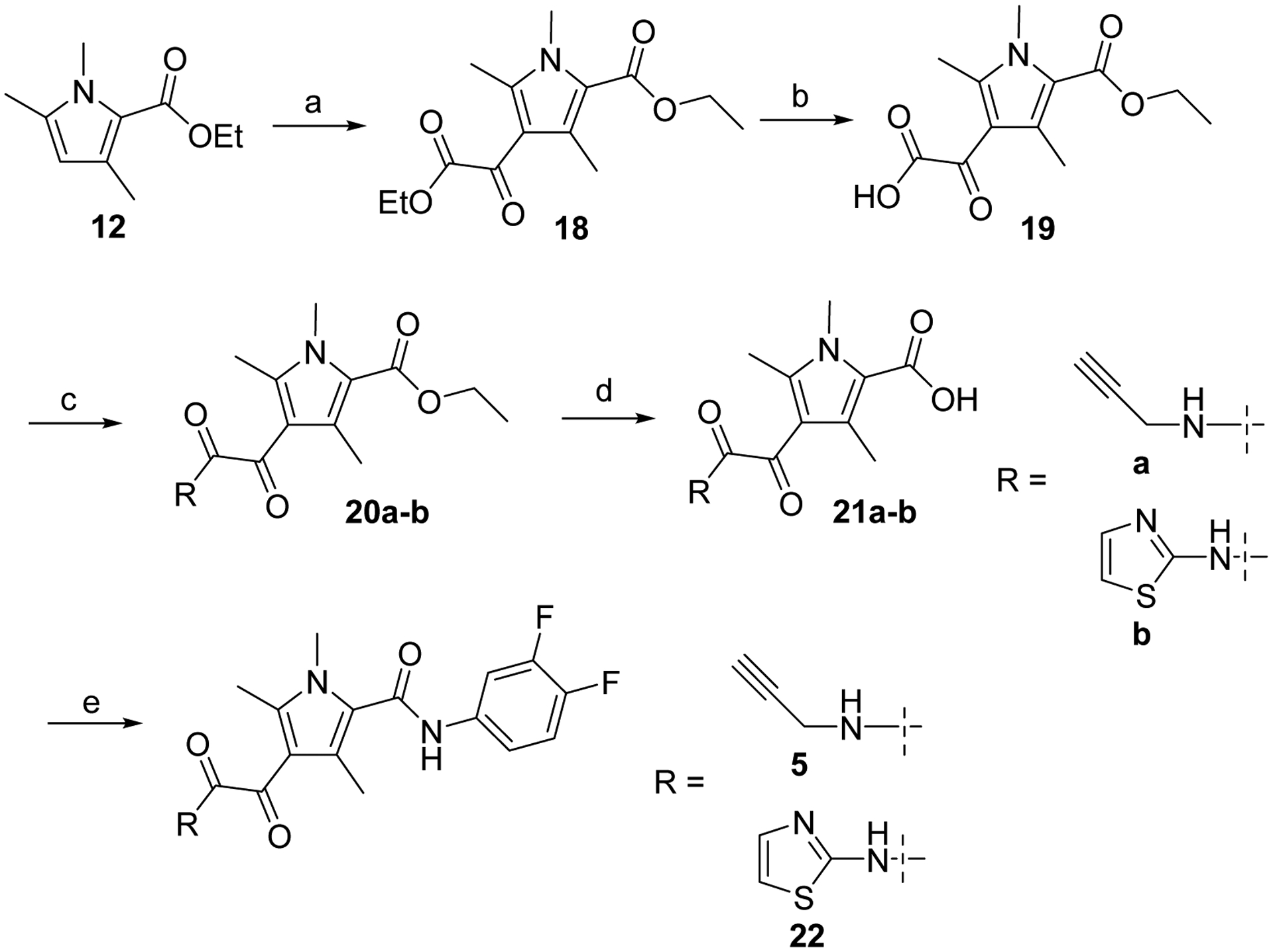
Reagents and conditions: a) ClC(O)COOEt, AlCl3, CH2Cl2; b) NaOH 5%, MeOH, rt, 10 min; c) CDI, amine, DMF, 3 h, rt; d) NaOH 5%, MeOH, 16 h, rt; e) 3,4-difluoroaniline, HATU, DIPEA, DMF, 16 h, rt.
2.2. Anti-HBV activity and cytotoxicity
The in vitro anti-HBV activity and safety profile of the synthesized compounds were assessed by real-time PCR in HepAD38 cells as previously described.11 The concentration of compound that inhibited HBV DNA replication by 50% (EC50) was determined by linear regression. The antiviral data are provided relative to the untreated control. Entecavir and (ETV) and lamivudine (3TC), two nucleoside analogs approved for the treatment of HBV along with GLS4, a well-known HBV CAM12 were used as positive controls. In addition, cytotoxicity was determined by using the CellTiter 96 non-radioactive cell proliferation colorimetric assay (Promega) in peripheral blood mononuclear (PBM), human T lymphoblast (CEM), African green monkey kidney (Vero), and human hepatocellular carcinoma (HepG2) cells. Our laboratory uses several cytotoxicity assays to determine routinely if our compounds are broadly safe in different cells. Toxicity levels were measured as the concentration of test compound that inhibited cell growth by 50% (CC50). All the data on 16 novel synthesized compounds are summarized in Table 1.
Table 1.
Anti-HBV activity and cytotoxicity of compounds 10a-i, 17a-f, 5 and 22.
| Compound | Anti-HBV activity (μM) | Cytotoxicity CC50 (μM) | ||||
|---|---|---|---|---|---|---|
| EC50 | EC90 | PBM | CEM | Vero | HepG2a | |
| 4 | 1.2 ± 0.6 | 6.3 ± 2.9 | 69.1 | 3.7 | 14.7 | 27.0 |
| 10a | 5.6 ± 1.60 | >10 | 57.9 | 70.3 | 89.0 | >100 |
| 10b | 0.09 ± 0.07 | 3.05 ± 1.83 | >100 | >100 | 68.1 | >100 |
| 10c | 0.47 ± 0.53 | 4.10 ± 2.42 | >100 | >100 | 31.2 | >100 |
| 10d | 3.8 ± 3.1 | 9.67 ± 0.47 | >100 | >100 | >100 | >100 |
| 10e | 1.1 ± 1.5 | 4.62 ± 4.50 | >100 | >100 | 41.9 | 93.9 |
| 10f | 0.56 ± 0.69 | 2.95 ± 2.51 | ≥100 | 37.9 | 80.1 | >100 |
| 10g | 0.08 ± 0.02 | 1.80 ± 1.23 | >100 | >100 | 13.4 | >100 |
| 10h | 0.41 ± 0.01 | 7.95 ± 0.07 | >100 | >100 | 62.4 | >100 |
| 10i | 5.16 ± 0.23 | 10.0 ± 0.03 | >100 | >100 | 91.4 | >100 |
| 17a | 0.004 ± 0.0001 | 0.06 ± 0.03 | >100 | 14.6 | >100 | 83.7 |
| 17b | 0.005 ± 0.010 | 0.10 ± 0.04 | ≥ 100 | 24.7 | ≥ 100 | 95.4 |
| 17c | 0.58 ± 0.08 | 5.64 ± 3.60 | 91.7 | 16.8 | >100 | 89.9 |
| 17d | 0.21 ± 0.74 | 3.44 ± 1.56 | 90.0 | 35.8 | 46.9 | 74.9 |
| 17e | 0.16 ± 0.09 | 2.02 ± 1.68 | >100 | 38.4 | 9.6 | 7.2 |
| 17f | 0.01 ± 0.01 | 0.17 ± 0.23 | >100 | 46.9 | 50.6 | 34.7 |
| 5 (GLP-26) | 0.003 ± 0.002 | 0.014 ± 0.002 | >100 | >100 | 46.4 | >100 |
| 22 | 0.006 ± 0.002 | 0.03 ± 0.003 | >100 | 26.8 | >100 | 83.9 |
| GLS-4 | 0.08 ± 0.02 | 0.28 ± 0.06 | 28.4 | 68.7 | 17.7 | >100 |
| 3TC | 0.41 ± 0.36 | 1.65 ± 0.92 | >100 | >100 | >100 | >100 |
| ETV | 0.0006 ± 0.0003 | 0.011 ± 0.002 | 15.7 | >100 | >100 | >100 |
ETV: Entecavir; 3TC: Lamivudine
AD38 cells are derived from HepG2 cells
Replacement of the phenyl sulfonyl moiety in our lead compound 4 with a diketo 1-methyl pyrrole moiety (compound 10d) did not improve the potency of the parent compound (1.3 μM VS 3.83 μM). However, further substitution of the difluoro azetidine in 10d, with an allylamine (10b), an amino thiazole (10c), a cyclopentylhydroxylamine (10f) or an aniline (10h) led to sub-micromolar HBV inhibitors (EC50s between 0.09 and 0.56 μM). Interestingly, all these compounds showed also a better toxicity profile in our panel of cell lines when compared to compound 4. Replacement of the phenyl sulfonyl moiety in our lead compound 4 with a diketo trimethyl pyrrole moiety was also evaluated (Scheme 2 and 3). In this series, introduction, at the position B of our molecules (Figure 2), of a diethyl amine, a benzoimidazole amine, a 2-picolylamine led to compounds 17c-e that did not showed better activities than compounds from the 1-methyl pyrrole series (EC50s of 0.58, 0.21 and 0.16 μM, respectively). However, introduction of a pyridine, an N-methylpiperazine, an N-methyl imidazole or a thiazole group gave highly active compounds 17a-b, 17f and 22 displaying EC50s in the single digit nanomolar range. However, it is worth noting that they all play slight to moderate toxicities in CEM and HEpG2 cells. More interestingly, introduction of a propargyl group on the part B of our diketo trimethyl pyrrole moiety proved to be beneficial. Indeed, GLP-26 (5) was \ our most promising compound in term of potency, with an EC50/90 of 0.003/0.014 μM and only displayed marginal toxicity in Vero cells. (Therapeutic Index, TI, in HepG2 cells > 33,333).
2.3. Modeling
Based on the structural similarity of GLP-26 and reference compound 4 and the fact that GLP-26 binds directly to HBV capsids, vide infra, we used the structure of 4 bound to the interfacial HBV capsid pocket as a template for modeling. By doing so, were able to observe that the docking of GLP-26 into this structure generated a pose similar to the co-crystallized ligand (Figure 4). The central amide bond acts as a hydrogen bond bridge between W102 of one subunit (chain B) and T128 of the other (chain C). The difluorobenzyl ring occupies a hydrophobic pocket formed by L30, T33, and I105 of one monomer and V124 of the other monomer. The trimethyl-pyrrole ring interacts with nonpolar residues F23, T118, and L140. The α-ketoamide amide accepts a hydrogen bond from the L140 backbone thereby orienting the alkyl group into solution.
Figure 4.
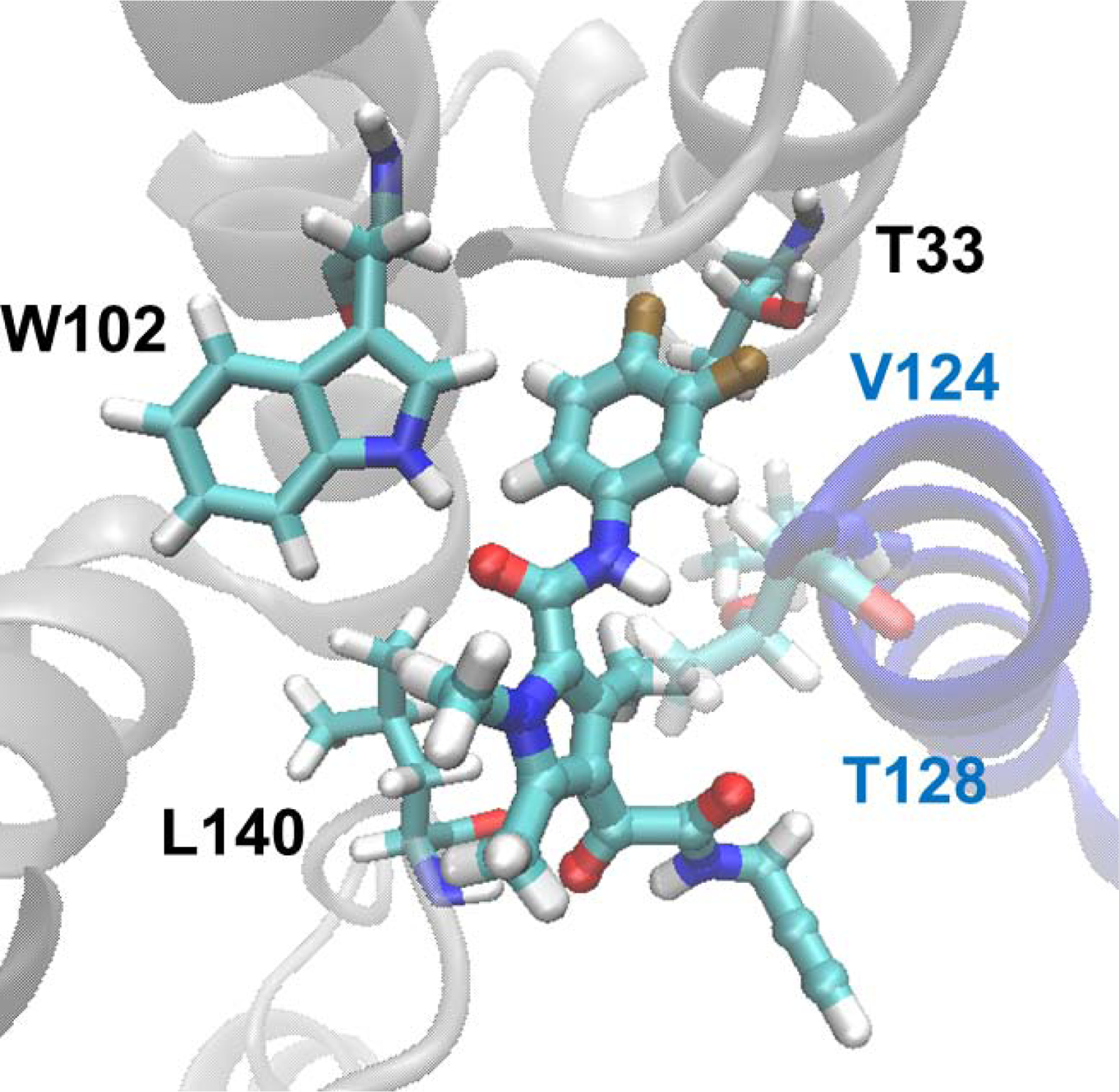
Molecular model of GLP-26 bound to the HBV capsid protein interface. GLP-26 was docked into the interfacial binding pocket of compound 4 in the reported crystal structure (PDBID 5T2P) using Glide. Chain B of the protein is grey, and chain C is colored blue. Key residues are shown as licorice, and the GLP-26 is rendered as a ball-and-stick.
3. Conclusion
Through in-depth SAR and chemical modifications of early lead compound 4, we identifed highly potent HBV CAM, GLP-26. Replacement of the phenyl sulfonyl moiety in compound 4 by a diketo trimethylpyrrole moiety was beneficial and lead to a better orientation of both the difluorophenyl amide group and the part B of the molecule within the HBV capsid pocket, while optimizing key hydrogen bindings. The unique in vitro and in vivo properties7 of GLP-26 make this compound an attractive option for the treatment of HBV infections.
4. Experimental Section:
4.1. Synthesis
Anhydrous solvents were purchased from Aldrich Chemical Company, Inc. (Milwaukee). Reagents were purchased from commercial sources. Unless noted otherwise, the materials used in the examples were obtained from readily available commercial suppliers or synthesized by standard methods known to one skilled in the art of chemical synthesis. 1H, 13C and 19F spectra were taken on a Bruker AscendTM 400 spectrometer at rt and reported in ppm downfield using residual solvent line as an internal standard for 1H-and 13C NMR. No standard was used for 19F spectra. NMR processing was performed with MestReNova version 10.0.2-15465. Deuterium exchange and decoupling experiments were utilized to confirm proton assignments. Signal multiplicities are represented by s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quadruplet), br (broad), bs (broad singlet), m (multiplet). All J-values are in Hz and calculated by Mnova or MestReNova programs. Mass spectra were determined on a Micromass Platform LC spectrometer using electrospray ionization. Analytic TLC was performed on Analtech GHLF silica gel plates, and preparative TLC on Analtech GF silica gel plates. Column chromatography was performed on Combiflash Rf200 or via reverse-phase high performance liquid chromatography.
4.1.1. 2-(5-((3, 4-Difluorophenyl) carbamoyl)-1-methyl-1H-pyrrol-3-yl)-2-oxoacetic acid (9).
To a solution of 1-methyl-1H-pyrrole-2-carboxylic acid 6 (4 g, 32 mmol), 3, 4-difluoroaniline (6.2 g, 48 mmol) and DIPEA (13 mL, 96 mmol) in DMF (150 mL) was added HATU (18.2 g, 48 mmol) at room temperature. The mixture was heated at 65 °C overnight and poured into a saturated solution of NH4Cl. After extraction with EtOAc (3 × 50 mL), the combined organic layers were dried over Na2SO4 and concentrated in vacuo. The resulting mixture was purified by flash chromatography (hexanes:EtOAc = 6:4 v/v), to give compound 7 as a white powder (82%, 6.2 g, 26.2 mmol). To a solution of N-(3,4-difluorophenyl)-1-methyl-1H-pyrrole-2-carboxamide 7 (3 g, 12.7 mmol) in CH2Cl2 (150 mL) was added dropwise, at 0 °C, a solution of ethyl 2-chloro-2-oxo-acetate (2.12 mL, 19.1 mmol) in CH2Cl2 (20 mL), followed by AlCl3 (5.1 g, 38.1 mmol) portion wise. The resulting mixture was stirred overnight at room temperature and then poured onto crushed ice. After addition of water (300 mL), the mixture was filtered on Celite and the aqueous layer was extracted with CH2Cl2 (3 × 100mL). The combined organic layers were washed with a saturated solution of sodium carbonate (100 mL), a saturated solution of NH4Cl (100 mL), dried over Na2SO4 and concentrated in vacuo. The resulting oil was diluted in MeOH /THF (v/v = 1/2, 30 mL) and a 5% solution of NaOH (30 mL) was added. After being stirred for 15 min at room temperature, the mixture was concentrated in vacuo. The mixture was washed with EtOAc (2 × 50 mL), acidified with a 1N HCl solution (pH = 1) and extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over Na2SO4, and concentrated in vacuo. The resulting solid 8 was washed with diethyl ether (20 mL) and hexanes (20 mL) to yield 2-(5-((3,4-difluorophenyl)carbamoyl)-1-methyl-1H-pyrrol-3-yl)-2-oxoacetic acid 9 (3.48 g, 11.3 mmol) as an off-white powder. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.02 (d, J = 1.9 Hz, 1H), 7.94 – 7.81 (m, 1H), 7.60 (d, J = 1.9 Hz, 1H), 7.56 – 7.47 (m, 1H), 7.44 – 7.24 (m, 1H), 3.96 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 181.1, 165.3, 159.5, 150.5 (d, J = 13.1 Hz), 148.0 (d, J = 13.1 Hz), 147.0 (d, J = 12.5 Hz), 144.6 (d, J = 12.6 Hz), 136.6, 136.4 (dd, J = 9.2, 2.8 Hz), 127.9, 119.0, 117.7 (d, J = 17.7 Hz), 116.8 (dd, J = 5.8, 3.2 Hz), 114.9, 109.4 (d, J = 21.7 Hz), 37.7. 19F NMR (377 MHz, DMSO-d6) δ −138.0 – −138.2 (m), −145.5 – −145.6 (m). HRMS (ESI): m/z [M+H]+ calcd for C14H11F2N2O4: 309.0687, found: 309.0673.
4.1.2. General procedure for the synthesis of compounds 10a-i
To a solution of 9 (0.3 g, 0.1 mmol) in DMF (4 mL) and CH2Cl2 (4 mL) were added CDI (243 mg, 0.15 mmol) and an amine (0.12 mmol) at room temperature. The mixture was stirred for 1–2 h at room temperature and poured into a solution of NH4Cl. After extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over Na2SO4 and concentrated in vacuo. The resulting residue was purified by flash chromatography (Hexanes:EtOAc = 7:3 v/v) to afford compounds 10a-i.
4.1.3. N-(3,4-Difluorophenyl)-1-methyl-4-(2-oxo-2-(prop-2-yn-1-ylamino)acetyl)-1H-pyrrole-2-carboxamide (10a).
Yield: 98%. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 9.21 (s, 1H), 8.20 (s, 1H), 7.92 – 7.87 (m, 1H), 7.67 (s, 1H), 7.59 – 7.51 (m, 1H), 7.30 (dd, J = 19.2 Hz, J = 9.2 Hz, 1H), 6.34 (s, 1H), 3.96 (s, 3H), 3.79 (d, J = 3.6 Hz, 1H), 3.15 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 181.9, 163.1, 159.6, 150.5 (d, J = 13.1 Hz), 148.1 (d, J = 13.1 Hz), 147.0 (d, J = 12.1 Hz), 144.6 (d, J = 13.1 Hz), 137.4, 136.5 (d, J = 6.1 Hz), 127.6, 118.9, 117.7 (d, J = 17.2 Hz), 116.8, 115.6, 109.4 (d, J = 21.2 Hz), 80.9, 73.5, 37.7, 28.4. 19F NMR (377 MHz, DMSO-d6) δ −137.4 (d, J = 7.5 Hz), −144.7 (d, J = 11.3 Hz). HRMS (ESI): m/z [M+H]+ calcd for C17H14F2N3O3: 346.1003, found: 346.0997.
4.1.4. 4-(2-(Allylamino)-2-oxoacetyl)-N-(3,4-difluorophenyl)-1-methyl-1H-pyrrole-2-carboxamide (10b).
Yield: 81%. 1H NMR (400 MHz, Acetone-d6) δ 9.60 (s, 1H), 8.23 (s, 1H), 8.14 (s, 1H), 8.04 – 7.90 (m, 1H), 7.58 (s, 1H), 7.57 – 7.49 (m, 1H), 7.30 (q, J = 9.6 Hz, 1H), 6.25 – 5.68 (m, 1H), 5.22 (d, J = 16.8 Hz, 1H), 5.11 (dt, J = 10.3, 1.4 Hz, 1H), 4.06 (s, 3H), 3.99 – 3.92 (m, 2H). 13C NMR (101 MHz, Acetone-d6) δ 182.7, 163.7, 161.1, 152.6 (d, J = 13.1 Hz), 150.2 (d, J = 12.9 Hz), 149.1 (d, J = 12.8 Hz), 146.7 (d, J = 12.9 Hz), 138.9, 137.9, 136.2, 129.0, 120.9, 118.8 (d, J = 18.0 Hz), 117.9 – 117.4 (m), 117.0, 116.6, 110.7 (d, J = 22.1 Hz), 42.9, 38.7. 19F NMR (377 MHz, Acetone-d6) δ −138.5 – −138.8 (m), −145.9 – −146.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C17H16F2N3O3: 348.1260, found: 348.1158.
4.1.5. N-(3,4-Difluorophenyl)-1-methyl-4-(2-oxo-2-(thiazol-2-ylamino)acetyl)-1H-pyrrole-2-carboxamide (10c).
Yield: 79%. 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H), 10.33 (s, 1H), 8.20 (s, 1H), 7.94 – 7.83 (m, 1H), 7.69 (s, 1H), 7.60 (d, J = 3.6 Hz, 1H), 7.56 – 7.49 (m, 1H), 7.48 – 7.38 (m, 1H), 3.98 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 180.6, 162.5, 159.5, 150.5 (d, J = 12.9 Hz), 148.1 (d, J = 12.5 Hz), 138.6, 137.2, 136.4 (d, J = 12.0 Hz), 127.9, 118.7, 117.8 (d, J = 17.6 Hz), 116.8, 109.5 (d, J = 21.7 Hz), 60.2, 37.8, 17.9 (d, J = 672.1 Hz). 19F NMR (377 MHz, DMSO-d6) δ −137.3 – −137.4 (m), −144.5 – −144.7 (m). HRMS (ESI): m/z [M+H]+ calcd for C17H13F2N4O3S: 391.0676, found: 391.0669.
4.1.6. 4-(2-(3,3-Difluoroazetidin-1-yl)-2-oxoacetyl)-N-(3, 4-difluorophenyl)-1-methyl-1H-pyrrole-2-carboxamide (10d).
Yield: 48%. 1H NMR (400 MHz, Acetone-d6) δ 9.60 (s, 1H), 8.06 (d, J = 1.7 Hz, 1H), 8.01 – 7.86 (m, 1H), 7.70 – 7.47 (m, 2H), 7.34 – 7.26 (m, 1H), 4.98 – 4.79 (m, 2H), 4.65 – 4.31 (m, 2H), 4.05 (s, 3H). 13C NMR (101 MHz, Acetone-d6) δ 182.6, 163.7, 161.1, 152.6 (d, J = 13.1 Hz), 150.2 (d, J = 13.6 Hz), 149.1 (d, J = 12.8 Hz), 146.7 (d, J = 13.1 Hz), 138.4, 138.2 – 137.6 (m), 129.2, 121.4, 121.0, 118.9 (d, J = 17.9 Hz), 118.3, 118.0 – 117.5 (m), 116.2, 115.6, 110.8 (d, J = 22.2 Hz), 65.8 (t, J = 28.6 Hz), 61.9 (t, J = 28.7 Hz), 38.8. 19F NMR (377 MHz, Acetone-d6) δ −102.1 (p, J = 12.3 Hz), −138.7 – −138.8 (m), −146.0 – −146.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C17H14F4N3O3: 384.0971, found: 384.0964.
4.1.7. N-(3,4-Difluorophenyl)-1-methyl-4-(2-morpholino-2-oxoacetyl)-1H-pyrrole-2-carboxamide (10e).
Yield: 80%. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.19 – 7.80 (m, 2H), 7.57 – 7.47 (m, 2H), 7.46 – 7.36 (m, 1H), 3.95 (s, 3H), 3.81 – 3.67 (m, 2H), 3.64 – 3.59 (m, 2H), 3.58 – 3.49 (m, 2H), 3.34 – 3.25 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 185.9, 165.8, 159.5, 150.5 (d, J = 13.1 Hz), 148.1 (d, J = 13.2 Hz), 147.1 (d, J = 12.8 Hz), 144.7 (d, J = 12.8 Hz), 136.4 (dd, J = 9.1, 3.0 Hz), 135.9, 128.1, 119.8, 117.8 (d, J = 17.7 Hz), 117.2 – 116.3 (m), 114.0, 109.5 (d, J = 21.7 Hz), 66.5 (d, J = 30.5 Hz), 46.3, 41.5, 37.8. 19F NMR (377 MHz, DMSO-d6) δ −137.2 – −137.4 (m), −144.5 – −144.6 (m). HRMS (ESI): m/z [M+H]+ calcd for C18H18F2N3O4: 378.1265, found: 378.1257.
4.1.8. 4-(2-((Cyclopentyloxy)amino)-2-oxoacetyl)-N-(3,4-difluorophenyl)-1-methyl-1H-pyrrole-2-carboxamide (10f).
Yield: 26%. 1H NMR (400 MHz, Acetone-d6) δ 10.83 (s, 1H), 9.63 (s, 1H), 8.14 (d, J = 1.7 Hz, 1H), 8.03 – 7.90 (m, 1H), 7.65 – 7.46 (m, 2H), 7.40 – 7.20 (m, 1H), 4.69 – 4.57 (m, 1H), 4.06 (s, 3H), 1.95 – 1.85 (m, 2H), 1.79 – 1.65 (m, 4H), 1.57 (s, 2H). 13C NMR (101 MHz, Acetone-d6) δ 181.0, 161.2 (d, J = 178.9 Hz), 159.7, 159.3, 150.8 (d, J = 13.2 Hz), 148.3 (d, J = 13.2 Hz), 147.3 (d, J = 12.8 Hz), 144.9 (d, J = 12.7 Hz), 136.8, 136.2 (dd, J = 9.1, 3.0 Hz), 127.4, 119.2, 117.0 (d, J = 18.0 Hz), 116.5 – 115.7 (m), 114.5, 109.1 (d, J = 22.2 Hz), 87.4, 87.0, 37.0, 30.9, 23.4. 19F NMR (377 MHz, Acetone-d6) δ −138.6 – −138.7 (m), −146.0 – −146.1 (m). HRMS (ESI): m/z [M+H]+ calcd for C19H20F2N3O4: 392.1422, found: 392.1415.
4.1.9. N-(3,4-Difluorophenyl)-1-methyl-4-(2-oxo-2-(phenylamino)acetyl)-1H-pyrrole-2-carboxamide (10g).
Yield: 32%. 1H NMR (400 MHz, Acetone-d6) δ 9.79 (s, 1H), 9.64 (s, 1H), 8.30 (s, 1H), 8.04 – 7.94 (m, 1H), 7.93 (s, 1H), 7.91 (s, 1H), 7.63 (d, J = 1.8 Hz, 1H), 7.60 – 7.52 (m, 1H), 7.44 – 7.37 (m, 2H), 7.36 – 7.27 (m, 1H), 7.22 – 7.14 (m, 1H), 4.10 (s, 3H). 13C NMR (101 MHz, Acetone-d6) δ 182.4, 162.0, 161.1, 152.6 (d, J = 13.0 Hz), 150.2 (d, J = 13.2 Hz), 149.1 (d, J = 12.5 Hz), 146.7 (d, J = 12.8 Hz), 139.7, 139.1, 138.0 (dd, J = 9.2, 2.9 Hz), 130.6, 129.2, 126.3, 121.8, 120.5, 118.9 (d, J = 17.9 Hz), 117.8 (dd, J = 5.9, 3.5 Hz), 116.7, 110.9 (d, J = 22.3 Hz), 38.8. 19F NMR (377 MHz, Acetone-d6) δ −138.6 – −138.9 (m), −146.1 – −146.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C20H16F2N3O3: 384.116, found: 384.1153.
4.1.10. N-(3,4-Difluorophenyl)-1-methyl-4-(2-oxo-2-(phenylamino)acetyl)-1H-pyrrole-2-carboxamide (10h).
Yield: 99%.1H NMR (400 MHz, Acetone-d6) δ 9.59 (s, 1H), 8.52 (s, 1H), 8.23 (d, J = 1.2 Hz, 1H), 7.99 – 7.93 (m, 1H), 7.59 (d, J = 1.7 Hz, 1H), 7.56 – 7.51 (m, 1H), 7.39 – 7.31 (m, 4H), 7.30 – 7.24 (m, 2H), 4.54 (s, 1H), 4.53 (s, 1H), 4.05 (s, 3H). 13C NMR (101 MHz, Acetone-d6) δ 182.7, 163.9, 161.1, 159.9, 152.6 (d, J = 13.0 Hz), 150.1 (d, J = 13.3 Hz), 149.1 (d, J = 12.7 Hz), 146.7 (d, J = 12.8 Hz), 142.8, 140.7, 139.0, 138.0 (d, J = 12.1 Hz), 130.2, 130.0, 129.4, 129.0, 128.9, 128.4, 120.9, 118.8 (d, J = 18.3 Hz), 118.3 – 117.5 (m), 116.7, 110.8 (d, J = 22.2 Hz), 45.3, 44.3, 38.8. 19F NMR (377 MHz, Acetone-d6) δ −138.7 – −138.8 (m), −146.1 – −146.4 (m). HRMS (ESI): m/z [M+H]+ calcd for C21H18F2N3O3: 398.1316, found: 398.1312.
4.1.11. 4-(2-((Cyanomethyl)amino)-2-oxoacetyl)-N-(3,4-difluorophenyl)-1-methyl-1H-pyrrole-2-carboxamide (10i).
Yield: 30%. 1H NMR (400 MHz, Acetone-d6) δ 9.43 (s, 1H), 8.07 (s, 1H), 8.06 – 7.71 (m, 1H), 7.54 – 7.40 (m, 1H), 7.37 – 7.12 (m, 1H), 6.93 (s, 1H), 6.88 – 6.84 (m, 1H), 3.93 (s, 3H), 3.66 – 3.43 (m, 1H), 1.99 – 1.93 (m, 3H), 1.79 – 1.68 (m, 2H), 1.67 – 1.55 (m, 2H). 13C NMR (101 MHz, Acetone-d6) δ 181.1, 164.1, 161.1, 139.0, 129.3, 120.5, 118.9 (d, J = 17.6 Hz), 118.0, 117.8 (dd, J = 6.0, 3.2 Hz), 116.6, 110.9 (d, J = 22.1 Hz), 38.8, 28.8. 19F NMR (377 MHz, Acetone-d6) δ −138.7 – −138.8 (m), −145.9 – −146.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C17H20F2N3O3S : 384.1193, found : 384.1186.
4.1.12. Ethyl 1,3,5-trimethylpyrrole-2-carboxylate (12).
Ethyl 3,5-dimethylpyrrole-2-carboxylate 11 (100.0 g, 0.59 mol) was added to a solution of potassium hydroxide (100.6 g, 1.79 mol) in DMSO (1 L) and stirred for 30 min under nitrogen at 0 °C. Methyl iodide (55.9 mL, 0.89 mol) was then added and the reaction mixture was allowed to warm up to room temperature and stirred for 4 h. The reaction was then extracted with diethyl ether (3 × 1 L) and the combined organic layers were finally washed with water (2 × 150 mL), dried over Na2S04 and concentrated in vacuo. The residue slowly crystallized to yield ethyl 1, 3, 5-trimethylpyrrole-2-carboxylate 12 (102.4 g, 0.56 mol, 94%) as a yellowish solid. 1H NMR (400 MHz, CDCl3) δ: 1.32 (t, 3H), 2.20 (s, 3H), 2.30 (s, 3H), 3.75 (s, 3H), 4.22 (q, 2H), 5.75 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 161.7, 136.1, 128.5, 118.8, 110.8, 59.3, 32.9, 14.8, 14.6, 12.5. MS (ESI): m/z [M+H]+ calcd for C9H14NO2: 182.2, found: 182.3.
4.1.13. 1,3,5-Trimethyl-1H-pyrrole-2-carboxylic acid (13).
To a solution of 11 (3 g, 72 mmol) in EtOH (100 mL) was added NaOH 20% (70 mL). The reaction was heated at 100 °C for 6 h. EtOH was evaporated under vacuum and the mixture was washed with DCM (3 × 30 mL). The aqueous layer was carefully acidified to pH 3–4 with 1M HCl. The mixture was extracted with DCM (3 × 30 mL). Combined organic layers were dried over MgSO4 and concentrated in vacuo. The resulting solid was washed with cold Et2O to afford 13 in 61% yield (6.7 g) as a pink solid. 1H NMR (400 MHz, DMSO-d6) δ 11.88 (s, 1H), 5.75 (s, 1H), 3.68 (s, 3H), 2.19 (s, 3H), 2.15 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.2, 135.6, 128.4, 119.3, 110.6, 32.9, 14.6, 12.6. HRMS (ESI): m/z [M+H]+ calcd for C8H12NO2: 154.0868, found: 154.0860.
4.1.14. N-(3,4-Difluorophenyl)-1,3,5-trimethyl-1H-pyrrole-2-carboxamide (14).
To a solution of 13 (2.9 g, 18.9 mmol) in DMF (20 mL) were added 3, 4-difluoroaniline (4.5 mL, 22.7 mmol), HATU (8.6 g, 22.7 mmol) and DIPEA (6.6 mL, 37.8 mmol) at 0 °C. The mixture was heated at 60 °C for 2 days. The reaction mixture was then diluted with EtOAc and washed with 1 M HCl, water and brine. The organic layers was dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography using hexanes/EtOAc (8:2) to afford 14 in 37% yield (1.85 g). 1H NMR (400 MHz, CDCl3) δ 7.76 – 7.65 (m, 1H), 7.19 – 7.07 (m, 2H), 5.80 (s, 1H), 3.77 (s, 3H), 2.38 (s, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.2, 149.4 (dd, J = 243.5 Hz, J = 13.1 Hz), 145.5 (dd, J = 241.6 Hz, J = 12.9 Hz), 137.0 (dd, J = 9.3 Hz, J = 2.7 Hz), 133.6, 124.6, 122.2, 117.7 (d, J = 17.8 Hz), 116.0 (dd, J = 5.7 Hz, 3.4 Hz), 109.5, 108.7 (d, J = 21.8 Hz), 32.0, 13.1, 12.3.MS (ESI): m/z [M+H]+ calcd for C14H14F2N2O: 264.1, found: 265.5.
4.1.15. 2-(5-((3,4-Difluorophenyl)carbamoyl)-1,2,4-trimethyl-1H-pyrrol-3-yl)-2-oxoacetic acid (16).
To a solution of 14 (860 mg, 3.26 mmol) in DCM (30 mL) were added ethyl oxalylchloride (980 μL, 8.80 mmol) and AlCl3 (1.08 g, 8.15 mmol) at 0 °C. The mixture was stirred at room temperature for 16 h and poured into crushed ice. The mixture was extracted with DCM and combined organic layers were filtered on Celite. The filtrate was concentrated and the resulting residue was used in the next step without further purification. To a solution of crude 15 in EtOH was added NaOH 10 % (25 mL). The mixture was stirred for 1 h at room temperature. EtOH was evaporated under vacuum and the mixture was extracted with EtOAc (3 × 10 mL). The aqueous layer was acidified with 1M HCl. The mixture was extracted with EtOAc (3 × 10 mL). Combined organic layers were dried over MgSO4 and concentrated in vacuo. The resulting solid was washed with Et2O to afford 16 in 59% yield (646 mg) over two steps. 1H NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 10.46 (s, 1H), 8.04 – 7.71 (m, 1H), 7.59 – 7.28 (m, 2H), 3.60 (s, 3H), 2.46 (s, 3H), 2.26 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 185.5, 168.5, 160.4, 149.4 (dd, J = 244.3 Hz, J = 13.2 Hz), 146.0 (dd, J = 242.9 Hz, J = 12.6 Hz), 141.5, 136.3 (dd, J = 8.9 Hz, J = 2.8 Hz), 127.5, 122.3, 117.9 (d, J = 17.8 Hz), 116.5 (dd, J = 5.8 Hz, 3.4 Hz), 115.4, 109.1 (d, J = 21.7 Hz), 32.4, 11.6, 11.5. MS (ESI): m/z [M+H]+ calcd for C16H15F2N2O4: 337.1, found: 337.5.
4.1.16. General procedure for the synthesis of compounds 17a-f
To a solution of 16 (40 mg, 0.119 mmol) in a DMF/DCM mixture (2 mL, 1:1) was added CDI (29 mg, 0.178 mmol) at room temperature. After 15 min, the amine (0.178 mmol) was added and the mixture was stirred for 1 h. The reaction mixture was diluted with EtOAc and washed with H2O (3 × 5 mL). The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography using DCM/MeOH (98:2) to afford compounds 17a-f.
4.1.17. N-(3,4-Difluorophenyl)-1,3,5-trimethyl-4-(2-oxo-2-(pyridin-2-ylamino)acetyl)-1H-pyrrole-2-carboxamide (17a).
Yield: 69%. 1H NMR (400 MHz, CDCl3) δ 9.33 (s, 1H), 8.45 – 8.38 (m, 1H), 8.30 (d, J = 8.3 Hz, 1H), 7.85 – 7.67 (m, 2H), 7.49 (s, 1H), 7.22 – 7.12 (m, 3H), 3.75 (s, 3H), 2.45 (s, 3H), 2.44 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 185.8, 161.4, 160.0, 150.4, 148.5, 142.3, 138.5, 126.0, 123.4, 120.7, 117.5 (t, J = 18.2 Hz), 115.3, 114.1, 109.7 (d, J = 22.2 Hz), 32.5, 12.7, 12.4. 19F NMR (377 MHz, DMSO-d6) δ −136.6 (m), −143.4 (m). HRMS (ESI): m/z [M+H] + calcd for C21H19F2N4O3: 413.1425, found: 413.1416.
4.1.18. N-(3,4-Difluorophenyl)-1,3,5-trimethyl-4-(2-(4-methylpiperazin-1-yl)-2 - oxoacetyl)-1H-pyrrole-2-carboxamide (17b).
Yield: 74%. 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.80 – 7.69 (m, 1H), 7.43 – 7.32 (m, 1H), 7.13 (dt, J = 10.0, 8.8 Hz, 1H), 3.72 – 3.67 (m, 2H), 3.66 (s, 3H), 3.32 (dd, J = 5.9, 4.1 Hz, 2H), 2.46 (t, J = 5.2 Hz, 2H), 2.42 (s, 3H), 2.35 (t, J = 5.1 Hz, 2H), 2.31 (s, 3H), 2.30 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 187.3, 167.2, 160.4, 151.2 (d, J = 13.1Hz), 148.8(d, J =13.1Hz), 148.2(d, J =12.1Hz), 145.7 (d, J = 13.1 Hz), 141.6, 134.8(t, J = 6.1 Hz), 127.2, 122.9, 117.1(d, J = 18.2 Hz), 116.5, 115.6(t, J = 4.0 Hz), 109.6(d, J = 21.2 Hz), 54.4(d, J = 50.5 Hz), 45.9 (d, J = 24.2 Hz), 41.0, 32.2, 11.8, 11.6. 19F NMR (377 MHz, DMSO-d6) δ −137.3– −137.4 (m), −144.0 – −144.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C21H25F2N4O3: 419.1895, found: 419.1886.
4.1.19. 4-(2-(Diethylamino)-2-oxoacetyl)-N-(3,4-difluorophenyl)-1,3,5-trimethyl-1H-pyrrole-2-carboxamide (17c).
Yield: 71%. 1H NMR (400 MHz, CDCl3) δ 9.07 (s, 1H), 7.87 – 7.73 (m, 1H), 7.45 – 7.36 (m, 1H), 7.13 (dt, J = 10.0, 8.8 Hz, 1H), 3.66 (s, 3H), 3.48 (q, J = 7.1 Hz, 2H), 3.22 (q, J = 7.0 Hz, 2H), 2.42 (s, 3H), 2.31 (s, 3H), 1.21 (t, J = 7.1 Hz, 3H), 1.14 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 187.7, 168.6, 160.5, 151.2 (d, J =13.1 Hz), 148.8 (d, J =13.1 Hz), 148.1 (d, J =13.1 Hz), 145.7 (d, J =13.1 Hz), 141.5, 134.9 (d, J =11.1 Hz), 127.1, 123.0, 117.2, 117.0, 116.5, 115.7(dd, J =5.1, 3.0 Hz), 109.8, 109.6, 42.1, 38.7, 32.2, 13.7, 12.3, 11.8, 11.6. 19F NMR (377 MHz, CDCl3) δ −137.4– −137.5 (m), −144.2– −144.3 (m). HRMS (ESI): m/z [M+H]+ calcd for C20H24F2N3O3: 392.1786, found: 392.1776.
4.1.20. 4-(2-((1H-Benzo[d]imidazol-2-yl)amino)-2-oxoacetyl)-N-(3,4-difluorophenyl)-1, 3, 5-trimethyl-1H-pyrrole-2-carboxamide (17d).
Yield: 46%. 1H NMR (400 MHz, DMSO-d6) δ 12.42 (s, 2H), 10.45 (s, 1H), 8.03 – 7.76 (m, 1H), 7.56 – 7.38 (m, 4H), 7.18 (dd, J = 5.9, 3.2 Hz, 2H), 3.61 (s, 3H), 2.45 (s, 3H), 2.27 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.5, 150.6 (d, J =13.1 Hz), 148.2 (d, J = 13.1Hz), 147.1, 144.8 (d, J =13.1 Hz), 141.2, 136.4, 127.3, 122.6(d, J =16.2 Hz), 118.0(d, J =18.2 Hz), 116.4, 109.1(d, J =22.2 Hz), 32.4, 11.8. 19F NMR (377 MHz, DMSO-d6) δ −138.5– −138.6 (m), −145.6 – −145.7 (m). HRMS (ESI): m/z [M+H] + calcd for C23H20F2N5O3: 452.1534, found: 452.1525.
4.1.21. N-(3,4-Difluorophenyl)-1,3,5-trimethyl-4-(2-oxo-2-((pyridin-2-ylmethyl)amino) acetyl)-1H-pyrrole-2-carboxamide (17e).
Yield: 55%. 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 9.28 (t, J = 6.1 Hz, 1H), 8.53 (d, J = 4.8 Hz, 1H), 7.94 – 7.77 (m, 2H), 7.46 – 7.41 (m, 2H), 7.38 (d, J = 8.0 Hz, 1H), 7.34 – 7.26 (m, 1H), 4.50 (d, J = 5.9 Hz, 2H), 3.59 (s, 3H), 2.38 (s, 3H), 2.21 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 188.2, 171.0, 167.9, 160.6, 158.1, 149.4, 146.3, 141.1, 137.2, 127.2, 122.8, 121.9, 120.1, 118.0, 117.9, 116.8, 116.4 (d, J = 4.0 Hz), 109.2, 109.0, 44.3, 32.4, 11.9, 11.8. 19F NMR (377 MHz, DMSO-d6) δ −138.5– −138.6 (m), −145.7 – −145.7 (m). HRMS (ESI): m/z [M+H] + calcd for C22H21F2N4O3: 427.1582, found: 427.1572.
4.1.22. N-(3,4-Difluorophenyl)-1,3,5-trimethyl-4-(2-(((1-methyl-1H-imidazol-2-yl) methyl)amino)-2-oxoacetyl)-1H-pyrrole-2-carboxamide (17f).
Yield: 63%. 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 2H), 7.77 – 7.66 (m, 1H), 7.26 – 7.20 (m, 1H), 7.13 (dt, J = 9.9, 8.7 Hz, 1H), 6.93 (d, J = 1.3 Hz, 1H), 6.85 (d, J = 1.3 Hz, 1H), 4.53 (d, J = 5.7 Hz, 2H), 3.72 (s, 3H), 3.64 (s, 3H), 2.30 (s, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 186.6, 165.3, 160.2, 151.4, 148.9(d, J = 13.1 Hz), 148.3, 145.8(d, J = 12.1 Hz), 143.9, 141.8, 134.5 (dd, J = 9.1, 3.0 Hz), 127.5, 126.2, 123.5, 121.7, 117.6, 117.4, 117.2, 115.6(dd, J = 6.1, 4.0 Hz), 109.7(d, J = 22.2 Hz), 35.1, 33.0, 32.3, 12.1, 11.9. 19F NMR (377 MHz, CDCl3) δ −136.9– −137.0(m), −143.7 – −143.8(m). HRMS (ESI): m/z [M+H]+ calcd for C21H22F2N5O3: 430.1691, found: 430.1681.
4.1.23. 1,3,5-trimethyl-4-(2-oxo-2-(prop-2-yn-1-ylamino)acetyl)-1H-pyrrole-2-carboxylic acid (21a).
To a solution of ethyl 1, 3, 5-trimethylpyrrole-2-carboxylate 12 (10.0 g, 55.2 mmol) in CH2Cl2 (250 mL) at 0°C was added dropwise a solution of ethyl 2-chloro-2-oxo-acetate (9.3 mL, 82.8 mmol) in CH2Cl2 (100 mL) followed by AlCl3 (22.1 g, 165.7 mmol) portion wise. The reaction mixture was then stirred overnight at room temperature and then quenched with ice. After addition of water (300 mL), the mixture was filtered on celite and extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were washed with a saturated solution of sodium carbonate (250 mL) and a saturated solution of NH4Cl (250 mL), dried over Na2SO4 and concentrated in vacuo. To the resulting oil were added MeOH (100 mL) and a 5% solution of NaOH (100 mL) and the mixture was stirred for 15 min at room temperature. After removal of the volatiles under vacuum, the mixture was washed with EtOAc (2 × 100 mL), acidified with a 1N HCl solution (pH = 1) and extracted with EtOAc (3 × 100 mL). The combined organic layers were dried over Na2SO4 and concentrated under vacuum. The resulting solid was washed with diethyl ether (100 mL) and hexanes (100 mL) to yield 2-(5-ethoxycarbonyl-1,3,5-trimethyl-pyrrol-3-yl)-2-oxo-acetic acid 19 (8.1 g, 32.0 mmol, 58%) as an off-white powder. To a solution of 2-(5-ethoxycarbonyl-1, 3, 5-trimethyl-pyrrol-3-yl)-2-oxo-acetic acid 19 (2.0 g, 7.9 mmol) in DMF (15 mL) and CH2Cl2 (10 mL) was added CDI (1.92 g, 11.8 mmol) and propargylamine (0.607 mL, 9.5 mmol). After being stirred for 2 h at room temperature, the reaction mixture was poured into a saturated solution of NH4Cl and extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to give 20a as a yellowish oil. To the crude ethyl 4-[2-(propargylamino)-2-oxo-acetyl]-1, 3, 5-trimethyl-pyrrole-2-carboxylate 20a dissolved in MeOH (10 mL) and THF (10 mL) was added a 5% solution of NaOH (10 mL). The reaction mixture was stirred at room temperature overnight and after evaporation of the volatiles in vacuo, the aqueous solution was washed with EtOAc (2 × 50 mL), acidified with a 1N HCl solution (pH = 1) and extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The resulting solid was washed with diethyl ether (50 mL) and hexanes (50 mL) to yield 4-[2-(propargylamino)-2-oxo-acetyl]-1,3,5-trimethyl-pyrrol-2-carboxylic acid 21a (1.8 g, 6.9 mmol, 87%) as a white powder. 1H NMR (400 MHz, DMSO-d6) δ 12.77 (s, 1H), 9.14 (t, J = 5.7 Hz, 1H), 3.99 (dd, J = 5.7, 2.6 Hz, 2H), 3.75 (s, 3H), 3.18 (t, J = 2.4 Hz, 1H), 2.37 (s, 6H). 13C NMR (101 MHz, DMSO) δ 188.0, 167.2, 163.0, 142.8, 129.7, 121.8, 117.5, 80.5, 73.8, 33.3, 28.1, 12.3, 12.0. HRMS (ESI): m/z [M+H]+ calcd for C13H15N2O4: 263.1032, found: 263.1025.
4.1.24. 4-[(Propargylamino)(oxo)acetyl]-N-(3,4-difluorophenyl)-1,3,5-trimethyl-1H-pyrrole-2-carboxamide (5).
To a solution of 4-[2-(propargylamino)-2-oxo-acetyl]-1,3,5-trimethyl-pyrrol-2-carboxylic acid 21a (750 mg, 2.7 mmol), 3,4-difluoroaniline (410 mg, 3.2 mmol) and DIPEA (746 μL, 4.3 mmol) in DMF (15 mL) was added HATU (1.63 g, 4.3 mmol) at room temperature. The mixture was stirred at 50 °C for 3 h. In order to reach completion, more 3, 4-difluoroaniline (410 mg, 3.2 mmol) was added and the mixture was further stirred overnight at 65 °C. The reaction mixture was then poured into a saturated solution of NH4Cl and extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (Hexanes:EtOAc = 6:4 v/v) to give compound 5 as a white powder (47%, 503 mg, 1.4 mmol). 1H NMR (400 MHz, Acetone-d6) δ 9.49 (s, 1H), 8.22 – 8.07 (m, 1H), 8.07 – 7.95 (m, 1H), 7.59 – 7.43 (m, 1H), 7.33 (q, J = 9.4 Hz, 1H), 4.21 – 4.07 (m, 2H), 3.69 (s, 3H), 2.73 (s, 1H), 2.43 (s, 3H), 2.29 (s, 3H). 13C NMR (101 MHz, Acetone) δ 188.9, 167.9, 162.1, 152.7, 152.6, 150.3, 150.2, 149.3, 149.2, 146.9, 146.8, 142.8, 137.9, 137.9, 128.5, 124.6, 119.1, 118.9, 118.8, 117.5, 117.5, 117.4, 117.4, 110.7, 110.5, 81.4, 73.2, 33.2, 29.7, 12.8, 12.7. 19F NMR (377 MHz, Acetone-d6) δ −139.8 – −140.0 (m), −147.1 – −147.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C19H18F2N3O3: 374.1316, found: 374.1309.
4.1.25. 1, 3, 5-Trimethyl-4-(2-oxo-2-(thiazol-2-ylamino) acetyl)-1H-pyrrole-2-carboxylic acid (21b).
To a solution of 2-(5-ethoxycarbonyl-1,3,5-trimethyl-pyrrol-3-yl)-2-oxo-acetic acid 19 (2.0 g, 7.9 mmol) in DMF (15 mL) and CH2Cl2 (10 mL) was added CDI (1.92 g, 11.8 mmol) and 2-aminothiazole (0.95 g, 9.5 mmol). After stirring for 2 h at room temperature, the reaction mixture was poured into a saturated solution of NH4Cl, filtered through a fritted funnel and finally dried under vacuum to give 20b as a yellow solid. To the crude ethyl 1,3,5-trimethyl-4-(2-oxo-2-(thiazol-2-ylamino)acetyl)-1H-pyrrole-2-carboxylate 20b dissolved in MeOH (10 mL) and THF (10 mL) was added a 5% solution of NaOH (10 mL). The reaction mixture was stirred at room temperature overnight and after evaporation of the volatiles in vacuo, the aqueous solution was washed with EtOAc (2 × 50 mL), acidified with a 1 N HCl solution (pH = 1) and extracted again with EtOAc (3 × 50 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The resulting solid was washed with diethyl ether (50 mL) and hexanes (50 mL) to yield 1,3,5-trimethyl-4-(2-oxo-2-(thiazol-2-ylamino)acetyl)-1H-pyrrole-2-carboxylic acid 21b (1.9 g, 6.1 mmol, 78%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H), 7.58 (s, 1H), 7.38 (s, 1H), 3.77 (s, 3H), 2.37 (s, 3H), 2.33 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 185.7, 165.8, 163.0, 157.5, 143.4, 138.4, 129.5, 122.3, 117.1, 115.3, 33.5, 12.1, 12.0. MS (ESI): m/z [M+H]+ calcd for C13H14N3O4: 308.3, found: 308.4.
4.1.26. N-(3,4-Difluorophenyl)-1,3,5-trimethyl-4-(2-oxo-2-(thiazol-2-ylamino)acetyl)-1H-pyrrole-2-carboxamide (22).
To a solution of 1,3,5-trimethyl-4-(2-oxo-2-(thiazol-2-ylamino)acetyl)-1H-pyrrole-2-carboxylic acid 21b (250 mg, 0.8 mmol), 3,4-difluoroaniline (158 mg, 1.2 mmol) and DIPEA (283 μL, 1.6 mmol) in DMF (15 mL) was added HATU (0.37 g, 1.0 mmol) at room temperature. The mixture was stirred at 50 °C for 3 h. In order to reach completion, more 3, 4-difluoroaniline (80 mg, 0.6 mmol) was added and the mixture was further stirred overnight at 65 °C. The reaction mixture was then poured into a saturated solution of NH4Cl and extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (DCM: MeOH = 98:2 v/v) to give compound 22 as a yellowish powder (55%, 187 mg, 0.4 mmol). 1H NMR (400 MHz, DMSO-d6) δ 13.01 (s, 1H), 10.47 (s, 1H), 7.88 (dd, J = 13.3, 7.5 Hz, 1H), 7.58 (d, J = 3.6 Hz, 1H), 7.54 – 7.35 (m, 3H), 3.62 (s, 3H), 2.41 (s, 3H), 2.19 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 185.5, 166.0, 160.4, 157.5, 150.6 (d, J = 13.4 Hz), 148.2 (d, J = 13.2 Hz), 147.2 (d, J = 12.7 Hz), 144.8 (d, J = 12.5 Hz), 141.9, 138.5, 136.3 (dd, J = 8.9, 2.4 Hz), 127.6, 122.6, 117.9 (d, J = 17.9 Hz), 116.5, 116.3, 115.3, 109.2 (d, J = 21.7 Hz). 32.5, 11.8, 11.7. 19F NMR (377 MHz, DMSO-d6) δ −137.2 (m), −144.2 (dd, J = 15.1, 7.5 Hz, 1H). HRMS (ESI): m/z [M+H]+ calcd for C19H17F2N4O3S: 419.0989, found: 419.0984.
4.2. HBV Assay in HepAd38.
The HBV 7-day assay was performed in HepAD38 wild type (HepAD38) cells as previously described.14,13 Briefly, HepAD38 cells were seeded onto 96-well plates and incubated for two days at 37°C in a humidified 5% CO2 atmosphere. On day two, medium was removed and cells were washed with 1X phosphate buffer saline (PBS). Forty mM stock solutions of the compounds were prepapared for the assay. The desired aliquot of the solution was diluted in medium without tetracycline and added in duplicate at various concentrations to the wells. On day seven, total DNA was extracted using DNeasy 96 Tissue kit (Qiagen), and HBV DNA was amplified by RT-PCR.14 Antiviral activity was measured by determining the average threshold cycle for the HBV amplification of the compounds (alone or in combination), which was subtracted from the average cycle of the untreated-tetracycline control (ΔCT). Drugs were first tested individually for effective concentration, which inhibited 50% and 90% of HBV DNA replication (EC50 and EC90) using CalcuSyn software program (Biosoft, Cambridge, UK).
4.3. Cytotoxicity assays.
In vitro cytotoxicity was determined using the CellTiter 96 non-radioactive cell proliferation colorimetric assay (MTT assay, Promega) in primary human peripheral blood mononuclear cells (PBMC), human T lymphoblast (CEM) and human hepatocellular carcinoma (HepG2) cell lines. Toxicity levels were measured as the concentration of test compound that inhibited cell proliferation by 50% (CC50).
Acknowledgements
This work was supported by NIH Grant 1-R01-AI-132833, and in part by 5P30-AI-50409 (CFAR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
Drs. Schinazi, Amblard and Bassit along with Emory University are entitled to equity and royalties related to products licensed to Aligos Therapeutics, Inc. being further evaluated in the research described in this paper. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict of interest policies.
References
- 1.World Health Organization (WHO) website, available at: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b. Accessed 02 June 2020.
- 2.Nijampatnam B; Liotta DC Recent advances in the development of HBV capsid assembly modulators. Curr. Opin. Chem. Biol 2019; 50, 73–79 [DOI] [PubMed] [Google Scholar]
- 3.Berke JM; Dehertogh P; Vergauwen K; Mostmans W; Vandyck K; Raboisson P; Pauwels F Antiviral properties and mechanism of action studies of the hepatitis B virus capsid assembly modulator JNJ-56136379 Antimicrob. Agents Chemother 2020, 64, e02439–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuen MF; Agarwal K; Gane EJ; Schwabe C; Ahn SH; Kim DJ; Lim Y-S; Cheng W; Sievert W; Visvanathan K; Ruby E; Liaw S; Yan R; Huang Q; Colonno R; Lopatin U Safety, pharmacokinetics, and antiviral effects of ABI-H0731, a hepatitis B virus core inhibitor: a randomised, placebo-controlled phase 1 trial. Lancet Gastroenterol. Hepatol 2020, 5, 152–166. [DOI] [PubMed] [Google Scholar]
- 5.Yuen MF; Agarwal K; Gane EJ; Nguyen TT; Hassanein TI; Kim DJ; Alves K; Zayed H; Qiang D; Ruby E; Evanchik M; Huang Q; Knox SJ; Colonno R The second-generation hepatitis B virus (HBV) core inhibitor (CI) ABI-H2158 is associated with potent antiviral activity in a 14-Day monotherapy study in HBeAg-positive patients with chronic hepatitis B (CHB) AASLD 2019. November 8–12 Boston [Google Scholar]
- 6. https://www.hepb.org/treatment-and-management/drug-watch/
- 7.Amblard F, Boucle S; Bassit L; Cox B; Sari O; Tao S; Chen Z; Ozturk T; Verma K; Ollinger Russell O; Rat V; de Rocquigny H; Fiquet O; Boussand M; Di Santo J; Strick-Marchand H; Schinazi RF Novel hepatitis B virus capsid assembly modulator induces potent antiviral responses in vitro and humanized mice, Antimicrob. Agents Chemother, 2020, 64, e01701–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sari O; Boucle S; Cox BD; Ozturk T; Ollinger Russell O; Bassit L; Amblard F; Schinazi RF; Synthesis of sulfamoylbenzamide derivatives as HBV capsid assembly effector, Eur. J. Med. Chem, 2017, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Z, Hu T, Zhou X, Wildum S, Garcia-Alcalde F, Xu Z, Wu D, Mao Y, Tian X, Zhou Y, Shen F, Zhang Z, Tang G, Najera I, Yang G, Shen HC, Young JA, Qin N Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Sci. Rep 2017, 7, 42374–42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.BIOvIA DS (2015). Discovery studio modeling environment. San Diego, Dassault Systemes, Release, 4. [Google Scholar]
- 11.Stuyver LJ; Lostia S; Adams M; Mathew JS; Pai BS; Grier J; Tharnish PM; Choi Y; Chong Y; Choo H; Chu CK; Otto MJ; Schinazi RF Antiviral activities and cellular toxicities of modified 2′,3′-dideoxy-2′,3′-didehydrocytidine analogues. Antimicrob. Agents Chemother 2002, 46, 3854–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu G; Liu B; Zhang Y; Li J; Arzumanyan A; Clayton MM; Schinazi RF, Wang Z; Goldmann S; Ren Q; Zhang F; Feitelson MA Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly. Antimicrob. Agents Chemother, 2013, 57, 5344–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ladner SK; Otto MJ; Barker CS; Zaifert K; Wang GH; Guo JT; Seeger C, King RW Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother 1997, 41, 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pas SD; Fries E; De Man RA; Osterhaus AD; Niesters HG Development of a quantitative real-time detection assay for hepatitis B virus DNA and comparison with two commercial assays. J. Clin. Microbiol 2000, 38, 2897–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]