

Abstract
The complement system consists of a collection of serum proteins, which act as the main frontline effector arm of the innate immune system. Activation of complement can occur through the individual induction pathways: the classical, mannose-binding lectin, and alternative pathways. Activation results in opsonization, recruitment of effector cells through potent immune mediators known as anaphylatoxins, and cell lysis via the formation of the membrane attack complex (MAC). Stringent regulation of complement is required to protect against the inappropriate activation of the complement cascade. Complement activation within the tumor microenvironment does not increase anti-tumoral action, instead it enhances tumor growth and disease progression. Radiation therapy (RT) is a staple in treatment of malignancies and controls tumor growth through direct DNA damage and the influx of immune cells, reshaping the makeup of the TME. The relationship between RT and complement activity in the TME is uncertain at best. The following review will focus on the complex interaction of complement activation and the immune modulating effects of RT, and the overall effect on tumor progression. The clinical implications of complement activation in cancer, and the use of therapeutics and potential biomarkers will also be covered.
Introduction
The complement system is a large collection of proteins which function as an arm of the innate immune system and first line of defense against invading microorganisms. It is made up of a tightly regulated network of plasma proteins, which function in triggering opsonization and phagocytosis, cell lysis, and inducing an effective immune response to assist in fighting invading pathogens [1][2]. Activation of complement leads to a cascade of enzymatic reactions, forming highly efficient immune mediators known as anaphylatoxins and ultimately culminates in the formation of a protein complex known as the membrane attack complex (MAC).
The complement system, and its role in mitigating infections caused by invasive pathogens is an extensively studied topic. The role of complement in cancer and its implications in tumor progression, on the other hand, is extraordinarily complex and the current understanding is still far from complete [3][4]. Due to its essential role in inflammation, it was originally suspected to have an equivalent role in the immune response against cancer [3]. Newfound knowledge, however, has debunked this thinking as more was discovered about the complex interplay during tumor pathogenesis, and increasing amount of evidence points to complement activation promoting tumor growth, rather than suppressing it [2][4]. Complement activation in tumors has been implicated in the influx of immunosuppressive cells, decreased production of inflammatory cytokines, and responsibility for early epithelial-mesenchymal transition (EMT) and metastasis of cancer cells [4]. Complement degradation products are commonly found in many tumors, indicating local activation of the complement cascade does not lead to direct tumor cell killing [5].
The effect of radiation therapy (RT), a mainstay treatment for cancer, and its impact on the function of the complement system is an understudied concept and will be the focus of this review. Through direct DNA damage causing tumor cell death and induced immunological changes via secreted danger signals and cytokines, RT reshapes the tumor microenvironment (TME) to allow for greater treatment efficacy and tumor control [6][7]. The intention of this review is not to act as an all-encompassing, comprehensive review of radiation induced immunogenicity, nor the multifaceted role complement plays in tumorigenesis. Rather, the focus is to examine the intersection of radiation and its effect on complement within the TME, as this is a topic that is largely understudied and the few studies that have explored RT’s effect on complement activation have provided contradictory results.
The Complement System
Complement activation can be induced through three distinct pathways: classical, alternative, and mannose binding lectin. While activated through different means, all three pathways converge at the molecule C3 and share the final steps of the pathway. An overview of complement induction pathways is summarized in Figure 1.
Figure 1.
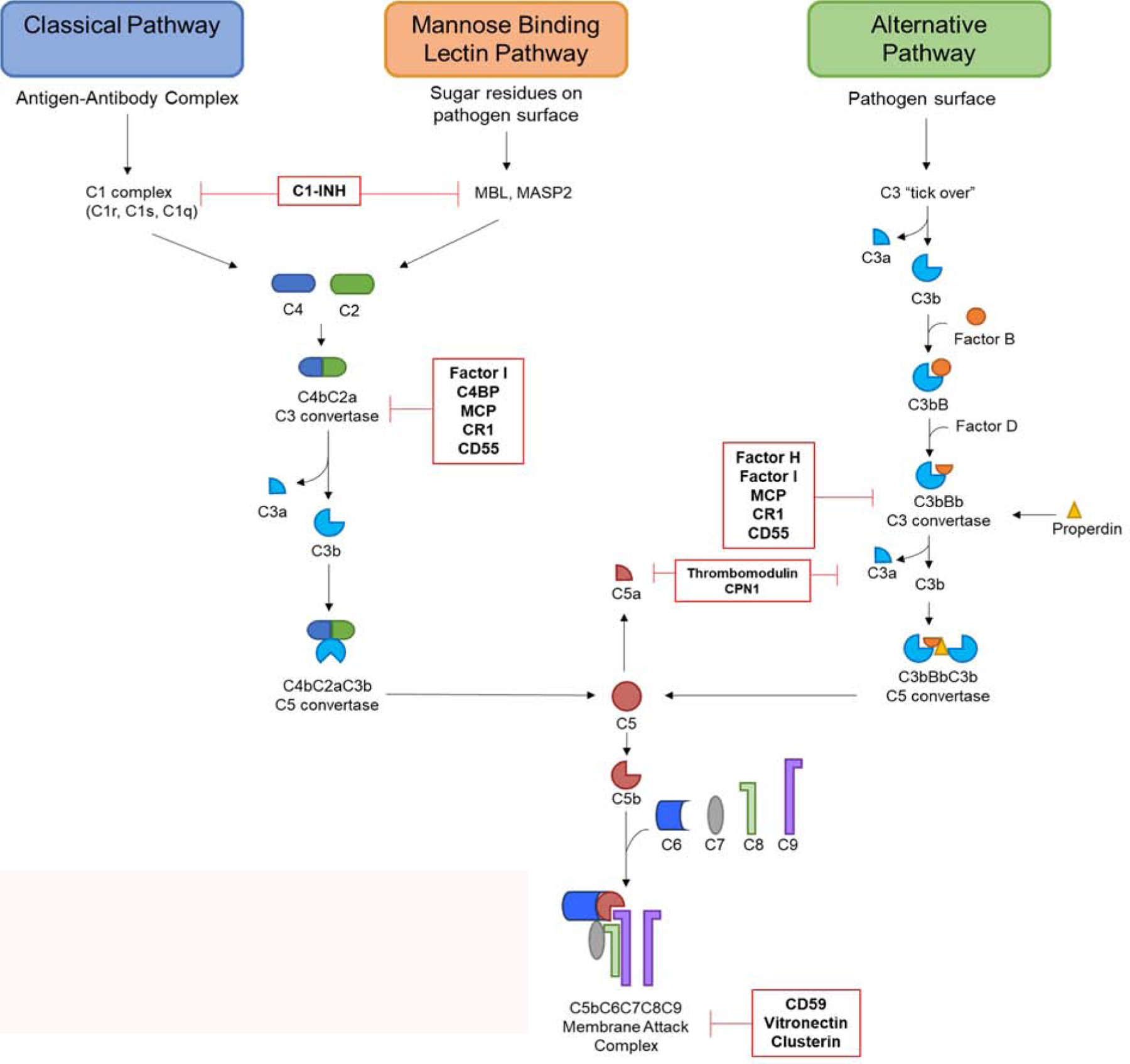
Overview of the three complement induction pathways. Terminating with the formation of the membrane attack complex. Regulatory proteins and where they act along the pathway are shown in red.
The classical pathway is activated through formation of immune complexes bound to antigens or other foreign material. The C1 complex binds the Fc portion of an IgG or IgM complex, cleaving C2 and C4 to form C3 convertase [5]. C3 convertase (C4bC2a) allows for cleavage of C3, the most abundant complement component, into biochemically active fragments C3a and C3b. C3a is an anaphylatoxin, a potent immune cell chemoattractant, while C3b deposits on the surface of cells and functions as an opsonizing agent [1][8]. C3b along with its degradation products, iC3b, C3dg, C3d, as well as C4b, plays an important effector role in the clearance of foreign bodies by interaction with complement receptors (CR) on immune populations, notably macrophages and neutrophils [8][9][10]. The mannose binding lectin pathway of complement begins when mannose binding lectin (MBL) molecule binds sugar residues on cell surfaces. MBL molecules complex with mannose associated serine proteases (MASPS) leading to autoactivation and cleavage of C2 and C4 and construction of C3 convertase [11]. The alternative pathway is activated by spontaneous hydrolysis of C3 in the blood which happens at consistent low levels, commonly referred to as “tick over”. C3b binding to pathogen or foreign bodies recruits factor B, which is further cleaved by factor D, to form the alternative pathway C3 convertase (C3bBb). Binding of the protein properdin, released by activated neutrophils, stabilizes the complex and resists degradation by complement regulatory proteins [12]. Due to the constant low level “tick over”, the alternative pathway can act to amplify the classical and MBL pathways to further potentiate the effects of complement activation.
It is worth noting that the activation and generation of certain, downstream components of the complement system can be create via the coagulation and fibrinolytic systems, the processes by which the body creates and subsequently breaks down clots after vascular trauma. These systems operate via a serine protease driven cascade similar to the complement system, and C3 and C5 can be cleaved into biologically active components, primarily by thrombin. Due to the crosstalk between systems, the coagulation pathway can be informally thought of as an additional complement induction pathway, though likely far more uncommon and less robust [13][14].
Although the initial steps of activation varies, all induction pathways converge on the C3 convertase step and follow the same path towards termination. Binding of C3b to the existing C3 convertases leads to formation of the C5 convertase, which cleaves C5 into the anaphylatoxin C5a and C5b. C5b binds to cell surfaces to set up the final steps of the cascade. C5b forms a complex with C6 and C7. The formation of this C5bC6C7 complex is the trigger that sets off MAC formation, C5bC6C7 complexes with C8 and polymerized C9 to form a pore in the cell membrane causing cell lysis and death [1].
Complement Regulation
The complement system is a potent effector arm of the innate immune system, one that is constantly working and surveying for pathogens in the host blood, poised to strike as soon as a threat is detected. As with any branch of the immune system, tight regulation and inhibition of complement activators is paramount to prevent inappropriate activation, which can cause a massive inflammatory response and host cell damage [15]. While control can be enacted at any step in the pathway, regulation of C3 generation is the most central control point due to C3 being the most critical step in all complement pathways. Regulator molecules are present in the plasma at higher concentrations than that of activator molecules, rendering inappropriate activation by non-pathogenic foreign particles almost nonexistent when functioning properly [1][16][17].
Dysregulation of the complement system and its components can result in primary and secondary immune deficiencies, manifesting in autoimmune diseases such as atypical hemolytic uremic syndrome (aHUS) [18] and systemic lupus erythematosus (SLE) [19]. Similarly, deficiencies in complement components including but not limited to C5-C9, can render individuals more susceptible to bacterial infections by Neisseria meningitidis, among others [8][20]. The presence of diseases linked to complement deficiencies highlights the vital role the complement system plays in maintaining homeostasis [21].
Complement regulation can occur through numerous mechanisms, with each having specific targets along the cascade pathways (Figure 1, Table 1). Serine proteases are present in the serum which oversee targeted cleavage of C3b and C4b, leading to the creation of protein fragments iC3b, C3dg, C3c, C4c, C4d [16]. C4 binding protein binds C4b, preventing formation of C3 convertase in classical and MBL pathways [22]. Cleavage of C3b and C4b into fragments can be carried out by Factor I with the assistance of the cofactors complement receptor 1 (CR1), membrane cofactor protein (MCP), and Factor H. CR1 and MCP are both membrane bound cofactors which act synergistically with Factor I to promote targeted cleavage of C3b and C4b into inactive fragments [23][24][25][26]. Factor H is a plasma glycoprotein which binds C3b located on host cell surfaces and triggers cleavage and inactivation of C3b [23]. C1 inhibitor (C1-INH), a plasma protease inhibitor, regulates the activation of the classical and MBL pathways through competitive inhibition by permanently inactivating C1r and C1s as well as MASP1 and MASP2 of the MBL pathway [27]. CD55, also referred to as decay-accelerating factor, prevents formation of C3 convertase by interacting with membrane bound C4b and C3b, negating the necessary polymerization [28]. Carboxypeptidase N (CPN1) is a plasma protein that degrades and inactivates C3a and C5a, preventing immunostimulatory action of anaphylatoxins [29]. Thrombomodulin increases Factor H activity and activates the degradation of C3a and C5a [30]. The C5a degradation product, C5a des-Arg, itself retains a fraction of it signaling capabilities and has a high affinity for C5aR2, also referred to as C5-like receptor 2 (C5L2) [31][32]. CD59, vitronectin, and clusterin all play roles in preventing the formation of the MAC through binding complement proteins C5b-C9 [15][33][34][35].
Table 1.
Complement system regulators, function, and location within the system
| Regulatory Protein | Regulatory Function | Type | References |
|---|---|---|---|
| CD59 | Block C9 complex with C5bC6C7C8 to prevent MAC formation | Membrane Bound | 33 |
| Complement Receptor 1 (CR1) | Membrane bound cofactor for Factor I | Membrane Bound | 24 |
| CD55 | Prevents C3 convertase formation by interaction with C3b and C4b | Membrane Bound | 28 |
| Membrane Cofactor Protein (MCP) | Membrane bound cofactor for Factor I | Membrane Bound | 25 |
| Thrombomodulin | Enchances Factor H. C3a C5a degredation | Membrane Bound | 30 |
| C1 Inhibitor (C1-INH) | Prevents induction of classical and MBL pathways by inactivating C1r and C1s, MASP’s | Plasma | 27 |
| C4 Binding Protein (C4BP) | Binds C4b. Prevents formation of C3 convertase in MBL and Classical Pathways | Plasma | 22 |
| Clusterin | Binds C5bC6C7 and inhibits binding of C9 | Plasma | 34 |
| Factor H | Binds C3b on cell surface and triggers degredatation, preventing AP activation | Plasma | 23 |
| Factor I | Proteolytic cleavage of C3b and C4b | Plasma | 26 |
| CPN1 | Degrades C3a and C5a | Plasma | 29 |
| Vitronectin | Binds C5bC6C7 and blocks C9 polymerization | Plasma | 35 |
Complement and Cancer
Due to its capacity to modulate activation of the innate and adaptive immune systems, complement was originally viewed with promise as promoting tumor cell death through complement mediated lysis via the MAC, as well as the recruitment of immune effectors through anaphylatoxin signaling and opsonization of tumor cells [36]. This, however, does not appear to be the case, as evidence now points to complement acting as a tumor promotor in most situations [37]. This leads to a series of complicated and confusing interactions that are only beginning to be understood, the full breadth of which is far too encompassing for this review.
Besides its pivotal role in modulating the immune response through signaling interactions with immune cells, anaphylatoxins have been linked to promoting tumor progression. C3a receptor (C3aR) and C5a receptor 1 (C5aR1) are G-protein coupled receptors and are widely expressed on many cell types, including myeloid cells, lymphocytes, hematopoietic cells, epithelial cells, and cancer cells [36], and the binding of these anaphylatoxins to receptor molecules induces a host of immunological effects. Unlike C5aR1, the previously mentioned C5aR2 is not a G-protein coupled receptor, and is much less characterized than its counterpart, though it appears neutrophils and immature dendritic cells (DC) express high levels of C5aR2. The overall functionality of C5aR2 and its relation to cancer is still undetermined [31][32]. C3a and C5a signaling is responsible for activation and release of histamine and cytokines from mast cells, basophils, and eosinophils, increasing tissue inflammation and smooth muscle contraction [38][39][40]. The expression of C3aR and C5aR1 on T-cells indicates that anaphylatoxins are potentially capable of regulating the differentiation of T-cell sub populations. Furthermore, recent evidence suggests that intracellular complement signaling is a vital component to maintaining T cell homeostasis, survival and regulation of IFN-γ secretion, challenging the axiom that complement is strictly a serum bound effector system [41][42].
In the context of cancer, C3a and C5a binding to cognate receptors can create a pro-tumor TME [3]. For example, C5a was found to increase migration of myeloid derived suppressor cells (MDSC’s) and production of TGF- β and IL-10, which correlated in a decrease in CD8+ T cell infiltration [43]. Furthermore, IL-10 and TGF-β were responsible for increased numbers of regulatory T cells (Tregs) and Th2 T cells both of which facilitate immunosuppression within the TME [44]. C5a-C5aR signaling has also been reported to polarize tumor associated macrophages (TAMs) towards a suppressive M2 phenotype [45]. Signaling through C3aR or C5aR resulted in decreased overall numbers of natural killer (NK) cells and cytotoxic activity, a reversal of this effect could be when complement activation was blocked [46]. Activation of the C3a-C3aR axis suppresses effective tumor control by inhibiting neutrophil and CD4+ T cell responses [5][47], and studies have shown that complement supports epithelial mesenchymal transition (EMT), an important part of early metastasis, through C3a signaling [2][3]. Similarly, C5aR activation on tumor cells can trigger enhanced expression of matrix metalloproteases (MMP’s), potentiating tumor cell dissemination and metastasis [48].
Complement regulators are shown to be overexpressed on the surface of cancer cells [49][50]. If cancer cells benefit from complement activation, what benefit would there be in increasing expression of proteins that actively control its activity? A hypothesis laid forth by Afshar-Kharghan states that anaphylatoxin activity and low, sublytic levels of MAC serve to promote tumor growth [3]. Whereas higher concentrations of MAC, cause tumor cell lysis. Therefore, cancer cells benefit from anaphylatoxin production during early activation of complement cascade but inhibit formation of MAC to prevent tumor cell lysis. CD59, a regulator inhibiting MAC formation, being the most overexpressed regulatory protein on cancer cell surfaces [4] supports the hypothesis that overexpression of regulators is an attempt to stave off complement mediated lysis via the MAC.
The Impact of Radiation Therapy on Complement Activation and Anti-Tumor Immunity
Radiation therapy is an essential tool in the arsenal against many malignancies, used alone or in tandem with chemotherapies or surgery. Radiation is known to affect immune cells [51][52]. RT aims to kill tumor cells, but it is now abundantly clear that RT is often not enough to suppress tumors and that activation of the immune system is a requirement for effective tumor control [52][53]. Radiation therapy is particularly useful for stimulating immunogenically poor tumors, increasing MHC-1 presentation and inducing tumor antigens, in turn generating a more robust anti-tumor response [52][54].
Different schemes and doses of radiation will elicit different effects on immune populations, and further understanding of the influence of RT is critical for determination of an “optimal” dose. These effects are summarized in Table 2. High dose RT has been shown to stimulate infiltration of CD8+ T cells [52][55], increase antigen presentation and infiltration of macrophages [54], and increased maturation of DC’s [53][54], both important aspects of anti-tumor immunity. However, high dose radiation also induces infiltration of Tregs [52][56][57] and a polarization of macrophages towards the immunosuppressive M2 phenotype [52]. Classical fractionated RT strategy has been shown to increase antigen release and has the benefit of minimizing the toxicity to non-cancer cells. Macrophage and DC infiltration was noted to increase during fractionation, resulting in increased antigen presentation [54][58]. Repeated irradiation, however, can potentially kill the infiltrating effector lymphocytes, which are highly radiosensitive cells, before they are able to enact their anti-tumoral effects [59]. Additionally, an increase in Tregs was also seen with fractionated radiation which tend to be more radio resistant than their effector lymphocyte counterparts [52][57][60][61]. Hypofractionated RT, breaking the total radiation dose into larger chunks with fewer treatments, suppresses myeloid derived suppressor cells (MDSC) [59][62] and results in the maturation of DC’s [58], subsequently causing activation of CD8+ T cells [59]. A late infiltration of Tregs has been seen following the influx of CD8+ T cells. Therefore, it is readily apparent that much work still needs to be done to determine an optimal dosing scheme [52][53].
Table 2.
The effect of differing RT schemes on immune cell populations. DC = Dendritic cells, Treg = Regulatory T cell, Ag = Antigen, MDSC = Myeloid Derived Suppressor Cell
| Classical Fractionated | Hypofractionated | Low Dose | High Dose |
|---|---|---|---|
| Macrophage and DC Infiltration (54) Maturation of DC’s (58) Increased Ag Presentation (54) |
Suppresses MDSC’s (59, 62) Maturation of DC’s (58, 59) CD8+ T Cell Activation (59) |
M1 Macrophage Phenotype (52) | CD8+T Cell Activation (52) Maturation of DC’s (53) Macrophage and DC Infiltration (54) Increased Ag Presentation (54) |
| CD8+ T Cell Inhibition (52) Infiltration of Tregs (52, 57) |
Late Infiltration of Tregs (59) |
Infiltration of Tregs (52, 56, 57) M2 Macrophage Phenotype (52) |
|
A primary effect of radiation therapy (RT) is to induce double-stranded DNA damage either through the direct contact with ionized particles, or through the creation of reactive oxygen species (ROS), resulting in tumor cell death by apoptosis, necrosis, senescence, and mitotic catastrophe (Figure 2) [51][56][63]. A non-targeted byproduct of the cell damage caused by RT is release of damage associated molecular patterns (DAMP’s) and cytokines which prime and activate the immune system, enhancing promotion of anti-tumor immunity (Figure 2) [56][64]. Another potential secondary cellular effect in response to RT is the local production of complement components, although the exact mechanism behind the production of complement is unclear, evidence links this with cell necrosis, apoptosis, and the recognition of DAMPs [65][66]. Few studies have been conducted exploring this link between complement activation and RT.
Figure 2.

Cellular effects of radiation induced cell death. Primary effects of radiation induced cell death are caused by direct effects of cellular DNA damage. Irradiation of tumor cells also causes secondary effects through release of DAMPs and cytokines from dying cell, triggering activation of innate and adaptive immune cell populations which reshape the TME phenotype. Local production and activation of complement pathways is a suspected secondary effect of RT.
Multiple studies have shown an association between radiation therapy and complement activation. Surace et al. described how irradiation (5Gy, 20Gy) was shown to induce a local, transient activation of classical and alternative complement pathways [66]. While the majority of complement factors are produced in the liver, it is known that complement components, most notably C3, can be produced locally by a various different tissues and cell types, including tumor cells [2][66][67]. This locally produced complement results in the generation of C3a and C5a which interact with cognate receptors on tumor associated dendritic cells to activate and induce maturation. In turn, mature DC’s are responsible for the induction of anti-tumor activity by cytotoxic CD8+ T lymphocytes. Additionally, locally produced complement components will also lead to greater C3b deposition on the surface of tumor cells, potentially increasing the effects of complement, including greater opsonization and mediated lysis via the MAC [66]. These findings are particularly interesting, considering C5a has been previously implicated in regulating the migration of MDSC’s, which are important for the modulating immune suppression and inhibition of C5a signaling with a C5aR antagonist showed reduction in tumor growth comparable to that of paclitaxel, a chemotherapeutic anti-cancer drug [43][65].
In juxtaposition, separate evidence has implicated complement activation in pro tumorigenic effects, with blockade of cleaved C3 components leading to increased efficacy during radiation therapy [65]. Complement activation, it was argued, increased the clearance of apoptotic tumor cells creating an anti-inflammatory environment in which the remaining tumor cells can thrive. Treatment with CR2-Crry, an inhibitor targeting C3 cleavage fragments, modulates the immune response after RT (1.5Gy x 10), inducing an influx of neutrophils, reducing the clearance of apoptotic cells and creating more available neo-antigen, driving it towards a pro-inflammatory tumor microenvironment. This had a profound effect which increased mature DC and CD8+ T cell infiltration, resulting in reduction of tumor growth and increased overall survivability [65]. These findings are in line with the general consensus that complement activation promotes tumor progression and inhibiting components of the complement cascade drives tumor regression and fuels anti-tumor immunity [3][4].
The question at hand is why there are these two opposing effects from complement activation in response to radiation therapy, and what are the underlying mechanisms that are causing this discrepancy? The answer to this question is likely not simple, and one that is highly multifactorial. One possibility is that the observed differences are due to different approaches to the delivery of RT in each study, fractionated vs single dose.
RT administered as a single high dose lead to elevated levels of necrotic cell death and release of DAMPS. A single, high dose, of RT was argued by the authors to be more beneficial, as fractionated radiation schemes have been implicated in destroying the infiltrating T cell populations shown to be necessary for anti-tumor immunity [66]. The resulting activation of complement corresponded with maturation and activation of DC’s [66] (Figure 2), in turn leading to an increase in the numbers of tumor infiltrating CD8+ T cells [52]. Upon stimulation by RT, DC’s were shown to upregulate the expression of C3a and C5a receptors [66]. This, mixed with the local production of complement caused by RT and complement activation in the TME, are argued to be the driving factors behind the pro-inflammatory response and subsequent tumor control. When C3a, C5a interaction with their cognate receptors on DC’s was blocked RT failed to provide an anti-tumor effect and an increase in Tregs in the tumor was seen, showcasing the importance of anaphylatoxin/RT synergy in this study [66].
On the other hand, fractionated treatment generated a pro tumor environment, featuring more apoptotic cell death [65]. Only when complement activity was negated through the use of the C3 complement inhibitor CR2-Crry did the scales tip back to a robust anti-tumor response [65]. The clearance of apoptotic cells is considered to be an anti-inflammatory response that has been attributed to maintaining immunological tolerance [68] and deposition of complement components on membranes is vital to the clearance of apoptotic bodies. By controlling complement mediated clearance, apoptotic cells progress to secondary necrosis [68], releasing DAMPS and inducing the maturation of DC’s, promoting activation and anti-tumor immunity in CD8+ T cells [66]. Repeated irradiation might therefore lead to a state with more apoptotic cells [65], increasing the clearance of apoptotic bodies via complement deposition, pushing the TME towards one that is more anti-inflammatory and immunotolerogenic [68], promoting tumor growth and reducing the efficacy of RT treatment.
Taken together, these details might explain why there are such stark differences in the activation of complement after administration of RT. As previously mentioned, RT will elicit different immunological effects based on the dosing scheme employed (Table 2). Complement activation is clearly induced by RT [66][65], although the variables behind whether the activation generates a pro or anti-tumor response remains a mystery and more research is needed to uncover these details.
Clinical Implications of Complement Manipulation
Efforts have been made to use complement to augment the immune response. Many existing therapies utilize complement fixation by monoclonal antibodies (mAbs) to initiate complement dependent cytotoxicity [69][70] via the classical pathway. However, the efficiency of complement mediated tumor cell killing, particularly in solid tumors, is uncertain and inconsistent [69]. Cetuximab [71], rituximab [72], ofatumumab [73], and pertuzumab [74] are examples of complement-fixing, tumor antigen specific mAb’s currently in use. Preclinical data indicate that complement inhibition may represent an additional target for cancer therapies. A study by Ajona et al. used a combination treatment of a C5aR1 inhibitor paired with anti-PD1 therapy. This treatment was highly synergistic and effectively reduced growth and metastasis of established tumors. This effect was mediated by the increased infiltration and activity of CD8 T cells, and a decrease in MDSC’s [75]. Regardless of their promising potential as anti-cancer therapies, clinical trials targeting complement components are sparse, with only one trial, targeting C5aR1, currently active according to clinicaltrials.gov [76].
As discussed previously, complement regulatory proteins are often overexpressed on tumor cells [49][50]. The upregulation and over expression of these complement regulators on the surface of tumor cells degrades active deposited complement fragments, thus reducing the ability of mAbs to fix complement, inhibiting MAC formation and cell death via complement dependent cytotoxicity [77]. Studies conducted by Zhao et al. and Mamidi et al. showed that the efficacy of trastuzumab and/or pertuzumab was enhanced when paired with neutralizing antibodies against CD55 and CD59 [78][74]. Therefore, targeted inhibition of surface bound regulators can indeed increase the efficacy of certain mAb therapies. However, as a side effect this could lead to systemic overactivation or dysregulation, which would need to be carefully monitored [16] [77]; and while effective in reducing tumor growth and other pro-tumor effects initiated by complement, blocking complement activation does not come without risks. Wholesale inhibition of complement activation leaves the host without the capacity for opsonization, recruitment of immune populations through C3a and C5a signaling, and MAC formation, all of which can result in increased susceptibility to septic infections [79].
Dexamethasone (DEX), a corticosteroid anti-inflammatory, is commonly prescribed by physicians to treat symptoms associated with RT and chemotherapies. It is also been shown to inhibit complement activation [80]. The study by Surace et al, demonstrated that administration of DEX after RT resulted in loss of treatment efficacy, indicating a pro tumor action of DEX through the inhibition of complement activation [66]. Interestingly, previous studies linked complement inhibition to improved treatment efficacy [65]. Administration of DEX to treat symptoms associated with RT could, as a secondary effect, further improve treatment efficacy. Whether or not the effect of DEX seen by Surace et. al is purely circumstantial, caused by the tumor model and/or dosing scheme, remains to be seen. This does however suggest the potential need for alternative prescriptions to help manage the adverse effects of treatment, though admittedly more work is needed to validate any potential negative effects of DEX.
Complement components have been identified as potential prognostic biomarkers. A study by Maher et al. identified elevated levels of C3a and C4a in serum of patients with esophageal cancer. Elevated serum levels of C3a and C4a in pretreatment patients was linked to a poorer response to neoadjuvant chemo and radiation therapy [81]. Admittedly, the relationship between serum C3a and C4a levels and treatment response requires further validation in a large independent patient cohort [81], though if this relationship does indeed hold true it would provide an excellent, minimally invasive, predictor of treatment response. Another study by Bouwens et al. showed elevated MBL and decreased factor B levels have been suggested as a biomarker for glioblastoma multiforme [82]. Furthermore, high C4d deposition and serum levels correlated to shorter survival in both advanced and early stage tumors, indicating that C4d is a potential biomarker for lung cancer survival [83]. CD59 has been reported as a potential prognostic biomarker for predicting the response of esophageal cancer to radiation therapy. Elevated expression of CD59 in esophageal cancer cell lines and clinical samples was shown to correlate with a lower overall and disease-free survival among patients who received RT [84].
Complement inhibition has also been shown to synergize with common chemotherapy drugs. A study by Medler et al. demonstrated that C5aR1 inhibition, in combination with paclitaxel, reprograms macrophages towards an M1 phenotype and leads to recruitment of activated effector CD8+ T cells [85]. This effect occurred independently of IFN-γ signaling and indicating the potential of complement inhibitors to increase the efficacy of chemotherapeutic agents [85]. Contradictory to this effect, a study conducted by Li et al. showed that complement activation and deposition inside the TME increased the effectiveness of temozolomide (TMZ) and RT, alongside an inhibitor of indoleamine 2,3-dioxygenase (IDO). The increased anti-tumor effect was mediated through complement, as C3 deficient mice exhibited none of the increased treatment efficacy showed by the combination treatment [86]. This once again highlights the dualistic nature of complement activation inside the TME.
Conclusion
The relationship between complement and cancer is a confusing, and controversial subject, and activation of complement has been implicated in endowing pro and anti-tumorigenic qualities. Numerous strategies are being developed to use complement components as prognostic biomarkers, and augment complement activation to assist in control and clearance of tumors. While RT appears to induce local complement activation, the overall effect on cancer progression shows opposing effects and the details behind such are still unclear. RT has been a staple of cancer treatment for decades and will continue to be for years to come due to its ability to stimulate immunogenically poor tumors. Separate studies have shown that complement activation can either enhance or negate treatment efficacy, highlighting the dire need for additional research in this field. Understanding more about the complex interactions between, complement, cancer, and RT will add another arrow in the quiver of combined immunotherapies against these deadly malignancies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sarma JV, Ward PA. The complement system. Cell and tissue research. 2011;343(1):227–235. doi: 10.1007/s00441-010-1034-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roumenina LT, Daugan MV, Petitprez F, Sautès-Fridman C, Fridman WH. Context-dependent roles of complement in cancer. Nature Reviews Cancer. 2019;19(12):698–715. doi: 10.1038/s41568-019-0210-0 [DOI] [PubMed] [Google Scholar]
- 3.Afshar-Kharghan V The role of the complement system in cancer. The Journal of clinical investigation. 2017;127(3):780–789. doi: 10.1172/JCI90962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mamidi S, Höne S, Kirschfink M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology. 2017/01/01/ 2017;222(1):45–54. doi: 10.1016/j.imbio.2015.11.008 [DOI] [PubMed] [Google Scholar]
- 5.Ajona D, Ortiz-Espinosa S, Pio R, Lecanda F. Complement in Metastasis: A Comp in the Camp. Front Immunol. 2019;10:669. doi: 10.3389/fimmu.2019.00669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manda K, Glasow A, Paape D, Hildebrandt G. Effects of ionizing radiation on the immune system with special emphasis on the interaction of dendritic and T cells. Frontiers in oncology. 2012;2:102–102. doi: 10.3389/fonc.2012.00102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orth M, Lauber K, Niyazi M, et al. Current concepts in clinical radiation oncology. Radiation and environmental biophysics. 2014;53(1):1–29. doi: 10.1007/s00411-013-0497-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walport MJ. Complement: First of Two Parts. New England Journal of Medicine. 2020/06/27 2001;344(14):1058–1066. doi: 10.1056/NEJM200104053441406 [DOI] [PubMed] [Google Scholar]
- 9.Nagar B, Jones RG, Diefenbach RJ, Isenman DE, Rini JM. X-ray crystal structure of C3d: a C3 fragment and ligand for complement receptor 2. Science. May 1998;280(5367):1277–81. doi: 10.1126/science.280.5367.1277 [DOI] [PubMed] [Google Scholar]
- 10.Walport MJ. Complement: Second of Two Parts. New England Journal of Medicine. 2020/06/27 2001;344(15):1140–1144. doi: 10.1056/NEJM200104123441506 [DOI] [PubMed] [Google Scholar]
- 11.Turner MW. Mannose-binding lectin: the pluripotent molecule of the innate immune system. Immunol Today. November 1996;17(11):532–40. doi: 10.1016/0167-5699(96)10062-1 [DOI] [PubMed] [Google Scholar]
- 12.Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. January 2006;281(4):2128–32. doi: 10.1074/jbc.M508928200 [DOI] [PubMed] [Google Scholar]
- 13.Amara U, Rittirsch D, Flierl M, et al. Interaction between the coagulation and complement system. Advances in experimental medicine and biology. 2008;632:71–79. doi: 10.1007/978-0-387-78952-1_6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber-Lang M, Sarma JV, Zetoune FS, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. June 2006;12(6):682–7. doi: 10.1038/nm1419 [DOI] [PubMed] [Google Scholar]
- 15.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nature Reviews Immunology. 2009/10/01 2009;9(10):729–740. doi: 10.1038/nri2620 [DOI] [PubMed] [Google Scholar]
- 16.Noris M, Remuzzi G. Overview of complement activation and regulation. Seminars in nephrology. 2013;33(6):479–492. doi: 10.1016/j.semnephrol.2013.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liszewski MK, Atkinson JP. Complement regulators in human disease: lessons from modern genetics. Journal of Internal Medicine. 2020/05/06 2015;277(3):294–305. doi: 10.1111/joim.12338 [DOI] [PubMed] [Google Scholar]
- 18.Mache CJ, Acham-Roschitz B, Frémeaux-Bacchi V, et al. Complement inhibitor eculizumab in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. August 2009;4(8):1312–6. doi: 10.2215/CJN.01090209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biesecker G, Katz S, Koffler D. Renal localization of the membrane attack complex in systemic lupus erythematosus nephritis. J Exp Med. December 1981;154(6):1779–94. doi: 10.1084/jem.154.6.1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. July 1991;4(3):359–95. doi: 10.1128/cmr.4.3.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. September 2010;11(9):785–97. doi: 10.1038/ni.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujita T, Gigli I, Nussenzweig V. Human C4-binding protein. II. Role in proteolysis of C4b by C3b-inactivator. The Journal of experimental medicine. 1978;148(4):1044–1051. doi: 10.1084/jem.148.4.1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zipfel PF, Skerka C. Complement factor H and related proteins: an expanding family of complement-regulatory proteins? Immunol Today. March 1994;15(3):121–6. doi: 10.1016/0167-5699(94)90155-4 [DOI] [PubMed] [Google Scholar]
- 24.Java A, Liszewski MK, Hourcade DE, Zhang F, Atkinson JP. Role of complement receptor 1 (CR1; CD35) on epithelial cells: A model for understanding complement-mediated damage in the kidney. Molecular immunology. 2015;67(2 Pt B):584–595. doi: 10.1016/j.molimm.2015.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liszewski MK, Post TW, Atkinson JP. Membrane Cofactor Protein (MCP or CD46): Newest Member of the Regulators of Complement Activation Gene Cluster. Annual Review of Immunology. 2020/04/30 1991;9(1):431–455. doi: 10.1146/annurev.iy.09.040191.002243 [DOI] [PubMed] [Google Scholar]
- 26.Roversi P, Johnson S, Caesar JJE, et al. Structural basis for complement factor I control and its disease-associated sequence polymorphisms. Proceedings of the National Academy of Sciences. 2011;108(31):12839. doi: 10.1073/pnas.1102167108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang H, Wagner E, Zhang H, Frank MM. Complement 1 inhibitor is a regulator of the alternative complement pathway. The Journal of experimental medicine. 2001;194(11):1609–1616. doi: 10.1084/jem.194.11.1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun X, Funk CD, Deng C, Sahu A, Lambris JD, Song WC. Role of decay-accelerating factor in regulating complement activation on the erythrocyte surface as revealed by gene targeting. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(2):628–633. doi: 10.1073/pnas.96.2.628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skidgel RA, Erdös EG. Structure and function of human plasma carboxypeptidase N, the anaphylatoxin inactivator. International immunopharmacology. 2007;7(14):1888–1899. doi: 10.1016/j.intimp.2007.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heurich M, Preston RJS, O’Donnell VB, Morgan BP, Collins PW. Thrombomodulin enhances complement regulation through strong affinity interactions with factor H and C3b-Factor H complex. Thrombosis Research. 2016;145:84–92. doi: 10.1016/j.thromres.2016.07.017 [DOI] [PubMed] [Google Scholar]
- 31.Ohno M, Hirata T, Enomoto M, Araki T, Ishimaru H, Takahashi TA. A putative chemoattractant receptor, C5L2, is expressed in granulocyte and immature dendritic cells, but not in mature dendritic cells. Molecular Immunology. 2000;37(8):407–412. doi: 10.1016/S0161-5890(00)00067-5 [DOI] [PubMed] [Google Scholar]
- 32.Zhang T, Garstka MA, Li K. The Controversial C5a Receptor C5aR2: Its Role in Health and Disease. J Immunol Res. 2017;2017:8193932. doi: 10.1155/2017/8193932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai B, Xie S, Liu F, et al. Rapid degradation of the complement regulator, CD59, by a novel inhibitor. The Journal of biological chemistry. 2014;289(17):12109–12125. doi: 10.1074/jbc.M113.547083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hochgrebe TT, Humphreys D, Wilson MR, Easterbrook-Smith SB. A Reexamination of the Role of Clusterin as a Complement Regulator. Experimental Cell Research. 1999;249(1):13–21. doi: 10.1006/excr.1999.4459 [DOI] [PubMed] [Google Scholar]
- 35.Singh B, Su Y-C, Riesbeck K. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Molecular Microbiology. 2020/04/30 2010;78(3):545–560. doi: 10.1111/j.1365-2958.2010.07373.x [DOI] [PubMed] [Google Scholar]
- 36.Sayegh ET, Bloch O, Parsa AT. Complement anaphylatoxins as immune regulators in cancer. Cancer Med. August 2014;3(4):747–58. doi: 10.1002/cam4.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol. January 2018;18(1):5–18. doi: 10.1038/nri.2017.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bischoff SC, de Weck Al Fau - Dahinden CA, Dahinden CA. Interleukin 3 and granulocyte/macrophage-colony-stimulating factor render human basophils responsive to low concentrations of complement component C3a. (0027–8424 (Print)) [DOI] [PMC free article] [PubMed]
- 39.Nilsson G, Johnell M Fau - Hammer CH HL Hammer Ch Fau - Tiffany, et al. C3a and C5a are chemotaxins for human mast cells and act through distinct receptors via a pertussis toxin-sensitive signal transduction pathway. (0022–1767 (Print)) [PubMed]
- 40.Ali H, Panettieri RA. Anaphylatoxin C3a receptors in asthma. Respir Res. February 2005;6:19. doi: 10.1186/1465-9921-6-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liszewski MK, Kolev M, Le Friec G, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. December 2013;39(6):1143–57. doi: 10.1016/j.immuni.2013.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arbore G, West EE, Spolski R, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4⁺ T cells. Science. June 2016;352(6292):aad1210. doi: 10.1126/science.aad1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Markiewski MM, DeAngelis RA, Benencia F, et al. Modulation of the antitumor immune response by complement. Nature immunology. 2008;9(11):1225–1235. doi: 10.1038/ni.1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vadrevu SK, Chintala NK, Sharma SK, et al. Complement C5a Receptor Facilitates Cancer Metastasis by Altering T-Cell Responses in the Metastatic Niche. Cancer Research. 2014;74(13):3454. doi: 10.1158/0008-5472.CAN-14-0157 [DOI] [PubMed] [Google Scholar]
- 45.Piao C, Zhang WM, Li TT, et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp Cell Res. May 2018;366(2):127–138. doi: 10.1016/j.yexcr.2018.03.009 [DOI] [PubMed] [Google Scholar]
- 46.Janelle V, Langlois MP, Tarrab E, Lapierre P, Poliquin L, Lamarre A. Transient complement inhibition promotes a tumor-specific immune response through the implication of natural killer cells. Cancer Immunol Res. March 2014;2(3):200–6. doi: 10.1158/2326-6066.CIR-13-0173 [DOI] [PubMed] [Google Scholar]
- 47.Kwak JW, Laskowski J, Li HY, et al. Complement Activation via a C3a Receptor Pathway Alters CD4. Cancer Res. January 2018;78(1):143–156. doi: 10.1158/0008-5472.CAN-17-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maeda Y, Kawano Y, Wada Y, et al. C5aR is frequently expressed in metastatic renal cell carcinoma and plays a crucial role in cell invasion via the ERK and PI3 kinase pathways. Oncol Rep. April 2015;33(4):1844–50. doi: 10.3892/or.2015.3800 [DOI] [PubMed] [Google Scholar]
- 49.Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Molecular Immunology. 2003/09/01/ 2003;40(2):109–123. doi: 10.1016/S0161-5890(03)00112-3 [DOI] [PubMed] [Google Scholar]
- 50.Kapka-Skrzypczak L, Wolinska E, Szparecki G, Wilczynski GM, Czajka M, Skrzypczak M. CD55, CD59, factor H and factor H-like 1 gene expression analysis in tumors of the ovary and corpus uteri origin. Immunol Lett. October 2015;167(2):67–71. doi: 10.1016/j.imlet.2015.06.017 [DOI] [PubMed] [Google Scholar]
- 51.Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol. August 2010;31(4):363–72. doi: 10.1007/s13277-010-0042-8 [DOI] [PubMed] [Google Scholar]
- 52.Karam SD, Raben D. Radioimmunotherapy for the treatment of head and neck cancer. Lancet Oncol. August 2019;20(8):e404–e416. doi: 10.1016/S1470-2045(19)30306-7 [DOI] [PubMed] [Google Scholar]
- 53.Shahabi V, Postow MA, Tuck D, Wolchok JD. Immune-priming of the tumor microenvironment by radiotherapy: rationale for combination with immunotherapy to improve anticancer efficacy. Am J Clin Oncol. February 2015;38(1):90–7. doi: 10.1097/COC.0b013e3182868ec8 [DOI] [PubMed] [Google Scholar]
- 54.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. June 2005;174(12):7516–23. doi: 10.4049/jimmunol.174.12.7516 [DOI] [PubMed] [Google Scholar]
- 55.Arnold KM, Flynn NJ, Raben A, et al. The Impact of Radiation on the Tumor Microenvironment: Effect of Dose and Fractionation Schedules. Cancer growth and metastasis. 2018;11:1179064418761639–1179064418761639. doi: 10.1177/1179064418761639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deloch L, Derer A, Hartmann J, Frey B, Fietkau R, Gaipl US. Modern Radiotherapy Concepts and the Impact of Radiation on Immune Activation. Frontiers in oncology. 2016;6:141–141. doi: 10.3389/fonc.2016.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schaue D, Ratikan JA, Iwamoto KS, McBride WH. Maximizing Tumor Immunity With Fractionated Radiation. International Journal of Radiation Oncology*Biology*Physics. 2012/07/15/ 2012;83(4):1306–1310. doi: 10.1016/j.ijrobp.2011.09.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kulzer L, Rubner Y, Deloch L, et al. Norm- and hypo-fractionated radiotherapy is capable of activating human dendritic cells. J Immunotoxicol. October 2014;11(4):328–36. doi: 10.3109/1547691X.2014.880533 [DOI] [PubMed] [Google Scholar]
- 59.Frey B, Rubner Y, Wunderlich R, et al. Induction of abscopal anti-tumor immunity and immunogenic tumor cell death by ionizing irradiation - implications for cancer therapies. Curr Med Chem. 2012;19(12):1751–64. doi: 10.2174/092986712800099811 [DOI] [PubMed] [Google Scholar]
- 60.Cao M, Cabrera R, Xu Y, Liu C, Nelson D. Different radiosensitivity of CD4(+)CD25(+) regulatory T cells and effector T cells to low dose gamma irradiation in vitro. Int J Radiat Biol. January 2011;87(1):71–80. doi: 10.3109/09553002.2010.518208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qu Y, Jin S, Zhang A, et al. Gamma-ray resistance of regulatory CD4+CD25+Foxp3+ T cells in mice. Radiat Res. February 2010;173(2):148–57. doi: 10.1667/RR0978.1 [DOI] [PubMed] [Google Scholar]
- 62.Chen J, Wang Z, Ding Y, et al. Hypofractionated Irradiation Suppressed the Off-Target Mouse Hepatocarcinoma Growth by Inhibiting Myeloid-Derived Suppressor Cell-Mediated Immune Suppression. Original Research. Frontiers in Oncology. 2020–February–11 2020;10(4)doi: 10.3389/fonc.2020.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ianzini F, Bertoldo A, Kosmacek EA, Phillips SL, Mackey MA. Lack of p53 function promotes radiation-induced mitotic catastrophe in mouse embryonic fibroblast cells. Cancer Cell Int. April 2006;6:11. doi: 10.1186/1475-2867-6-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Darragh LB, Oweida AJ, Karam SD. Overcoming Resistance to Combination Radiation-Immunotherapy: A Focus on Contributing Pathways Within the Tumor Microenvironment. Front Immunol. 2018;9:3154. doi: 10.3389/fimmu.2018.03154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elvington M, Scheiber M, Yang X, et al. Complement-dependent modulation of antitumor immunity following radiation therapy. Cell reports. 2014;8(3):818–830. doi: 10.1016/j.celrep.2014.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Surace L, Lysenko V, Fontana AO, et al. Complement is a central mediator of radiotherapy-induced tumor-specific immunity and clinical response. Immunity. April 2015;42(4):767–77. doi: 10.1016/j.immuni.2015.03.009 [DOI] [PubMed] [Google Scholar]
- 67.Kolev M, Markiewski MM. Targeting complement-mediated immunoregulation for cancer immunotherapy. Seminars in immunology. 2018;37:85–97. doi: 10.1016/j.smim.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nauta AJ, Daha MR, van Kooten C, Roos A. Recognition and clearance of apoptotic cells: a role for complement and pentraxins. Trends Immunol. March 2003;24(3):148–54. doi: 10.1016/s1471-4906(03)00030-9 [DOI] [PubMed] [Google Scholar]
- 69.Macor P, Capolla S, Tedesco F. Complement as a Biological Tool to Control Tumor Growth. Frontiers in immunology. 2018;9:2203–2203. doi: 10.3389/fimmu.2018.02203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rogers LM, Veeramani S, Weiner GJ. Complement in monoclonal antibody therapy of cancer. Immunologic research. 2014;59(1–3):203–210. doi: 10.1007/s12026-014-8542-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hsu YF, Ajona D, Corrales L, et al. Complement activation mediates cetuximab inhibition of non-small cell lung cancer tumor growth in vivo. Mol Cancer. June 2010;9:139. doi: 10.1186/1476-4598-9-139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bologna L, Gotti E, Manganini M, et al. Mechanism of action of type II, glycoengineered, anti-CD20 monoclonal antibody GA101 in B-chronic lymphocytic leukemia whole blood assays in comparison with rituximab and alemtuzumab. J Immunol. March 2011;186(6):3762–9. doi: 10.4049/jimmunol.1000303 [DOI] [PubMed] [Google Scholar]
- 73.Beurskens FJ, Lindorfer MA, Farooqui M, et al. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J Immunol. April 2012;188(7):3532–41. doi: 10.4049/jimmunol.1103693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mamidi S, Cinci M, Hasmann M, Fehring V, Kirschfink M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol Oncol. June 2013;7(3):580–94. doi: 10.1016/j.molonc.2013.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ajona D, Ortiz-Espinosa S, Moreno H, et al. A Combined PD-1/C5a Blockade Synergistically Protects against Lung Cancer Growth and Metastasis. Cancer Discov. July 2017;7(7):694–703. doi: 10.1158/2159-8290.CD-16-1184 [DOI] [PubMed] [Google Scholar]
- 76.Innate Pharma. IPH5401 (Anti- C5aR ) in Combination With Durvalumab in Patients With Advanced Solid Tumors (STELLAR-001). Accessed June, 27, 2020 https://clinicaltrials.gov/ct2/show/NCT03665129?term=C5aR&recrs=a&cond=Cancer&draw=2&rank=1
- 77.Kourtzelis I, Rafail S. The dual role of complement in cancer and its implication in anti-tumor therapy. Annals of translational medicine. 2016;4(14):265–265. doi: 10.21037/atm.2016.06.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao WP, Zhu B, Duan YZ, Chen ZT. Neutralization of complement regulatory proteins CD55 and CD59 augments therapeutic effect of herceptin against lung carcinoma cells. Oncol Rep. June 2009;21(6):1405–11. doi: 10.3892/or_00000368 [DOI] [PubMed] [Google Scholar]
- 79.Ward PA. Sepsis, apoptosis and complement. Biochem Pharmacol. December 2008;76(11):1383–8. doi: 10.1016/j.bcp.2008.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Engelman RM, Rousou JA, Flack JE, Deaton DW, Kalfin R, Das DK. Influence of steroids on complement and cytokine generation after cardiopulmonary bypass. The Annals of thoracic surgery. 1995/09// 1995;60(3):801–804. doi: 10.1016/0003-4975(95)00211-3 [DOI] [PubMed] [Google Scholar]
- 81.Maher SG, McDowell DT, Collins BC, Muldoon C, Gallagher WM, Reynolds JV. Serum proteomic profiling reveals that pretreatment complement protein levels are predictive of esophageal cancer patient response to neoadjuvant chemoradiation. Ann Surg. November 2011;254(5):809–16; 10.1097/SLA.0b013e31823699f2 [DOI] [PubMed] [Google Scholar]
- 82.Bouwens TA, Trouw LA, Veerhuis R, Dirven CM, Lamfers ML, Al-Khawaja H. Complement activation in Glioblastoma multiforme pathophysiology: evidence from serum levels and presence of complement activation products in tumor tissue. J Neuroimmunol. January 2015;278:271–6. doi: 10.1016/j.jneuroim.2014.11.016 [DOI] [PubMed] [Google Scholar]
- 83.Ajona D, Okrój M, Pajares MJ, et al. Complement C4d-specific antibodies for the diagnosis of lung cancer. Oncotarget. 2017;9(5):6346–6355. doi: 10.18632/oncotarget.23690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou Y, Chu L, Wang Q, et al. CD59 is a potential biomarker of esophageal squamous cell carcinoma radioresistance by affecting DNA repair. Cell Death Dis. August 2018;9(9):887. doi: 10.1038/s41419-018-0895-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Medler TR, Murugan D, Horton W, et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell. October 2018;34(4):561–578.e6. doi: 10.1016/j.ccell.2018.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li M, Bolduc AR, Hoda MN, et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. Journal for immunotherapy of cancer. 2014;2:21–21. doi: 10.1186/2051-1426-2-21 [DOI] [PMC free article] [PubMed] [Google Scholar]