

Abstract
Gα proteins promote dynamic adjustments of cell shape directed by actin-cytoskeleton reorganization via their respective RhoGEF effectors. For example, Gα13 binding to the RGS-homology (RH) domains of several RH-RhoGEFs allosterically activates these proteins, causing them to expose their catalytic Dbl-homology (DH)/pleckstrin-homology (PH) regions, which triggers downstream signals. However, whether additional Gα proteins might directly regulate the RH-RhoGEFs was not known. To explore this question, we first examined the morphological effects of expressing shortened RH-RhoGEF DH/PH constructs of p115RhoGEF/ARHGEF1, PDZ-RhoGEF (PRG)/ARHGEF11, and LARG/ARHGEF12. As expected, the three constructs promoted cell contraction and activated RhoA, known to be downstream of Gα13. Intriguingly, PRG DH/PH also induced filopodia-like cell protrusions and activated Cdc42. This pathway was stimulated by constitutively active Gαs (GαsQ227L), which enabled endogenous PRG to gain affinity for Cdc42. A chemogenetic approach revealed that signaling by Gs-coupled receptors, but not by those coupled to Gi or Gq, enabled PRG to bind Cdc42. This receptor-dependent effect, as well as CREB phosphorylation, was blocked by a construct derived from the PRG:Gαs-binding region, PRG-linker. Active Gαs interacted with isolated PRG DH and PH domains and their linker. In addition, this construct interfered with GαsQ227L's ability to guide PRG's interaction with Cdc42. Endogenous Gs-coupled prostaglandin receptors stimulated PRG binding to membrane fractions and activated signaling to PKA, and this canonical endogenous pathway was attenuated by PRG-linker. Altogether, our results demonstrate that active Gαs can recognize PRG as a novel effector directing its DH/PH catalytic module to gain affinity for Cdc42.
Keywords: PDZ-RhoGEF (PRG), ARHGEF11, DH/PH catalytic module, Rho guanine nucleotide exchange factor (RhoGEF), Rho GTPases, Cdc42, Gαs, cell signaling, GPCR, Rho (Rho GTPase), guanine nucleotide exchange factor (GEF), heterotrimeric G protein, G protein–coupled receptor (GPCR), cell signaling, Galpha-s, PDZ-RhoGEF
Migrating cells follow extracellular cues that guide dynamic protrusions and contractions (1, 2). At the plasma membrane, phosphoinositides and signaling proteins allosterically activate Rho guanine nucleotide exchange factors (RhoGEFs) exposing their catalytic DH/PH modules, composed of Dbl-homology and Pleckstrin-homology domains in tandem (3–5). RhoGEFs stimulate their cognate GTPases to exchange GDP for GTP, orchestrating cytoskeleton remodeling pathways (6, 7). RhoA promotes the assembly of stress fibers and contractile actomyosin structures, whereas Rac and Cdc42 lead the extension of actin-driven plasma membrane protrusions known as lamellipodia and filopodia, respectively (8). Although these Rho GTPases exhibit contrasting effects, they can be alternatively activated at edges of moving cells (9). Therefore, fine-tuning mechanisms are likely involved.
Several RhoGEFs are effectors of heterotrimeric G proteins (3, 10, 11). GTP-bound Gα13 and Gαq proteins stimulate RhoA, whereas Gβγ activates Rac and Cdc42 (3, 10, 12). Gβγ signaling to Rac is itself directly regulated by Gα13 and Gαq (13). GTP-Gα13 proteins allosterically activate RH-RhoGEFs (p115RhoGEF, PDZ-RhoGEF (PRG), and LARG) (3, 14, 15). The mechanism involves direct interaction 0of Gα13 with the RhoGEF RGS-homology (RH) domain, which in consequence exposes the catalytic DH/PH cassette (16, 17). The DH domain activates RhoA, and it is positively modulated by the PH domain (6, 18). Here, we investigated whether PRG DH/PH catalytic module is directly targeted by additional Gα proteins.
Results
Membrane-anchored PRG-DH/PH promotes cell contraction and formation of filopodia-like protrusions
RH-RhoGEFs are potent activators of RhoA, which generally counteracts cell protruding processes led by Rac and Cdc42 (3, 9). To characterize potential regulatory mechanisms directly targeting RH-RhoGEF DH/PH domains, we first analyzed the cellular effects of membrane-anchored, EGFP-tagged, p115RhoGEF-DH/PH, PRG-DH/PH, and LARG-DH/PH constructs (Fig. 1A). Their morphological effects, assessed by confocal microscopy of transfected endothelial cells (Fig. 1B), revealed the assembly of actin stress fibers, as expected for RhoA activity (8). Intriguingly, ∼50% of cells expressing PRG-DH/PH also induced actin-based thin protrusions (Fig. 1, B and C), reminiscent of Cdc42-induced filopodia (19).
Figure 1.

In addition to its canonical effect on RhoA, PRG DH/PH activates Cdc42 and promotes filopodia formation. A, hypothetical effects of membrane-anchored RH-RhoGEF DH/PH catalytic modules. B, confocal images of PAE cells showing EGFP-RH-RhoGEF DH/PH-CAAX constructs (green) and their effects on F-actin (red). Zoomed-in areas are shown in the bottom row. Scale bar, 20 μm. C, graph shows the percentage of cells exhibiting filopodia-like structures. Each dot represents the means ± S.E. of at least 30 cells per experiment (n = 3). ***, p < 0.0001; n.s., no significance, one-way ANOVA followed Tukey. D, representative blot shows expression of EGFP-RH-RhoGEF DH/PH-CAAX constructs. E and F, activation of Cdc42 (E) and RhoA (F) by EGFP-RhoGEF DH/PH-CAAX constructs transfected into HEK293T cells was detected by PAK-CRIB and Rhotekin-RBD pulldown (PD), respectively. The graphs represent the means ± S.E. densitometric values (n = 3). **, p = 0.005; ***, p < 0.001; ****, p < 0.0001, one-way ANOVA followed Tukey. G, pulldown of active EGFP-RhoGEF DH/PH-CAAX constructs based on their affinity for nucleotide-free recombinant Cdc42-G15A (left panel) and RhoA-G17A (middle panel). Total cell lysates (TCL) are shown in the right panel. H, interaction between PRG DH/PH and Cdc42-T17N was assayed in transfected HEK293T cells subjected to pulldown assays (PD:GST). D–H, protein expression is confirmed in total cell lysates.
PRG-DH/PH catalytic module stimulates Cdc42
Consistent with its morphological effects, PRG-DH/PH significantly stimulated Cdc42 (Fig. 1E). In contrast, all three RH-RhoGEF EGFP-DH/PH-CAAX constructs stimulated RhoA (Fig. 1F). Furthermore, recombinant nucleotide-free Cdc42-G15A, used to isolate active Cdc42-GEFs (20), pulled down PRG-DH/PH, and to a lesser extent p115RhoGEF-DH/PH (Fig. 1G, left panel), whereas RhoA-G17A pulled down the three constructs (Fig. 1G, middle panel). Furthermore, cotransfected into HEK293T cells, GST-PRG-DH/PH interacted with Cdc42-T17N, a dominant-negative mutant (Fig. 1H).
Constitutively active Gαs-Q227L increases PRG-DH/PH activity toward Cdc42
The mechanistic basis of p63RhoGEF and TRIO activation by Gαq (21, 22) inspired us to assess whether GTPase-deficient Gα-QL mutants bind PRG DH/PH to stimulate Cdc42 (Fig. 2A). With pulldown assays, we revealed that Gαs-Q227L significantly increased PRG-DH/PH interaction with Cdc42-G15A (Fig. 2B) without affecting its interaction with RhoA-G17A (Fig. 2C). Gαs-Q227L was also detected in the pulldown assay (Fig. 2B), depending on the presence of PRG-DH/PH (Fig. 2D). In HEK293T cells, Gαs-QL interacted with PRG-DH/PH (Fig. 2E), as well as with other RH-RhoGEFs (Fig. 2F), as revealed by pulldown assays using GST-DH/PH constructs. In contrast, Gα13-QL, known to interact with RGS-like domain of RH-RhoGEFs (14, 23), was absent in the GST-DH/PH pulldowns (Fig. 2F). We also found that Gαq-QL interacted with PRG-DH/PH (Fig. 2E); however, it did not affect PRG-DH/PH:Cdc42 binding (Fig. 2B). Consistent with a functional effect, Gαs-QL stimulated PRG-DH/PH to activate Cdc42 (Fig. 2G, top panel and graph) and remained bound to the PRG-DH/PH construct pulled down with nucleotide-free Cdc42 (Fig. 2, B, D, and G, middle panel). Furthermore, the interaction of PRG DH/PH with Cdc42-T17N was further stimulated by Gαs-QL (Fig. 2H).
Figure 2.

Gαs-Q227L binds PRG DH/PH enabling this prototypical RhoA-specific GEF to directly activate Cdc42. A, hypothetic model postulating Gα subunits as potential regulators of PRG DH/PH catalytic module. B and C, the effect of GTPase-deficient Gα subunits on the interaction of EGFP-PRG-DH/PH-CAAX with Cdc42-G15A (B) and RhoA-G17A (C) was analyzed by pulldown (PD) using lysates from HEK293T cells transfected with HA-tagged Gαs, Gαi, Gαq, or Gα13 QL mutants and EGFP-PRG-DH/PH-CAAX. The graph in B represents the means ± S.E. (n = 3). ***, p < 0.001; n.s., no significance, one-way ANOVA followed Tukey. D, to address whether Gαs-QL detected in the PRG-DH/PH·Cdc42-G15A pulldown was part of a ternary complex, pulldown experiments were done in the presence or absence of PRG-DH/PH. The graph represents the means ± S.E. (n = 3). **, p < 0.0001, t test. E, the potential interaction between active Gα subunits and PRG DH/PH was analyzed in HEK293T cells transfected with GST-PRG-DH/PH and HA-tagged GTPase-deficient Gα subunits subjected to pulldown assays. F, interaction between Gαs-QL and the catalytic domain of the three RH-RhoGEFs was assayed by pulldown using HEK293T cells transfected with HA-Gαs-QL and GST-p115-DH/PH, GST-PRG-DH/PH, or GST-LARG-DH/PH. GST and HA-Gα13-QL served as negative controls. G, the effect of Gαs-QL on the activation of Cdc42 by PRG-DH/PH was assessed by pulldown using lysates of transfected HEK293T cells. The graph represents the means ± S.E. (n = 3). **, p = 0.01, t test. Representative blots show the fraction of active Cdc42 (top panel) and the active fraction of PRG-DH/PH with affinity for Cdc42-G15A (middle panel). H, the effect of Gαs-QL on the interaction between PRG-DH/PH and Cdc42-T17N was analyzed by pulldown using lysates of transfected HEK293T cells. The graph represents the means ± S.E. (n = 3). *, p = 0.01, t test. I and J, the effect of Gαs-QL on full-length PRG affinity for Cdc42 was analyzed in HEK293T cells that were transfected with full-length AU1-PRG (I) without or with HA-Gαs-QL or only with HA-Gαs-QL to address its effect on endogenous PRG (J). The active fraction of full-length PRG with affinity for Cdc42-G15A was isolated by pulldown and revealed by immunoblotting with anti-PRG antibodies. The graphs represents the means ± S.E. (n = 3). *, p = 0.01 in H and 0.04 in I, t test.
Gαs-Q227L drives full-length PRG to interact with Cdc42
To address whether full-length PRG is sensitive to be driven by Gαs-QL to gain affinity for Cdc42, we used lysates from transfected HEK293T cells. We found that Gαs-QL stimulated full-length PRG, either transfected (Fig. 2I) or endogenous (Fig. 2J), to bind Cdc42. Furthermore, Gαs-QL remained bound to PRG pulled down with nucleotide-free Cdc42. Expression of transfected and endogenous proteins was confirmed in total cell lysates (Fig. 2, C–J, TCL).
Gαs-Q227L interaction interface at PRG involves the DH and PH domains and the linker region joining them
To characterize how Gαs-Q227L binds PRG-DH/PH guiding this prototypic RhoA-specific GEF to interact with Cdc42, we cotransfected Gαs-Q227L with different PRG constructs spanning the DH/PH module, fused to GST (Fig. 3A), and addressed by pulldown their potential interaction. As shown in Fig. 3B, Gαs-QL interacted with the three PRG-DH/PH constructs that had in common the linker region that joins the DH and PH domains. When PRG-DH, PRG-linker, and PRG-PH were used as independent constructs, the three of them interacted with Gαs-QL, indicating that PRG-linker is critical to strengthen the interaction (Fig. 3C). Consistent with previous results, Gα13-QL served as negative control (Fig. 3B). Because the PRG-linker sequence is conserved among the three RH-RhoGEFs (Fig. 3D) but lacks homology with any other protein, we used an EGFP-tagged PRG-linker construct as a potential inhibitor of Gαs-dependent PRG·Cdc42 interaction. Based on previous results and the structure of the PRG-DH/PH·RhoA complex (Fig. 3E) (24), we postulated that Gαs-Q227L interacts with PRG-DH/PH, forming a complex with affinity for Cdc42 (Fig. 3F). Then we tested the potential inhibitory effect of PRG-linker on the interaction between Gαs-Q227L and PRG (Fig. 3G). As predicted, the PRG-linker construct not only inhibited Gαs-QL·PRG-DH/PH interaction (Fig. 3H) but also interfered on the effect of Gαs-QL to drive full-length PRG to be pulled down as an active Cdc42-GEF (Fig. 3I); EGFP served as negative control.
Figure 3.

Gαs-Q227L binds PRG DH and PH domains and the linker region joining them. A, model showing GST-tagged PRG-DH/PH constructs used to map Gαs·PRG-DH/PH interaction. B and C, interaction between HA-Gαs-QL and the indicated GST-PRG-DH/PH constructs was analyzed by pulldown using lysates of transfected HEK293T cells. HA-Gαs-QL and HA-Gα13-QL (used as control) were revealed with anti-HA antibodies. D, multiple alignment of p115RhoGEF, LARG, and PRG-linker regions. E, structure of PRG-DH/PH·RhoA complex (24). F, model showing Gαs-GTP·PRG-DH/PH complex; hypothetically, active Gαs constrains PRGDH/PH to bind Cdc42. G, model showing the potential inhibitory effect of the EGFP–PRG-linker construct on PRG activation by Gαs-QL. H, HEK293T cells transfected with EGFP-tagged PRG-linker construct (or EGFP) together with HA-Gαs-QL and GST-PRG-DH/PH (or GST) were subjected to GST pulldown assays. I, Gαs-dependent PRG·Cdc42 interaction was analyzed in HEK293T cells transfected with HA-Gαs-QL (or control plasmid) and AU1-PRG together with EGFP–PRG-linker or EGFP and subjected to Cdc42-G15A pulldown. The graph represents the means ± S.E. (n = 3). **, p = 0.001; ***, p = 0.0001; ns, no significance, one-way ANOVA followed Tukey.
Agonist-dependent stimulation of Gs-coupled receptors drives PRG to gain affinity for Cdc42
To investigate whether Gs-coupled receptors stimulate PRG to acquire affinity for Cdc42, we first used PAE and HT29 cells as models of endogenous prostaglandin-dependent Gs signaling. As an initial readout of agonist-driven Gs-dependent effect on PRG, we stimulated PAE and HT29 cells with PGE2 and butaprost, respectively, and assessed PRG recruitment to membrane fractions. In both cases, PRG exhibited a significant time-dependent association to membrane fractions (Fig. 4, A and B, respectively). We then used COS7 cells expressing Gs-DREADDs to test the effect of endogenous Gs on PRG·dc42 interaction. In these cells, clozapine N-oxide (CNO), the agonist of Gs-DREADDs, enabled PRG to bind nucleotide-free Cdc42 in a time-dependent manner (Fig. 4C). This effect was elicited by Gs-coupled, but not by Gi- or Gq-coupled DREADDs (Fig. 4D), and was inhibited by the PRG-linker peptide (Fig. 4E), which also interfered on CREB phosphorylation (Fig. 4F). Endogenous Gs-coupled endothelial EP2 receptors signaling to cAMP/PKA pathway was also inhibited by the PRG-linker construct, as indicated by a decrease on butaprost-dependent phosphorylation of PKA substrates (Fig. 4G).
Figure 4.
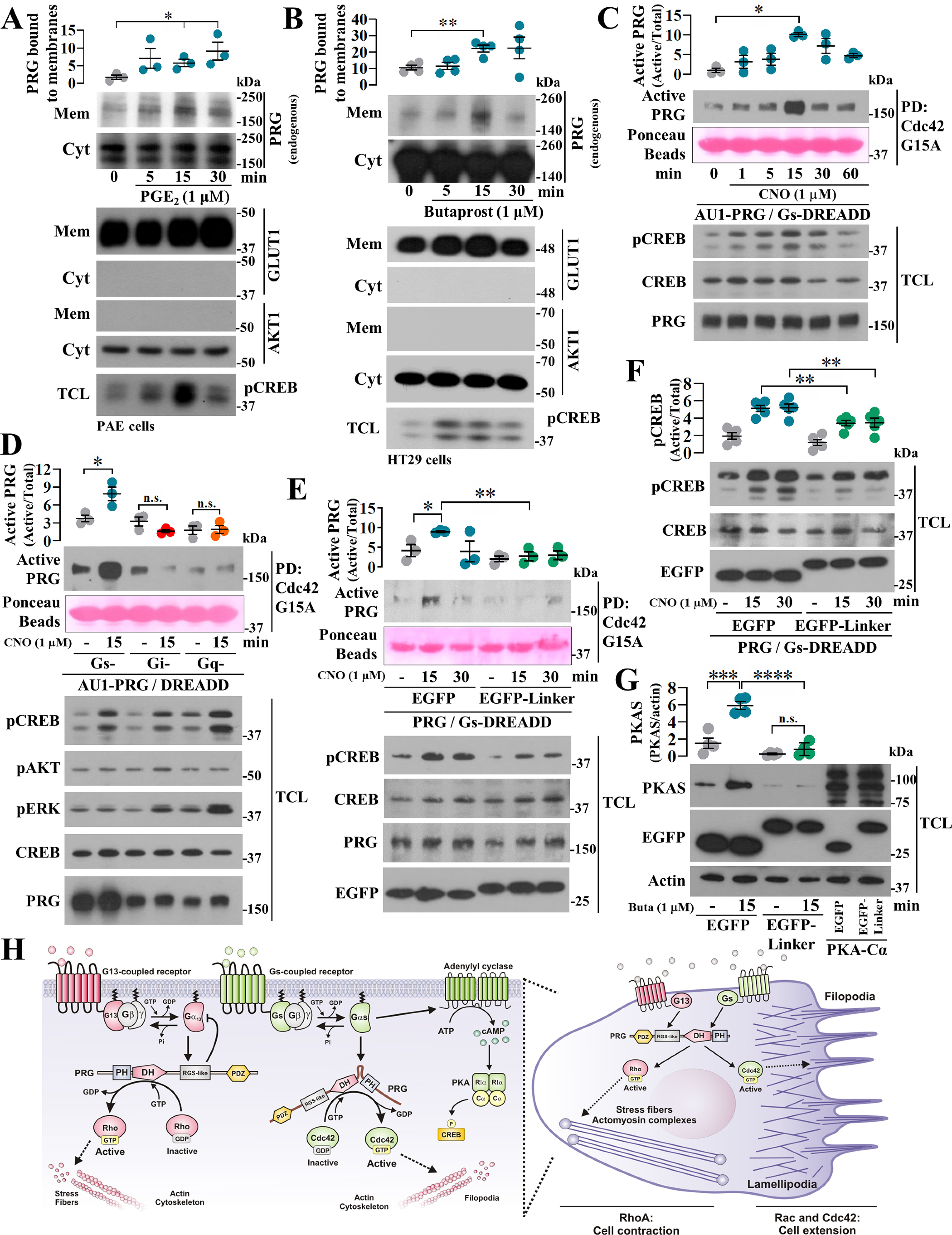
Agonist-dependent stimulation of Gs-coupled receptors enables PRG to bind Cdc42. A and B, membrane recruitment of endogenous PRG promoted by Gs-coupled GPCR signaling was assessed in PAE (A) and HT29 (B) cells stimulated with 1 μm PGE2 or butaprost, respectively. PRG in membrane fractions was revealed by Western blotting. GLUT1 and AKT1 were used as membrane and cytosolic markers, respectively. The graphs represent the means ± S.E. (n = 3, *, p < 0.05 in A; and n = 4, **, p < 0.01 in B; t test). C, time course of PRG·Cdc42 interaction was assessed in COS7 expressing Gs-DREADD receptors. The cells were stimulated with 1 µm CNO and subjected to Cdc42-G15A pulldown. The graph represents the means ± S.E. (n = 3). *, p < 0.05, one-way ANOVA followed Tukey. D, the effect of different endogenous heterotrimeric G proteins on PRG affinity for Cdc42 was studied in COS7 cells transfected with AU1-PRG and Gs-, Gi-, or Gq-DREADDs. The cells were stimulated with CNO for 15 min and subjected to Cdc42-G15A pulldown assays. The graph represents the means ± S.E. (n = 3). *, p < 0.05, one-way ANOVA followed Tukey. E, effect of the PRG-linker construct on agonist-stimulated interaction between PRG and Cdc42 was assessed in COS7 cells transfected with Gs-DREADD, AU1-PRG, and EGFP–PRG-linker or EGFP. The cells were stimulated with CNO for 15 and 30 min and subjected to Cdc42-G15A pulldown. The graph represents the means ± S.E. (n = 3). *, p = 0.0342; **, p = 0.0056, t test. F, effect of PRG-linker on agonist-dependent phosphorylation of CREB was assessed in COS7 cells expressing Gs-DREADDs and stimulated with CNO. The graph represents the means ± S.E. (n = 5). **, p < 0.01, t test. G, agonist-dependent phosphorylation of PKA substrates was assessed using PAE cells expressing EGFP or EGFP–PRG-linker and stimulated with butaprost. Lysates from EGFP-PKA-Cα−transfected cells served as control to detect PKA substrates. The graph represents the means ± S.E. (n = 4). ***, p = 0.0009; ****, p < 0.0001; n.s., no significance, one-way ANOVA followed Tukey. H, model depicts the canonical G13-PRG signaling axis to Rho and the emerging GPCR–Gαs–PRG–Cdc42 pathway based on the current findings. In cells, both systems putatively guide dynamic adjustments on actin-cytoskeleton reorganization.
Discussion
RH-RhoGEFs link heterotrimeric G proteins to Rho GTPases (25–27). They are activated by Gα12/13 proteins, which bind the RH domains unleashing the catalytic DH/PH region, known as specific for RhoA (14, 23). Here we demonstrate that active Gαs directly constrains the PRG DH/PH catalytic module to activate Cdc42, whereas its effect on RhoA is unaltered. Although future work using purified proteins is guaranteed, our results suggest that RhoGEF DH/PH domains can be allosterically controlled to expand their specificity.
Direct activation of RhoGEF DH/PH domains by active Gα subunits of heterotrimeric G proteins has been described. Specifically, Gαq stimulates p63RhoGEF and TRIO (21, 28). Physiological control of this system is lost by GNAQ mutation, causing the Gαq·TRIO signaling system to drive uveal melanoma progression (28, 29). Similarly, mutant GNAS is a driving oncogene in neuroendocrine cancers (30); however, a pathological link to Rho GTPases has not been established. Our results are reminiscent of the regulation of p63RhoGEF and TRIO by Gαq (21, 22, 28) but differ in the fact that Gαs expands PRG specificity directing the DH/PH module to gain affinity for Cdc42 without an apparent effect on RhoA. We speculate that Gαs pulls the PRG DH/PH module to accommodate Cdc42. Consistent with this possibility, conserved residues that directly bind the GTPase are more distant in intersectin-1, a Cdc42-specific GEF compared with the PRG DH/PH module in complex with RhoA (31–33).
Consistent with their reported effects (25, 27, 32, 34–36), RH-RhoGEF DH/PH catalytic modules strongly activated RhoA and promoted the assembly of actin stress fibers and cell contraction. These results confirmed that DH/PH constructs maintain catalysis and specificity (36–44). However, we found that PRG DH/PH also exhibited a previously unrecognized ability to stimulate Cdc42 and filopodia formation. Thus, we addressed the possibility that PRG directly activates Cdc42. We used pulldown assays to isolate active RhoGEFs based on their affinity for nucleotide-free GTPases (20, 45) and revealed that Gαs stimulates PRG to gain affinity for Cdc42, pointing to a direct effect (attenuated by PKA; Fig. S1). We demonstrated that GTPase-deficient Gαs binds the DH/PH module. The linker region joining these domains strengthen their interaction with active Gαs. Our evidence arguing for a functional relevance of this interaction derives from the inhibitory effect of the PRG-linker construct, which prevented PRG response to agonist-dependent stimulation of Gs-DREADDs and to GTPase-deficient Gαs coexpression. Our results not only indicate that Gαs guides PRG to bind Cdc42 but also suggest that this effector competes with other Gαs-dependent effectors. The Gs/PKA pathway activates Rho GTPases and regulates cytoskeletal dynamics at multiple levels. Recent evidence documented a role for PKA R1α subunit as a cAMP-dependent activator of P-REX1, a RacGEF (46), whereas kinase activity of PKA is linked to cytoskeletal dynamics at cell edges and is reciprocally regulated during cell migration (47–49). Our findings showing that Gαs activates a PRG/Cdc42 pathway expand the mechanisms of Gs signaling to Rho GTPases.
Although further experiments are needed to define the spatiotemporal conditions in which PRG is guided to activate Cdc42, our current model (Fig. 4H) illustrates the potential of Gs-coupled receptors to activate this pathway. Our work raises new questions and research avenues on how Gs and G13 signaling pathways are integrated to fine-tune Cdc42 activity in the context of strong RhoA activation to regulate cytoskeletal dynamics and set the basis to further investigate how the Gs/PRG/Cdc42 pathway guides polarized cell migration and its potential pathological implications, particularly in cancers in which mutant GNAS is a driving oncogene.
Experimental procedures
Plasmids and cDNA constructs
RH-RhoGEF DH/PH catalytic modules and PRG DH-PH fragments were amplified by PCR and cloned into pCEFL-EGFP-CAAX, pCEFL-EGFP, and pCEFL-GST. Primer sequences are available upon request. Other constructs have been previously described (13, 46).
Cell culture, transfection, immunoblotting, and GST pulldown
HEK293T, PAE, HT29, and COS7 cells were maintained and transfected as described (46). The cells were serum-starved for 16 h before experiments and were all done 48 h after transfection. GST fusion proteins and their interactors were detected by pulldown (13, 46). The cell lysates and pulldowns were analyzed by Western blotting using the following antibodies: EGFP SC-9996, GST B-14 SC-138, Cdc42 SC-8401, RhoA SC-418, p-AKT1/2/3 Ser473 SC-7985-R, and ERK2 SC-154 from Santa Cruz Biotechnology; HA from Covance; p-CREB Ser-133 9191, p-ERK1/2 T202/Y204 9191, CREB 9197S, and PKAS 9624 from Cell Signaling Technology; PRG/ARHGEF11 and HPA014658 from Atlas Antibodies; and AKT1 and P2482 from Sigma. The secondary antibodies were goat anti-mouse (Zymed Laboratories Inc., Invitrogen, or KPL) or goat anti-rabbit (Rockland Immunochemicals or KPL).
GTPase (Cdc42 and RhoA) activation and active GEF capture assays
Activation of RhoA and Cdc42, using cells grown in 10-cm dishes, was assessed by pulldown using recombinant Rhotekin and PAK effector domains (13, 46). Active RhoGEFs were detected by pulldown with nucleotide-free GTPases (RhoA G17A and Cdc42 G15A) fused to GST (20, 45). Before lysis, the cells were washed with PBS containing 10 mm MgCl2 and lysed with 1 ml of ice-cold lysis buffer (50 mm Tris, 150 mm NaCl, pH 7.5, containing 1% Triton X-100, 5 mm EDTA, and protease and phosphatase inhibitors (13)). Lysates were subjected to GST pulldowns as described (13, 46). COS7 cells were transfected with DREADDs exclusively coupled to Gs, Gi, or Gq and stimulated with CNO (Tocris) (13).
Membrane and cytoplasmic fractionation of PAE and HT29 cells
Serum-starved PAE and HT29 cells, grown in 10-cm Petri dishes, were stimulated with 1 μm prostaglandin E2 or butaprost, as indicated in Fig. 4. The cells were washed with cold PBS, scraped into 1 ml of cold PBS containing protease and phosphatase inhibitors, and subjected to three freeze/thaw cycles. The lysates were centrifuged at low speed (1,400 rpm for 10 min at 4 °C). Supernatants were centrifuged at 13,000 rpm for 10 min at 4 °C. Cytosol-enriched supernatants were prepared with Laemmli buffer. The pellets were washed once with cold PBS, centrifuged again, incubated with 250 μl of lysis buffer containing 1% Triton X-100 for 20 min, and centrifuged at 13,000 rpm for 10 min at 4 °C. Supernatants containing solubilized membranes were prepared with Laemmli buffer. PRG was analyzed by Western blotting, together with GLUT1 and AKT1, as membrane and cytosol markers, respectively.
Cytoskeletal effects of RH-RhoGEF DH/PH constructs
PAE cells were seeded at low density on gelatin-coated coverslips. Transfected cells were starved for 16 h with serum-free medium. Subsequently, the cells were fixed in 4% paraformaldehyde in PBS for 20 min, washed twice with PBS, and prepared for conventional phalloidin staining. The cell images were visualized in a Leica confocal laser scanning microscope TCS SP8 using a 63× 1.4 oil immersion objective. The images were analyzed with FIJI-ImageJ software. The cells were counted as having filopodia-like structures when they had at least nine of these finger-like protrusions containing F-actin (19).
Statistical analysis
The data are presented as means ± S.E. of at least three independent experiments. Densitometric quantitation of Western blots was done with ImageJ. Active proteins and interactions in pulldowns were normalized respect to total proteins and pulldown efficiency. Statistical analysis was performed using Sigma Plot 11.0, and graphs were prepared with Prism software V8.0. Statistical tests are indicated at the figure legends.
Data availability
All the described data are contained within this article.
Supplementary Material
Acknowledgments
We acknowledge technical assistance provided by Estanislao Escobar-Islas, Margarita Valadez, Jaime Escobar Herrera, David Pérez, Omar Hernández, and Jaime Estrada Trejo.
Author contributions—A. C.-K., I. G.-J., and J. V.-P. conceptualization; A. C.-K., I. G.-J., R. D. C.-V., S. R. A.-G., Y. M. B.-N., J. S. G., G. R.-C., and J. V.-P. formal analysis; A. C.-K., I. G.-J., R. D. C.-V., S. R. A.-G., Y. M. B.-N., and J. V.-P. investigation; A. C.-K., I. G.-J., R. D. C.-V., S. R. A.-G., and J. V.-P. methodology; J. S. G., G. R.-C., and J. V.-P. resources; J. S. G., G. R.-C., and J. V.-P. writing-review and editing; G. R.-C. and J. V.-P. supervision; G. R.-C. and J. V.-P. funding acquisition; J. V.-P. data curation; J. V.-P. validation; J. V.-P. writing-original draft; J. V.-P. project administration.
Funding and additional information—This work was supported by Consejo Nacional de Ciencia y Tecnología (Mexico) Grants 286274 (to J. V.-P.) and 1794 (to G. R.-C.) and fellowships awarded to A. C.-K., I. G.-J., R. D. C.-V., S. R. A.-G., and Y. M. B.-N.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- RH
- RGS homology
- DH
- Dbl homology
- PH
- pleckstrin homology
- GEF
- guanine nucleotide exchange factor
- PRG
- PDZ-RhoGEF
- EGFP
- enhanced GFP
- CNO
- clozapine N-oxide
- GST
- glutathione S-transferase
- HA
- hemagglutinin
- ANOVA
- analysis of variance
- GPCR
- G protein–coupled receptor.
References
- 1. Ridley, A. J., Schwartz, M. A., Burridge, K., Firtel, R. A., Ginsberg, M. H., Borisy, G., Parsons, J. T., and Horwitz, A. R. (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709 10.1126/science.1092053 [DOI] [PubMed] [Google Scholar]
- 2. Pollard, T. D., and Borisy, G. G. (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 10.1016/S0092-8674(03)00120-X [DOI] [PubMed] [Google Scholar]
- 3. Aittaleb, M., Boguth, C. A., and Tesmer, J. J. (2010) Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol. Pharmacol. 77, 111–125 10.1124/mol.109.061234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cook, D. R., Rossman, K. L., and Der, C. J. (2014) Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 33, 4021–4035 10.1038/onc.2013.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lawson, C. D., and Ridley, A. J. (2018) Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 217, 447–457 10.1083/jcb.201612069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rossman, K. L., Der, C. J., and Sondek, J. (2005) GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- 7. Etienne-Manneville, S., and Hall, A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 10.1038/nature01148 [DOI] [PubMed] [Google Scholar]
- 8. Ridley, A. J. (2015) Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 36, 103–112 10.1016/j.ceb.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Machacek, M., Hodgson, L., Welch, C., Elliott, H., Pertz, O., Nalbant, P., Abell, A., Johnson, G. L., Hahn, K. M., and Danuser, G. (2009) Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99–103 10.1038/nature08242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vazquez-Prado, J., Bracho-Valdes, I., Cervantes-Villagrana, R. D., and Reyes-Cruz, G. (2016) Gβγ pathways in cell polarity and migration linked to oncogenic GPCR signaling: potential relevance in tumor microenvironment. Mol. Pharmacol. 90, 573–586 10.1124/mol.116.105338 [DOI] [PubMed] [Google Scholar]
- 11. Hernández-Vásquez, M. N., Adame-García, S. R., Hamoud, N., Chidiac, R., Reyes-Cruz, G., Gratton, J. P., Côté, J. F., and Vázquez-Prado, J. (2017) Cell adhesion controlled by adhesion G protein-coupled receptor GPR124/ADGRA2 is mediated by a protein complex comprising intersectins and Elmo-Dock. J. Biol. Chem. 292, 12178–12191 10.1074/jbc.M117.780304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Welch, H. C. (2015) Regulation and function of P-Rex family Rac-GEFs. Small GTPases 6, 49–70 10.4161/21541248.2014.973770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cervantes-Villagrana, R. D., Adame-García, S. R., García-Jiménez, I., Color-Aparicio, V. M., Beltrán-Navarro, Y. M., König, G. M., Kostenis, E., Reyes-Cruz, G., Gutkind, J. S., and Vázquez-Prado, J. (2019) Gβγ signaling to the chemotactic effector P-REX1 and mammalian cell migration is directly regulated by Gαq and Gα13 proteins. J. Biol. Chem. 294, 531–546 10.1074/jbc.RA118.006254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kozasa, T., Hajicek, N., Chow, C. R., and Suzuki, N. (2011) Signalling mechanisms of RhoGTPase regulation by the heterotrimeric G proteins G12 and G13. J. Biochem. 150, 357–369 10.1093/jb/mvr105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vázquez-Prado, J., Miyazaki, H., Castellone, M. D., Teramoto, H., and Gutkind, J. S. (2004) Chimeric Gαi2/Gα13 proteins reveal the structural requirements for the binding and activation of the RGS-like (RGL)–containing Rho guanine nucleotide exchange factors (GEFs) by Gα13. J. Biol. Chem. 279, 54283–54290 10.1074/jbc.M410594200 [DOI] [PubMed] [Google Scholar]
- 16. Bodmann, E. L., Krett, A. L., and Bünemann, M. (2017) Potentiation of receptor responses induced by prolonged binding of Gα13 and leukemia-associated RhoGEF. FASEB J. 31, 3663–3676 10.1096/fj.201700026R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meyer, B. H., Freuler, F., Guerini, D., and Siehler, S. (2008) Reversible translocation of p115-RhoGEF by G12/13-coupled receptors. J. Cell. Biochem. 104, 1660–1670 10.1002/jcb.21732 [DOI] [PubMed] [Google Scholar]
- 18. Chen, Z., Medina, F., Liu, M. Y., Thomas, C., Sprang, S. R., and Sternweis, P. C. (2010) Activated RhoA binds to the pleckstrin homology (PH) domain of PDZ-RhoGEF, a potential site for autoregulation. J. Biol. Chem. 285, 21070–21081 10.1074/jbc.M110.122549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nobes, C. D., and Hall, A. (1995) Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 10.1016/0092-8674(95)90370-4 [DOI] [PubMed] [Google Scholar]
- 20. García-Mata, R., Wennerberg, K., Arthur, W. T., Noren, N. K., Ellerbroek, S. M., and Burridge, K. (2006) Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 406, 425–437 10.1016/S0076-6879(06)06031-9 [DOI] [PubMed] [Google Scholar]
- 21. Lutz, S., Shankaranarayanan, A., Coco, C., Ridilla, M., Nance, M. R., Vettel, C., Baltus, D., Evelyn, C. R., Neubig, R. R., Wieland, T., and Tesmer, J. J. (2007) Structure of Gαq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318, 1923–1927 10.1126/science.1147554 [DOI] [PubMed] [Google Scholar]
- 22. Rojas, R. J., Yohe, M. E., Gershburg, S., Kawano, T., Kozasa, T., and Sondek, J. (2007) Gαq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology–associated pleckstrin homology domain. J. Biol. Chem. 282, 29201–29210 10.1074/jbc.M703458200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fukuhara, S., Chikumi, H., and Gutkind, J. S. (2001) RGS-containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene 20, 1661–1668 10.1038/sj.onc.1204182 [DOI] [PubMed] [Google Scholar]
- 24. Bielnicki, J. A., Shkumatov, A. V., Derewenda, U., Somlyo, A. V., Svergun, D. I., and Derewenda, Z. S. (2011) Insights into the molecular activation mechanism of the RhoA-specific guanine nucleotide exchange factor, PDZRhoGEF. J. Biol. Chem. 286, 35163–35175 10.1074/jbc.M111.270918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hart, M. J., Jiang, X., Kozasa, T., Roscoe, W., Singer, W. D., Gilman, A. G., Sternweis, P. C., and Bollag, G. (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science 280, 2112–2114 10.1126/science.280.5372.2112 [DOI] [PubMed] [Google Scholar]
- 26. Fukuhara, S., Murga, C., Zohar, M., Igishi, T., and Gutkind, J. S. (1999) A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J. Biol. Chem. 274, 5868–5879 10.1074/jbc.274.9.5868 [DOI] [PubMed] [Google Scholar]
- 27. Fukuhara, S., Chikumi, H., and Gutkind, J. S. (2000) Leukemia-associated Rho guanine nucleotide exchange factor (LARG) links heterotrimeric G proteins of the G12 family to Rho. FEBS Lett. 485, 183–188 10.1016/S0014-5793(00)02224-9 [DOI] [PubMed] [Google Scholar]
- 28. Bandekar, S. J., Arang, N., Tully, E. S., Tang, B. A., Barton, B. L., Li, S., Gutkind, J. S., and Tesmer, J. J. G. (2019) Structure of the C-terminal guanine nucleotide exchange factor module of Trio in an autoinhibited conformation reveals its oncogenic potential. Sci. Signal. 12, eaav2449 10.1126/scisignal.aav2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feng, X., Degese, M. S., Iglesias-Bartolome, R., Vaque, J. P., Molinolo, A. A., Rodrigues, M., Zaidi, M. R., Ksander, B. R., Merlino, G., Sodhi, A., Chen, Q., and Gutkind, J. S. (2014) Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 25, 831–845 10.1016/j.ccr.2014.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Hayre, M., Vázquez-Prado, J., Kufareva, I., Stawiski, E. W., Handel, T. M., Seshagiri, S., and Gutkind, J. S. (2013) The emerging mutational landscape of G proteins and G-protein–coupled receptors in cancer. Nat. Rev. Cancer 13, 412–424 10.1038/nrc3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kapp, G. T., Liu, S., Stein, A., Wong, D. T., Remenyi, A., Yeh, B. J., Fraser, J. S., Taunton, J., Lim, W. A., and Kortemme, T. (2012) Control of protein signaling using a computationally designed GTPase/GEF orthogonal pair. Proc. Natl. Acad. Sci. U.S.A. 109, 5277–5282 10.1073/pnas.1114487109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Derewenda, U., Oleksy, A., Stevenson, A. S., Korczynska, J., Dauter, Z., Somlyo, A. P., Otlewski, J., Somlyo, A. V., and Derewenda, Z. S. (2004) The crystal structure of RhoA in complex with the DH/PH fragment of PDZRhoGEF, an activator of the Ca(2+) sensitization pathway in smooth muscle. Structure 12, 1955–1965 10.1016/j.str.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 33. Hernández-García, R., Iruela-Arispe, M. L., Reyes-Cruz, G., and Vázquez-Prado, J. (2015) Endothelial RhoGEFs: a systematic analysis of their expression profiles in VEGF-stimulated and tumor endothelial cells. Vascul. Pharmacol. 74, 60–72 10.1016/j.vph.2015.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rümenapp, U., Blomquist, A., Schwörer, G., Schablowski, H., Psoma, A., and Jakobs, K. H. (1999) Rho-specific binding and guanine nucleotide exchange catalysis by KIAA0380, a dbl family member. FEBS Lett. 459, 313–318 10.1016/S0014-5793(99)01270-3 [DOI] [PubMed] [Google Scholar]
- 35. Dubash, A. D., Wennerberg, K., García-Mata, R., Menold, M. M., Arthur, W. T., and Burridge, K. (2007) A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. J. Cell Sci. 120, 3989–3998 10.1242/jcs.003806 [DOI] [PubMed] [Google Scholar]
- 36. Jaiswal, M., Gremer, L., Dvorsky, R., Haeusler, L. C., Cirstea, I. C., Uhlenbrock, K., and Ahmadian, M. R. (2011) Mechanistic insights into specificity, activity, and regulatory elements of the regulator of G-protein signaling (RGS)-containing Rho-specific guanine nucleotide exchange factors (GEFs) p115, PDZ-RhoGEF (PRG), and leukemia-associated RhoGEF (LARG). J. Biol. Chem. 286, 18202–18212 10.1074/jbc.M111.226431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Solski, P. A., Wilder, R. S., Rossman, K. L., Sondek, J., Cox, A. D., Campbell, S. L., and Der, C. J. (2004) Requirement for C-terminal sequences in regulation of Ect2 guanine nucleotide exchange specificity and transformation. J. Biol. Chem. 279, 25226–25233 10.1074/jbc.M313792200 [DOI] [PubMed] [Google Scholar]
- 38. Worthylake, D. K., Rossman, K. L., and Sondek, J. (2000) Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature 408, 682–688 10.1038/35047014 [DOI] [PubMed] [Google Scholar]
- 39. Reuther, G. W., Lambert, Q. T., Booden, M. A., Wennerberg, K., Becknell, B., Marcucci, G., Sondek, J., Caligiuri, M. A., and Der, C. J. (2001) Leukemia-associated Rho guanine nucleotide exchange factor, a Dbl family protein found mutated in leukemia, causes transformation by activation of RhoA. J. Biol. Chem. 276, 27145–27151 10.1074/jbc.M103565200 [DOI] [PubMed] [Google Scholar]
- 40. Rossman, K. L., Worthylake, D. K., Snyder, J. T., Siderovski, D. P., Campbell, S. L., and Sondek, J. (2002) A crystallographic view of interactions between Dbs and Cdc42: PH domain-assisted guanine nucleotide exchange. EMBO J. 21, 1315–1326 10.1093/emboj/21.6.1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Snyder, J. T., Worthylake, D. K., Rossman, K. L., Betts, L., Pruitt, W. M., Siderovski, D. P., Der, C. J., and Sondek, J. (2002) Structural basis for the selective activation of Rho GTPases by Dbl exchange factors. Nat. Struct. Biol. 9, 468–475 10.1038/nsb796 [DOI] [PubMed] [Google Scholar]
- 42. Kristelly, R., Gao, G., and Tesmer, J. J. (2004) Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J. Biol. Chem. 279, 47352–47362 10.1074/jbc.M406056200 [DOI] [PubMed] [Google Scholar]
- 43. Cash, J. N., Davis, E. M., and Tesmer, J. J. (2016) Structural and biochemical characterization of the catalytic core of the metastatic factor P-Rex1 and its regulation by PtdIns(3,4,5)P3. Structure 24, 730–740 10.1016/j.str.2016.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lucato, C. M., Halls, M. L., Ooms, L. M., Liu, H. J., Mitchell, C. A., Whisstock, J. C., and Ellisdon, A. M. (2015) The phosphatidylinositol (3,4,5)-trisphosphate-dependent Rac exchanger 1·Ras-related C3 botulinum toxin substrate 1 (P-Rex1·Rac1) complex reveals the basis of Rac1 activation in breast cancer cells. J. Biol. Chem. 290, 20827–20840 10.1074/jbc.M115.660456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guilluy, C., Dubash, A. D., and García-Mata, R. (2011) Analysis of RhoA and Rho GEF activity in whole cells and the cell nucleus. Nat. Protoc. 6, 2050–2060 10.1038/nprot.2011.411 [DOI] [PubMed] [Google Scholar]
- 46. Adame-García, S. R., Cervantes-Villagrana, R. D., Orduña-Castillo, L. B., Del Rio, J. C., Gutkind, J. S., Reyes-Cruz, G., Taylor, S. S., and Vázquez-Prado, J. (2019) cAMP-dependent activation of the Rac guanine exchange factor P-REX1 by type I protein kinase A (PKA) regulatory subunits. J. Biol. Chem. 294, 2232–2246 10.1074/jbc.RA118.006691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McKenzie, A. J., Svec, K. V., Williams, T. F., and Howe, A. K. (2020) Protein kinase A activity is regulated by actomyosin contractility during cell migration and is required for durotaxis. Mol. Biol. Cell 31, 45–58 10.1091/mbc.E19-03-0131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Howe, A. K., Baldor, L. C., and Hogan, B. P. (2005) Spatial regulation of the cAMP-dependent protein kinase during chemotactic cell migration. Proc. Natl. Acad. Sci. U.S.A. 102, 14320–14325 10.1073/pnas.0507072102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lim, C. J., Han, J., Yousefi, N., Ma, Y., Amieux, P. S., McKnight, G. S., Taylor, S. S., and Ginsberg, M. H. (2007) α4 integrins are type I cAMP-dependent protein kinase-anchoring proteins. Nat. Cell Biol. 9, 415–421 10.1038/ncb1561 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the described data are contained within this article.