

Abstract
Deregulation of noncoding RNAs, including microRNAs (miRs), is implicated in the pathogenesis of many human cancers, including breast cancer. Through extensive analysis of The Cancer Genome Atlas, we found that expression of miR-22-3p is markedly lower in triple-negative breast cancer (TNBC) than in normal breast tissue. The restoration of miR-22-3p expression led to significant inhibition of TNBC cell proliferation, colony formation, migration, and invasion. We demonstrated that miR-22-3p reduces eukaryotic elongation factor 2 kinase (eEF2K) expression by directly binding to the 3′ untranslated region of eEF2K mRNA. Inhibition of EF2K expression recapitulated the effects of miR-22-3p on TNBC cell proliferation, motility, invasion, and suppression of phosphatidylinositol 3-kinase/Akt and Src signaling. Systemic administration of miR-22-3p in single-lipid nanoparticles significantly suppressed tumor growth in orthotopic MDA-MB-231 and MDA-MB-436 TNBC models. Evaluation of the tumor response, following miR-22-3p therapy in these models using a novel mathematical model factoring in various in vivo parameters, demonstrated that the therapy is highly effective against TNBC. These findings suggest that miR-22-3p functions as a tumor suppressor by targeting clinically significant oncogenic pathways and that miR-22-3p loss contributes to TNBC growth and progression. The restoration of miR-22-3p expression is a potential novel noncoding RNA-based therapy for TNBC.
Keywords: triple-negative breast cancer, breast cancer, miR-22, Src, eEF2K, EF2K, PI3K, Akt, nanoparticles, liposomes, tumor suppressor, gene regulation, gene silencing, therapy
Graphical abstract
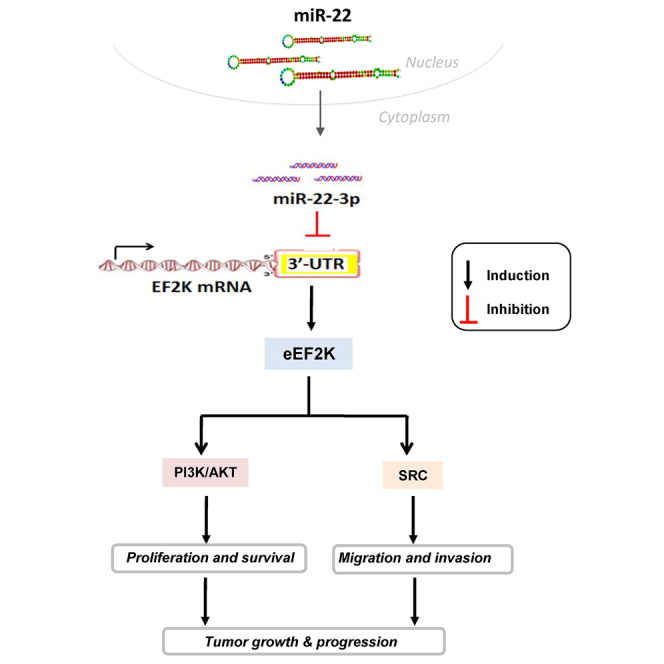
Our study demonstrates that microRNA-22-3p (miR-22-3p) acts as a tumor suppressor in which its expression is markedly reduced in TNBC tumors. Restoration of its expression by miR-22-3p nanotherapy suppress tumor growth and progression by directly inhibiting of eEF2K-PI3K/AKT and Src signaling. miR-22-3p-based RNA therapy may be novel treatment for TNBC.
Introduction
Diagnosis of about 280,000 new breast cancer cases in the United States was expected by the American Cancer Society in 2021.1 Gene-expression profiles have led to the classification of breast cancer into five main groups: two estrogen receptor (ER) positive (luminal A and B) and three ER negative (normal breast like, human epidermal growth factor receptor 2 positive, and basal like).2 Triple-negative breast cancer (TNBC), which constitutes 20% of breast cancer cases, is characterized by a lack of expression of ER, progesterone receptor, and human epidermal growth factor receptor 2 (HER2/Neu). TNBC has the highest mortality rates of all breast cancer subtypes due to its highly aggressive and metastatic nature and insensitivity to chemotherapeutics. High TNBC mortality rates are also attributed to its significant tumor heterogeneity, with six genetically defined subgroups, and quick development of resistance to standard chemotherapy.3 Patients with advanced, metastatic disease have a median survival time of only about 12 months. Currently, US Food and Drug Administration-approved targeted therapy for TNBC is lacking. Therefore, TNBC is an unmet therapeutic challenge, and better understanding of the biology of TNBC and identification of novel targets for this disease are critical.3
MicroRNAs (miRNAs) are small-noncoding RNAs, 20−25 nt in length, that silence their target genes by binding to the specific binding sites at 3′ untranslated regions (UTRs), regulating their post-transcriptional expression via either inhibition of translation or degradation of mRNA.4 Functional studies have confirmed that many cases of cancer are directly linked to miRNA deregulation, with miRNAs acting as tumor suppressors or oncogenes. Currently, more than 2,500 human mature miRNA sequences are cataloged in the miRBase miRNA registry (http://www.mirbase.org). Some miRNAs function as tumor-suppressor miRNAs and are downregulated in tumors, whereas oncomiRs are overexpressed in cancer cells. The miRNAs miR-10b, miR-21, miR-146a, miR-155, and miR-182 are overexpressed in TNBCs, whereas miR-34a, miR-603, miR-221/222, and tumor-suppressor miRNAs are under expressed in them. Recently, miRNA-based therapeutics using miRNA mimics and inhibitors (antimiRs) entered a phase 1 clinical trial in patients with advanced carcinomas, suggesting a new era of cancer therapy with a new class of therapeutics.5
miR-22 is a poor prognostic factor in breast cancer patients and overexpressed in TNBC cell lines, and it promotes cell invasion and metastasis.6 The role of miR-22-3p and the underlying molecular mechanism contributing to TNBC tumorigenesis remain to be elucidated. In this study, our evaluation of miRNA expression profiles in The Cancer Genome Atlas patient database demonstrated that miR-22-3p is significantly downregulated in TNBCs. Also, we demonstrated for the first time that miR-22-3p expression is significantly reduced in human TNBCs and acts as a tumor suppressor in TNBC cells by inhibiting eukaryotic elongation factor 2 (EF2) kinase (eEF2K) oncogenic signaling by directly binding to the 3′ UTR of eEF2K mRNA, leading to inhibition of TNBC cell proliferation, colony formation, migration, and invasion. More importantly, in vivo delivery of miR-22-3p in single lipid-based nanoparticles suppresses tumor growth in TNBC models, suggesting that miR-22-3p acts as a tumor suppressor and that delivery of this miRNA into tumors is a potential novel therapy for TNBC.
Results
miR-22-3p is downregulated in human TNBCs and TNBC cell lines
Our integrated genomic/transcriptomic analysis of breast cancers from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) database, which includes long-term clinical outcomes, showed that miR-22-3p expression was significantly lower in TNBCs (n = 159) than in normal breast tissue samples (n = 93; p < 0.0007) (Figure 1A). Similarly, the expression of miR-22-3p in basal-like tumors (n = 122), which are often classified as TNBC, was lower than that in normal breast tissue (n = 93, p < 0.0014) (Figure 1B). To validate this biological effect of miR-22-3p, we investigated the relative expression of miR-22-3p in various TNBC cell lines and the human normal immortalized mammary epithelial cell line HMEC. We found that expression of miR-22-3p was considerably lower in all TNBC cell lines tested, including MDA-MB-231, MDA-MB-436, and BT-20, than in noncancerous HMEC cells (Figure 1C), suggesting that miR-22-3p is downregulated in TNBCs.
Figure 1.

miR-22-3p expression is downregulated in human TNBCs and TNBC cell lines
(A and B) METABRIC gene-expression analysis of miR-22-3p levels in TNBCs (n = 159) (A) and basal-like breast tumors (n = 122) (B) compared to normal breast tissue samples (n = 93). Results of quantitative RT-PCR analysis of miR-22-3p expression in normal breast epithelial cells (HMECs) and TNBC cell lines using specific primers for miR-22-3p (C). ∗∗p < 0.01; ∗∗∗p < 0.001.
miR-22-3p suppresses the proliferation and clonogenicity of TNBC cells
Based on the findings described above, we hypothesized that reduced miR-22-3p expression in TNBC cells may cause increased proliferation. To test this hypothesis, we subjected MDA-MB-231 and MDA-MB-436 cells transiently transfected with control (cont) miRNA or a miR-22-3p mimic to a colony-formation assay. As shown in Figures 2A−2F, 10 days after transfection, miR-22-3p inhibited the proliferation of TNBC cells markedly more than control miRNA did. In addition, MDA-MB-231, MDA-MB-436, and BT-20 cells transfected with miR-22-3p had lower colony-formation capacity (both size and number of colonies) than these cells transfected with control miRNA (p < 0.05).
Figure 2.

Ectopic expression of miR-22-3p suppresses the clonogenic capacity of TNBC cells in vitro
(A–F) Effects of overexpression of miR-22-3p on the colony-forming capacity of MDA-MB-231 (A and B), MDA-MB-436 (C and D), and BT-20 (E and F) cells. Left panels, representative culture dishes from a colony-formation assay. Right panels, the numbers of colonies formed by the cells transfected with miR-22-3p normalized according to the numbers of colonies formed by the cells transfected with control miRNA (Cont-miR). The data are presented as mean (±SD) (p < 0.05). ∗∗p < 0.01; ∗∗∗p < 0.001.
miR-22-3p expression impairs TNBC cell motility, migration, and invasion
To better understand the functionality of miR-22-3p in the progression of aggressive TNBC, we tested the effect of miR-22-3p on the invasion of TNBC cells. We then performed an in vitro Transwell assay with Matrigel to determine the effect of miR-22-3p expression on the invasiveness of TNBC cells. MDA-MB-231 and MDA-MB-436 cells transfected with miR-22-3p were significantly less invasive than cells transfected with control miRNA (mean [± standard deviation (SD)] number of invaded cells: MDA-MB-231 transfected with miR-22-3p, 254.50 ± 20.46; MDA-MB-231 transfected with control miRNA, 538.00 ± 18.58; MDA-MB-436 transfected with miR-22-3p, 237 ± 46; MDA-MB-436 transfected with control miRNA, 620.90 ± 33.18; p < 0.0001) (Figures 3A and 3B). We also examined the effect of miR-22-3p on the motility and migration of TNBC cells. We transfected MDA-MB-231 and MDA-MB-436 cells with a miR-22-3p mimic or control miRNA. In a wound-healing assay, the migration of cells transfected with the miR-22-3p mimic was markedly impaired when compared with that of cells transfected with the control miRNA. Specifically, the migration of cells transfected with the control miRNA was inhibited by 1% and 5%, whereas that of cells transfected with miR-22-3p was inhibited by 86% (MDA-MB-231) and 80% (MDA-MB-436) (Figures 3C and 3D). Similarly, the numbers of invaded and migrated BT-20 and BT-549 TNBC cells transfected with miR-22-3p were substantially lower than those of cells transfected with control miRNA (Figures S1A−S1D). These results demonstrated that miR-22-3p expression plays an important role in TNBC cells by suppressing the migration and invasion.
Figure 3.

The expression of miR-22-3p in TNBC cells suppresses their migration and invasion in vitro
(A and B) The invasiveness of MDA-MB-231 (A) and MDA-MB-436 (B) cells was assessed after miR-22-3p or control miRNA transfection or did not undergo transfection (NT) on a Matrigel Transwell assay. The cells were cultured for 72 h after transfection and then transferred to Transwell chambers and incubated for 24 h. The invading cells were counted, and the mean (±SD) results of triplicate experiments are shown on the right. (C and D) The migration capacity of MDA-MB-231 (C) and MDA-MB-436 (D) cells transfected with miR-22-3p or control miRNA or those did not undergo transfection were assessed using a wound-healing assay (at 0 h and 48 h). These representative images show the percentage inhibition or number of invaded cells, which were calculated compared to control treatment. ∗∗∗∗p < 0.0001.
eEF2K is a predicted target for miR-22-3p
To identify the molecular mechanisms by which miR-22-3p mediates its effects on inhibition of cell proliferation, migration, and invasion, we used four prediction algorithms (TargetScan, DIANA Tools, miRDB, and microRNA.org) to identify the mRNA targets of miRNAs. We identified a miR-22-3p-binding site in the 3′ UTR sequence of eEF2K. This analysis suggested that eEF2K is a target gene in which its expression is suppressed by miR-22-3p (Figure 4A).
Figure 4.

miR-22-3p expression suppresses eEF2K expression in TNBC cells by binding directly to the 3′ UTR of eEF2K mRNA
(A) Prediction of binding sites on the 3′ UTR region of eEF2K mRNA that is bound by miR-22-3p by using four different algorithms (TargetScan, miRDB, DIANA Tools, and microRNA.org). (B) Protein levels of eEF2K upon ectopic miR-22-3p expression in MDA-MB-231 cells. The MDA-MB-231 cells were transfected with the miR-22-3p mimic or control miRNA, and eEF2K protein levels were analyzed using western blotting 72 h after transfection. β-actin was used as a loading control. Band intensities were quantified using densitometry. (C) miR-22-3p transfection decreased eEF2K mRNA expression in MDA-MB-231 cells. The relative eEF2K mRNA expression levels in these MDA-MB-231 cells were analyzed using qRT-PCR with specific primers 48 h after transfection with miR-22-3p or a control miRNA. Data were normalized to the expression of β-actin and are presented as mean (±SD) from three independent experiments. ∗∗p < 0.001. (D) Two predicted miR-22-3p-binding sites in the 3′ UTR of human wild-type eEF2K mRNA and their sequences. Mutations that were introduced in the seed miR-22-3p-binding sequence of the full-length eEF2K 3′ UTR are shown in red. (E) Luciferase reporter assay results showing that miR-22-3p directly targets the eEF2K 3′ UTR-luciferase reporter pEZX-MT06 (with wild-type or mutant [MT] miR-22-3p-binding sites) in cells incubated with a miR-22-3p mimic for 48 h before analysis. The firefly luciferase activity of the reporter was normalized to the internal Renilla luciferase activity. The data are presented as mean (±SD) from three independent experiments. ∗∗∗∗p < 0.0001.
miR-22-3p downregulates eEF2K in TNBC cells
To characterize the relationship of miR-22-3p with eEF2K in TNBCs, we performed a western blot analysis of eEF2K protein expression in MDA-MB-231 cells after transfection with a miR-22-3p mimic using the HiPerFect transfection reagent (72 h). As shown in Figure 4B, miR-22-3p expression significantly suppressed (by 62.1%) eEF2K protein expression in these cells (p < 0.001). Furthermore, quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) demonstrated that ectopic expression of miR-22-3p led to significantly reduced eEF2K mRNA expression in MDA-MB-231 cells (p < 0.001) (Figure 4C).
miR-22-3p binds directly to the 3′ UTR of eEF2K
To confirm that miR-22-3p directly targets eEF2K in human embryonic kidney HEK293 cells, we inspected the 3′ UTR of eEF2K mRNA and found a site that could be recognized by miR-22-3p (Figure 4D). We used luciferase reporter assays in which we cotransfected miR-22-3p or scrambled negative control miRNA and luciferase constructs containing the eEF2K 3′ UTR or a control eEF2K 3′ UTR clone into HEK293 cells. After 48 h, the luciferase activity in the cells cotransfected with miR-22-3p, and the eEF2K 3′ UTR clone decreased by about 60% compared with the control group (Figure 4E), whereas we found no change in the luciferase activity in cells transfected with a plasmid containing a mutated eEF2K-binding site and miR-22-3p. These results demonstrated that miR-22-3p directly and specifically recognizes and binds to the 3′ UTR to regulate expression of an eEF2K mRNA transcript.
Knocking down eEF2K inhibits TNBC cell proliferation, migration, and invasion
Based on the findings described above, we hypothesized that the effects of miR-22-3p on TNBC cells are mediated by the downregulation of eEF2K. To test this hypothesis, we transiently transfected MDA-MB-231 cells with control small interfering RNA (siRNA) or eEF2K siRNA and subjected them to a colony-formation assay. As shown in Figure 5A, MDA-MB-231 cells transfected with eEF2K siRNA exhibited significantly less proliferation and lower colony-formation capacity than did cells transfected with the control siRNA (p = 0.0157). To define the effect of eEF2K on TNBC cell migration, we assessed wound healing in MDA-MB-231 cells transfected with eEF2K siRNA, which showed less inhibition of migration (73% inhibition) than that of cells transfected with the control siRNA (Figure 5B). To determine whether knockdown of eEF2K has a similar impact on cell invasion, we performed a Matrigel invasion assay. Knockdown of eEF2K resulted in considerably fewer invading MDA-MB-231 cells (mean [±SD], 210.80 ± 17.87) than did treatment with control siRNA (mean [±SD], 301.00 ± 15.26), an effect similar to that of miR-22-3p transfection (Figure 5C).
Figure 5.

eEF2K knockdown recapitulates the effects of miR-22-3p expression and leads to inhibition of TNBC cell proliferation, colony formation, invasion, and migration in vitro
(A) Knockdown of eEF2K by siRNA (50 nM) significantly reduced the number of colonies formed by MDA-MB-231 cells. ∗p = 0.0157. (B) Invasiveness of the eEF2K-depleted MDA-MB-231 cell transfected with eEF2K siRNA or control siRNA or that did not undergo transfection were assessed using a Matrigel Transwell assay. Cells that invaded the Matrigel and passed through the membrane were counted under a light microscope. ∗∗p < 0.01. (C) The migration capacity of eEF2K-knockdown MDA-MB-231 cells was assessed using a wound-healing assay, and a wound was formed in each cell monolayer via scraping. The area of the wound was measured at 0 h and 48 h. ∗∗∗∗p < 0.0001.
Inhibition of eEF2K expression recapitulates the effects of miR-22-3p expression by regulating phosphatidylinositol 3-kinase (PI3K)/Akt and Src signaling
We used western blot analysis to detect the mechanism underlying this effect and found that eEF2K expression was dramatically lower in miR-22-3p mimic-transfected MDA-MB-231 cells than in control miRNA-transfected cells (Figure 6A). Levels of eEF2K, phosphorylated Akt (p-Akt; Ser473), and p-Src (Tyr416) were all reduced following ectopic expression of miR-22-3p in these TNBC cells. Western blot analysis showed that siRNA-mediated knockdown of eEF2K also markedly reduced the expression of p-Akt (Ser473) and p-Src (Tyr416) in MDA-MB-231 cells (Figure 6B). As expected, eEF2K knockdown inhibited phosphorylation of the previously identified target p-EF2 (Thr56), which we used as a positive control.
Figure 6.

miR-22-3p inhibits PI3K/Akt and Src signaling by suppressing eEF2K expression
(A) Western blot analysis showing that ectopic expression of miR-22-3p inhibited the expression of eEF2K, p-Akt (Ser473), p-Src (Tyr416), and p-EF2 (Thr56) in cells treated with miR-22-3p. TNBC cells were analyzed following transfection with a miR-22-3p mimic (72 h) or control miRNA. (B) Western blot analysis of p-Akt (Ser473), p-Src (Tyr416), and p-EF2 (Thr56) protein levels in MDA-MB-231 cells after siRNA-mediated knockdown of eEF2K. (C and D) Effect of miR-22-3p transfection in eEF2K-overexpressed MDA-MB-231 cells on cell proliferation and colony formation (10 days). ∗p < 0.05; n.s, not significant.
Expression of eEF2K reverses the effects of miR-22-3p on TNBC cells
To further validate that the effects of miR-22-3p are mediated by targeting eEF2K in TNBC cells, we performed a rescue experiment. To this end, we transduced MDA-MB-231 cells with a lentiviral expression vector containing eEF2K to induce overexpression of eEF2K in MDA-MB-231 cells. We found that overexpression of eEF2K completely reversed the effect of miR-22-3p of inhibiting cell proliferation and colony formation. However, miR-22-3p significantly inhibited the proliferation of and colony formation by mock control vector-transduced MDA-MB-231 cells (Figures 6C and 6D), suggesting that eEF2K plays a key role in miR-22-3p-induced effects on TNBC cells.
In vivo therapeutic systemic administration of miR-22-3p inhibits the growth of orthotopic TNBC xenografts in mice
To determine the role of miR-22-3p in TNBC growth and progression in vivo, as well as its therapeutic potential for this cancer, we injected MDA-MB-231 and MDA-MB-436 orthotopic xenograft mouse models a formulation of miR-22-3p or control miRNA in single-lipid liposomal nanoparticles (SLNPs), once a week for 4 weeks. The volumes of the xenograft tumors were measured every week for the 4 weeks of treatment. At the end of week 4, tumors were excised and weighed and analyzed for protein expression, proliferation, angiogenesis, and apoptosis markers. We measured the volumes of the xenograft tumors in both groups every week during the treatment period. In both the MDA-MB-231 (Figure 7A) and MDA-MB-436 (Figure 8A) TNBC models, the tumor growth in the mice given miR-22-3p was significantly delayed when compared with that in the mice given control miRNA (p < 0.05). After 4 weeks of treatment, we observed no toxic effects in the mice given SLNP-miR-22-3p or those given SLNP-control miRNA. The mice appeared healthy and did not lose weight during the treatment.
Figure 7.

Systemic administration of single lipid nanoparticle (SLNP)-miR-22-3p inhibits the growth of MDA-MB-231 TNBC xenografts in nude mice
MDA-MB-231 TNBC cells were orthotopically injected into the mammary fat pads of female athymic nude mice. The mice were randomized to two treatment groups: SLNPs incorporating miR-22-3p or SLNP-control miRNA (0.15 mg/kg administered intravenously twice a week for 4 weeks; five mice/group). (A) MDA-MB-231 tumor xenograft volumes were measured weekly and are shown as mean (±SD). The tumor tissues of TNBC xenografts in MDA-MB-231 collected from nude mice that received SLNP-miR-22-3p or control miRNA. (B and C) miR-22-3p (B) and eEF2K mRNA (C) levels were detected by qRT-PCR. ∗p < 0.05 and ∗∗p < 0.01, respectively. (D−G) Protein expressions of p-eEF2 (Thr56) (D), eEF2 (E), and eEF2K (F) were evaluated by western blot. ∗∗∗∗p < 0.0001. (H−J) Immunohistochemistry analysis of Ki-67 proliferation marker (H) and CD31 angiogenesis marker (I) and TUNEL assay showing apoptotic cells (green) were evaluated in in MDA-MB-231 tumors collected from mice treated with SLNP-miR22-3p or SLNP-control miRNA (J). ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Figure 8.

Systemic administration of SLNP-miR-22-3p inhibits the growth of MDA-MB-436 TNBC xenografts in nude mice
(A) MDA-MB-436 TNBC cells were orthotopically injected into the mammary fat pads of female athymic nude mice. The mice were randomized to two treatment groups: SLNP-miR-22-3p or SLNP-control miRNA (0.15 mg/kg administered intravenously twice a week for 4 weeks; five mice/group). MDA-MB-436 tumor xenograft volumes were measured weekly. (B−E) Western blot analyses of p-eEF2 (B), eEF2 (C), and eEF2K (D) protein levels in MDA-MB-436 tumors. ∗∗p < 0.01; ∗∗∗p < 0.001. (F and G) Immunohistochemistry analysis of Ki-67 (F) and CD31 (G) in MDA-MB-436 tumor xenograft after SLNP-miR22-3p or control-miRNA treatments. (H) TUNEL assay showing apoptotic cells (green) in tumors after SLNP-miR22-3p or SLNP-control miRNA in an MDA-MB-436 orthotopic model. ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. (I) A mathematical model was applied to evaluate the in vivo antitumor efficacy of treatment with miR-22-3p in the MDA-MB-231 and MDA-MB-436 TNBC orthotopic xenograft mouse models. Tumor growth rates (r) were first estimated for MDA-MB-231 and MDA-MB-436 cells (solid lines), and their tumor growth-inhibition (α) and decay (β) rates were estimated under treatment with miR-22-3p nanoparticles (dashed lines). Estimated parameter values are shown at the top of the graph.
To confirm the systemic delivery of miR-22-3p by the SLNP nanoliposomal carriers, using qRT-PCR, we evaluated the relative expression of miR-22-3p in tumor samples obtained from MDA-MB-231 tumor-bearing mice after treatment with SLNP-miR-22-3p or SLNP-control miRNA. Our results showed that the expression of miR-22-3p was significantly higher in the SLNP-miR-22-3p-treated mice than in the SLNP-control miRNA-treated mice (Figure 7B; p = 0.0102). We also analyzed the tumor samples for the effects of miR-22-3p gene delivery and evaluated the expression of eEF2K mRNA in tumor samples obtained from MDA-MB-231 tumor-bearing mice using qRT-PCR. Tumor samples from miR-22-3p-treated mice had significantly lower expression of eEF2K mRNA than did samples from control miRNA-treated mice (Figure 7C; p = 0.0098). We further analyzed tumor samples obtained from these mice using western blotting to determine the effects of miR-22-3p on p-EF2 (Thr56), the only known downstream target of eEF2K, total eEF2, and eEF2K protein expression. We observed that miR-22-3p significantly decreased the expression of p-eEF2 (Thr56) (Figure 7D) and eEF2K (Figure 7F) but no significant differences in total eEF2 (Figure 7E) levels in tumors compared to those given control miRNA (p < 0.0001 and p = not significant, respectively). Similar findings were observed in MDA-MB-436 tumors with regard to p-eEF2 (Thr56), eEF2K, and eEF2 expressions after SLNP-miR22-3p treatments compared to SLNP-miR-control (Figures 7D−7G and 8B−8E) (p = 0.0002 and p = not significant, respectively).
In vivo treatment with SLNP-miR-22-3p inhibits tumor-cell proliferation and microvessel density and induces apoptosis in MDA-MB-231 and MDA-MB-436 TNBC tumor models
We assessed the intratumoral-proliferative effect of miR-22-3p in TNBC tumors obtained from MDA-MB-231 and MDA-MB-436 tumor-bearing mice by evaluating the expression of the proliferation marker Ki-67 using immunohistochemistry. The numbers of Ki-67-positive tumor cells were significantly lower in miR-22-3p-treated mice than in control miRNA-treated mice (MDA-MB-231 tumors, p = 0.0006, Figure 7H; MDA-MB-436 tumors, p = 0.0001, Figure 8F). Also, immunohistochemical analyses of microvessel density as a marker of angiogenesis showed that the number of CD31-positive endothelial cells was dramatically lower in the miR-22-3p group than in the control miRNA group (MDA-MB-231 tumors, p = 0.0002, Figure 7I; MDA-MB-436 tumors, p = 0.0175, Figure 8G). Finally, a terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick end-labeling (TUNEL) assay demonstrated that treatment with miR-22-3p resulted in a significantly greater number of TUNEL-positive cells (green; indicating apoptosis) than did treatment with control miRNA in both TNBC models (MDA-MB-231 tumors, p = 0.0348, Figure 7J; MDA-MB-436 tumors, p = 0.0165, Figure 8H). Taken together, these results revealed that systemic treatment with miR-22-3p inhibits TNBC growth in vivo in association with significant inhibition of tumor-cell proliferation and microvessel density and induction of apoptosis.
Analysis of the antitumor efficacy of miR-22-3p therapy in TNBC using a mathematical model
To assess the efficacy of miR-22-3p therapy for TNBC, we used a mathematical model for the TNBC orthotopic xenograft mouse models (Figure 8I). Researchers designed the mathematical model to incorporate various parameters, including tumor growth, intratumoral proliferation rates, induction of apoptosis, and molecular changes, such as angiogenesis.7, 8, 9, 10, 11, 12 We first fit the model to the data for the control group (i.e., in the absence of actual treatment, where both α = 0 and β = 0 at all times) to determine the tumor growth rate (r). We then fit the model to the data for mice given a miR-22-3p mimic to determine the tumor growth-inhibition rate (α) and tumor decay rate (β). The estimated parameter values are shown in Figure 8I. Next, we used the mathematical model to predict treatment outcomes in vivo, as indicated by tumor volume change over time. The model successfully predicted treatment outcome and reproduced experimentally obtained values with acceptable accuracy under both treatment conditions and for both xenograft models. With and without treatment, tumor size increased monotonically over the entire treatment course (in reality, a tumor cannot grow indefinitely in a confined environment), but the results demonstrated the impact of SLNP-miR-22-3p on the growth of both MDA-MB-231 and MDA-MB-436 tumors. Of note, the r and β for both tumor types were similar, but the α for MDA-MB-436 tumors was almost 40 times that for MDA-MB-231 tumors. This demonstrated that miR-22-3p therapy has greater effects on suppression of the growth of MDA-MB-436 tumors than in MDA-MB-231 tumors by lowering the cell proliferation potential but has less of an apoptosis-inducing effect.
Discussion
Our results suggest for the first time that miR-22-3p is a clinically significant tumor suppressor in which its expression is significantly reduced in TNBCs. Our data demonstrated that miR-22-3p acts as a tumor suppressor in TNBCs by directly regulating eEF2K expression, thereby inhibiting cell proliferation, migration, and invasion and tumorigenesis. More importantly, our findings also demonstrated for the first time that in vivo therapeutic delivery of miR-22-3p in SLNPs leads to significant inhibition of tumor growth in multiple TNBC models. Evaluation of the effects of SLNP-miR-22-3p in TNBC models using a mathematical model suggested that treatment with miR-22-3p has antitumor efficacy with highly reproducible accuracy.
eEF2K is one of a group of unusual protein kinases termed α-kinases and is the only calcium/calmodulin-dependent member of that group.13, 14, 15 It regulates the elongation stage of protein synthesis by phosphorylating and inhibiting its only known substrate, eEF2. Phosphorylation of serine and threonine residues (such as Thr56, Ser366, Ser78, and Ser398) can regulate the activity of eEF2K. When phosphorylated on Thr56 through its amino acid sequence, eEF2 cannot engage with ribosomes and is essentially inactive during translation.16, 17, 18 eEF2K is highly overexpressed in TNBCs and promotes proliferation, invasion, tumorigenesis, and chemotherapy resistance of highly aggressive tumors such as TNBC13 and pancreatic cancer by inducing oncogenic signaling pathways related to cell growth, survival, and invasion.13,19,20 eEF2K was only relatively recently proposed as a potential therapeutic target for cancer.13 Inhibition of eEF2K signaling suppresses tumor growth and significantly enhances the efficacy of most the widely used chemotherapeutic agents in TNBC models.13 Similarly, in pancreatic cancer cases, eEF2K is involved in regulation of the transglutaminase 2/urokinase-type plasminogen activator receptor/matrix metalloproteinase-2 axis and epithelial-mesenchymal transition.20 eEF2K is involved in inducing effects on multiple signaling pathways, including PI3K/Akt and Src, to promote several processes associated with cell migration and invasion.13
The results of the present study demonstrated that miR-22-3p expression is significantly reduced in TNBC cells. Reduced expression of miR-22-3p, a direct regulator of eEF2K, may be one of the mechanisms that leads to upregulation of eEF2K in TNBC cells. Furthermore, in vitro and in vivo restoration of miR-22-3p expression recapitulates the effects of eEF2K suppression on TNBC, regulation of PI3K/Akt and Src, and inhibition of tumor-cell proliferation, migration, and invasion, further demonstrating that regulation of these pathways by miR-22-3p is mediated by inhibition of eEF2K expression.
Notably, as demonstrated by the mathematical model, the inhibition of MDA-MB-436 tumors induced by treatment with SLNP-miR-22-3p was superior to that of MDA-MB-231 tumors, demonstrating that MDA-MB-231 cells are less responsive than MDA-MB-436 cells to treatment with miR-22-3p. Although both MDA-MB-436 and MDA-MB-231 cells are considered TNBC-basal B/mesenchymal cells, these two cell types have similar levels of miR-22-3p and eEF2K expression and harbor p53 mutations. MDA-MB-231 cells also have a K-RAS mutation, which may reduce their sensitivity in in vivo studies, demonstrating that genetic differences in tumors may alter the response of TNBC to treatment with miR-22-3p. Also, miR-22-3p may have suppressed other potential target genes, which may have contributed to tumor growth and progression in the MDA-MB-436 model. Therefore, future studies should investigate the genes potentially regulated by miR-22-3p.
Recent studies suggested that miR-22 acts as an oncogene by targeting ten-eleven-translocation (TET) in hematopoietic stem cells21 and breast cancer cells.6 In particular, Pandey et al.6 suggested a role for miR-22 in promoting stemness and metastasis. However, they did not indicate whether they investigated the effects of miR-22-3p or miR-22-5p. They did report that miR-22 is upregulated in some cell lines and inhibits the expression of TIP60, a lysine acetyltransferase. In their study, they showed that blocking endogenous miR-22 can restore TIP60 levels, which in turn decreases the migration and invasive capacity of MDA-MB-231 cells, suggesting that miR-22 induces breast cancer invasion and progression. In contrast, our findings described herein suggest that miR-22-3p acts as a tumor suppressor and inhibits proliferation, migration, and invasion by targeting eEF2K in three TNBC cell lines (MDA-MB-231, MDA-MB-436, and BT-20). More importantly, our studies demonstrated that knockdown of eEF2K recapitulated the effects of miR-22-3p in inhibiting the PI3K/Akt axis and Src activity. However, other studies suggested that miR-22 acts as a tumor suppressor for osteosarcoma and prostate, cervical, and lung cancer by targeting ATP citrate lyase.22 Because miR-22-3p expression was markedly lower in TNBCs (n = 159) than in normal breast tissue samples (n = 93), restoring miR-22-3p expression via in vivo delivery of SLNPs containing an miR-22-3p mimic provided significant antitumor effects through inhibition of tumor-cell proliferation and angiogenesis and induction of apoptosis. Overall, the in vivo experiment in the present study demonstrated that therapeutic targeting of eEF2K designed to induce miR-22-3p overexpression or reduce eEF2K expression is a potential monotherapeutic approach to controlling TNBC growth. However, studies combining miR-22-3p therapy with standard chemotherapy should be evaluated to determine whether miR-22-3p sensitizes TNBC to chemotherapy and enhances its therapeutic efficacy.
Materials and methods
Cell lines and culture conditions
HMEC; TNBC cell lines MDA-MB-231, MDA-MB-436, BT-20, BT-549, and MDA-MB-361; and HEK cell line HEK293 were purchased from the ATCC. MDA-MB-231, MDA-MB-436, and HEK293 cells were cultured in a mixture of Dulbecco’s modified Eagle’s medium and F12 medium supplemented with 10% fetal bovine serum and a 100-U/mL penicillin-streptomycin solution (Sigma-Aldrich). HMEC cells were maintained in a nutrient mixture consisting of Dulbecco’s modified Eagle’s medium/F12 medium supplemented with 5% horse serum, epidermal growth factor, hydrocortisone, insulin, and cholera toxin. BT-549 cells were cultured in RPMI-1640 medium, and BT-20 and MDA-MB-361 cells were cultured in Eagle’s minimum essential medium. Cell lines were authenticated in a core laboratory at The University of Texas MD Anderson Cancer Center. All cultured cells were incubated at 37°C in a water-saturated 95% air-5% CO2 atmosphere.
Expression of miR-22 in breast cancer patient samples and database analyses
The expression profile for miR-22 in breast cancers was defined by using a miRNA profiling system (Agilent Technologies) and the METABRIC database. The assay uses 100 ng of nonfractionated total RNA directly labeled via ligation of a Cy3-labeled pCp molecule to the 3′ end of the RNA. The labeled cytosine interacts with the guanidine at the 5′ end of the probe, which adds stability to the hybridization complex. The probes used in this assay are designed to provide both sequence and size discrimination, generally resulting in highly specific detection of closely related mature miRNAs. The assay’s labeling and probe design strategies allow for precise, accurate measurement that spans a linear dynamic range greater than four orders of magnitude from at least 0.2 amol to 2 fmol of miRNA with a detection limit of less than 0.1 amol. For miRNA sequencing, we derived reads per million miRNA mapped values for the mature form of miR-22 from the illuminahiseq_mirnaseq-miR_isoform_expression file available from the Broad Institute (http://gdac.broadinstitute.org/). The METABRIC analysis encompassed 93 normal breast tissue samples obtained from women without breast cancer, 159 tumor samples from patients with TNBC, and 122 tumor samples from patients with basal-like breast cancer.
Proliferation and colony-formation assay
A clonogenic assay was used to determine the effect of miR-22-3p on colony formation by TNBC cell lines. Briefly, cells were gently mixed and plated on six-well plates (500 cells/well). After incubation for 24 h, the cells were transfected with control miRNA or miR-22-3p, eEF2K, or control siRNA and cultured for 10−14 days. The cells were then stained with crystal violet. Colonies with a diameter of more than 50 cells were counted. This experiment was repeated three times independently.
Transient transfection of a miRNA mimic or eEF2K siRNA
MDA-MB-231 and MDA-MB-436 TNBC cell lines were plated in six-well plates (1.0 × 105 or 1.3 × 105 cells/well in 2 mL of medium) and allowed to adhere overnight. The plated cells were transfected with either a miR-22-3p mimic or control miRNA (100 nM) for 72 h using HiPerFect transfection reagent (QIAGEN) in Opti-MEM Reduced Serum Medium (Life Technologies), according to the manufacturer’s protocol. Other cells were transfected with either eEF2K siRNA or control siRNA (final concentration, 50 nM), according to the same protocol.
RNA isolation and RT-PCR
Total cellular RNA, including miRNAs, was isolated from the treated HMEC, MDA-MB-231, MDA-MB-436, BT-20, and MDA-MB-361 cells using a miRNeasy Mini Kit (QIAGEN), according to the manufacturer’s recommended protocol. The concentration and purity of the RNA were measured according to ultraviolet absorbance at 260 and 280 nm using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Complementary DNA was obtained from 1 μg samples of total RNA using a qScript microRNA cDNA Synthesis Kit (Quantabio). The RNA was converted to complementary DNA using a RT procedure under the following conditions: 37°C for 60 min, 70°C for 5 min, 42°C for 20 min, and 85°C for 5 min. The relative amounts of gene products were verified by the detection of an internal control glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript. miR-22-3p expression was detected using a PerfeCTa microRNA Assay kit (Quantabio) with Quanta miRNA primers. The miR-22-3p expression level was normalized to that for the endogenous control RNU6 (Quantabio).
Protein extraction and western blot analysis
MDA-MB-231 and MDA-MB-436 were seeded in six-well plates (1 × 105 cells/well) in 2 mL of medium. After transfection with miR-22-3p, control miRNA, eEF2k siRNA, or control siRNA, the cells were cultured for 72 h, centrifuged, washed once in ice-cold phosphate-buffered saline solution, and suspended in a lysis buffer that contained protease and phosphatase inhibitors at 4°C to obtain whole-cell lysates. The lysates were centrifuged at 13,000 × g for 10 min at 4°C, and the supernatant fractions were collected. The total protein concentration for each sample was determined using a Pierce Bicinchoninic Acid (BCA) Protein Assay Kit (Thermo Fisher Scientific). 40 μg of each sample was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis with a 4%–15% gradient to separate the proteins, which were then electrotransferred to polyvinylidene difluoride membranes. The membranes were probed with primary antibodies against eEF2K, eEF2, p-EF2 (Thr56), Src, p-Src (Tyr416), Akt, p-Akt (Ser473), focal adhesion kinase (FAK), p-FAK (Tyr397; Cell Signaling Technology), and β-actin (Sigma-Aldrich), diluted in Tris-buffered saline (TBS)-Tween 20 containing 2.5% dry milk and incubated at 4°C overnight. The membranes were washed with TBS-Tween 20 and incubated with a horseradish peroxidase-conjugated secondary anti-rabbit or anti-mouse antibody (Cell Signaling Technology). Chemiluminescence was detected using the HyGLO Chemiluminescent HRP Antibody Detection Reagent (Denville Scientific). The blots were visualized using a FluorChem 8900 imager and quantified using a densitometer with the Alpha Imager software (Alpha Innotech). All experiments were repeated independently three times.
In vitro invasion assay
MDA-MB-231 and MDA-MB-436 cells were treated with 100 nM indicated control miRNA or miR-22-3p or with eEF2K siRNA or control siRNA. Equal numbers of viable-treated cells (5 × 104) were diluted in serum-free medium and seeded onto Matrigel-coated Transwell filters (8 μm pores) in the upper chambers of a Matrigel invasion plate (BD Biosciences). The plate was incubated for 24 h to permit the cells in the upper chambers to invade toward the lower chambers, which contained medium with 10% fetal bovine serum. The cells that invaded the lower chamber were fixed and stained with Hema 3 (Thermo Fisher Scientific), whereas noninvading cells on the upper surface of the filter were removed using cotton swabs. The cells that invaded the lower side of the membrane were counted under a light microscope in a minimum of five randomly selected areas. The experiment was performed in triplicate, and the results were reported as mean (±SD).
Cell motility and migration assay
An in vitro wound-healing assay was used to assess cell motility and migration capacity. MDA-MB-231 and MDA-MB-436 cells were plated in six-well plates (1 × 105 cells/well) and cultured in medium containing 10% fetal bovine serum to achieve a nearly confluent cell monolayer. After 24 h of incubation, the cells were transfected with control miRNA or miR-22-3p or with eEF2K siRNA or control siRNA. After 72 h, the surface of the cell layer was carefully scratched with a 200-μL sterile pipette tip, and the medium was replaced with a fresh medium. The cells were immediately photographed (time point, 0 h) using a phase-contrast microscope (Nikon Instruments) to determine the wound width. The cells were photographed again at the 24-, 48-, and 72-h time points. The photographs were compared, and the distance of the cell migration at the leading edge of the wound was analyzed at each time point. The values obtained were then expressed as percentage migration, setting the gap width at 0 h as 100%. All experiments were done in triplicate.
Luciferase reporter assay of miR-22-3p expression
HEK293 cells were transfected with two PEZX-MT06 miRNA reporter vectors: one expressing the 3′ UTR of eEF2K with the miR-22-3p-binding site and the other expressing the 3′ UTR of eEF2K with a mutant miR-22-3p-binding site, a point mutation, and the luciferase gene (GeneCopoeia, Rockville, MD, USA). HEK293 cells were seeded in 96-well plates. After incubation for 24 h, cells were cotransfected with one of the two reporter vectors (50 ng) together with 50 nM miR-22-3p mimic or control miRNA using HiPerFect transfection reagent in Opti-MEM reduced-serum medium, according to the manufacturer’s instructions. 48 h after transfection, the cells were assayed using both firefly and Renilla luciferase with a dual luciferase assay system (GeneCopoeia, Rockville, MD, USA), according to the manufacturer’s instructions. For each sample, firefly luciferase activity was normalized according to Renilla luciferase activity. All experiments were conducted in triplicate and repeated three times independently.
Preparation of nanoliposomal miRNAs
miR-22-3p and control miRNA were incorporated into SLNPs (mean size, 70 nm) composed of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids), as we described previously.18 Briefly, control miRNA or miR-22-3p was mixed with the lipid at a ratio of 1:10 (w/w). Tween 20 was added to the lipid and miRNA mixture at a ratio of 1:20 in the presence of excess tertiary butanol. After vortexing, the mixture was frozen in an acetone/dry ice bath and lyophilized. Before in vivo administration to mice, the preparation was hydrated with normal 0.9% saline (100 μL/mouse) for intravenous injection at a dose of 0.15 mg/kg equivalent (4 μg/mouse) once a week in a volume of 100 μL. The mean size of the nanoliposomes incorporating the siRNAs was measured using a Zetasizer Nano ZS (Malvern Panalytical) and found to be about 70 nm.
Orthotopic xenograft TNBC models
Athymic female nu/nu mice were obtained from the Department of Experimental Radiation Oncology at MD Anderson. All studies were conducted according to an experimental protocol approved by the MD Anderson Institutional Animal Care and Use Committee. 5-week-old mice were injected with MDA-MB-231 or MDA-MB-436 TNBC cells suspended in 20% Matrigel (1 × 106 cells/mouse) into the mammary fat pads. Tumor size was measured weekly using calipers, and the xenografts were allowed to grow to a maximum diameter of about 5 mm. 2 weeks after injection, when the tumor size reached 3−5 mm, SLNP-miRNA treatments were initiated. The treatment group received miR-22-3p (0.15 mg/kg body weight or ∼4 μg per 25 g of body weight), whereas the control group received control miRNA (volume equivalent of 4 μg/mouse). The treatments, in 100 μL of a saline solution, were administered via tail-vein injection once a week for 4 weeks (total, four injections). After completion of the treatment, the mice were killed humanely via CO2 inhalation and weighed to assess tumor growth. Tumor samples were explanted and subjected to analysis.
Immunohistochemistry
Representative 5 μm sections of tumors obtained from MDA-MB-231 and MDA-MB-436 tumor-bearing mice were stained with hematoxylin and eosin. Sections of formaldehyde-fixed, paraffin-embedded tumors were prepared, de-waxed in xylene, and rehydrated in ethanol. CD31 (Abcam; catalog number [Cat. #]ab28364) and Ki-67 expression in the sections were analyzed by using rabbit polyclonal primary antibody (Thermo Scientific; Cat. #RB-9043-P1) and peroxidase-AffiniPure F (ab′)2 fragment goat anti-rabbit immunoglobulin G (IgG; Jackson ImmunoResearch Laboratories; Cat. #111-036-047) kits, according to the manufacturers’ instructions. Briefly, tumor sections were incubated in an antigen-retrieval solution (Promega, Dako) at 95°C for 40 min. After blocking endogenous peroxidases with methanol containing 3% hydrogen peroxide for 15 min, the sections were incubated with a primary antibody against Ki-67 or CD31 at 4°C overnight and then incubated with secondary antibodies at room temperature for 1 h. The sections were then counterstained with hematoxylin for 30 s and examined under a microscope.
Detection of apoptosis in vivo using a TUNEL assay
Apoptosis in TNBC samples after treatment with miR-22-3p or control miRNA was detected using a TUNEL assay (Promega), according to the manufacturer’s protocol with histological slides prepared from formalin-fixed, paraffin-embedded tumor sections. Briefly, tumor sections from control miRNA- and miR-22-3p-treated mice were incubated with biotin-dUTP and terminal deoxynucleotidyl transferase for 1 h. Fluorescein-conjugated avidin was added to the tissue sections and incubation continued for another 30 min in the dark. The slides were then covered with Hoechst 33342 dye (Thermo Fisher Scientific) to counterstain DNA. Positively stained, fluorescein-labeled, and Hoechst 33342-counterstained cells were examined under an inverted fluorescence microscope. The apoptosis rate in tumor sections was calculated by determining the average number of TUNEL-positive cells in each section in five microscopic fields.
Mathematical model of TNBC treatment response
The following mathematical model used for predicting the TNBC response to treatment with miR-22-3p was based on the mathematical modeling of cancer treatment:7, 8, 9, 10, 11, 12,23
where V is the tumor volume, i represents the treatment group (control or miR-22-3p), ri is the tumor growth rate, αi is the growth-inhibition rate due to treatment, βi is the tumor decay rate due to treatment, and K is a constant that represents the carrying capacity of the tumor system. Of note is that K varies across different tumor systems and different in vivo animal models, but its value is not important in interpreting our model results. Hence, we set K to 2,090 mm3 as recommended in the literature.24
Statistical analyses
Data are expressed as mean (±SD) values. Analysis of variance was used to compare the control and treated groups. All comparisons were analyzed using a two-tailed test. The Student’s t test was used to determine p values. p values less than 0.05 were considered significant. Statistical analyses were performed using Prism software (version 6.02; GraphPad Software).
Acknowledgments
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors. The studies described herein were supported by the MD Anderson Center for RNA Interference and Non-Coding RNA; NIH/NCI under award number P30CA016672 using the NIH/NCATS grant UH3TR00943-01 through the NIH Common Fund; and Office of Strategic Coordination. G.C. is the Felix L. Haas Endowed Professor in Basic Science and is supported by NCI grants 1R01 CA182905-01 and 1R01 CA222007-01A1, an NIGMS 1R01 GM122775-01 grant, U54 grants CA096297/CA096300, a University of Puerto Rico/MD Anderson Partnership for Excellence in Cancer Research 2016 Pilot Project, a Team DOD (CA160445P1) grant, a Sister Institution Network Fund 2017 grant, and the Estate of C.G. Johnson, Jr.
Author contributions
A.G. and B.O. conceived and coordinated the study and wrote the manuscript. R.B. and P.K. performed the luciferase assay and prepared figures. C.I. analyzed overall survival. B.O. and N.K. prepared nanoliposomal particles incorporating miRNAs and performed in vivo animal studies. E.B. and D.K. performed immunohistochemical analysis of in vivo tissue samples and contributed to preparation of figures. H.A.M., S.K., N.N.K., and B.A. provided technical assistance. Z.W. and V.C. performed mathematical remodeling of miR-22-3p and control miRNA for TNBC tumors of MDA-MB-231 and MDA-MB-436. G.L.B., G.C., and L.T. contributed to writing the manuscript. All authors analyzed the results and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2021.01.016.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2016. CA Cancer J. Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Reis-Filho J.S., Tutt A.N. Triple negative tumours: a critical review. Histopathology. 2008;52:108–118. doi: 10.1111/j.1365-2559.2007.02889.x. [DOI] [PubMed] [Google Scholar]
- 3.Fornier M., Fumoleau P. The paradox of triple negative breast cancer: novel approaches to treatment. Breast J. 2012;18:41–51. doi: 10.1111/j.1524-4741.2011.01175.x. [DOI] [PubMed] [Google Scholar]
- 4.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 6.Pandey A.K., Zhang Y., Zhang S., Li Y., Tucker-Kellogg G., Yang H., Jha S. TIP60-miR-22 axis as a prognostic marker of breast cancer progression. Oncotarget. 2015;6:41290–41306. doi: 10.18632/oncotarget.5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pascal J., Ashley C.E., Wang Z., Brocato T.A., Butner J.D., Carnes E.C., Koay E.J., Brinker C.J., Cristini V. Mechanistic modeling identifies drug-uptake history as predictor of tumor drug resistance and nano-carrier-mediated response. ACS Nano. 2013;7:11174–11182. doi: 10.1021/nn4048974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koay E.J., Truty M.J., Cristini V., Thomas R.M., Chen R., Chatterjee D., Kang Y., Bhosale P.R., Tamm E.P., Crane C.H., et al. Transport properties of pancreatic cancer describe gemcitabine delivery and response. J. Clin. Invest. 2014;124:1525–1536. doi: 10.1172/JCI73455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frieboes H.B., Smith B.R., Wang Z., Kotsuma M., Ito K., Day A., Cahill B., Flinders C., Mumenthaler S.M., Mallick P., et al. Predictive Modeling of Drug Response in Non-Hodgkin’s Lymphoma. PLoS ONE. 2015;10:e0129433. doi: 10.1371/journal.pone.0129433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z., Kerketta R., Chuang Y.L., Dogra P., Butner J.D., Brocato T.A., Day A., Xu R., Shen H., Simbawa E., et al. Theory and Experimental Validation of a Spatio-temporal Model of Chemotherapy Transport to Enhance Tumor Cell Kill. PLoS Comput. Biol. 2016;12:e1004969. doi: 10.1371/journal.pcbi.1004969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cristini V., Koay E., Wang Z. CRC Press; 2017. An Introduction to Physical Oncology: How Mechanistic Mathematical Modeling Can Improve Cancer Therapy Outcomes. [Google Scholar]
- 12.Brocato T.A., Coker E.N., Durfee P.N., Lin Y.S., Townson J., Wyckoff E.F., Cristini V., Brinker C.J., Wang Z. Understanding the Connection between Nanoparticle Uptake and Cancer Treatment Efficacy using Mathematical Modeling. Sci. Rep. 2018;8:7538. doi: 10.1038/s41598-018-25878-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tekedereli I., Alpay S.N., Tavares C.D., Cobanoglu Z.E., Kaoud T.S., Sahin I., Sood A.K., Lopez-Berestein G., Dalby K.N., Ozpolat B. Targeted silencing of elongation factor 2 kinase suppresses growth and sensitizes tumors to doxorubicin in an orthotopic model of breast cancer. PLoS ONE. 2012;7:e41171. doi: 10.1371/journal.pone.0041171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nairn A.C., Bhagat B., Palfrey H.C. Identification of calmodulin-dependent protein kinase III and its major Mr 100,000 substrate in mammalian tissues. Proc. Natl. Acad. Sci. USA. 1985;82:7939–7943. doi: 10.1073/pnas.82.23.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryazanov A.G., Pavur K.S., Dorovkov M.V. Alpha-kinases: a new class of protein kinases with a novel catalytic domain. Curr. Biol. 1999;9:R43–R45. doi: 10.1016/s0960-9822(99)80006-2. [DOI] [PubMed] [Google Scholar]
- 16.Redpath N.T., Price N.T., Proud C.G. Cloning and expression of cDNA encoding protein synthesis elongation factor-2 kinase. J. Biol. Chem. 1996;271:17547–17554. [PubMed] [Google Scholar]
- 17.Carlberg U., Nilsson A., Nygård O. Functional properties of phosphorylated elongation factor 2. Eur. J. Biochem. 1990;191:639–645. doi: 10.1111/j.1432-1033.1990.tb19169.x. [DOI] [PubMed] [Google Scholar]
- 18.Hamurcu Z., Ashour A., Kahraman N., Ozpolat B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget. 2016;7:16619–16635. doi: 10.18632/oncotarget.7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashour A.A., Abdel-Aziz A.A., Mansour A.M., Alpay S.N., Huo L., Ozpolat B. Targeting elongation factor-2 kinase (eEF-2K) induces apoptosis in human pancreatic cancer cells. Apoptosis. 2014;19:241–258. doi: 10.1007/s10495-013-0927-2. [DOI] [PubMed] [Google Scholar]
- 20.Ashour A.A., Gurbuz N., Alpay S.N., Abdel-Aziz A.A., Mansour A.M., Huo L., Ozpolat B. Elongation factor-2 kinase regulates TG2/β1 integrin/Src/uPAR pathway and epithelial-mesenchymal transition mediating pancreatic cancer cells invasion. J. Cell. Mol. Med. 2014;18:2235–2251. doi: 10.1111/jcmm.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song S.J., Ito K., Ala U., Kats L., Webster K., Sun S.M., Jongen-Lavrencic M., Manova-Todorova K., Teruya-Feldstein J., Avigan D.E., et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. 2013;13:87–101. doi: 10.1016/j.stem.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xin M., Qiao Z., Li J., Liu J., Song S., Zhao X., Miao P., Tang T., Wang L., Liu W., et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget. 2016;7:44252–44265. doi: 10.18632/oncotarget.10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu D., Takeshita F., Hino Y., Fukunaga S., Kudo Y., Tamaki A., Matsunaga J., Takahashi R.U., Takata T., Shimamoto A., et al. miR-22 represses cancer progression by inducing cellular senescence. J. Cell Biol. 2011;193:409–424. doi: 10.1083/jcb.201010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W., Chen R., Li J., Fu Y., Yang L., Su H., Yao Y., Li L., Zhou T., Lu W. Pharmacokinetic/Pharmacodynamic Modeling of Schedule-Dependent Interaction between Docetaxel and Cabozantinib in Human Prostate Cancer Xenograft Models. J. Pharmacol. Exp. Ther. 2018;364:13–25. doi: 10.1124/jpet.117.243931. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.