

Abstract
We recently established a role for the stretch-activated two-pore-domain K+ (K2P) channel TREK-1 (K2P2.1) in inflammatory cytokine secretion using models of hyperoxia-, mechanical stretch–, and TNF-α–induced acute lung injury. We have now discovered the expression of large conductance, Ca2+-activated K+ (BK) channels in human pulmonary microvascular endothelial cells and primary human alveolar epithelial cells using semiquantitative real-time PCR, IP and Western blot, and investigated their role in inflammatory cytokine secretion using an LPS-induced acute lung injury model. As expected, LPS induced IL-6 and CCL-2 secretion from pulmonary endothelial and epithelial cells. BK activation with NS1619 decreased LPS-induced CCL-2 but not IL-6 secretion from endothelial cells and had no effect on epithelial cells, although fluorometric assays revealed that BK activation hyperpolarized the plasma membrane potential (Em) of both cell types. Interestingly, BK inhibition (Paxilline) did not alter cytokine secretion or the Em in either cell type. Furthermore, LPS treatment by itself did not affect the Em or intracellular Ca2+ concentrations. Therefore, we propose BK channel activation as a novel targeted approach to counteract LPS-induced CCL-2 secretion from endothelial cells. This protective effect appears to occur via Em hyperpolarization but independent of intracellular Ca2+ concentrations.
Keywords: acute lung injury, large conductance potassium channels, LPS, cytokines, inflammation
Clinical Relevance
Our data show for the first time the functional expression of large conductance K+ (BK) channels in human pulmonary microvascular endothelial cells and primary human alveolar epithelial cells. Furthermore, we propose a role for the BK channel activator NS1619 in inflammatory cytokine secretion in an LPS-induced lung injury model. These results will contribute to the development of new therapeutic approaches against LPS-induced lung injury by highlighting BK channels as potentially new targets.
Infectious stimuli are the most common triggers for lung injury in humans, which in its most severe form may progress to acute respiratory distress syndrome (ARDS). Decades of intense research have focused on creating experimental acute lung injury (ALI) models to dissect the underlying mechanisms and identify potential therapeutic targets. LPS-induced ALI models are routinely used to experimentally mimic gram-negative infections, and the LPS-mediated proinflammatory effects on cytokine secretion from immune and inflammatory cells are well documented (1). However, less is known about the mechanism of LPS-induced injury in lung resident cells, including alveolar epithelial and endothelial cells. Nevertheless, upregulation of inflammatory mediators such as CCL-2 and IL-6 is consistently observed in the serum and BAL fluid from lung injury patients, and in multiple experimental ALI models (2). Besides antibacterial therapies to treat underlying infections, currently no targeted interventions exist against ALI/ARDS that translate into improved patient outcomes.
Using different in vivo and in vitro ALI models, we have previously shown that regulation of inflammatory cytokine secretion by K+ channels plays an important role in the development of alveolar inflammation (3–6). In general, one of the main functions of K+ channels is the regulation and stabilization of the plasma membrane potential (Em), and our lab is particularly interested in exploring mechanisms that regulate the Em as a tool to counteract alveolar inflammation and injury. We previously found that TREK-1 K+ channels, which are characterized by a single channel conductance in the range of 40–150 pS (7), participate in the regulation of inflammatory cytokine secretion from alveolar epithelial cells (3, 4, 8). Based on these findings, it is intriguing to speculate that manipulation of channels with a larger unitary conductance, such as the large conductance, Ca2+- and voltage-activated K+ channels (BK, KCa1.1, MaxiK), may constitute an even more powerful tool to regulate alveolar epithelial and endothelial inflammation.
The functional BK channel is a homotetramer of four pore-forming α-subunits (BK-α) associated with modulatory β-subunits (BK-β), which can modify the Ca2+- and voltage dependence of the channels and its gating properties (9). Four types of BK-β auxiliary subunits have been identified thus far in mammals, of which BK-β2 and -β3 are expressed in lung tissue (10). Interestingly, the vascular BK-β1-subunit has been implicated in LPS-induced hypotension, organ damage, and mortality (11). Overall, despite differences in its subunit composition, BK channels are ubiquitously expressed in the body and represent a unique class of K+ channels, not only because of their large single channel conductance (∼250 pS in symmetrical K+ solutions) but also given their individual and synergistic sensitivity to intracellular Ca2+ (iCa2+) levels and membrane depolarization (12). Consequently, BK channels are involved in key physiological roles, including regulation of neurotransmitter release, neuronal and cardiac excitability, smooth muscle tone, endocrine secretion, cell proliferation, and renal electrolyte regulation, and dysregulation of BK channels has been linked to various disease processes such as schizophrenia, cardiac arrhythmias, and diabetes mellitus (13).
It is unknown whether human pulmonary microvascular endothelial cells (HULEC5a) and human primary alveolar epithelial cells (HPAEpiC) express BK channels, and thus, their potential role in inflammatory cytokine secretion from these cells remains unclear. This study was designed to determine the expression and contribution of BK channels to LPS-induced cytokine secretion from alveolar epithelial and microvascular endothelial cells, and to establish the underlying mechanisms as potential targets for future therapeutic interventions. We hypothesize that pharmacological BK activation counteracts LPS-induced IL-6 and CCL-2 secretion through Em- and iCa2+-dependent mechanisms. Of note, some of the results of this study have been previously reported in the form of an abstract (14).
Methods
Cells
HULEC5a (ATCC-CRL-3244), human alveolar epithelial A549 (ATCC-CRM-CCL-185), and NCI-H441 (ATCC-HTB-174) cells were purchased from American Type Culture Collection. HPAEpiC were purchased from ScienCell. HEK293 cells were obtained from Dr. Olcese (University of California Los Angeles). All cells were used at passages 6–15.
Gene Expression
Total RNA was isolated using a Qiagen RNeasy Mini Kit; 1–8 μg RNA were reverse transcribed with a High Capacity cDNA Reverse Transcription kit (Applied Biosystems) and amplified by semiquantitative real-time PCR (TaqMan) with primers for KCNMA1 (BK-α1), KCNMB1–4 (BK-β1–4), or GAPDH (Applied Biosystems).
Protein Expression
BK-α1 protein was immunoprecipitated in cell lysates with 10 μg mouse monoclonal anti-BK-α antibody (UC Davis/NeuroMab) or nonspecific rabbit polyclonal IgG isotype (negative control; Cell Signaling). Immunocomplexes were captured with 100 μl A/G PLUS agarose beads (Santa Cruz Biotechnology). Dissociated samples were separated on a NuPAGE Bis-Tris 10% gel, transferred onto 0.45-μm Immuno-Blot PVDF membranes (BioRad), and probed with a primary mouse monoclonal anti–BK-α antibody (1:200; UC Davis/NeuroMab) and a goat anti-mouse-Cy3 secondary antibody (1:1000; Millipore).
As a positive control for Western blot, the BK-α1-subunit (KCNMA1) was overexpressed in HEK293 cells using the human plasmid BK-α1-YFP-pcDNA3.1.
Em Measurements
Em changes were analyzed by fluorescence-based plate reader (FLIPR) Membrane Potential Assay (Molecular Devices) (15–17). Relative fluorescence of Em red dye–loaded cells was recorded every 7 seconds using a Synergy-2 Multi-Mode Microplate Reader (Biotek; excitation 525 nm, emission 590 nm, at 37°C) after adding LPS (2 μg/ml; Cell Signaling; E. coli serotype O111:B4), NS1619 (30 μM; Millipore), NS19504 (30 μM; Alomone Labs), Paxilline (10 μM; Alomone Labs), or equimolar vehicle controls.
Cytokine Measurements
CCL-2 and IL-6 concentrations in 5% FBS-containing supernatants were quantified using ELISA assays (BD Biosciences) after treatment of cells with 2 μg/ml LPS, NS1619 (30 μM), Paxilline (10 μM), or equimolar vehicle controls.
Ca2+ Measurements
iCa2+ concentrations were quantified by Fluo-4 NW Calcium Assay (Fisher Scientific; excitation 488 nm, emission 516 nm). Baseline fluorescence was recorded for 1 minute before adding LPS (2 μg/ml), NS1619 (30 μM), Paxilline (10 μM), Ionomycin (5 μM; Adipogen), or equimolar vehicle controls. Relative fluorescence was recorded every 11 seconds. All buffers contained 1.8 mM CaCl2.
Gene Microarray Experiments
NS1619-mediated alterations in LPS signaling were analyzed using a TaqMan 94-gene Array Human Inflammation kit (#4414074; Fisher Scientific). RNA isolation and DNA synthesis were performed as above. Target gene amplification was performed using a QuantStudio-5 system (Fisher Scientific) and normalized to GAPDH. Canonical pathway analysis was performed by Ingenuity Pathway Analysis (Qiagen) (18) with a threshold for significance set at ≥1.2-fold change (FC).
Statistics
Data are expressed as mean ± SEM, or median with minimum and maximum values. Comparisons were performed using Mann-Whitney U or Student’s t tests (GraphPad 8.3), with P ≤ 0.05 considered significant.
Results
BK Channels Are Expressed in Human Pulmonary Microvascular Endothelial and Primary Human Alveolar Epithelial Cells
Using semiquantitative real-time PCR, we measured mRNA expression of the pore-forming α1-subunit KCNMA1 and the auxiliary β-subunits KCNMB1, KCNMB2, KCNMB3, and KCNMB4 in cultured HULEC5a and human alveolar type-2 (A549 and H441), and in HPAEpiC cells (Figures 1A and E1A). Interestingly, KCNMA1, KCNMA3, and KCNMA4 subunit expression was detected in all cell types tested, whereas the KCNMB2 was only detected in HPAEpiC and H441 but not HULEC5a and A549 cells. Because BK channel mRNA is expressed in both transformed (A549, H441) and primary HPAEpiC cells, for the rest of the study, we focused on HPAEpiC cells.
Figure 1.
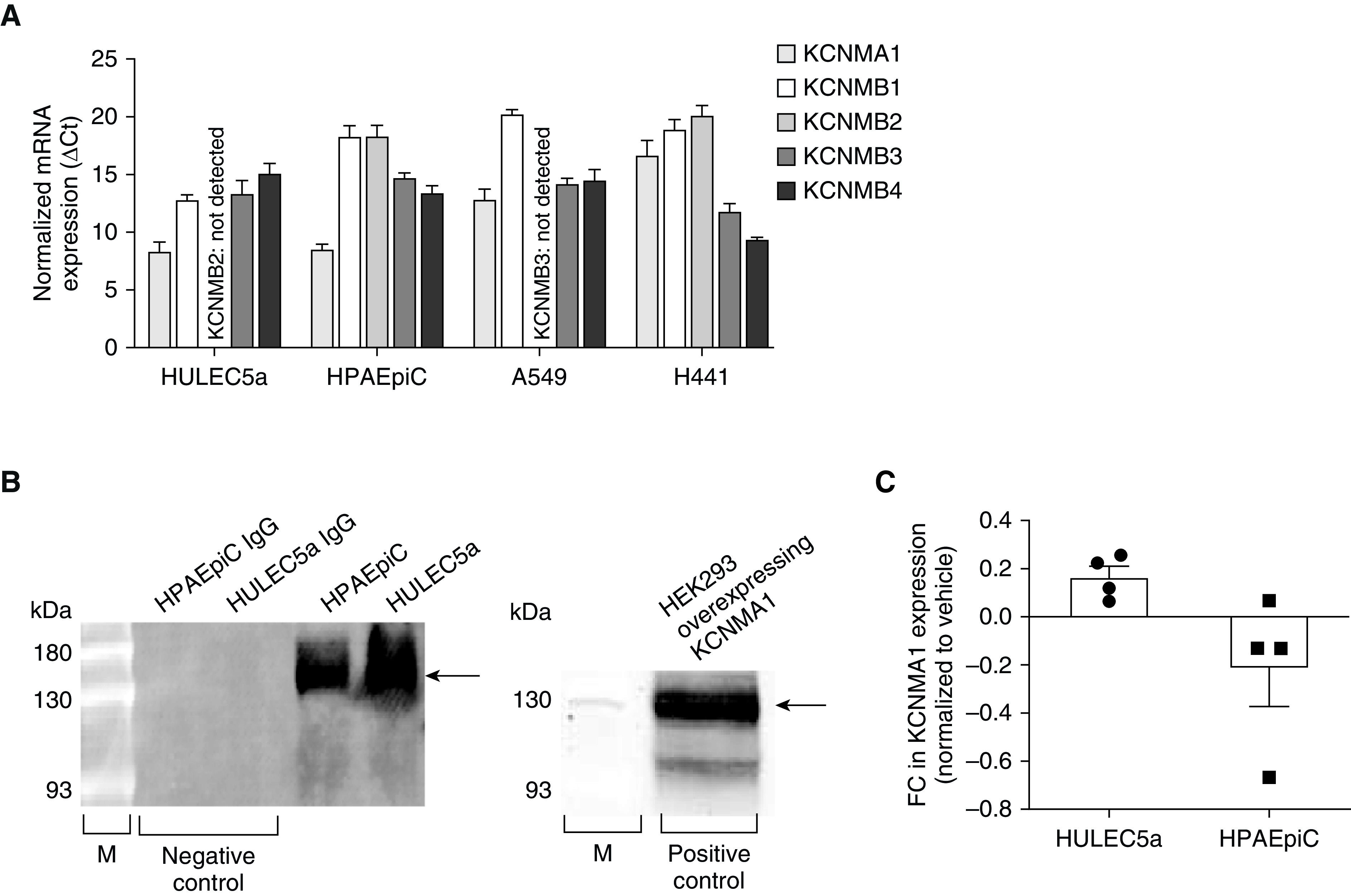
Large conductance potassium (BK) channels are expressed in human pulmonary microvascular endothelial cells (HULEC5a) and human primary alveolar epithelial cells (HPAEpiC). (A) Relative mRNA expression (ΔCt) of the pore-forming α-1 subunit KCNMA1 and the auxiliary KCNMB1, -2, -3, -4 β-subunits in HULEC5a (n = 6) and HPAEpiC (A549 and H441; n = 4–7). (B) On the left: a representative IP and Western blot experiment: pore-forming KCNMA1 protein expression in HULEC5a and HPAEpiC cells (n = 3). On the right: a representative Western blot experiment showing that the anti-KCNMA1 BK-α1 antibody detected the protein at the expected size (100–130 kD; right lane) in HEK293 cells overexpressing the KCNMA1 BK-α1-subunit. Arrows depict expected band size. (C) Fold change in KCNMA1 gene expression in LPS-treated (6 h, 2 μg/ml) HULEC5a and HPAEpiC cells (n = 4). FC = fold change; M = molecular weight marker.
Similar to gene expression, we also detected protein expression of the pore-forming KCNMA1 subunit by IP and Western blot in endothelial HULEC5a and epithelial HPAEpiC cells (Figure 1B). To control for nonspecific antibody binding, we replaced the primary antibody with a rabbit polyclonal IgG isotope as a negative control. As a positive control for Western blot analysis, we used HEK293 cells overexpressing the BK-α1-subunit (KCNMA1). Treatment of endothelial HULEC5a and epithelial HPAEpiC cells with LPS (2 μg/ml) did not affect KCNMA1 expression levels (Figure 1C). Taken together, these findings demonstrate that human microvascular endothelial cells and alveolar epithelial cells express BK channels, and that BK expression in these cells is not altered by LPS exposure.
BK Channel Activity Regulates the Em of Human Pulmonary Microvascular Endothelial Cells during LPS-induced Inflammation
To determine the functional role of BK channels in regulating the Em in human endothelial and epithelial cells, we activated or inhibited BK channels with NS1619 (19) and NS19504 (20), or Paxilline (21), respectively, and measured Em changes using fluorometric FLIPR assays. We also performed an internal validation of the assay for both endothelial and epithelial cells by incrementally increasing extracellular K+ concentrations using 2- to 45-mM K2SO4 to gradually depolarize the Em (Figures E2A–E2D).
Interestingly, LPS treatment by itself did not alter the Em in either endothelial or epithelial cells (Figures 2A–2E). In endothelial cells, activation of BK channels with both BK agonists, NS1619 (30 μM) and NS19504 (30 μM), resulted in Em hyperpolarization (reflected by a decrease in relative fluorescence) in both untreated and LPS-treated (2 μg/ml) cells (Figures 2A and 2B), suggesting that both drugs target BK channels and indicating the functional presence of BK channels in these cells. Despite the difference between the two drugs not reaching statistical significance, the hyperpolarizing effect of NS1619 appeared stronger than with NS19504, and for this reason NS1619 was used for the rest of this study. Furthermore, inhibition of BK channels with Paxilline (10 μM) did not alter the Em in endothelial cells, suggesting a low level of BK activity in untreated cells (Figure 2C).
Figure 2.
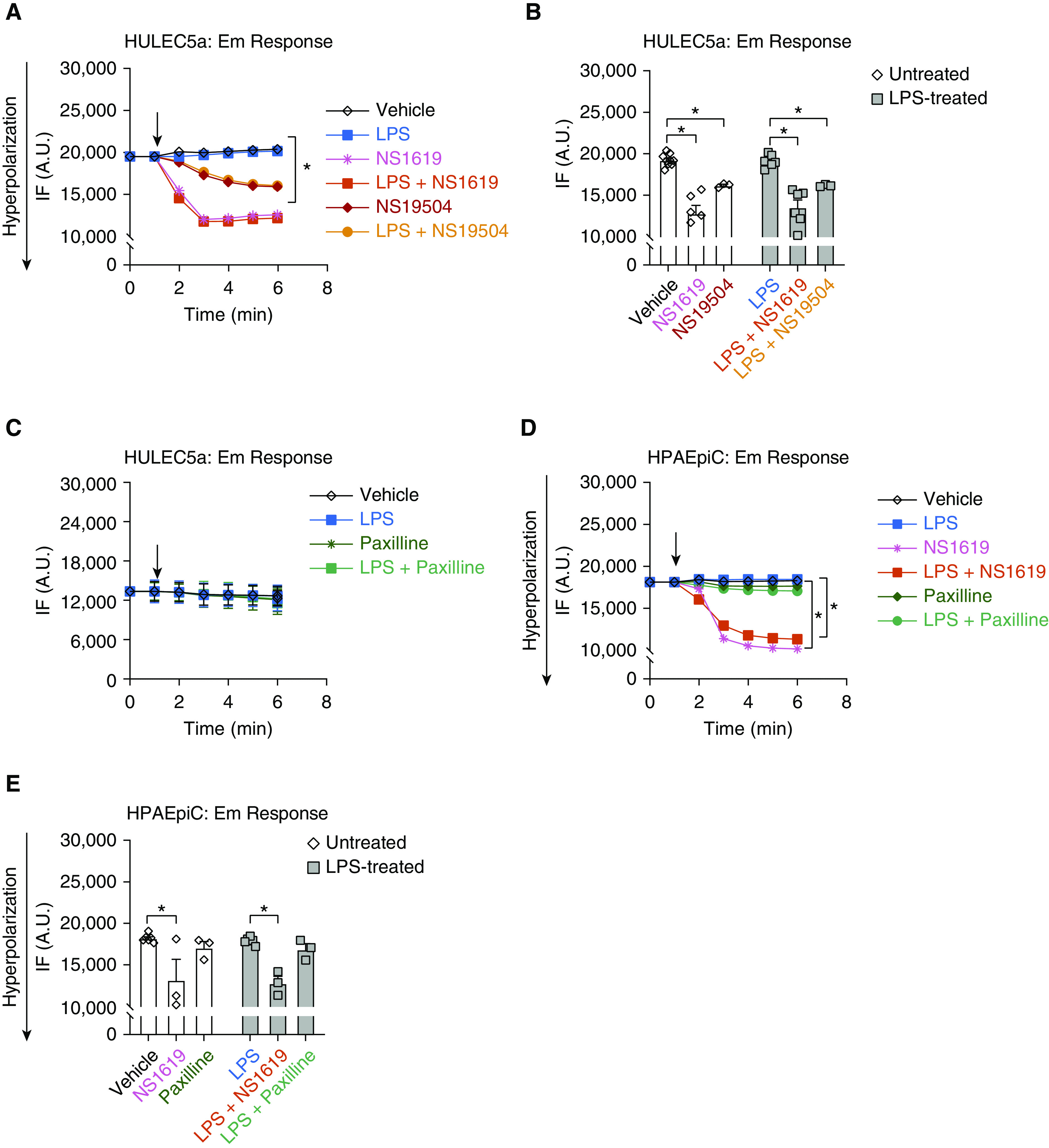
BK channels regulate the plasma membrane potential (Em) in HULEC5a during LPS-induced inflammation. Fluorescence intensity (IF) measured by fluorescence-based plate reader (FLIPR) assay depicts Em changes in endothelial (HULEC5a) and epithelial (HPAEpiC) cells. Cells were treated with LPS (2 μg/ml), the BK activators NS1619 (30 μM) and NS19504 (30 μM), the BK inhibitor Paxilline (10 μM), or a combination of LPS with these agents. (A) A representative recording showing NS1619- and NS19504-induced Em hyperpolarization. (B) NS1619 and NS19504 cause a decrease in IF in HULEC5a cells representing Em hyperpolarization, both at baseline and after LPS treatment (2 μg/m, n = 6, *P ≤ 0.05). (C) Treatment of endothelial cells with LPS, Paxilline, or LPS+Paxilline did not change the Em (n = 6). (D and E) Treatment of epithelial cells with LPS, Paxilline, or LPS+Paxilline had no effect on the Em, whereas NS1619 alone and in combination with LPS caused Em hyperpolarization (n = 3–6, *P ≤ 0.05). Black arrows indicate the timing of drug delivery. A.U. = arbitrary units.
Similarly to endothelial cells, activation of BK channels with NS1619 also hyperpolarized the Em of HPAEpiC, whereas BK channel inhibition with Paxilline had no effect on the Em (Figures 2D and 2E), suggesting a similarly low level of BK activity at baseline as seen in endothelial cells.
Taken together, these findings demonstrate 1) the functional presence of BK channels on endothelial and epithelial cells, 2) the potential for pharmacological hyperpolarization of both cell types, and 3) a lack of direct LPS effects on the Em of endothelial and epithelial cells.
BK Channel Activation with NS1619 Decreases LPS-induced CCL-2 but Not IL-6 Secretion from HULEC5a Cells
To determine whether BK channel activation regulates inflammatory cytokine secretion from HULEC5a and HPAEpiC cells, we quantified LPS-induced CCL-2 and IL-6 secretion using ELISA assays (Figure 3). Neither BK channel activation with NS1619 (30 μM) nor inhibition with Paxilline (10 μM) affected baseline CCL-2 or IL-6 secretion from these two cell types (Figures 3A–3D). Treatment of endothelial cells with LPS (2 μg/ml) increased both CCL-2 and IL-6 secretion compared with untreated controls (Figures 3A and 3B). Pilot experiments revealed that 2 μg/ml and 10 μg/ml of LPS evoke a similar increase in CCL-2 and IL-6 levels (data not shown), and therefore, we used the lower 2 μg/ml LPS dose for the rest of this study. Specifically, LPS treatment increased mean endothelial CCL-2 concentrations from 166 ± 13 to 855 ± 196 pg/ml (P ≤ 0.001), and mean IL-6 concentrations from 170 ± 24 to 2,753 ± 694 pg/ml (P ≤ 0.001), representing an 81% and 94% increase over baseline, respectively (Figures 3A and 3B; red horizontal bars). Importantly, BK channel activation with NS1619 inhibited LPS-induced CCL-2 secretion, as indicated by a decrease in mean LPS-induced CCL-2 levels decreased from 855 ± 196 to 322 ± 59 pg/ml after BK activation with NS1619 (P ≤ 0.05), representing a 62% decrease (Figure 3A, red horizontal bars). Interestingly, LPS-induced IL-6 secretion was not affected by BK channel activation with NS1619 (Figure 3B). In contrast to BK channel activation, BK channel inhibition with Paxilline did not affect LPS-induced CCL-2 or IL-6 secretion from endothelial cells (Figures 3A and 3B), supporting the idea that most BK channels are closed at baseline.
Figure 3.
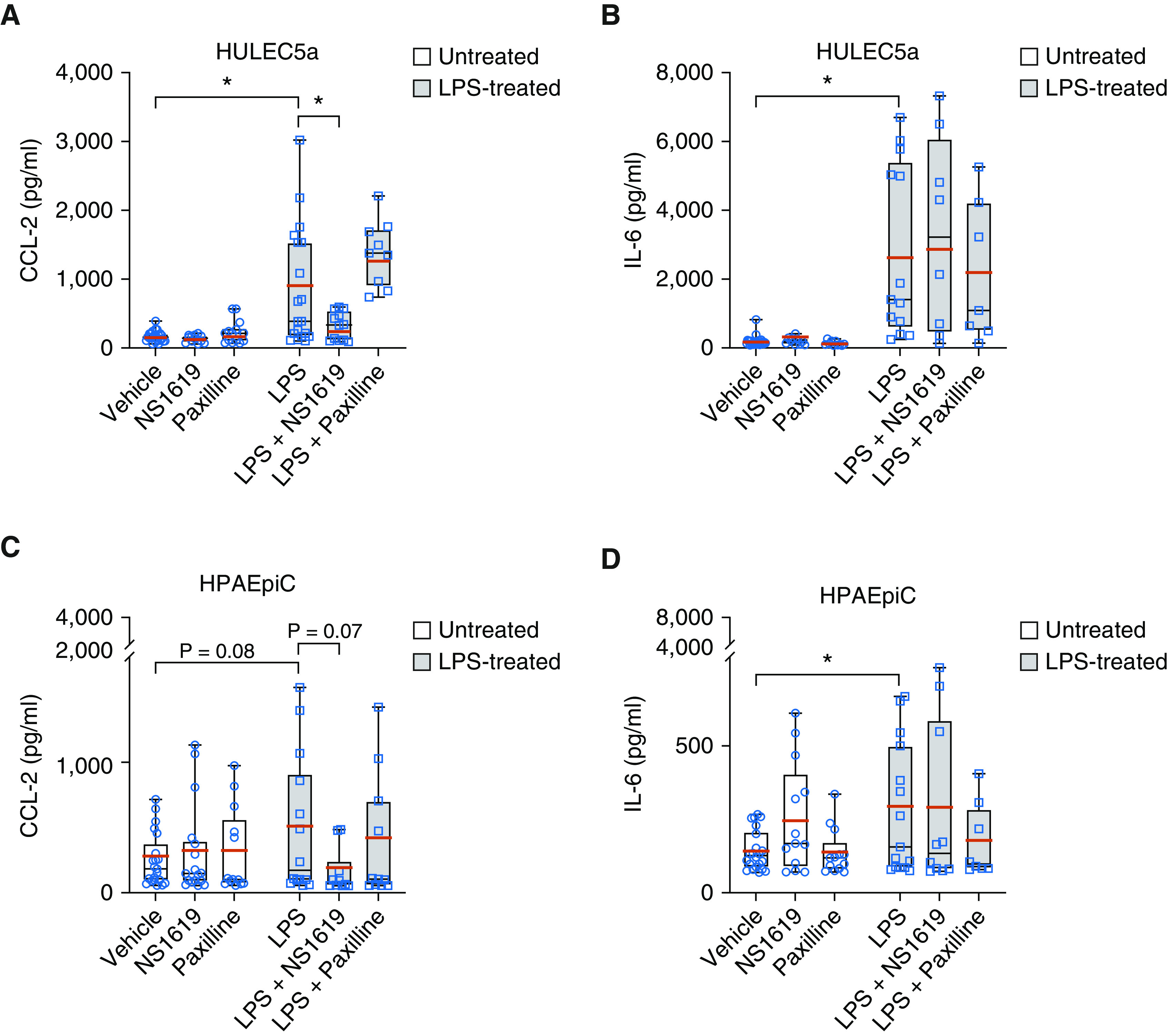
BK activation with NS1619 decreased CCL-2 but not IL-6 secretion from HULEC5a cells during LPS-induced inflammation. (A and B) Treatment of HULEC5a cells with LPS (2 μg/ml; 24 h) increased CCL-2 and IL-6 secretion. BK activation with NS1619 decreased LPS-induced CCL-2 but not IL-6 section, whereas BK inhibition with Paxilline had no effect on LPS-induced CCL-2 or IL-6 secretion (n = 7–26, *P ≤ 0.05) from HULEC5a cells. (C and D) LPS treatment (2 μg/ml; 24 h) also increased CCL-2 and IL-6 secretion from HPAEpiC cells, but neither BK activation with NS1619 nor BK inhibition with Paxilline affected CCL-2 or IL-6 secretion from these cells (n = 8–22, *P ≤ 0.05). Mean values are represented in red horizontal bars; black bars indicate medians.
Similar to endothelial cells, LPS treatment also increased IL-6 secretion from HPAEpiC cells from 144 ± 14 pg/ml to 277 ± 57 pg/ml, representing a 48% increase compared with untreated cells (P ≤ 0.05), but had no effect on CCL-2 secretion from epithelial cells (Figures 3C and 3D, red horizontal bars). In contrast to endothelial cells, neither BK channel activation (NS1619) nor inhibition (Paxilline) affected LPS-induced CCL-2 or IL-6 secretion (Figures 3C and 3D). Taken together, these data suggest a novel link between BK-mediated regulation of the endothelial Em and downstream LPS-induced CCL-2 but not IL-6 secretion, which is not observed in alveolar epithelial cells.
LPS-induced CCL-2 and IL-6 Secretion from HULEC5a Cells Occurs Independently of iCa2+ Concentrations
To determine whether BK channel–mediated Em alterations (Figure 2) and downstream inflammatory cytokine secretion (Figure 3) were dependent on Ca2+ influx via voltage-gated Ca2+ channels (CaV), we quantified LPS-induced CCL-2 and IL-6 secretion from HULEC5a cells after blocking L-type CaV channels with Nifedipine (10 μM; Figure 4). Interestingly, in unstimulated endothelial cells, Nifedipine decreased baseline secretion of CCL-2 but not IL-6 (Figures 4A and 4B), but following LPS stimulation, CaV inhibition with Nifedipine did not affect CCL-2 or IL-6 secretion.
Figure 4.
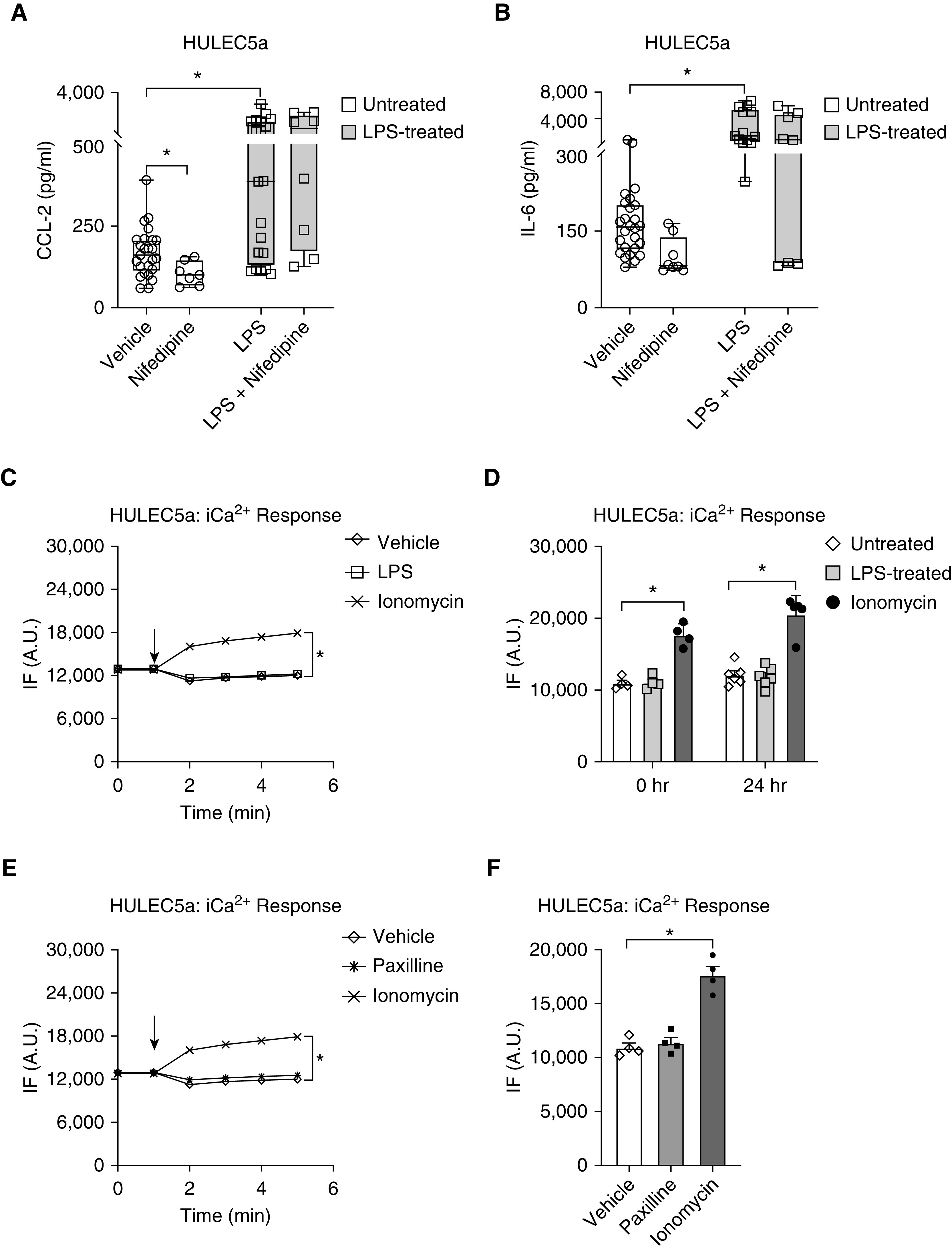
LPS-induced CCL-2 and IL-6 secretion from HULEC5a cells does not require intracellular Ca2+ (iCa2+). (A and B) Inhibition of L-type CaV channels with Nifedipine (10 μM; 24 h) decreased baseline CCL-2 but not IL-6 levels. Although LPS (2 μg/ml; 24 h) induced both CCL-2 and IL-6 secretion, these effects were not inhibited by Nifedipine (n = 7–26, *P ≤ 0.05). (C) Representative recording of iCa2+ kinetics represented by changes in relative IF using a Fluo-4 assay. Treatment of endothelial cells with LPS (2 μg/ml) did not affect iCa2+ concentrations. (D) iCa2+ levels were recorded either immediately after 2 μg/ml LPS treatment (time 0 h) or after 24 hours of LPS treatment (n = 4–6). BK inhibition with Paxilline (10 μM) did not affect iCa2+ levels as shown in a representative recording in E and as a summary of n = 4 in F. Ionomycin served as a positive control for the iCa2+ signal (n = 4–6, P ≤ 0.05). Black arrows indicate the timing of drug delivery.
Because LPS-induced CCL-2 and IL-6 secretion appears to occur independently of extracellular Ca2+ influx via CaV channels, we determined whether LPS stimulation itself affects iCa2+ concentrations in HULEC5a cells (Figures 4C and 4D). Using fluorometric Fluo-4 assays, we measured the immediate effect of LPS treatment on endothelial iCa2+ concentrations, as well as iCa2+ concentrations after 24 hours of LPS exposure (Figure 4D). These studies showed that LPS treatment had neither short- nor long-term effects on endothelial iCa2+ concentrations. Furthermore, BK inhibition with Paxilline did not increase iCa2+ concentrations, supporting the idea that in endothelial cells at baseline most BK channels are closed, and do not mediate any changes in Em or iCa2+ homoeostasis. The Ca2+ ionophore Ionomycin was used as positive control to validate the responsiveness of cells and the sensitivity of the Fluo-4 assay (Figures 4C–4F). Taken together, these findings suggest that baseline CCL-2 secretion from pulmonary endothelial cells is dependent on CaV activity, but LPS-induced CCL-2 and IL-6 secretion occurs independently of alterations in iCa2+ concentrations.
BK-mediated Changes in LPS-induced Cytokine Secretion Alter Specific Downstream Signaling Pathways in HULEC5a Cells
To further explore the electrochemical coupling processes and signaling mechanisms linking BK-mediated Em alterations to downstream LPS-induced cytokine secretion, we used a human inflammatory 94-gene microarray and confirmatory real-time PCR (Tables 1 and E1). For this purpose, we compared gene expression patterns between endothelial cells stimulated with LPS alone (2 μg/ml) for 8 hours and cells treated with LPS+ NS1619 (BK activator). We calculated fold changes in relative gene expression (FC; a quantitative parameter for scored genes, indicative of the ratio between the experimental and control values) to quantify the effects of BK activation with NS1619 on specific signaling pathways in LPS-treated cells. As shown in Table 1, using an FC ≥1.2 as the cutoff criterion, our PCR results identified top gene candidates based on the highest FC values: NS1619 treatment downregulated two proinflammatory genes (TNFSF13B, FC −3.3; CASP1, FC −2.0) and upregulated one proinflammatory gene (PTGS2, FC 2.2) and one antiinflammatory gene (IL1R1, FC 1.2). Importantly, JNK-1 (FC 1.4), a kinase that has both pro- and antiinflammatory properties, was also upregulated by NS1619. Importantly, real-time PCR also revealed that TLR-4 expression (not included in the microarray panel) was downregulated by NS1619 (FC −1.2). In total, in the microarray assay, 8 genes were downregulated and 21 upregulated (FC ≥1.2 or ≤–1.2; Table E1). Canonical pathway analysis revealed that the Eicosanoid signaling pathway was particularly affected by BK activation.
Table 1.
Summary of Top Gene Candidates Affected by NS1619 in LPS-treated Human Pulmonary Microvascular Endothelial Cells
| Gene Symbol | Gene Description | FC by Microarray | FC by Confirmatory RT-PCR |
|---|---|---|---|
| TNFSF-13B | Tumor necrosis factor ligand superfamily member 13B/B-cell activating factor (BAFF) | −3.1 | −3.3 |
| CASP-1 | Caspase-1 | −2.3 | −2.0 |
| NF‐κB-1 | Nuclear factor kappa B subunit-1 | 2.0 | 1.0 |
| ALOX-5 | Arachidonate 5-lipoxygenase | −1.5 | 0.1 |
| TNF | Tumor necrosis factor | 1.3 | −0.8 |
| PTGS-2 | Prostaglandin-endoperoxide synthase-2, cyclooxygenase-2 or COX-2 | 4.3 | 2.2 |
| IL1R-1 | Interleukin 1 receptor, type I/Cluster of differentiation 121a (CD121a) | 2.4 | 1.2 |
| TLR-4 | Toll-like receptor-4 | Not tested | −1.2 |
| MAPK-8 | Mitogen-activated protein kinase-8 (JNK-1) | 1.2 | 1.4 |
| MAPK-14 | Mitogen-activated protein kinase-14 (p38) | −0.1 | 0.0 |
| MAPK-3 | Mitogen-activated protein kinase-3 (ERK-1) | 0.1 | −0.2 |
| MAPK-1 | Mitogen-activated protein kinase-1 (ERK-2) | 0.2 | 0.4 |
Definition of abbreviation: FC = fold change in gene expression.
Collectively, these data identify signaling pathways with a high likelihood of involvement in BK-mediated regulation of LPS-induced cytokine secretion by reducing proinflammatory and promoting antiinflammatory signals.
Discussion
In this study, we provide the first evidence for the expression of functional BK channels (pore-forming α1 subunit KCNMA1 and the auxiliary KCNMB1, -3, -4 β-subunits) in human microvascular endothelial cells and in primary human alveolar epithelial cells by demonstrating pharmacological hyperpolarization with two BK activators (NS1619 and NS19504), and propose a potential role for BK channels in LPS-induced cytokine secretion.
In tissues other than the lung, a role for BK channels in LPS-induced inflammation has previously been implicated by a few other groups. In fact, in LPS-infected mouse urinary bladders, inhibition of BK with Iberiotoxin prevents inflammatory cytokines secretion, including IL-6 (22), and in rat hippocampal neurons, BK activation promotes IL-1-β release (23). BK-mediated regulation of cytokine secretion from macrophages appears dependent of the cellular phenotype. In RAW264.7 murine macrophages, genetic (siRNA) or pharmacological (Iberiotoxin) inhibition of BK channels increases TNF-α release (24), whereas in bone marrow–derived macrophages, BK inhibition suppresses TNF-α and IL-6 release (25, 26).
Our findings expand our knowledge on the role of BK channels in regulation of inflammatory processes by demonstrating an important role for BK activity in cytokine secretion from human lung resident cells, such as primary alveolar epithelial cells and pulmonary microvascular endothelial cells. Remarkably, BK channel activation with NS1619, the most commonly used BK agonist, inhibited CCL-2 but not IL-6 secretion from endothelial but not epithelial cells. Conversely, BK channel inhibition with a classic BK blocker, Paxilline, had no effect on LPS-induced cytokine secretion from either cell type (27), suggesting that most BK channels are likely closed under those conditions. It was also interesting to observe a larger variability in baseline CCL-2 and IL-6 secretion from epithelial cells than endothelial cells, which could be explained by the fact that the HULEC5a cells consist of a fairly homogenous cell population, whereas the primary alveolar epithelial cells represent a mixture of type-1 and type-2 cells and may derive from more than one donor.
Based on current knowledge, we initially anticipated that LPS stimulation would increase iCa2+ levels, resulting in an increase in inflammatory cytokine secretion, and that we could counteract this process by hyperpolarizing the Em with a BK activator (NS1619 and NS19504). Of note, NS1619, a benzimidazolone, was introduced as a synthetic BK opener in 1994 and is the most commonly used pharmacological tool to activate functional BK channels (19). To our surprise, we found no effect of LPS exposure on the Em in either endothelial or epithelial cells. Previous literature reports suggest that the effects of LPS on the Em may vary among different cell types. In fact, LPS activates BK channels in murine vascular smooth muscle cells (28) but causes a delay in rectifier potassium currents in rabbit left atrial myocytes (29). Importantly, both cell types were susceptible to pharmacological Em hyperpolarization with two BK activators, NS1619 and NS19504, suggesting the presence of functional BK channels in both cell types and supporting the specificity of both drugs toward BK channels. Previous multiple parameter optimization analysis, which predicts drug-like properties of small molecules (30), revealed that NS19504 stands out from earlier reported compounds because of its low molecular weight and absence of an acidic function and, therefore, represents an interesting lead in the search for new BK channel modulators. Importantly, NS19504 (5-[(4-bromophenyl) methyl]-1,3-thiazol-2-amine), introduced in 2014 by Nausch and colleagues in HEK293 cells, has very little effect on IK, SK3, and Nav1.2 channels, and virtually no effect on high-threshold Cav channels (20). Furthermore, NS19504 also lacks any effects on 65 other channels and receptors, including L- and N-type Ca2+-channels; Nav, KATP, and hERG channels; muscarinic M1, M2, and M3 receptors; neuropeptide Y1 and Y2 receptors; NK1 receptor; and purinergic P2X and P2Y receptors (20). Together with our NS1619 data, these observations provide strong experimental support for the selectivity profile of NS19504 for BK channels and limit concerns for off-target effects. Nevertheless, as with all ion channel modulators, it is always important to remain vigilant for potential off-target effects. In fact, inhibitory effects of NS1619 have been reported on L-type Ca2+ channels in rat ventricular myocytes (31) and human neuroblastoma cells (SH-SY5Y) (32), but the lack of effect of the specific L-type Ca2+ channel inhibitor Nifedipine on cytokine secretion in our study argues against a contribution of such an off-target effect to our results. Another potential off-target effect of NS1619 observed in cardiac and neuronal cells is inhibition of the mitochondrial electron transport chain and ROS production, resulting in protection from ischemia and reperfusion injury (33). In an initial set of experiments, we found no effect of NS1619 or LPS on endothelial ROS production, and therefore, we chose to rather explore in more detail the effects of NS1619 on CCL-2 and IL-6 signaling cascades (Tables 1 and E1).
One of our most interesting findings is that BK activation (NS1619) decreases LPS-induced CCL-2 secretion (Figure 3A). CCL-2 is a proinflammatory mediator found in high concentrations in the BAL fluid and plasma of patients with ARDS and LPS-induced ALI mouse models (34). Secretion of CCL-2 by LPS-activated endothelial and epithelial cells results in the selective recruitment of leukocytes to inflammatory foci, thus promoting inflammation (35), but can also accelerate bacterial clearance during sepsis (36).
Although multiple studies have linked ion channel activity to changes in cytokine secretion (37), the molecular mechanisms by which K+ channels regulate secretory events in alveolar epithelial and microvascular endothelial cells are still unclear. Our data highlight important differences in the Em dependency of CCL-2 and IL-6 secretion between epithelial and endothelial cells, by showing that once LPS-stimulated endothelial cells are exposed to a hyperpolarized environment (BK channel activation), CCL-2 secretion is impaired, whereas IL-6 secretion appears to occur unharmed. In contrast, in epithelial cells, neither CCL-2 nor IL-6 secretion are altered by Em hyperpolarization with NS1619. Therefore, in the case of endothelial CCL-2 secretion, it is reasonable to assume that at least one voltage-dependent step exists in the LPS signaling cascade. Because classical LPS signaling occurs via the Toll-like receptor-4 (TLR-4), NF‐κB and MAPKs pathways (38), it is intriguing to speculate that activation of one of these molecules is voltage-dependent and plays a key role in the electrochemical signal conversion process. Such a finding would be a very important, novel, and potentially paradigm-shifting concept and needs to be further explored. Notably, TLR-4 stimulation can also activate BK channels directly by elevating intracellular iCa2+ concentrations because BK is highly Ca2+ sensitive (39). However, such an iCa2+-dependent, LPS-TLR-4–mediated effect on BK channels is unlikely in endothelial cells because we saw no changes in iCa2+ levels with LPS treatment. Furthermore, although LPS is known to increase iCa2+ concentrations in several cell types (22, 40, 41), the lack of such an effect in HULEC5a cells helps explain why inhibition of voltage-gated CaV channels with Nifedipine had no effect on CCL-2 or IL-6 secretion (Figure 4). Notably, expression of Nifedipine-sensitive, L-type (CaV 1.2) channels in pulmonary endothelial cells has previously been reported (42). We also found that the BK inhibitor Paxilline had no effect on the Em (Figure 2C) or iCa2+ levels (Figures 4E and 4F), suggesting that in both endothelial and epithelial cells, most BK channels are constitutively closed.
Interestingly, and in contrast to our findings, in certain cell types, BK channel expression and activity appear to be upregulated by LPS. In RAW264.7 murine macrophages, LPS upregulates BK channel expression (24), and in mouse bladder epithelium (umbrella cells), BK channel activity is increased after LPS exposure (43). In murine microglia, plasma membrane BK currents can be activated by LPS at a very early stage through TLR-4, leading to nuclear translocation of NF-κB-1 and production of proinflammatory cytokines such as TNF-α and IL-6 (44). Similarly, studies in murine monocytes/macrophages found that BK channels are opened by LPS, leading to inhibition of NF-κB-1 translocation to the nucleus, and a decrease in TNF-α and IL-8 secretion (45).
Our microarray and real-time PCR results in HULEC5a cells highlight several pro- and antiinflammatory genes involved in LPS-induced signaling pathways that are regulated by the BK activator NS1619 and could be responsible for the observed decrease in LPS-induced CCL-2 secretion (Table 1). Interestingly, although our top candidates (TNFSF-13B, CASP-1, PTGS-2, IL1R-1) confirm NF-κB-1–dependent signaling cascades, NF-κB-1 expression itself was not changed by NS1619. This is a particularly interesting finding because NF-κB is known for both its pro- and antiinflammatory properties (46). Of note, TNFSF13B (BAFF), a member of the TNF ligand superfamily, acts downstream of the IL1R1 and TLR-pathway through an NF-κB pathway (47), whereas CASP-1 is recruited by the NLRP3-inflammasome and promotes secretion of proinflammatory cytokines (“proinflammatory” caspase) (48), and IL1R-1 facilitates TLR-driven antiinflammatory cytokine production (49, 50). The effects of BK activation (NS1619) on members of the eicosanoid signaling pathway are also interesting (upregulation of PTGS-2) and need further investigation. Furthermore, we also found that NS1619 downregulates TLR-4 expression, the main LPS-binding receptor, which might contribute to the observed decrease in endothelial CCL-2 secretion. Similarly, it appears that among the numerous MAP kinases, the JNK-1 pathway is particularly involved in the regulation of CCL-2 secretion, whereas other main pathway candidates (p38 and ERK-1/2) are less likely contributors.
Taken together, our results demonstrate that BK channel activation reduces LPS-induced CCL-2 release from human pulmonary microvascular endothelial cells but not alveolar epithelial cells, and that these events occur independently of Ca2+ influx. Based on our microarray and real-time PCR data, we propose a model where NS1619 downregulates TLR-4 expression and subsequently (or simultaneously) activates JNK-1-mediated signaling cascades that ultimately result in a decrease in LPS-induced CCL-2 secretion (Figure 5). For future studies, it is particularly important to focus on Em-dependent versus Em-independent mechanisms of inflammatory cytokine secretion. These processes remain poorly understood but may constitute novel targets for rational antiinflammatory drug design.
Figure 5.

(A) Functional protein association network (STRING) model based on our microarray and confirmatory real-time PCR data on HULEC5a cells treated with LPS and NS1619. (B) Putative model of NS1619-mediated LPS signaling downstream pathways in HULEC5a cells. More-likely pathways are labeled in black, while less-likely pathways are labeled in gray.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank Dr. Liana Basova (San Diego Biomedical Research Institute) for her help with interpretation of the microarray data.
Footnotes
Supported by the U.S. National Institutes of Health grants: 7K08HL118118-03 (A.S.), R01HL134346 (R.O.), and R01HL130308 (M.O.).
Author Contributions: T.Z., M.O., R.O., and A.S.: conception and design of the research. T.Z., B.L., A.L., C.G., and L.W.: experimental execution. T.Z., B.L., A.L., M.O., R.O., and A.S.: data analysis and interpretation. T.Z. and A.S.: figure and manuscript preparation. All authors approved the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0228OC on November 20, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Nova Z, Skovierova H, Calkovska A. Alveolar-capillary membrane-related pulmonary cells as a target in endotoxin-induced acute lung injury. Int J Mol Sci. 2019;20:E831. doi: 10.3390/ijms20040831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, et al. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS: plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995;107:1062–1073. doi: 10.1378/chest.107.4.1062. [DOI] [PubMed] [Google Scholar]
- 3.Schwingshackl A, Teng B, Ghosh M, Waters CM. Regulation of monocyte chemotactic protein-1 secretion by the two-pore-domain potassium (K2P) channel TREK-1 in human alveolar epithelial cells. Am J Transl Res. 2013;5:530–542. [PMC free article] [PubMed] [Google Scholar]
- 4.Schwingshackl A, Roan E, Teng B, Waters CM. Trek-1 regulates cytokine secretion from cultured human alveolar epithelial cells independently of cytoskeletal rearrangements. PLoS One. 2015;10:e0126781. doi: 10.1371/journal.pone.0126781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwingshackl A, Teng B, Ghosh M, West AN, Makena P, Gorantla V, et al. Regulation and function of the two-pore-domain (K2P) potassium channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2012;302:L93–L102. doi: 10.1152/ajplung.00078.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwingshackl A, Teng B, Makena P, Ghosh M, Sinclair SE, Luellen C, et al. Deficiency of the two-pore-domain potassium channel TREK-1 promotes hyperoxia-induced lung injury. Crit Care Med. 2014;42:e692–e701. doi: 10.1097/CCM.0000000000000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lengyel M, Czirják G, Enyedi P. Formation of functional heterodimers by trek-1 and trek-2 two-pore domain potassium channel subunits. J Biol Chem. 2016;291:13649–13661. doi: 10.1074/jbc.M116.719039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwingshackl A, Teng B, Ghosh M, Lim KG, Tigyi G, Narayanan D, et al. Regulation of interleukin-6 secretion by the two-pore-domain potassium channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;304:L276–L286. doi: 10.1152/ajplung.00299.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dopico AM, Bukiya AN, Jaggar JH. Calcium- and voltage-gated BK channels in vascular smooth muscle. Pflugers Arch. 2018;470:1271–1289. doi: 10.1007/s00424-018-2151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Contreras GF, Castillo K, Enrique N, Carrasquel-Ursulaez W, Castillo JP, Milesi V, et al. A BK (Slo1) channel journey from molecule to physiology. Channels (Austin) 2013;7:442–458. doi: 10.4161/chan.26242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu H, Wang Y, Garver H, Galligan JJ, Fink GD. Vascular BK channel deficiency exacerbates organ damage and mortality in endotoxemic mice. J Cardiovasc Pharmacol. 2012;59:207–214. doi: 10.1097/FJC.0b013e31823b493b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carvacho I, Gonzalez W, Torres YP, Brauchi S, Alvarez O, Gonzalez-Nilo FD, et al. Intrinsic electrostatic potential in the BK channel pore: role in determining single channel conductance and block. J Gen Physiol. 2008;131:147–161. doi: 10.1085/jgp.200709862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morera FJ, Saravia J, Pontigo JP, Vargas-Chacoff L, Contreras GF, Pupo A, et al. Voltage-dependent BK and Hv1 channels expressed in non-excitable tissues: new therapeutics opportunities as targets in human diseases. Pharmacol Res. 2015;101:56–64. doi: 10.1016/j.phrs.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Zyrianova T, Lopez B, Liao A, Gu C, Wong L, Ottolia M, et al. Large conductance Ca2+-activated k+ channels regulate lps-induced cytokine secretion from alveolar epithelial and endothelial cells. 64th Annual meeting of the Biophysical Society. 2020, San Diego, CA. [Google Scholar]
- 15.Chen MX, Gatfield K, Ward E, Downie D, Sneddon HF, Walsh S, et al. Validation and optimization of novel high-throughput assays for human epithelial sodium channels. J Biomol Screen. 2015;20:242–253. doi: 10.1177/1087057114552399. [DOI] [PubMed] [Google Scholar]
- 16.Gadgaard C, Jensen AA. Functional characterization of 5-HT1A and 5-HT1B serotonin receptor signaling through G-protein-activated inwardly rectifying K+ channels in a fluorescence-based membrane potential assay. Biochem Pharmacol. 2020;175:113870. doi: 10.1016/j.bcp.2020.113870. [DOI] [PubMed] [Google Scholar]
- 17.Falk-Petersen CB, Søgaard R, Madsen KL, Klein AB, Frølund B, Wellendorph P. Development of a robust mammalian cell-based assay for studying recombinant α4 β1/3 δ GABAA receptor subtypes. Basic Clin Pharmacol Toxicol. 2017;121:119–129. doi: 10.1111/bcpt.12778. [DOI] [PubMed] [Google Scholar]
- 18.Krämer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olesen SP, Munch E, Moldt P, Drejer J. Selective activation of Ca(2+)-dependent K+ channels by novel benzimidazolone. Eur J Pharmacol. 1994;251:53–59. doi: 10.1016/0014-2999(94)90442-1. [DOI] [PubMed] [Google Scholar]
- 20.Nausch B, Rode F, Jørgensen S, Nardi A, Korsgaard MP, Hougaard C, et al. NS19504: a novel BK channel activator with relaxing effect on bladder smooth muscle spontaneous phasic contractions. J Pharmacol Exp Ther. 2014;350:520–530. doi: 10.1124/jpet.113.212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y, Lingle CJ. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J Gen Physiol. 2014;144:415–440. doi: 10.1085/jgp.201411259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeh J, Lu M, Alvarez-Lugo L, Chai TC. Bladder urothelial BK channel activity is a critical mediator for innate immune response in urinary tract infection pathogenesis. Am J Physiol Renal Physiol. 2019;316:F617–F623. doi: 10.1152/ajprenal.00554.2018. [DOI] [PubMed] [Google Scholar]
- 23.Zhang B, Zhang Y, Wu W, Xu T, Yin Y, Zhang J, et al. Chronic glucocorticoid exposure activates BK-NLRP1 signal involving in hippocampal neuron damage. J Neuroinflammation. 2017;14:139. doi: 10.1186/s12974-017-0911-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida M, Willis D. Evidence for a role for BK channels in the regulation of ADAM17 activity [preprint] bioRxiv. 2019 [accessed 2019 Oct 19]. Available from: https://www.biorxiv.org/content/10.1101/811000v1. [Google Scholar]
- 25.Essin K, Gollasch M, Rolle S, Weissgerber P, Sausbier M, Bohn E, et al. BK channels in innate immune functions of neutrophils and macrophages. Blood. 2009;113:1326–1331. doi: 10.1182/blood-2008-07-166660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wissuwa B, Heinemann SH, Bauer M, Coldewey SM. Studies into Slo1 K+ channels and their ligand docosahexaenoic acid in murine sepsis to delineate off-target effects of immunonutrition. Life Sci. 2018;203:112–120. doi: 10.1016/j.lfs.2018.04.031. [DOI] [PubMed] [Google Scholar]
- 27.Imlach WL, Finch SC, Dunlop J, Dalziel JE. Structural determinants of lolitrems for inhibition of BK large conductance Ca2+-activated K+ channels. Eur J Pharmacol. 2009;605:36–45. doi: 10.1016/j.ejphar.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 28.Yakubovich N, Eldstrom JR, Mathers DA. Lipopolysaccharide can activate BK channels of arterial smooth muscle in the absence of iNOS expression. Biochim Biophys Acta. 2001;1514:239–252. doi: 10.1016/s0005-2736(01)00378-9. [DOI] [PubMed] [Google Scholar]
- 29.Tai BY, Wen ZH, Cheng PY, Yang HY, Duh CY, Chen PN, et al. Lemnalol modulates the electrophysiological characteristics and calcium homeostasis of atrial myocytes. Mar Drugs. 2019;17:619. doi: 10.3390/md17110619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park WS, Kang SH, Son YK, Kim N, Ko JH, Kim HK, et al. The mitochondrial Ca2+-activated K+ channel activator, NS 1619 inhibits L-type Ca2+ channels in rat ventricular myocytes. Biochem Biophys Res Commun. 2007;362:31–36. doi: 10.1016/j.bbrc.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 32.Curci A, Mele A, Camerino GM, Dinardo MM, Tricarico D. The large conductance Ca(2+) -activated K(+) (BKCa) channel regulates cell proliferation in SH-SY5Y neuroblastoma cells by activating the staurosporine-sensitive protein kinases. Front Physiol. 2014;5:476. doi: 10.3389/fphys.2014.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gáspár T, Katakam P, Snipes JA, Kis B, Domoki F, Bari F, et al. Delayed neuronal preconditioning by NS1619 is independent of calcium activated potassium channels. J Neurochem. 2008;105:1115–1128. doi: 10.1111/j.1471-4159.2007.05210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao J, Yu H, Liu Y, Gibson SA, Yan Z, Xu X, et al. Protective effect of suppressing STAT3 activity in LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2016;311:L868–L880. doi: 10.1152/ajplung.00281.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bierhaus A, Chen J, Liliensiek B, Nawroth PP. LPS and cytokine-activated endothelium. Semin Thromb Hemost. 2000;26:571–587. doi: 10.1055/s-2000-13214. [DOI] [PubMed] [Google Scholar]
- 36.Gomes RN, Teixeira-Cunha MG, Figueiredo RT, Almeida PE, Alves SC, Bozza PT, et al. Bacterial clearance in septic mice is modulated by MCP-1/CCL2 and nitric oxide. Shock. 2013;39:63–69. doi: 10.1097/SHK.0b013e31827802b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamacher J, Hadizamani Y, Borgmann M, Mohaupt M, Männel DN, Moehrlen U, et al. Cytokine-ion channel interactions in pulmonary inflammation. Front Immunol. 2018;8:1644. doi: 10.3389/fimmu.2017.01644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-like receptors: significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol. 2018;37:20–36. doi: 10.1080/08830185.2017.1380200. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann A, Kann O, Ohlemeyer C, Hanisch UK, Kettenmann H. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J Neurosci. 2003;23:4410–4419. doi: 10.1523/JNEUROSCI.23-11-04410.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tauseef M, Knezevic N, Chava KR, Smith M, Sukriti S, Gianaris N, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med. 2012;209:1953–1968. doi: 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang K, Wang P, Huang S, Wang X, Li T, Jin Y, et al. Different mechanism of LPS-induced calcium increase in human lung epithelial cell and microvascular endothelial cell: a cell culture study in a model for ARDS. Mol Biol Rep. 2014;41:4253–4259. doi: 10.1007/s11033-014-3296-1. [DOI] [PubMed] [Google Scholar]
- 42.De Proost I, Brouns I, Pintelon I, Timmermans JP, Adriaensen D. Pulmonary expression of voltage-gated calcium channels: special reference to sensory airway receptors. Histochem Cell Biol. 2007;128:301–316. doi: 10.1007/s00418-007-0318-2. [DOI] [PubMed] [Google Scholar]
- 43.Lu M, Li JR, Alvarez-Lugo L, Li Y, Yu S, Li X, et al. Lipopolysaccharide stimulates BK channel activity in bladder umbrella cells. Am J Physiol Cell Physiol. 2018;314:C643–C653. doi: 10.1152/ajpcell.00339.2017. [DOI] [PubMed] [Google Scholar]
- 44.Yang X, Wang G, Cao T, Zhang L, Ma Y, Jiang S, et al. Large-conductance calcium-activated potassium channels mediate lipopolysaccharide-induced activation of murine microglia. J Biol Chem. 2019;294:12921–12932. doi: 10.1074/jbc.RA118.006425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blunck R, Scheel O, Müller M, Brandenburg K, Seitzer U, Seydel U. New insights into endotoxin-induced activation of macrophages: involvement of a K+ channel in transmembrane signaling. J Immunol. 2001;166:1009–1015. doi: 10.4049/jimmunol.166.2.1009. [DOI] [PubMed] [Google Scholar]
- 46.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim DH, Do MS. BAFF knockout improves systemic inflammation via regulating adipose tissue distribution in high-fat diet-induced obesity. Exp Mol Med. 2015;47:e129. doi: 10.1038/emm.2014.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.González-Navajas JM, Law J, Nguyen KP, Bhargava M, Corr MP, Varki N, et al. Interleukin 1 receptor signaling regulates DUBA expression and facilitates Toll-like receptor 9-driven antiinflammatory cytokine production. J Exp Med. 2010;207:2799–2807. doi: 10.1084/jem.20101326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCann B, Miaskowski C, Koetters T, Baggott C, West C, Levine JD, et al. Associations between pro- and anti-inflammatory cytokine genes and breast pain in women prior to breast cancer surgery. J Pain. 2012;13:425–437. doi: 10.1016/j.jpain.2011.02.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.