

Abstract
Newborn screening for severe combined immunodeficiency (SCID), the most profound form of primary immune system defects, has long been recognized as a measure that would decrease morbidity and improve outcomes by helping patients avoid devastating infections and receive prompt immune restoring therapy. The TREC test, developed in 2005, proved to be successful in pilot studies starting in 2008-2010, and by 2019 all states in the USA had adopted versions of it in their public health programs. Introduction of newborn screening for SCID, the first immune disorder accepted for population-based screening, has drastically changed the presentation of this disorder while providing important lessons for public health programs, immunologists and transplanters.
Keywords: primary immunodeficiency; T cell receptor excision circle (TREC); severe combined immunodeficiency (SCID); lymphocytopenia; dried blood spot, newborn screening
Introduction
In 2010, severe combined immunodeficiency (SCID) was added to the national list of conditions for which newborn screening (NBS) should be conducted, called the Recommended Uniform Screening Panel (RUSP), based on findings of the Advisory Committee for Heritable Disorders of Newborns and Children to the Department of Health and Human Services.1 This marked a watershed in the diagnosis and treatment of SCID in the United States. Indeed, by the end of 2018, NBS for SCID had been adopted by public health programs in all 50 states, the Navajo Nation and Puerto Rico. With NBS in place, the overwhelming majority of infants with SCID are identified by screening instead of by the classical presentation with infectious complications.
The case for SCID NBS arose from data showing improved survival of infants recognized by a positive family history and treated with life-saving hematopoietic cell transplantation (HCT) therapy within the first 3.5 months of life, compared to infants diagnosed and treated later, after suffering increasingly severe and debilitating infections.2 SCID is caused by mutations in any of over 20 genes governing the differentiation and proliferation of mature lymphocyte lineages derived from hematopoietic stem cells (HSC) in the bone marrow.3 However, a large majority of SCID cases are sporadic, with no recognized family history to prompt early diagnosis.4 Infants with SCID appear healthy as newborns and, prior to SCID NBS, were diagnosed only after frequent medical encounters for recurrent and persistent infections, infections with opportunistic organisms, and/or failure to thrive. These non-specific indications led to delays in recognition of the underlying cause and subsequent delays in treatment, with HCT often performed only after the patient had acquired devastating infections that compromised outcomes.5
In 2014, the newborn screening programs from 10 states and the Navajo Nation, which were early to adopt SCID NBS, reported their pooled experience.6 They found that the incidence of SCID was 1/58,000, nearly double what had been estimated from previous studies. Further, the survival of infants who received HCT, gene therapy (GT) or enzyme replacement therapy, for adenosine deaminase (ADA) deficient SCID, was 92%, in contrast with prior reports and current data of around 70% in a large European study of SCID HCT without NBS.7 An additional recent publication of results from screening over 3.2 million infants in California confirmed a similar incidence of 1/65,000 births, with 96% survival.8 These and other publications have also addressed a range of non-SCID conditions that can be found in infants who have an abnormal SCID NBS result, but do not have SCID.
As of this writing, multiple nations around the world have instituted population-wide NBS for SCID, including New Zealand, Israel, Sweden and Germany, while others offer SCID screening in limited areas or have published pre-implementation analyses and pilot studies, all with enthusiastic support.9-14 Some programs point to particular local issues that make TREC screening even more important, such as founder mutations or high rates of consanguineous marriage. We review here what has been learned to date from the development and implementation of SCID NBS.
The TREC assay
SCID screening was made possible by the fortuitous circumstance that a stable circular DNA molecule, a T cell receptor excision circle (TREC), is generated during the normal process of T cell receptor (TCR) diversification in the thymus.15 TRECs are a byproduct of recombination of the genes encoding the cell surface receptors for antigen, which are responsible for recognizing molecules held by major histocompatibility complexes on the surface of antigen presenting cells. As thymocytes become mature T cells, the genes encoding TCR subunits, which contain arrays of alternate variable (V) diversity (D) and joining (J) segments undergo a process of DNA rearrangement to bring unique combinations of segments together to be joined with a constant region; when expressed, these VDJ recombinant TCR genes encode highly diverse receptor molecules, generating T cells that can bind to vast numbers of molecules encountered by the host. In the immature thymocytes, VDJ recombination generates approximately 1011 different in-frame splice products that express TCRs, but further recombination and selection reduces this population to around 3-9 x 109 different antigenic specificities among T cells in peripheral blood.
The excised DNA fragments of TCR genes that are not destined to be part of the mature TCR locus are joined at their ends to form circular TRECs (Figure 1). Late in the maturation program, 70% of the thymocytes that will ultimately express αβ TCRs form one specific circular DNA TREC, the δRec-ψJa signal joint TREC, from the excised TCRδ gene (TCRD) that lies within the TCRα gene (TCRA) locus. TRECs are stable and are maintained following cell divisions; but because they do not replicate, they become diluted as T cells proliferate by mitotic division. A quantitative polymerase chain reaction (qPCR) across the joint of the TREC, using primers indicated by the short arrows in Figure 1, provides the TREC copy number, an indicator of new thymic emigrant T cells being produced. Absent TRECs suggest the inability to produce T cells due to any of a number of mutations in genes required for the differentiation of T cells from hematopoietic stem cells. The peripheral blood of infants with SCID will be devoid of T cells and have very low or undetectable TRECs.16 Testing laboratories empirically select a cutoff value for TRECs below which referral to an immunologist for follow-up testing is indicated. This cutoff must be set to include nearly all typical SCID and even leaky or hypomorphic SCID cases, while also minimizing false positives. Now that a large number of programs have instituted TREC testing we have learned that initial cutoff values were invariably too high, resulting in excessive referrals; reduction of cutoffs to achieve optimal screening test specificity can take place without losing actual SCID cases.8,17
Figure 1. Generation of the δRec-ψJα TREC.
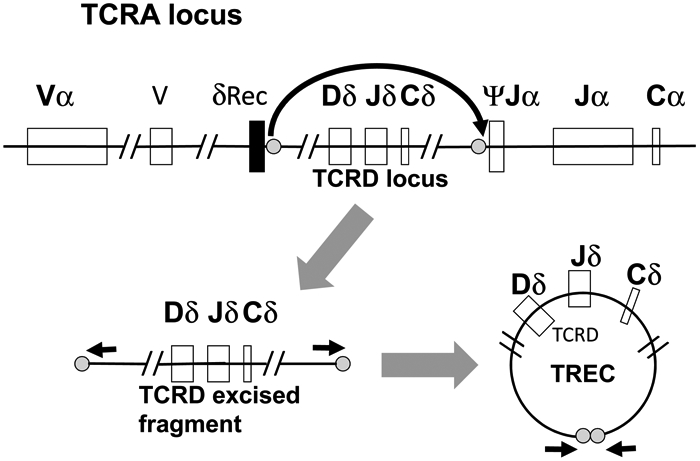
The germ line configuration of the TCRA locus, with TCRD embedded, is shown at the top of the Figure, which also shows the signal and joining sequences (gray circles) at which the DNA is cut to excise the TCRD locus. The DNA segment excised is shown in its linear form, lower left, and following ligation at the fragment ends, lower right, to form a T cell receptor excision circle (TREC). PCR primers (black arrows) can amplify a short DNA segment containing the joined ends of the circle.
TREC suitability as a newborn screening test
NBS tests in public health programs, like all population screening tests, need to meet established criteria regarding public health need, performance, laboratory methodology, and economic impact. The basic principles of population screening, outlined by Wilson and Jungner in 1968,18 were formulated to guide attempts to identify the small number of individuals within a large population who are at sufficiently high risk for a disease to justify referral for diagnostic testing. A successful screening test must find almost all of the target disease within the population; that is, it will have a very high sensitivity. At the same time, it is necessary not to refer too large a percentage of the general population for diagnostic testing; the specificity must also be high. From the perspective of health system utilization, it is important that the diagnostic specialists’ time not be overrun with false positive cases. Ideally, the positive predictive value of the test should be very high.
The laboratory criteria for NBS testing are similarly based on the need for streamlined processing. The specimens need to be transported from the birth hospitals to the central laboratory, so the specimen needs to be sufficiently stable for the analysis to be accurate. NBS programs have predominantly relied on dried blood spots (DBS) on filter paper (Guthrie cards) as the specimen of choice. In addition to uniformity and ease of sample collection, the DBS is easy to transport and stable essentially indefinitely if stored under desiccation at −20° C. The largest NBS programs need to test hundreds, or even thousands, of specimens each day. Due to the high throughput, appropriate tests need to be capable of automation with rapid run times. An economic criterion is that the expense of the test, both in fixed costs for personnel and equipment and incremental, per-test costs, must be very low, since the overall test volume is large.
The TREC assay is based on qPCR for the DNA sequence representing the joining of the circular TCR excision fragment. After excluding tests for which the DNA amplification fails entirely, as demonstrated by the failure of control genomic DNA (either an actin or RNaseP gene segment) to amplify, this assay is highly specific. The technology is sufficiently mature that commercially available kits are available, for use either with existing PCR equipment or with equipment provided.
While TREC testing required introduction of DNA analysis as a primary screening modality for the first time, we have learned that this platform is not limited to TRECs. A benefit of the adoption of DNA extraction and qPCR technology by NBS laboratories has been the ease of further expansion to other disorders that can be detected by well-characterized sequences and are also deemed appropriate for NBS.19,20 Recently, spinal muscular atrophy (SMA), caused by homozygous deletion of exon 7 of the SMN1 (survival motor neuron one) gene, was added to the RUSP for NBS. The test uses DNA from DBS to detect the two single nucleotide differences between the sequences of SMN1, the functionally impaired gene in SMA, and SMN2, a very close homologue in which one modified nucleotide causes the omission of exon 7 in the spliced transcript.
Follow-up for abnormal TREC test results
The interpretation of TREC results in the California NBS program are summarized in Table 1; every program has developed individual interpretation algorithms and cutoffs, but overall approaches are similar. In California and many programs, the first TREC qPCR run does not include the control gene (actin or RNaseP), primarily for cost-saving reasons. If the initial TREC value is below the program cutoff, the analysis is repeated using the same DBS specimen, this time including the control gene. In this second analysis, if the control gene does not amplify, it is necessary to get a new DBS specimen. If the control gene does amplify but the TREC value remains low, the infant will be flagged for further testing, which in California is performed as a follow-on test within the program. Most NBS programs also have an “Urgent positive” cutoff for DBS with undetectable or very low TREC numbers, but satisfactory amplification of control DNA (Table 1). Infants with Urgent positive values are at very high risk for SCID, and consequently, their follow-up is expedited. Infants who appear healthy, but who have Urgent positive TREC results, are referred immediately for lymphocyte subset determination by flow cytometry and evaluation by a specialist in pediatric immunology. Apparently healthy infants with a positive, but not urgent TREC result are also referred.
Table 1:
TREC test results, interpretation and actions required in the California SCID NBS program.a
| TREC copy number |
Actin control copy number |
TREC test interpretation |
Action required |
|---|---|---|---|
| >18/μlb | N/Ac | Normal | No further action. |
| Undetectable or <4/μl | >35/μlb | Urgent positive | Immediate callback for liquid blood for lymphocyte subsets.d |
| 4-18/μl, infant not in NICUe | >35/μl | Positive | Callback for liquid blood for lymphocyte subsets.d |
| 4-18/μl, infant in NICU | N/A | Incomplete | Second DBS test in 2 weeks or at discharge from nursery; after 2 incomplete tests, liquid blood for lymphocyte subsets. |
Modified from ref. 8, Amatuni et al.
TREC and actin copies/μl of peripheral blood equivalent, using EnLite™ TREC kit (PerkinElmer).
Not applicable; actin not reported if initial TREC value normal.
Lymphocyte subsets included as part of the NBS program in California.
Neonatal intensive care unit.
Low birthweight, preterm and ill infants cared for in a neonatal intensive care unit (NICU) have higher rates of false positives or unacceptable results for all NBS tests for a variety of reasons; in the recent California report NICUs accounted for only 9% of births, but were the source of 47% of TREC tests that were positive or had poor control DNA amplification, even after obtaining a repeat DBS.8 Many NBS programs have a policy of routine collection of a second NBS specimen from infants in the NICU after two weeks (Table 1). Some also collect a third specimen at 28 days. Waiting to collect the second specimen allows the infant’s immune system time to mature, with the possibility of normalized TREC values. Extremely preterm and low birth weight infants have lower numbers of lymphocytes than term infants, so low TREC values in this group may simply indicate prematurity.
The follow-on test for infants who fail to have normal TREC results is a complete blood count, differential count and lymphocyte subsets by flow cytometry, including naïve and memory phenotype helper and cytotoxic T cells as well as B and NK cells. Because a liquid blood sample is required, infants who are no longer hospitalized must be recalled to a phlebotomy center for this level of testing. Flow cytometry may diagnose SCID immediately, but further testing of T cell function with lymphocyte proliferation assays and determination of maternal engraftment of T cells, as well as gene sequencing, are generally required to establish a specific form of SCID.
Ideally, flow cytometry testing should be coordinated by the NBS program, as is done in California. We have learned that there are several advantages of this, compared to screening programs that stop being involved after reporting out abnormal TREC results. Most important, because NBS should be viewed not merely as a test, but as a program to identify infants with serious, treatable disease, there is a public health interest to make sure all infants with SCID receive care from appropriate specialists without delay. If programs do not include a standard level of flow cytometry testing, some infants, notably those from disadvantaged families or who live in rural areas, may encounter delays for insurance authorization or may receive testing that is not optimally informative and requires further iterations and more time to make a diagnosis. It is also important for NBS programs to work closely with a network of pediatric immunologists who have expertise in newborns and who understand the importance of controlling health care costs. Some physicians may feel it necessary to order thousands of dollars’ worth of tests at a first visit, most of which will prove unnecessary since at least half of infants with positive TREC tests will have normal flow cytometry. Uniformity of testing and interpretation of results across the program is essential not only to assure that infants with abnormal TREC screens receive prompt, high quality care, but also to allow the program to track results and outcomes, making adjustments as needed for quality improvement.
The fraction of TREC-positive infants determined to have insufficient T cells has ranged from 35% to nearly 100%,6 depending on both the NBS program TREC cutoffs and the definitions of T cell leukopenia (TCL) applied by specialists. While some consider any T cell number below the reference range for term infants (~2250 to 2500/μl) to represent TCL, the California program restricts significant TCL to be 1500 or fewer T cells/μl as long as naïve T cells are present. The referral rate for this degree of TCL in California has been only 1 per 15,000 births. Experience has proven this cutoff, linked to the health intervention of live vaccine avoidance, to work very well. Live attenuated rotavirus vaccine, an otherwise important public health measure for infants in the first six months of life, has caused severe diarrhea in infants with SCID;21 therefore, the California immunologists working with the SCID NBS program consider it medically prudent to withhold this vaccine for all infants with fewer than 1500 T cells/μl and to evaluate these infants for the presence of not only SCID, but other conditions with T cell insufficiency.
After several years of recording lymphocyte data from the NBS program, we have been able to analyze data from over 300 infants with initial false positive TREC screens who proved to have no immune disorder upon follow-up. This permitted us to establish reference ranges for lymphocyte subsets of otherwise healthy preterm and low birthweight infants.22 We found that the absolute numbers of lymphocytes, T cells, T cell subsets, and B cells, increase with increasing gestational age and with birth weight, while NK cells rise only modestly. The relative percentages of the subsets remain essentially constant, making percentages less informative for tracking progress than absolute numbers. Notably, the multivariate analysis that was made possible by the large number of infants in this study revealed that after accounting for gestational age and birth weight there were no differences in lymphocyte values between California infants of different racial or ethnic backgrounds.22
Etiology of T-cell lymphopenia in TREC-positive newborns
The primary target of TREC screening is SCID, but the TREC test identifies infants with low TRECs and low T cells from any cause, summarized in Figure 2. In addition to SCID, categories of infants with medically significant non-SCID TCL identified by TREC screening include genetic syndromes associated with T cell impairment; excess losses of T cells or suppression of T cell maturation; preterm birth alone; and idiopathic TCL, with no identified cause.
Figure 2. Causes of T cell lymphopenia in 212 infants.
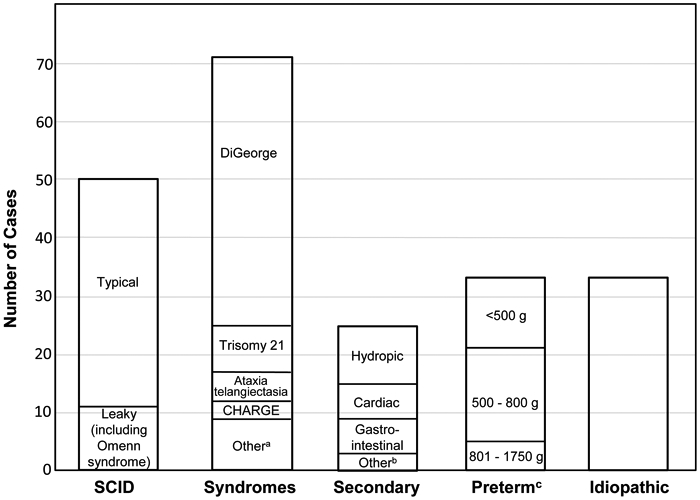
Included were 50 with typical or leaky SCID (including Omenn syndrome); 72 with a final diagnosis of a syndrome or single gene disorder associated with T cell impairment (of whom 22 were undiagnosed for several months or more); 25 with congenital conditions associated with secondary TCL; 33 with preterm birth alone; and 33 for whom no underlying defect was identified.
a Includes 3 cases of diabetic embryopathy, and one each of CLOVES syndrome (congenital lipomatous, overgrowth, vascular malformations, epidermal nevi and skeletal anomalies), EXTL3 deficiency (immunoskeletal dysplasia with neurodevelopmental abnormalities), Fryns syndrome (multiple anomalies with diaphragmatic defects), Nijmegen breakage syndrome (DNA repair defect in NBN causing immune defects, microcephaly, short stature and malignancy), Noonan syndrome (congenital heart disease, characteristic facial and physical features, short stature), and RAC2 deficiency (a non-SCID primary immunodeficiency affecting neutrophils and lymphocytes).
b Includes 2 cases of maternal immunosuppressive medication (azathioprine, fingolimod) and one case of neonatal teratoma of the thymus.
c Infants with TCL associated with preterm birth alone, grouped by birth weight.
SCID
The occurrence of the different SCID genotypes among newborn screened infants in the California experience is shown in Table 2 with a summary of their treatments and outcomes.8 While all genotypes of typical SCID have had absent or very low TRECs and T cells, B cells and NK may be present, with distinctive gene-associated lymphocyte profiles according to their respective damaged lymphocyte differentiation pathways, as reviewed elsewhere.23 In about 20% of SCID cases (Figure 2, left) hypomorphic mutations in SCID genes led to leaky SCID, including Omenn syndrome. Spontaneous engraftment of maternal T cells was also associated with higher T cell numbers than seen in typical SCID, but leaky SCID and maternal engraftment are almost always associated with positive TREC screens because the T cells have undergone antigen stimulation and expansion, diluting their TRECs exponentially with each division.6,15,16,23,24 Very rare cases of late-onset SCID have been missed by the TREC newborn screen, including two among 3.2 million California infants screened. Their initial TREC values were over three times the cutoff level,8 so no upward adjustment of the TREC cutoff would have allowed their detection. There are known hypomorphic mutations in several SCID genes that cause these late-onset, incomplete deficiencies. Individuals with the most mild ADA, IL2RG, RAG1 and RAG2 mutations may not present until adulthood, and presumably may have sufficient TRECs at birth to escape identification by NBS.25-7
Table 2.
Forty-nine infants with SCID detected by TREC screening in CA. Genotypes, treatments and outcomes
| SCID genotype (%) |
Number of typical and leaky casesa |
Outcome after treatmentb | ||
|---|---|---|---|---|
| Died post- HCT, cause |
T and B cell recovery |
T but not B cell recovery, still on IgG |
||
| IL2RG (28%) | 14 Typical | 1 (CMVc, SOSd) | 5 (2 Cond MUD, 2 GT, 1 Cond MUD after GT) | 8 (6 MMRD, 1 Cond MUDe, 1 GT after MMRD) |
| ADA (18%) | 8 Typical, 1 Leaky | 0 | 9 (8 GT, 1 MSD) | 0 |
| RAG1 (16%) | 2 Typical, 6 Leaky (3 Omenn) | 1 (SOS) | 6 (1 Cond MSD, 5 Cond MMRD) | 1 (Cond MMRD) |
| IL7R (12%) | 6 Typical | 0 | 6 (4 MMRD, 1 MUD, 1 Cond MSD after MSD) | 0 |
| JAK3 (6%) | 3 Typical | 0 | 2 (1 Cond MSD, 1 Cond MUD) | 1 (MMRD) |
| RAG2 (6%) | 2 Typical, 1 Leaky | 0 | 3 (1 MSD, 2 Cond MUD) | 0 |
| BCL11B (2%) | 1 Leaky | 0 | 1 (Cond MUD) | 0 |
| RMRP (2%) | 1 Leaky | 0 | 1 (Cond MUD) | 0 |
| Unknown (8%) | 3 Typical, 1 Leaky | 1 (CMV) | 2 (Cond MUD) | 1 (Cond MUD) |
Criteria from PIDTC for typical SCID and leaky SCID; Omenn syndrome, a subset of leaky SCID, includes erythroderma rash, adenopathy, oligocloal T cell expansion, eosinophilia, and elevated IgE.
Treatments: GT, gene therapy with low dose busulfan followed by autologous CD34+ bone marrow cells transduced with a correct copy of the mutated gene (IL2RG or ADA); MMRD, mismatched related donor (usually haploidentical parent); MSD, matched sibling donor; MUD, matched adult or cord blood unrelated donor. Cond, pretransplant chemotherapy conditioning with busulfan and in some instances fludarabine or other agents.
Cytomegalovirus infection.
Post-transplant hepatic sinusoidal obstruction syndrome after busulfan chemotherapy.
One patient left USA after conditioned MUD HCT and reconstitution; not known if still on IgG.
The 94% survival rate of California NBS SCID following treatment is outstanding compared to historical controls or to populations without newborn screening,7 but the three deaths in Table 2 remind us that challenges remain. In two fatalities, infection with cytomegalovirus (CMV) was involved, most likely acquired postnatally from intermittent virus shed in breastmilk of mothers who had experienced remote CMV infection. Even the short time between the NBS sample collection in the nursery and documentation of SCID by flow cytometry was sufficient for CMV infection to be established, and despite aggressive treatment this virus is difficult or impossible to control in infants lacking T cell immunity. Two fatalities were also linked to hepatic sinusoidal obstruction syndrome (SOS), a complication of chemotherapy with busulfan given for pre-transplant conditioning (one infant who died had both CMS and SOS). SOS can be minimized by pharmacokinetic targeting of exposure, essential for treating young infants with this agent because of its highly variable metabolism in those under 6 months of age.28,29 Important lessons from these deaths are twofold: 1) SCID should be regarded as a condition that requires most urgent management, including immediate measures to avoid CMV infection; and 2) for NBS to be fully effective, very young, very small infants with SCID are best treated in highly specialized pediatric transplant centers with experience and access to the latest information regarding SCID treatment.
Only 5 (10%) of the patients had a matched sibling donor (MSC), the ideal donor for HCT. Therefore, definitive lifesaving treatment for SCID is most often HCT from an alternative allogeneic donor, or, if available, autologous transplantation of gene corrected cells, to date available only in experimental clinical trials in the USA. Of the many genes that, when mutated, are associated with SCID3 the most commonly implicated (28% of California SCID cases, Table 2) is the X-linked interleukin-2 receptor (gamma) chain (IL2RG), in which T cells are absent, but intrinsically dysfunctional B cells are present.8,30-32 In agreement with the multicenter findings of the Primary Immune Deficiency Treatment Consortium (PIDTC),30,31 T cell recovery in X-linked SCID was regularly achieved with HCT from a variety of donors, but recovery of B cell function was uncommon after mismatched related donor HCT (usually with T-depleted cells from a parent without chemotherapy); all of 6 California infants receiving this treatment are likely to be dependent on lifelong IgG infusion therapy. In contrast, HCT from a matched unrelated donor (MUD) after preparative chemotherapy could lead to both B and T cell recovery, but this approach had the risk of toxicity from high dose chemotherapy, which proved fatal in one infant. The newer gene therapy option of targeted low-exposure busulfan followed by transplantation with autologous HSC transduced with a self-inactivating gene therapy vector, appears safe and promising, with at least two California infants to date fully immune reconstituted and off of IgG infusions.8,33 The vectors used in current trials have been redesigned for safety following the initial trials in France and England that were marred by vector-related T cell leukemias.34,35
The next most common genetic form of SCID after X-linked SCID was ADA deficiency (18% of California cases). Consistent with the systemic nature of this metabolic defect of purine salvage, infants with ADA SCID had, in addition to very low T, B, and NK cell numbers, neutropenia and low monocytes; further, they tended to have lower birth weights than other infants with SCID or healthy infants.8,36 Only with NBS for SCID did these very early features become apparent, as past reports lacked data close to the time of birth and were confounded in many instances by infectious complications that had occurred prior to diagnosis. Gene therapy for ADA SCID has become successful, as shown by the full immune reconstitution of all 8 infants given this treatment, generally after a “bridge” treatment period with injections of the ADA enzyme conjugated to polyethylene glycol. ADA was the original SCID genotype for which retroviral gene addition to autologous cells was attempted over four decades ago. With extensive technological evolution and the realization that low exposure chemotherapy was needed for durable, multi-lineage lymphocyte correction, gene therapy is now the treatment of choice for ADA SCID, particularly because success with HCT using donors other than matched siblings has not been satisfactory.36
The third most common defective gene in SCID cases is recombination activating gene 1 (RAG1), instrumental for the generation of the immune receptor repertoire. Infants with this form of SCID lack T and B cells, but have NK cells. Many of these infants have some level of T cell production; that is, they have leaky SCID. In Omenn syndrome, most commonly due to RAG1/2 defects, a small oligoclonal T cell pool expands dramatically, causing adenopathy, infiltrative erythroderma rashes and hepatosplenomegaly. Nonetheless, because TRECs are not replicated when T cells undergo mitosis, the number of TRECs in these infants is small, giving a positive NBS result.
Genetic syndromes
More than 14 genetic syndromes have been identified in newborns with positive TREC screening results, accounting for a third of all positive SCID NBS results (Figure 2).6,8 The most common by far is DiGeorge syndrome, usually due to heterozygous interstitial deletion of chromosome 22q11.2,6,8,37,38 found in 47 (66%) of the 71 syndromic TCL infants in the California series (Figure 2). Four of these 47 had complete DiGeorge syndrome treated by allogeneic thymus implantation.8 The next most common syndrome was trisomy 21 in eight infants (38%). CHARGE syndrome (coloboma of the eye, heart defect, atresia choanae, restricted growth and development, genital abnormality, and ear abnormality) and diabetic embryopathy8,39 (each with three cases), were also among the multisystem syndromes detected by NBS as having TCL. These syndromes have variable immune presentation, and many cases are identified due to congenital heart disease or other clinical features before the TREC result is available, although one study found that over half of DiGeorge syndrome cases identified by NBS had no prior recognition of the diagnosis.37 In contrast, ataxia telangiectasia, diagnosed in five California infants (7%) with positive SCID NBS, was never suspected before the clinical follow-up of the positive TREC result.8,40 This progressive disorder due to mutations of a DNA repair enzyme is characterized by cerebellar ataxia, neurodegeneration, ocular telangiectasia, variable T cell dysfunction and increased risk of lymphoma and other malignancies, but manifestations are absent in early infancy. Even the early indicator of increased alpha fetoprotein cannot be measured meaningfully until fetal levels wane, at around 6 months of age; after positive NBS, however, sequencing of the ataxia mutated gene (ATM) in an otherwise healthy appearing infant with low TRECs on NBS can establish the diagnosis of ataxia telangiectasia and permit early avoidance of X-ray exposure, anti-infectious prophylaxis, and family counseling regarding reproductive and cancer risks.40,41 Other extremely rare syndromes identified in California and elsewhere as having low TRECs on NBS include CLOVES syndrome (congenital lipomatous overgrowth, vascular and skeletal malformations, epidermal nevi); Cornelia de Lange syndrome;42 DOCK8 deficiency;43 EXTL3 deficiency with TCL, immuno-skeletal dysplasia and neurodevelopmental abnormalities;44 Fryns syndrome (multiple anomalies with diaphragmatic defects and lung maldevelopment); Kabuki syndrome;45 Nijmegen breakage syndrome;46 Noonan syndrome (congenital heart disease, characteristic facial and physical features, short stature); and RAC2 immunodeficiency (affecting neutrophils and lymphocytes).47
In some instances, these primary immune diseases were unknown before NBS for SCID and were identified only upon whole exome sequencing of newborns and their parents, followed by functional confirmatory studies in vitro or with animal models.44,47,48 Furthermore, because NBS is a population-level assessment, it can shed light on the proportion of individuals with these syndromes who have low T cell numbers at birth.40
T cell lymphopenia secondary to medical problems
The low T cells detected by the SCID NBS screen may be caused by anatomical or tissue abnormalities leading to vascular leakage, including congenital heart disease, hydrops, chylous effusion, gastroschisis, third-space fluid leakage, intestinal atresia and meconium ileus.6,8,49,50 Surgical or medical correction, or spontaneous resolution of the underlying defect leads to improvement of secondary T cell lymphopenia, which accounts for 12% of positive TREC screens (Figure 2). Teratoma of the thymus or thymoma in the newborn can present with a peripheral blood picture resembling SCID, but can resolve to normal after removal of the tumor. Similarly, neonatal leukemia with extensive infiltration of the bone marrow can suppress normal maturation of lymphocytes as well platelets and red blood cells, but recovery of the bone marrow can occur following successful treatment.
Maternal conditions and drugs given during pregnancy can also lead to positive TREC NBS and secondary TCL in the newborn. Maternal immunosuppressive therapy may be required during pregnancy to manage autoimmune diseases such as ulcerative colitis or Chron disease, lupus, rheumatoid arthritis and multiple sclerosis; maternal malignancy may also require urgent treatment before the infant can be delivered safely. Mycophenylate mofetil, methotrexate, azathioprine or 6-mercaptopurine given to mothers during pregnancy can cause transient TCL in infants.8,51 One infant, whose pregnant mother had received treatment for multiple sclerosis with fingolimod, a sphingosine-1-phosphate receptor modulator that sequesters lymphocytes in lymph nodes, had one TREC on NBS and a lymphocyte profile of T- B+ NK+ SCID with only 126 T cells/μl.8 However, the infant’s cells had normal proliferation to phytohemagglutinin, and with no treatment there were 1049 T cells/μl one week later and 2263 T cells/μl at 8 weeks of age. Fingolimod is a class C medication, meaning it is not to be given to women unless they are using effective contraception; indeed, the history was not immediately revealed to this baby’s immunology team, emphasizing the importance of building relationships between medical providers and parents of infants identified through NBS and of not rushing to HCT for SCID without first assessing T cell function. With increasing use of monoclonal antibody biologics to treat autoimmune disease, providers caring for infants with TCL need to recognize which ones may cross the placenta and affect the immune system of the fetus and newborn infant.
Prematurity
T cell lymphopenia can be associated with preterm birth, particularly in infants born before 33 weeks’ gestation and/or with birth weights under 800 g (16% of positive TREC screens, Figure 2). In general, the T cell numbers of these infants improve with time, approaching a normal level when the effective gestational age (gestational age at birth plus postnatal age) reaches 40 weeks.22 Serial measurements of lymphocytes, including naïve and memory CD4 and CD8 T cell subsets and total T and B cells showed steady increases over time, with less maturational rise of NK cells. However, we do not suggest that NBS programs modify their interpretation of TREC values or lymphocyte subsets based on gestational age at birth, birth weight, or NICU status.
Idiopathic T-cell lymphopenia
The final category of non-SCID TCL includes cases for which no explanation can be found during the follow-up evaluation. The California series had 55 cases of idiopathic TCL upon first report to the screening program (Table 3), but further information obtained over time often revealed an underlying diagnosis. Figure 2 shows the status of infants at the time of the report by Amatuni et al.8 with 33 cases (only 15% of all NBS positives) remaining without a diagnosis; but even further study, including whole exome and whole genome sequencing (WES and WGS) followed by functional studies of genetic variants is expected to explain further cases in the future. For example, WES in a few infants with abnormal TREC screens and TCL has revealed heterozygous variants in the FOXN1 (forkhead box N1) transcription factor gene, in which homozygous pathogenic mutations have caused a SCID phenotype with athymia as well as alopecia.52-54 Full understanding of which variants are damaging and the mechanism of their interference with T cell development will require further study. Moreover, the clinical course of the infants with idiopathic TCL has been instructive. Of those with available follow-up, T cells in approximately half improved or normalized with time, while half remained T lymphopenic. One child with persistent TCL and transfusion-dependent anemia with no proven underlying genotype required allogeneic HCT at age 3, and has achieved engraftment with functional T cells.55 Others, however, have remained healthy without recurrent infections. Continued follow-up is recommended for this group of children. They have already taught us about several previously unknown genes that are critical for lymphocyte development, leading to better understanding of immune system developmental processes.
Table 3:
Outcomes of 55 NBS cases of T cell lymphopenia initially classified as idiopathic.8
| Outcome | Number of infants |
Diagnosis, Comments |
|---|---|---|
| Received a diagnosis of a syndrome or single gene disorder affecting T cells | 22 | 9 DiGeorge 5 AT 3 diabetic embryopathy 2 CHARGE 1 each NBS, EXTL3, RAC2 |
| TCL resolved to normal range for age | 11 | In good health at last contact 1 in utero stress; maternal viral infection in late pregnancy |
| TCL improving at last contact | 2 | In good health |
| Persistent TCL | 13 | 1 HCT for low T cells and anemia 1 healthy but with nail dystrophy, heterozygous FOXN1 variant Others in good health |
| Died | 1 | Respiratory failure |
| Status unknown | 6 | Lost to follow-up |
Future directions
SCID NBS has proven to be an effective means to improve outcomes for affected infants, but several issues demand further attention. Many countries have not yet implemented population-wide TREC NBS, and even in the USA, where all state public health programs now include SCID in their panels of NBS diseases, there is no consensus on best practices for screening and follow-up despite over 10 years of experience. Centralized recording of outcomes is lacking in many programs, and many geographical areas do not have a nearby pediatric immunology center with expertise in treatment of these rare diseases. Even experts disagree about the best approaches to HCT, including how to make space in the bone marrow niches for donor HSCs with minimal toxicity from chemotherapy. The PIDTC, having already established that different SCID genotypes must be treated individually,56 aims to establish best treatments with natural history studies and current and future clinical trials.57
Infections, most notably CMV, have emerged as a significant cause of morbidity and mortality in infants with SCID despite NBS. Shortening the time required for screening and confirmatory testing may decrease the risk of acquiring CMV from breastmilk and allow for cessation of nursing and institution of prophylactic treatment for infants of CMV seropositive mothers. Although there are antiviral agents with activity against CMV, some have significant toxicities while others are not yet approved for use in very young infants. Furthermore, drug resistant strains are recognized, and resistance can develop during therapy, which must be continued until T cell immune reconstitution is achieved. More information about CMV prevention and treatment in SCID is needed.58
Finally, SCID NBS with TRECs fails to detect 100% of the infants with immune defects who would benefit from presymptomatic diagnosis. As mentioned above, there are very rare hypomorphic variants in SCID genes (IL2RG, ADA, RAG1/2) that result in a sufficiently leaky phenotype to escape detection by the TREC assay. Individuals with these variants come to clinical attention in childhood or even early adulthood with immune deficiency and/or dysregulation. Delayed onset or late onset ADA SCID may be identified by a biochemical assay of adenosine and 2’-deoxyadenosine,25 and individuals with RAG hypomorphic variants may have low or absent B cell kappa chain receptor excision circles (KRECs). B cell immunoglobulin genes undergo recombination analogous to that in T cells, also producing circular DNA byproducts. A qPCR assay for KRECs can be multiplexed with the TREC assay, as demonstrated by several pilot studies and adoption of TREC/KREC screening in Israel, Sweden, Iran and Switzerland.59-62 KREC screening could also detect primary immunodeficiencies affecting B cells at birth. The best known of these is X-linked agammaglobulinemia (XLA) resulting from a defect in the BTK gene, but autosomal recessive agammaglobulinemia (ARA) is also recognized, for which there are several genotypes.3 XLA and ARA are even more rare than SCID, and although they are not as lethal in early life if untreated, it is widely considered that early diagnosis and institution of immunoglobulin replacement therapy would lessen their morbidity and mortality. KREC NBS will not identify the most common B cell defects that are collectively known as common variable immunodeficiency, which develops later in life. Like all new screening tests, KREC NBS requires further studies to manage cutoff points for optimal detection of disease while minimizing false positive results.
There are many additional serious primary immunodeficiency and immune dysregulatory diseases that are not detectable by absence of TRECs or KRECs, but would benefit from early diagnosis and intervention if a suitable newborn screening test could be devised. Unfortunately, biomarkers leading to tests for these disorders have not been discovered to date. The increased utilization of WES and WGS as diagnostic tests for individuals who are already symptomatic raises the question of whether deep sequencing could also be applied in a screening context. However, at this time sequencing without the hint of a phenotype is unlikely to be successful due to: 1) the extreme rarity of individual primary immune defects; 2) the plethora of rare and private mutations that cause disease combined with our inability to distinguish them from the vast numbers of inconsequential genomic variants; and 3) our incomplete knowledge of the gene expression landscape underlying disease pathogenesis. A recent study comparing WES to the current biochemical testing for metabolic inborn errors showed that sequencing and analysis to have unacceptable false negative and false positive rates for use as a primary screening tool.63
What Do We Know about Newborn Screening for SCID?
TRECs are a highly relevant biomarker for indicating insufficiency of a diverse repertoire of new T cells.
TRECs are readily assayed using DNA extracted from dried blood spots obtained for newborn screening.
Absent or low TRECs in newborns can identify essentially all cases of typical SCID.
Population based newborn screening for SCID optimizes ascertainment and survival, giving affected infants prompt access to specialized care.
SCID newborn screening decreases incidence of infections prior to definitive therapy.
In addition to finding SCID, TREC based newborn screening flags infants with significant syndromic or secondary non-SCID T cell insufficiencies and a small proportion of infants with idiopathic T lymphopenia.
As with all newborn screened disorders, very rare infants (2 in over 3 million births in California) with leaky or hypomorphic mutations in SCID genes have normal TREC results at birth and present with symptoms only later in life.
What Is Still Unknown about Newborn Screening for SCID?
Best methods to provide SCID infants with complete, durable immune function and avoid treatment-related toxicity are not agreed upon and vary according to genotype.
New, promising approaches, including gene addition therapy, gene editing and targeted conditioning with anti-stem-cell monoclonal antibodies are promising, but not yet standard of care.
Avoiding exposure to infections, particularly CMV, remains challenging, with best practices and prophylactic medications not defined.
Management of non-SCID T lymphopenic conditions is highly variable and requires standardization.
Many other primary immune defects that could benefit from screening lack suitably sensitive and specific tests.
Acknowledgement
This work is dedicated to the memory of Fred Lorey, a friend and pioneer in newborn screening and public health, whose dedication and leadership helped SCID become a newborn screened disease. The authors also thank their collaborators in the California Genetic Disease Screening Program and the physicians who diagnose and treat infants with abnormal SCID screens. Dr Puck received support from cooperative agreement U54-AI082973 for the Primary Immune Deficiency Treatment Consortium from the National Institute of Allergy and Infectious Diseases (NIAID) and the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH), Bethesda, MD; the Jeffrey Modell Foundation; the Lisa and Douglas Goldman Fund; and the Michelle Platt-Ross Foundation.
Abbreviations used:
- ADA
Adenosine deaminase
- CMV
cytomegalovirus
- DBS
Dried blood spot
- GT
(gene therapy)
- KREC
Kappa light chain excision circle
- HCT
Hematopoietic cell transplantation
- NBS
Newborn screening
- NICU
Neonatal intensive care unit
- PIDTC
Primary Immune Deficiency Treatment Consortium, an NIH funded collaboration of 47 sites in the USA and Canada to study best treatments for primary immune defects
- qPCR
quantitative polymerase chain reaction
- RUSP
Recommended Uniform Screening Panel
- SCID
Severe combined immunodeficiency
- SMA
Spinal muscular atrophy
- TCR
T cell receptor
- TREC
T cell receptor excision circle
- WES
whole exome sequence
- WGS
whole genome sequence
- XLA
X-linked agammaglobulinemia
Footnotes
Conflict of Interest Statement:
Dr. Puck is a recipient of research grants from the National Institutes of Health and the California Institute of Regenerative Medicine. She receives royalties from Up-To-Date. Her spouse is an employee and stockholder in Invitae, Inc., a genetic diagnostics company. Dr. Currier has no conflicts to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Buckley RH. The long quest for neonatal screening for severe combined immunodeficiency. J Allergy Clin Immunol 2012;129:597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Ann Rev Immunol 2004;22:625–55. [DOI] [PubMed] [Google Scholar]
- 3.Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2020;40:24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan A, Scalchunes C, Boyle M, Puck JM. Early vs. delayed diagnosis of severe combined immunodeficiency: a family perspective survey. Clin Immunol 2011;138:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. New Engl J Med 2014;371:434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014;312:729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: Entering a new century, do we do better? J Allergy Clin Immunol 2010;126:602–10. [DOI] [PubMed] [Google Scholar]
- 8.Amatuni GS, Currier RJ, Church JA, Bishop T, Grimbacher E, Nguyen AA-C, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California, 2010-2017. Pediatrics 2019;143(2) e20182300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rechavi E, Lev A, Simon AJ, Stauber T, Daas S, Saraf-Levy T, et al. First year of Israeli newborn screening for severe combined immunodeficiency: clinical achievements and insights. Front Immunol 2017;8:1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barbaro M, Ohlsson A, Borte S, Jonsson S, Zetterström RH, King J, Winiarski J, et al. Newborn screening for severe primary immunodeficiency diseases in Sweden: a 2-year pilot TREC and KREC screening study. J Clin Immunol 2017;37:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blom M, Pico-Knijnenburg I, Sijne-van Veen M, Boelen A, Bredius RGM, van der Burg M, Schielen PCJI. An evaluation of the TREC assay with regard to the integration of SCID screening into the Dutch newborn screening program. Clin Immunol 2017;180:106–10. [DOI] [PubMed] [Google Scholar]
- 12.Thomas C, Durand-Zaleski I, Frenkiel J, Mirallié S, Léger A, Cheillan D, et al. Clinical and economic aspects of newborn screening for severe combined immunodeficiency: DEPiStREC study results. Clin Immunol 2019;202:33–9. [DOI] [PubMed] [Google Scholar]
- 13.Richards S, Pitt J, Choo S. Newborn screening for severe combined immunodeficiency: Evaluation of a commercial T-cell receptor excision circle-based method in Victorian dried blood spots. J Paediatr Child Health 2018;54:14–19 [DOI] [PubMed] [Google Scholar]
- 14.Argudo-Ramírez A, Martín-Nalda A, Marín-Soria JL, López-Galera RM, Pajares-García S, González de Aledo-Castillo JM, et al. First universal newborn screening program for severe combined immunodeficiency in Europe. Two-years' experience in Catalonia (Spain). Front Immunol 2019;10:2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hazenberg MD, Verschuren MC, Hamann D, Miedema F, van Dongen JJ. T cell receptor excision circles as markers for recent thymic emigrants: basic aspects, technical approach, and guidelines for interpretation. J Mol Med 2001; 79:631–40. [DOI] [PubMed] [Google Scholar]
- 16.Chan K, Puck J. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol 2005;115:391–8. [DOI] [PubMed] [Google Scholar]
- 17.Albin-Leeds S, Ochoa J, Mehta H, Vogel BH, Caggana M, Bonagura V, et al. Idiopathic T cell lymphopenia identified in New York State Newborn Screening. Clin Immunol 2017;183:36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson J, Jungner G. The Principles and practice of screening for disease. Geneva:World Health Organisation; (1968). https://apps.who.int/iris/handle/10665/37650 (accessed 9/6/2020) [Google Scholar]
- 19.Kemper AR, Comeau AM, Grosse S, Jones E, Lam KK, Kwon J, et al. Evidence-based review of newborn screening for spinal muscular atrophy (SMA): Final report (v5.2) (13 March 2018), commissioned by the ACHDNC. https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/previous-nominations.html (accessed 9/6/2020) [Google Scholar]
- 20.Scholrling DC, Pechmann A, Kirschner J. Advances in treatment of spinal muscular atrophy – New phenotypes, new challenges, new implications for care. J Neuromuscl Dis 2020;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patel NC, Hertel PM, Estes MK, de la Morena M, Petru AM, Noroski LM. Vaccine-acquired rotavirus in infants with severe combined immunodeficiency. N Engl J Med 2010;362:314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amatuni GS, Sciortino S, Currier RJ, Naides SJ, Church JA, Puck JM. Reference intervals for lymphocyte subsets in preterm and term neonates without immune defects. J Allergy Clin Immunol 2019;144:1674–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Immunol 2015;1(29 October):15061 https://www.nature.com/articles/nrdp201561 (accessed 9/6/2020) [DOI] [PubMed] [Google Scholar]
- 24.Kwan A, Puck JM. History and current status of newborn screening for severe combined immunodeficiency. Semin Perinatol 2015;39:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.la Marca G, Canessa C, Giocaliere E, Romano F, Duse M, Malvagia S, Lippi F, et al. Tandem mass spectrometry, but not T-cell receptor excision circle analysis, identifies newborns with late-onset adenosine deaminase deficiency. J Allergy Clin Immunol 2013;131:1604–10. [DOI] [PubMed] [Google Scholar]
- 26.Dorna MB, Barbosa PFA, Rangel-Santos A, Csomos K, Ujhazi B, Dasso JF, et al. Combined immunodeficiency with late-onset progressive hypogammaglobulinemia and normal B cell count in a patient with RAG2 deficiency. Front Pediatr 2019;7:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita M, Wakatsuki R, Kato T, Okano T, Yamanishi S, Mayumi N, et al. A synonymous splice site mutation in IL2RG gene causes late-onset combined immunodeficiency. Int J Hematol. 2019;109:603–11. [DOI] [PubMed] [Google Scholar]
- 28.Savic RM, Cowan MJ, Dvorak CC, Pai S-Y, Pereira L, Bartelink IH, et al. Effect of weight and maturation on busulfan clearance in infants and small children undergoing hematopoietic cell transplantation. Biol Blood Marrow Transplant 2013;19:1608–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dvorak CC, Long-Boyle J, Dara J, Melton A, Shimano KA, Huang JN, et al. Low exposure busulfan conditioning to achieve sufficient multilineage chimerism in patients with severe combined immunodeficiency. Biol Blood Marrow Transplant, 2019;25:1355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. New Engl J Med. 2014. July 31;371(5):434–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood 2018;132:1737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miggelbrink AM, Logan BR, Buckley RH, Parrott RE, Dvorak CC, Kapoor N, et al. B-cell differentiation and IL-21 response in IL2RG/JAK3 SCID patients after hematopoietic stem cell transplantation. Blood 2018;131:2967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mamcarz E, Zhou S, Lockey T, Abdelsamed H, Cross SJ, Kang G, et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N Engl J Med 2019;380:1525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pai S-y, Thrasher AJ. Gene therapy for X-linked severe combined immunodeficiency: Historical outcomes and current status. J Allergy Clin Immunol 2020;146:258–61. [DOI] [PubMed] [Google Scholar]
- 35.Fischer A, Hacein-Bey-Abina SH. Gene therapy for severe combined immunodeficiencies and beyond. J Exp Med 2020;217:e20190607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kohn DB, Hershfield MS, Puck JM, Aiuti A, Blincoe A, Gaspar HB, et al. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J Allergy Clin Immunol 2019;143:852–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barry JC, Crowley TB, Jyonouchi S, Heimall J, Zackai EH, Sullivan KE, McDonald-McGinn DM. Identification of 22q11.2 deletion syndrome via newborn screening for severe combined immunodeficiency. J Clin Immunol. 2017;37:476–85. [DOI] [PubMed] [Google Scholar]
- 38.Lingman Framme J, Borte S, von Döbeln U, Hammarström L, Oskarsdóttir S. Retrospective analysis of TREC based newborn screening results and clinical phenotypes in infants with the 22q11 deletion syndrome. J Clin Immunol 2014;34:514–9. [DOI] [PubMed] [Google Scholar]
- 39.Tangsinmankong N, Day NK, Nelson RP Jr, Puck J, Good RA. Severe combined immunodeficiency in an infant with multiple congenital abnormalities. J Allergy Clin Immunol 1999:103:1222–3. [DOI] [PubMed] [Google Scholar]
- 40.Mallott J, Kwan A, Church J, Gonzalez-Espinosa D, Lorey F, Tang LF, et al. Newborn screening for SCID identifies patients with ataxia telangiectasia. J Clin Immunol 2013;33:540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schoenaker MHD, Blom M, de Vries MC, Weemaes CMR, van der Burg M, Willemsen MAAP. Early diagnosis of ataxia telangiectasia in the neonatal phase: a parents’ perspective. Euro J Pediatr 2020;179:251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jyonouchi S, Orange J, Sullivan KE, Krantz I, Deardorff M. Immunologic features of Cornelia de Lange syndrome. Pediatrics. 2013;132:e484–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dasouki M, Okonkwo KC, Ray A, Folmsbeel CK, Gozales D, Keles S, et al. Deficient T cell receptor excision circles (TRECs) in autosomal recessive hyper IgE syndrome caused by DOCK8 mutation: implications for pathogenesis and potential detection by newborn screening. Clin Immunol 2011;141:128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Volpi S, Yamazaki Y, Brauer PM, van Rooijen E, Hayashida A, Slavotinek A, et al. EXTL3 mutations cause skeletal dysplasia, immune deficiency and developmental delay. J Exp Med 2017;214:623–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chrzanowska KH, Krajewska-Walasek M, Kus J, Michalkiewicz J, Maziarka D, Wolski JK, et al. Kabuki (Niikawa-Kuroki) syndrome associated with immunodeficiency. Clin Genet 1998;53:308–12. [DOI] [PubMed] [Google Scholar]
- 46.Patel JP, Puck JM, Srinivasan R, Brown C, Sunderam U, Kundu K, et al. Nijmegen breakage syndrome detected by newborn screening for T cell receptor excision circles (TRECs). J Clin Immunol 2015;35:227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu AP, Donkó A, Arrington ME, Swamydas M, Fink D, Das A et al. Dominant activating RAC2 mutation with lymphopenia, immunodeficiency, and cytoskeletal defects. Blood 2019;133:1977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Punwani D, Zhang Y, Yu J, Cowan MJ, Rana S, Kwan A, et al. Multisystem anomalies in severe combined immunodeficiency with mutant BCL11B. N Engl J Med 2016;375:2165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fullerton BS, Velazco CS, Hong CR, Carey AN, Jaksic T. High rates of positive severe combined immunodeficiency screening among newborns with severe intestinal failure. J Parenter Enteral Nutr 2018;42:239–46. [DOI] [PubMed] [Google Scholar]
- 50.Davey BT, Elder RW, Cloutier MM, Bennett N, Lee JH, Wang Z, et al. T-Cell Receptor Excision Circles in Newborns with Congenital Heart Disease. J Pediatr. 2019. October;213:96–102.e2. [DOI] [PubMed] [Google Scholar]
- 51.Kuo CY, Garcia-Lloret MI, Slev P, Bohnsack JF, Chen K. Profound T-cell lymphopenia associated with prenatal exposure to purine antagonists detected by TREC newborn screening. J Allergy Clin Immunol Pract 2017;5:198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D’Assante R, Fusco A, Palamaro L, Giardino G, Gallo V, Cirillo E, Pignata C. Unraveling the link between ectodermal disorders and primary immunodeficiencies. Int Rev Immunol 2016;35:25–38. [DOI] [PubMed] [Google Scholar]
- 53.Bosticardo M, Yamazaki Y, Cowan J, Giardino G, Corsino C, Scalia G, et al. Heterozygous FOXN1 variants cause low TRECs and severe T cell lymphopenia, revealing a crucial role of FOXN1 in supporting early thymopoiesis. Am J Hum Genet 2019;105:549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du Q, Huynh LK, Coskun F, Molina E, King MA, Raj P, et al. FOXN1 compound heterozygous mutations cause selective thymic hypoplasia in humans. J Clin Invest 2019;129:4724–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bifsha P, Leiding JW, Pai S-Y, Colamartino ABL, Hartog N, Church JA, et al. Diagnostic assay to assist clinical decisions for unclassified severe combined immune deficiency. Blood Advances 2020;4:2606–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dvorak CC, Haddad E, Buckley RH, Cowan MJ, Logan B, Griffith LM, et al. The genetic landscape of SCID in the US and Canada in the current era (2010-2018). J Allergy Clin Immunol 2019;143:405–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conditioning SCID Infants Diagnosed Early (CSIDE), Michael Pulsipher, Sponsor; https://clinicaltrials.gov/ct2/show/NCT03619551 (accessed 9/6/2020) [Google Scholar]
- 58.Heimall J, Puck J, Buckley R, Fleisher TA, Gennery AR, Neven B, et al. Current knowledge and priorities for future research in late effects after hematopoietic stem cell transplantation (HCT) for severe combined immunodeficiency patients: A consensus statement from the second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects after Pediatric HCT. Biol Blood Marrow Transplant 2017;23:379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Somech R, Lev A, Simon AJ, Korn D, Garty BZ, Amariglio N, et al. Newborn screening for severe T and B cell immunodeficiency in Israel: a pilot study. Isr Med Assoc J 2013;15:404–9. [PubMed] [Google Scholar]
- 60.Barbaro M, Ohlsson A, Borte S, Jonsson S, Zetterström RH, King J, et al. Newborn screening for severe primary Immunodeficiency diseases in Sweden--a 2-year pilot TREC and KREC screening study. J Clin Immunol 2017;37:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nourizadeh M, Shakerian L, Borte S, Fazlollahi M, Badalzadeh M, Houshmand M, et al. Newborn screening using TREC/KREC assay for severe T and B cell lymphopenia in Iran. Scand J Immunol 2018;June 26:e12699. [DOI] [PubMed] [Google Scholar]
- 62.Trück J, Prader S, Natalucci G, Hagmann C, Brotschi B, Kelly J, et al. Swiss newborn screening for severe T and B cell deficiency with a combined TREC/KREC assay – management recommendations. Swiss Med Wkly 2020;150:w20254. [DOI] [PubMed] [Google Scholar]
- 63.Adhikari AN, Gallagher RC, Wang Y, Currier RJ, Amatuni G, Bassaganyas L, et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat Med 2020(August 10); 10.1038/s41591-020-0966-5. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]