
Figure 3. PacBio and ONT long-read data types.
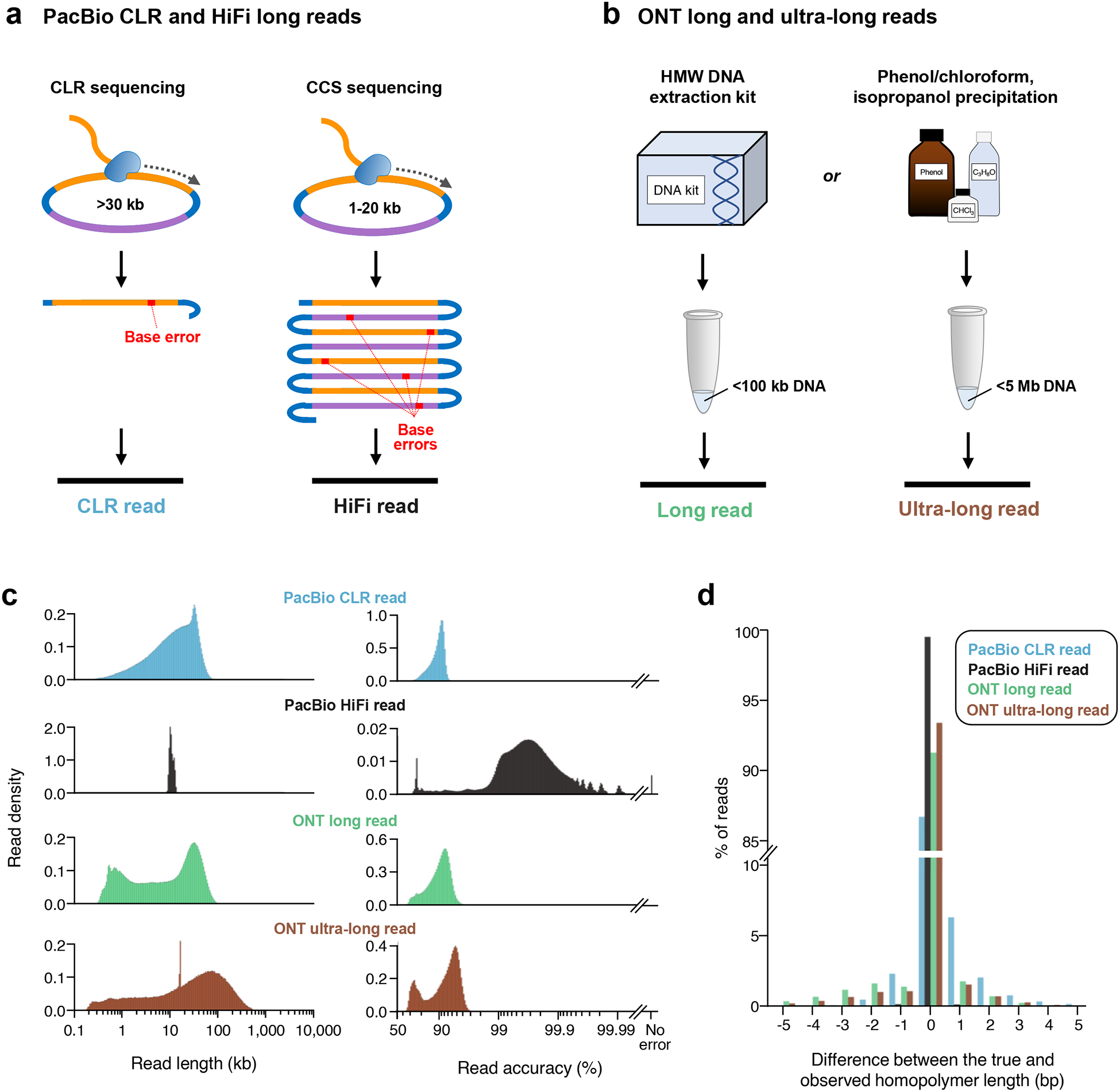
a) The PacBio platform can generate continuous long reads (CLR) or high-fidelity (HiFi) reads. CLR data is generated by sequencing a SMRTbell template containing a >30 kb DNA insert (yellow for forward strand, dark blue for reverse strand). Because of the large insert size, the polymerase often only completes a single pass through one strand of the template. A base is incorrectly called in about 1 out of every 10 bases, resulting in an error rate of 8–15% in the CLR. HiFi reads are generated by circular consensus sequencing (CCS) of a SMRTbell template containing a 10–30 kb DNA insert. The smaller insert size allows the polymerase to make several passes around the SMRTbell template. A consensus sequence is produced from the subreads, resulting in an error rate of ≤1% in the HiFi read. b) The ONT platform can generate long or ultra-long reads. To generate long and ultra-long ONT reads, high-molecular-weight (HMW) DNA is first extracted from cells or tissue. This extraction is commonly performed using either a commercially available DNA extraction kit, such as Qiagen’s Puregene kit or Genomic-tip 500/G kit, or via traditional methods, such as a phenol-chloroform extraction followed by either an ethanol or isopropanol precipitation. Kit-extracted DNA most often generates long (10–100 kb) reads, whereas high-molecular-weight DNA extracted by phenol-chloroform generates ultra-long (>100 kb) reads. c) Read length distributions and base accuracies of PacBio and ONT long-read data types differ. Shown are plots of the read length and accuracy distributions for: PacBio HG002 CLR data generated on the Sequel II platform; PacBio CHM13 HiFi data generated on the Sequel II platform; ONT CHM13 long-read data generated on the PromethION; and ONT ultra-long reads generated on the MinION and GridION. Read accuracy was estimated by aligning raw reads from each data type to GRCh38 and counting alignment differences as errors in the reads. Links to the publicly available datasets, a description of the methods used, and the code required to reproduce the analysis are provided in a Supplementary Note. A similar analysis was also performed in which raw reads were aligned to the T2T CHM13 assembly34, and differences in alignment between the reads and the highly curated ChrX were counted to estimate read accuracy. PacBio HiFi reads have a visibly higher read accuracy distribution when aligned to the CHM13 T2T assembly than GRCh38 because the high accuracy of the HiFi reads (>99%) is sufficient to detect differences between the two genome assemblies, which are interpreted as base errors. The other long-read data types are not accurate enough to detect differences between the two genome assemblies. Consequently, the accuracy distribution for these other data types are similar (Supplementary Figure 1a and Supplementary Note). d) Homopolymer accuracy differs between PacBio and ONT long-read data types. Shown is a plot of the homopolymer accuracy for the PacBio CLR, PacBio HiFi, ONT long, and ONT ultra-long datasets used in panel c. Homopolymer error was estimated by aligning raw reads from each data type to GRCh38 and comparing the observed homopolymer length in the reads to the homopolymer length. A similar analysis was performed where raw reads were aligned to the T2T CHM13 assembly34, and homopolymer error was estimated based on the comparison between the observed homopolymer length in the reads and the true homopolymer length in the highly curated ChrX assembly. In both cases, homopolymers ≥5 bases were assessed for accuracy (Supplementary Figure 1b and Supplementary Note).