

Abstract
We report the development of in situ (online) EPR and coupled EPR/NMR methods to study redox flow batteries, which are applied here to investigate the redox-active electrolyte, 2,6-dihydroxyanthraquinone (DHAQ). The radical anion, DHAQ3–•, formed as a reaction intermediate during the reduction of DHAQ2–, was detected and its concentration quantified during electrochemical cycling. The fraction of the radical anions was found to be concentration-dependent, the fraction decreasing as the total concentration of DHAQ increases, which we interpret in terms of a competing dimer formation mechanism. Coupling the two techniques—EPR and NMR—enables the rate constant for the electron transfer between DHAQ3–• and DHAQ4– anions to be determined. We quantify the concentration changes of DHAQ during the “high-voltage” hold by NMR spectroscopy and correlate it quantitatively to the capacity fade of the battery. The decomposition products, 2,6-dihydroxyanthrone and 2,6-dihydroxyanthranol, were identified during this hold; they were shown to undergo subsequent irreversible electrochemical oxidation reaction at 0.7 V, so that they no longer participate in the subsequent electrochemistry of the battery when operated in the standard voltage window of the cell. The decomposition reaction rate was found to be concentration-dependent, with a faster rate being observed at higher concentrations. Taking advantage of the inherent flow properties of the system, this work demonstrates the possibility of multi-modal in situ (online) characterizations of redox flow batteries, the characterization techniques being applicable to a range of electrochemical flow systems.
1. Introduction
Redox flow batteries are a promising large-scale energy storage technology. The catholyte and anolyte are stored in two separate tanks and, when required, flow through a connected electrochemical cell where they undergo redox reactions, either storing or releasing charge. This allows energy storage and power generation to be decoupled. Organic redox-active molecules have emerged in recent years as promising electrolyte molecules for flow battery applications.1−3 Since these molecules are comprised of earth-abundant elements, a high sustainability and low cost are expected.4,5 However, for deep market penetration of these types of flow battery systems, the bottlenecks are the relatively low energy densities and short lifetimes when compared to those of other battery systems.6 Therefore, understanding the reaction mechanisms involved is vital for further improvement of both the energy density and lifetime.
In a previous study, taking advantage of the inherent flow properties of a redox flow battery, we demonstrated two in situ NMR techniques to study these systems: online and operando NMR. In the online experiment, one of the electrolyte solutions is flowed through the NMR probe to study either the catholyte or anolyte while the cell is situated outside of the detection region. In contrast, the operando experiment consists of positioning a miniaturized cell within the detection region of the NMR probe to facilitate simultaneous study of both the catholyte and anolyte. By analyzing the bulk magnetic susceptibility shifts of the electrolyte peaks, we quantified the concentrations of radicals present during cycling and showed that the electrochemical reaction of 2,6-dihydroxyanthraquinone (DHAQ) proceeds through two single-electron transfers (DHAQ2– → DHAQ3–• and DHAQ3–• → DHAQ4–), accompanied by intermolecular electron transfer between the anions themselves. The radical (i.e., semiquinone, DHAQ3–•) concentration was shown to be set by the comproportionation reaction:
| 1 |
This equilibrium also dictates the difference in couples between the two one-electron-transfer reactions, allowing this difference to be estimated from a determination of the DHAQ3–• concentration. Finally, we demonstrated that one of the electrolyte degradation reactions is electrochemical in nature.7
The property of flow within these battery systems allows for simple implementation of multi-modal characterization. Here we move beyond our previous work and utilize this concept to couple EPR and NMR measurements. EPR spectroscopy has been applied to study in situ free radicals generated electrochemically since 1959.8−12 Simultaneous electrochemical–EPR measurements allow not only for the identification of reaction intermediates and products but also for the elucidation of the reaction mechanism and kinetics. Note that in situ EPR studies of an operating flow battery have not been previously demonstrated, although EPR has previously been used to study the crossover of vanadyl ions.13
Here, we report the coupled use of online EPR and NMR techniques to study anthraquinone-based flow batteries. They represent one of the most promising families of organic molecules for flow battery applications, offering fast electron transfer and excellent stability.14−17 Two anthraquinone molecules, DHAQ and 4,4′-((9,10-anthraquinone-2,6-diyl)dioxy)dibutyrate (DBEAQ), are studied. We directly detect and quantify the anthraquinone radical species, reveal the electronic structures of the radicals, and determine the rate constant for the electron transfer between the singly and doubly reduced DHAQ anions—a parameter that we were unable to determine in the NMR studies.7 The radical concentration is found to correlate with the overall DHAQ concentration (10, 100, and 200 mM), which we interpret in terms of a competing dimer formation mechanism. These overall concentrations are relevant for flow battery applications, where concentrations ranging from 0.01 to 3 M are generally used.6 Finally, the coupled NMR spectroscopy allows for the decomposition of DHAQ to be followed and for the degradation to be quantitatively correlated to the capacity fade of the flow battery.
2. Results and Discussion
A lab-scale flow battery was positioned outside the EPR and NMR magnets. Aqueous solutions of anthraquinone and potassium hexacyanoferrate(II and III) were used as the anolyte and catholyte, respectively. The evolution of anthraquinone was followed by EPR and NMR spectroscopies. The electrolyte solution was pumped from the reservoir to the electrodes, then through a flow EPR tube and an NMR tube, and back to the electrolyte reservoir (see Figure S1 for the setup). The round-trip time was 64 s. The residence time in the EPR detection region was 0.13 s, which is much longer than the electron spin–lattice relaxation time that is typically on the timescale of microseconds for semiquinones.18
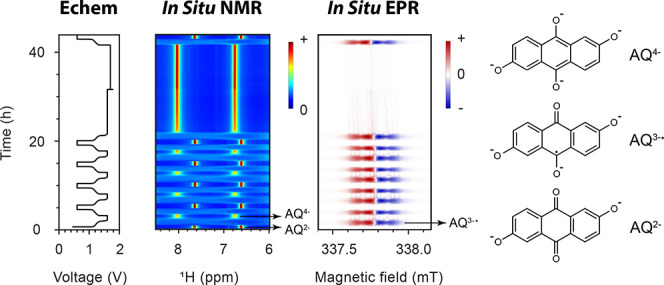
Two different concentrations of DHAQ were studied in full-cell batteries, namely 10 and 100 mM, with coupled in situ NMR and EPR experiments being performed for both concentrations. Since a large amount of data was generated from these in situ experiments, Figure 1 serves as a guideline to relate the data analyses presented in the following figures to the voltage profiles of the batteries.
Figure 1.

Cycling regimes used to analyze the various phenomena occurring in the DHAQ flow battery. The regions of interest are highlighted by dotted lines or solid arrows, and the voltage profiles are labeled with the subsequent figures that provide a more detailed analysis of the various processes. (a) Voltage of a 10 mM DHAQ versus 15 mM K4[Fe(CN)6] and 3.75 mM K3[Fe(CN)6] full cell as a function of time. During charge, a constant current of 10 mA was applied, followed by a voltage hold at 1.6 V. During discharge, a constant current of −10 mA (where the sign indicates the polarity of the electrodes) was applied, followed by a potential hold at 0.6 V. The charge–discharge cycles were repeated four times, then potential holds of 10 h each were applied at 1.6 and 1.7 V. (b) Voltage of a 100 mM DHAQ versus 150 mM K4[Fe(CN)6] and 50 mM K3[Fe(CN)6] full cell. During charge, a constant current of 150 mA was applied to a cutoff voltage of 1.7 V. During discharge, a constant current of −150 mA was applied to a cutoff voltage of 0.6 V. The 7th charge was followed by a voltage hold at 1.7 V for 20 h and another 20 charge/discharge cycles.
Coupled In Situ NMR and EPR Experiments
Figure 2 presents the in situ1H NMR and EPR spectra of 10 mM DHAQ as a function of electrochemical cycling. On charging (reduction of DHAQ2– anions) at 10 mA, the voltage of the battery increases from 1.15 V to a cutoff voltage of 1.6 V. In the NMR spectra, the proton signals A (defined in Figure 2a) have disappeared by the time the third spectrum has been acquired (i.e., between 192 and 288 s) after turning on the charging current and when 4% of DHAQ3–• radical anions have formed. Signal B is significantly broadened and only just visible above the baseline under the conditions used here; this is in contrast to our prior work where this signal was resolved more clearly throughout the cycling,7 which is ascribed to the lower concentration of DHAQ used in the present study. The loss of signals A (and B) is due to electron delocalization over the semiquinone DHAQ3–• radical anion and the intermolecular electron transfer between the DHAQ2– anion and the DHAQ3–•. Note that signal C is barely observable due to a H-D exchange reaction with the solvent D2O molecule during prior electrochemical cycling.
Figure 2.

In situ1H NMR and EPR spectra acquired during electrochemical cycling. (a) Structures of the DHAQ2–, DHAQ3–•, and DHAQ4– anions showing the labeling of the proton. (b) Voltage of a 10 mM DHAQ versus 15 mM K4[Fe(CN)6] and 3.75 mM K3[Fe(CN)6] full cell as a function of time. During charge, a constant current of 10 mA was applied, followed by a potential hold at 1.6 V. During discharge, a constant current of −10 mA (where the sign indicates the polarity of the electrodes) was applied, followed by a potential hold at 0.6 V. (c) NMR spectra of the anolyte in the aromatic region (full spectra are shown in Figure S2). The color bar (right) indicates the intensity of resonances in positive arbitrary units. The acquisition time per NMR spectrum is 95 s. (d) EPR spectra of the anolyte. The stack plot on the right shows every 10th spectrum. The acquisition time per EPR spectrum is 95 s, with a scanning time of 60 s, a coupling time of 30 s (time for automatic tuning), and a delay time of 5 s. Note that a typical continuous-wave EPR spectrum is detected and displayed as the first derivative of the absorption, and hence has negative and positive values. The color bar indicates the intensity of the resonance in arbitrary units. A different color scale is applied here because of the presence of negative peak intensities.
In the EPR spectra, an EPR resonance centered at 337.78 mT develops (corresponding to a g-factor of 2.0046), which coincides with the disappearance of NMR signals A and B. This signal is assigned to the DHAQ3–• radical anion. As charging continues, the EPR signal increases in intensity and broadens, reaching a maximum breadth and intensity at 50% state-of-charge (SOC), whereafter it decreases in intensity and sharpens. The broadening of signals is caused by Heisenberg spin exchange, i.e., the “flip-flop”, dipolar-driven (zero-quantum) spin-exchange between two unpaired electrons when they come into close proximity, and the intermolecular electron transfer.19,20 As the voltage reaches 1.6 V, the EPR signal is still observable, but it continues to drop in intensity, reaching its minimum, and the voltage is held; this is accompanied by the appearance of the NMR signals of A″ and B″ from the doubly reduced DHAQ4–. Note that a second voltage plateau is observed at 1.5 V. This plateau appears only after a voltage hold at 1.6 V for 2 h, and it was previously observed after a more prolonged voltage hold (see Extended Data Figure 8e in ref (7)). Since there are no noticeable changes in the NMR and EPR signals that are observed, this voltage plateau is likely to be caused by ferrocyanide decomposition or water oxidation at the catholyte side. The study of the catholyte is outside the scope of the current study but certainly warrants future investigation. During discharge, reversible changes in the NMR and EPR spectra were observed. When the voltage decreases to and is held at 0.6 V, the EPR signal of DHAQ3–• drops to below the detectable level (10 nM for the Magnettech benchtop spectrometer or 9 × 108 spins/G at a S/N ratio of 3), in contrast to the detectable amount of residual DHAQ3–• that remains throughout the high-voltage hold. This is due to the continual chemical oxidation of DHAQ4–, as discussed in our previous study,7 where the oxidant may be either trace amounts of oxygen gas that have permeated into the battery system or the solvent water itself.7 The charge–discharge cycle was repeated five times, followed by longer potential holds of 10 h at 1.6 V and then 1.7 V (Figure S2).
Determining the Radical Anion Concentration
The concentration of radical anions can be readily estimated by measuring the changes in the bulk magnetization from the solvent water resonance in the NMR spectra (Figure S2, eq 7),7 and with EPR, it can very simply be estimated by spin counting. In the latter method, the radical concentration is proportional to the double integral of the EPR signal, so the DHAQ3–• radical anion concentration can be readily determined by calibrating the signal integral to that of a sample with a known concentration. We used 4-OH-TEMPO dissolved in water as the reference (see Figure S3 for the EPR spectra; the method of spin counting is described in detail in the SI). The concentration of DHAQ3–• radical anions as a function of electrochemical cycling, estimated by both the NMR (b) and EPR (c) methods, is shown in Figure 3, with good consistency being seen between the two approaches. During charge, the concentration of DHAQ3–• radical anions increases from 0 to 7 mM and then decreases to 0.2 mM. The changes are reversible during discharge. Larger fluctuations can be seen in the concentrations estimated by NMR than by EPR. This is due to errors from the phase and linewidth of the NMR signals, where a FWHM of 11 Hz corresponds to an error of ±3 mM. However, the similar numbers determined by the two methods for both 10 and 100 mM concentrations (Figure 3d) help validate the previous NMR approach and our assumption that the effect of changes in pH and in water–quinone–salt interactions during cycling can be ignored when analyzing the bulk magnetic susceptibility shifts in the water resonance.7
Figure 3.
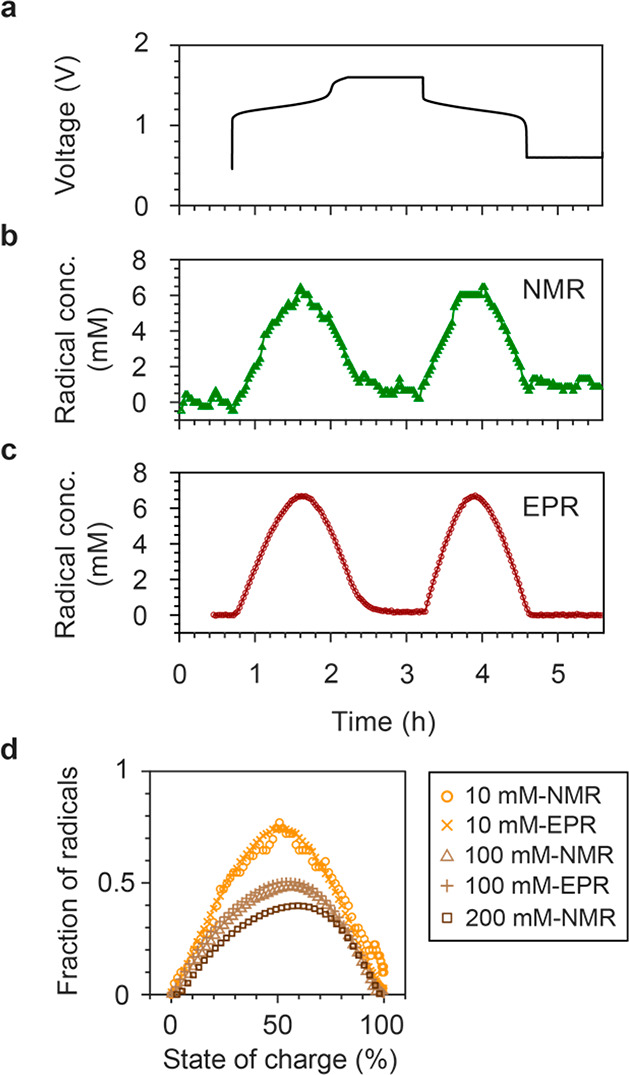
Concentrations of DHAQ3–• radical anions. (a) Voltage of a 10 mM DHAQ versus 15 mM K4[Fe(CN)6] and 3.75 mM K3[Fe(CN)6] full cell. (b, c) Concentrations of DHAQ3–• radical anions as a function of time, estimated from the bulk magnetic susceptibility shift of the water resonance in the NMR spectra and by spin counting in the EPR experiments, respectively. (d) Fractions of radicals as a function of state of charge for total concentrations of 10, 100, and 200 mM DHAQ. The data for 10 and 100 mM were obtained by EPR and NMR, while the 200 mM dataset was measured by NMR only. The data for 200 mM DHAQ are taken from the plot in the extended data in Figure 4c in ref (7).
The concentrations of radical anions were measured for three total concentrations of DHAQ, as shown in Figure 3d, the maximum DHAQ3–• fractional concentrations decreasing noticeably from 0.74 to 0.51 and 0.40 for the 10, 100, and 200 mM DHAQ solutions, respectively. The point at which the maximum value is observed shifts slightly toward higher SOCs: from 50.9, 56.1, and 59.6% SOC for 10, 100, and 200 mM DHAQ, respectively, where the SOC is defined by the oxidation state of DHAQ; i.e., 0% DHAQ4– anions corresponds to 0% SOC and 100% DHAQ4– corresponds to 100% SOC. This effect suggests that, besides the comproportionation reaction (eq 1) proposed in previous work,7,15 additional reaction equilibria exist, which result in lower AQ3–• fractional concentrations at higher concentrations. The strongest candidates for this are the dimerization equilibria,
| 2 |
| 3 |
which have been shown to be important in quinone-based systems.21−26 This overall concentration effect on the maximal radical concentration of quinones was studied as early as the 1930s.27 As the EPR spectra are consistent with a single EPR-active spin system, the dimer does not appear to be paramagnetic—i.e., it does not consist of two weakly coupled (but still paramagnetic) spin S = 1/2 AQ3–• monomers. Thus, the orientation and distance between two AQ3–• monomers must be such that a stronger coupling between the two unpaired electrons results, leading to a total spin of S = 0, i.e., a dimer that is EPR inactive. Indeed, the early study reported an overall decrease in the magnetic susceptibility of a solution with concentration as a result of dimerization.27 Overall, this suggests that reaction (2) is the predominant dimerization mechanism probed here: if dimers are formed via reaction (3), we might expect increased overall concentration to increase the dimer concentration and either to have little effect on radical concentration or, if reaction (2) also operates, to decrease radical concentration.
The implication of the dimerization for our study is that the apparent comproportionation reaction (eq 1) equilibrium constant—i.e., that determined by measuring the radical concentration—now depends on the initial AQ2– concentration. (Note that the inherent equilibrium constant defined by eq 1 is independent of the initial AQ2– concentration.) Thus, the calculated (apparent) voltage separation between the two one-electron couples will also depend on the initial concentration. However, as the AQ2– concentration tends to zero, the influence of the dimerization tends to zero, and the calculated voltage separation tends to its true (inherent) value. In our previous work,7 a voltage separation of 33 mV for the two one-electron couples was determined for a 100 mM DHAQ concentration (Table S1). It drops to 3.7 mV for the 200 mM solution, the marked asymmetry in the radical concentration with state of charge seen in Figure 3d also suggesting a second competing reaction associated with different kinetics. For the 10 mM DHAQ system reported here, the apparent voltage separation for the two one-electron couples has now increased (as expected) to 76 mV, which is close to the value of 60 mV reported for a low-concentration (5 mM) solution of DHAQ by simulating the cyclic voltammetry.15 Further details of this calculation can be found in the SI, together with calculations performed to explore the effect of dimerization on the apparent voltage profiles. The calculations indicate that the extracted voltage separation at a 10 mM concentration is close to the limiting voltage separation (i.e., the voltage separation of the extrapolated to zero concentration) of approximately 79 mV. Further measurements and modeling are in progress to explore these coupled equilibria in more detail. However, we note that, while more measurements at much lower concentrations may provide further insight, side reactions involving oxidation of the DHAQ4– anion further complicate the analysis.
Measuring the Rate Constant of Electron Transfer between DHAQ3–• and DHAQ4– Anions
In our previous study, we estimated the rate constant of intermolecular electron transfer between DHAQ2– and DHAQ3–• radical anions by measuring the line-broadening of NMR resonances A and C (see Figure 2a for species/resonances A and C), where the radical concentration was estimated from the charging current. However, we were unable to determine the rate constant for the electron transfer between DHAQ3–• and DHAQ4– anions (Figure 4a) due to the lack of a reliable estimation of the radical concentration at high voltages, where the decomposition reactions and chemical oxidation of DHAQ4– occur. Now, with the incorporation of a more sensitive EPR technique, the radical concentration at high voltages can be readily estimated by spin counting, even in the presence of these side reactions, and thus the rate constant of intermolecular electron transfer between the DHAQ3–• radical anion and the DHAQ4– anion can be determined unambiguously.
Figure 4.

(a) Self-exchange electron transfer reactions between DHAQ3–• and DHAQ4–, with rate of kex. (b) 1H spectra as a function of time during the voltage hold at 1.6 V are shown on the left. The corresponding concentrations of DHAQ3–• radical anions are shown on the right, as estimated by spin counting from the EPR experiments. (c) kex calculated at different radical concentrations during the voltage hold as a function of time.
In the slow exchange regime,7 when the transverse relaxation rate of the paramagnetic species, R2P, is much faster than the exchange rate, τP–1, i.e., R2P ≫ τP–1, the transverse relaxation rate of the diamagnetic species, R2ex, arising from the chemical exchange with the paramagnetic species, is given by
| 4 |
where kex is the bimolecular rate constant (or the electron-transfer rate constant) and [P] is the concentration of the paramagnetic species, i.e., the concentration of DHAQ3–• radical anions. The exchange rate is related to the rate constant by
| 5 |
where [D] is the concentration of the diamagnetic species, i.e., the concentration of DHAQ2– and DHAQ4– anions. The electron transfer was previously shown to be in the slow-exchange regime for resonances A and C of the DHAQ2– anion in a solution of 100 mM DHAQ.7 Here the concentration of DHAQ was 10 mM, so following eq 5, the exchange rate τP–1 is 10 times smaller than that at 100 mM, meaning that the slow-exchange regime is more easily achieved. Therefore, eq 4 can be used to determine the rate constant for the electron transfer.
Figure 4b presents the NMR spectra and the concentrations of DHAQ3–• radical anions during the voltage hold at 1.6 V. The linewidths of peaks A″ and B″ decrease as the concentration of DHAQ3–• radical anion decreases. Following eq 4, the rate constant kex is calculated and plotted as a function of time in Figure 4c. The values of kex range from 1.5 × 105 to 2.4 × 105 M–1 s–1, with an average of 2.0 × 105 M–1 s–1. The values of kex for the electron transfer between DHAQ3–• and DHAQ4– anions are on the same order of magnitude, i.e., 105 M–1 s–1, as the electron transfer between DHAQ2– and DHAQ3–• radical anions (kex ≈ 1.0 × 105 M–1 s–1).7
Unpaired Spin Densities and Consequences for H/D Exchange
EPR spectroscopy can be used to quantify the (unpaired) electron density distribution over the radical anions by extracting the hyperfine coupling constants for the unpaired electron to the neighboring nuclear spins, since a linear relationship between hyperfine coupling constant A and the unpaired π-electron populations on the isotropic proton of the carbon atoms in a π-type organic radical has been shown previously.28 This linearity should be applicable to the two anthraquinone radical anions studied here, i.e., DHAQ and DBEAQ. The values of A for the protons, as shown in Figure 5, were obtained by fitting the experimental spectra to the simulated ones. The simulation gives a set of hyperfine coupling constants but does not correlate the values to the specific protons. Therefore, the assignment was aided by the NMR observations and DFT calculations.7
Figure 5.
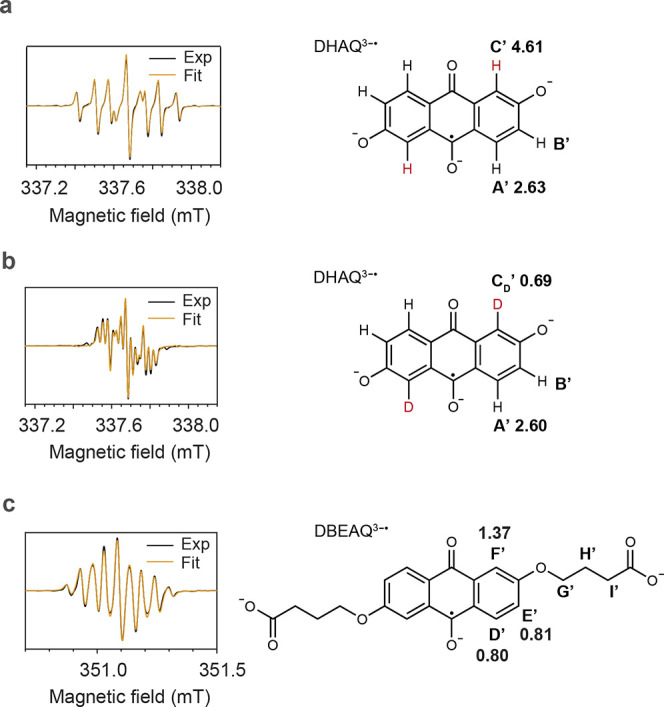
EPR spectra of DHAQ3–• and DBEAQ3–• radical anions. (a) Spectrum of the anolyte from a 1 mM DHAQ versus 20 mM K4[Fe(CN)6] and 10 mM K3[Fe(CN)6] full cell after 10 galvanostatic cycles. The solvent was H2O with 1 M KOH as the supporting electrolyte. The spectrum was acquired at 50% SOC during the last discharge cycle. The linewidth used in the final fit is 0.0121 mT with Gaussian broadening. (b) Spectrum of the anolyte from a full cell acquired under the same conditions as in (a) except that D2O was used as the solvent. The linewidth used in the final fit is 0.0116 mT with Gaussian broadening. (c) Spectrum of the anolyte from a 1 mM DBEAQ versus 20 mM K4[Fe(CN)6] and 10 mM K3[Fe(CN)6] full cell acquired at 50% SOC. D2O and 1 M KOH were used as the solvent and supporting electrolyte, respectively. The linewidth used in the final fit is 0.0100 mT with Gaussian broadening. The molecular structures used for the fits are shown to the right to each spectrum, together with the proton or deuterium labels and their associated hyperfine coupling constants (in MHz) extracted from the fit. The values of the g-factors for DHAQ3–• and DEBAQ3–• are 2.0046 and 2.0049, respectively.
For DHAQ, the values of A for the protons are as follows: AC′ (|4.61| MHz) > AA′ (|2.63| MHz) > AB′. For DBEAQ, they are AF′ (|1.37| MHz) > AE′ (|0.81| MHz) > AD′ (|0.80| MHz) ≫ AG′, AH′, and AI′. Note that the values of AB′, AG′, AH′, and AI′ extracted from the spectral fitting are smaller than the linewidths (0.34 MHz for DHAQ and 0.33 MHz for DBEAQ) used in the fits. Therefore, the electron spin densities on these protons are very small.
The H-D exchange can be seen in the in situ EPR spectrum when D2O was used as the solvent. As shown in Figure S4, during the galvanostatic cycling of 1 mM DHAQ at a current of 1 mA, EPR signals at 337.42 and 337.92 mT gradually decrease, while new EPR signals in the range of 337.53–337.85 mT develop. The best fit to the spectrum acquired at the end of electrochemical cycling (Figure 5b) was obtained by replacing the proton (I = 1/2) on the C′ position by a deuterium (I = 1). In contrast, when H2O was used as the solvent, there are no distinguishable changes in the in situ EPR spectra. These observations confirm that proton C′ is labile and undergoes an exchange reaction with the deuterium (or proton) from solvent water. In contrast, no H-D exchange occurs on DBEAQ molecules (Figure S5).
Identifying and Quantifying Decomposition Products
To detect the decomposition products following electrochemical cycling of the battery with 10 mM DHAQ electrolyte, the battery voltage was held for 10 h either at 1.6 or 1.7 V, the latter being shown in Figure 6a. Holding the voltage is a commonly implemented cycling condition to ensure the complete reduction or oxidation of the electrolyte.6 During the voltage hold, no decomposition products were detected in both the NMR and EPR spectra. As shown in Figure 6a, only 1H NMR signals A″ and B″ of the DHAQ4– anions are visible. However, in our previous study, decomposition products were observed from a 100 mM solution of DHAQ,7 which suggests that the decomposition reaction is concentration-dependent. In another experiment, we cycled a battery with 100 mM DHAQ at 150 mA four times, followed by a voltage hold at 1.7 V for 20 h followed by a further 20 continuous cycles. Figure 6b presents the NMR spectra acquired during the voltage hold. New signals at 6.55, 6.65, 6.84, 7.14, 7.50, 7.88, and 8.02 ppm were observed, consistent with our earlier studies.7 These signals are assigned to 2,6-dihyhroxyanthrone (DHA) and 2,6-dihydroxyanthranol (DHAL). Since the formation of DHAL2– is favored at high pH values,29 the signals of slightly higher intensity at 6.55, 6.84, 7.50, and 7.88 ppm were tentatively assigned to DHAL2–, as the pH value of the electrolyte solution is 14. The pH dependence of the ratio of these two species is still under systematic investigation. The in situ NMR spectra acquired before the voltage hold are shown in Figure S6. The concentration of DHA/DHAL was then quantified: after a voltage hold for 10 h, 28% of the DHAQ decomposed. Assuming that decomposition occurs at a similar rate for the 10 mM DHAQ solution, 2.8 mM DHAQ should have decomposed and therefore 1.4 mM DHA or DHAL should have been produced, assuming a 1:1 ratio. These concentrations are above the detection limit of the NMR technique, but since no new NMR signals were observed, it suggests that no decomposition products were formed when 10 mM was used.
Figure 6.

NMR analyses of electrolyte decomposition at two different concentrations. (a) 1H NMR spectra of 10 mM DHAQ during a voltage hold at 1.7 V for 10 h. (b) 1H NMR spectra of 100 mM DHAQ during a voltage hold at 1.7 V for 20 h. The green and red triangles highlight the signals of the decomposition products, DHA2– or DHAL2– anions, respectively. (c) In situ1H NMR spectrum acquired immediately after the voltage hold was stopped and the discharge started (top spectrum, red), and ex situ1H NMR spectrum of 10 mM as-synthesized DHA2– or DHAL2– dissolved in D2O with 1 M KOH (bottom spectrum, blue). The DHA2– and DHAL2– signals are highlighted by the green triangles. (d) COSY spectrum of the 10 mM as-synthesized DHA2– or DHAL2–. (e) Molecular structures of the DHA2– or DHAL2– anions.
To confirm the assignment of the new peaks at 6.55, 6.65, 6.84, 7.14, 7.50, 7.88, and 8.02 ppm in Figure 6b, we synthesized DHA and DHAL and acquired their 1H NMR spectra (blue color; Figure 6c). The expected eight doublets of the DHA and DHAL signals were observed, as highlighted by the green and red triangles. The eight doublets come from four pairs of closely bonded protons, which is further substantiated by the 2D 1H homonuclear correlation spectrum (COSY), as shown by the four off-diagonal signals in Figure 6d. This is consistent with the bonding relationship of the protons in the DHA2– and DHAL2– anions, as shown in Figure 6e, where there are four pairs of closely bonded aromatic protons. Note that DHA3– and DHAL3–, which are resonance structures, were proposed in our previous work.7 Since eight doublets were observed in the work presented in this paper, we propose that DHA3– and DHAL3– are in fact deuterated, forming DHA2– and DHAL2– anions which are tautomers.30 The 2D NMR spectrum of the synthesized DHA and DHAL matches the 2D spectrum of the decomposition products (extended data Figure 8b in ref (7)). The signals at 6.45, 6.50, 6.75, 7.20, 7.51 ppm, which are not highlighted in the 1D spectrum, likely come from the protons on the central ring and impurities from the synthesis. It is also possible that H-D exchange occurred on these protons after the sample was dissolved in D2O with 1 M KOH to acquire the NMR spectrum. The red spectrum in Figure 6c corresponds to the in situ spectrum acquired immediately after the voltage hold was stopped and the discharge was commenced. At this point, approximately 1% DHAQ3–• radical anions were present, as estimated by the current passed. As discussed in the preceding section, the fast electron transfer between DHAQ3–• and DHAQ4– anions causes broadening of peaks A″ and B″, allowing the decomposition product signals (again, marked with green and red triangles) to be revealed as they remain unaffected by this broadening mechanism. The chemical shifts of the decomposition products at 6.55, 6.65, 6.84, 7.14, 7.50, 7.88, and 8.02 ppm match reasonably well to the chemical shifts of the as-synthesized DHA2– and DHAL2– anions at 6.59, 6.65, 6.86, 7.19, 7.51, 7.94, and 8.02 ppm.
Having observed and quantified the degradation, we next wanted to understand how much capacity fade is caused by the decomposition of the DHAQ4– anions and whether the decomposition products are electrochemically active. After the five galvanostatic cycles and the voltage hold, the battery was discharged and charged at 150 mA for 20 cycles. At the end of the first discharge cycle, as shown in Figure 7a, a second voltage plateau appears at 0.7 V. Deducting this value from the redox potential of 0.5 V of [Fe(CN)6]4–/[Fe(CN)6]3–, gives a redox potential of −0.2 V vs SHE. Note that an irreversible oxidation peak at −0.2 V vs SHE has been reported for DHA at pH 14 in a cyclic voltammetry experiment,31 consistent with our observation. Furthermore, this second oxidation plateau disappears during the subsequent cycles, suggesting that the oxidation reaction is irreversible. In the NMR spectra corresponding to the second plateau, as shown on the right in Figure 7a, the intensity of signals at 6.64 and 6.84 ppm decrease, while the intensity of signal at 7.49 ppm increases, which suggests that the DHA2– or DHAL2– anions have reacted further. In the subsequent cycles, no significant changes in the decomposition product signals were observed, the observed shift of the signals from the decomposition products being instead caused by the change of bulk magnetization of the sample during the electrochemical cycling.
Figure 7.

NMR analysis after the potential hold. (a) 1H NMR spectra of 100 mM DHAQ during galvanostatic cycling at 150 mA after a voltage hold at 1.7 V. The signals of the decomposition products are highlighted by blue triangles. The voltage of the battery is shown on the left. The expanded view of the NMR spectra corresponding to the second voltage plateau at 0.7 V during the first discharge cycle is shown on the right. (b) Concentrations of DHAQ4– and DHA2– + DHAL2– as a function of time during the voltage hold at 1.7 V, calculated based on the analysis of the in situ NMR spectra in Figure 6b. Note that we cannot differentiate the signals of DHA and DHAL, so the concentration calculated is for the total of the two molecules. (c) Capacity of the battery as a function of cycle number.
The NMR measurements have been shown to be in the quantitative regime.7 This enables us to track the concentration changes of the redox-active electrolyte and the decomposition products. During the voltage hold at 1.7 V for 100 mM DHAQ, a signal at 7.48 ppm grows in and is well separated from other signals. This signal is used to calculate the concentrations of DHA2– or DHAL2– anions. Signal A″ at 7.96 ppm is used to calculate the concentration of DHAQ4– anions. However, it overlaps with the two signals of DHA2– or DHAL2– anions at 7.88 and 8.02 ppm; thus, the difference of signal integrals is taken and used for the calculation (see Methods for details). As shown in Figure 7b, the total concentration of DHA2– and DHAL2– anions increases from 0 to 43.0 mM, while the concentration of DHAQ4– anions decreases from 90 to 54.0 mM.
Figure 7c presents the capacity of the battery as a function of cycle numbers before and after the voltage hold. The capacity decreases from 93.5 mA h (the theoretical capacity is 96.4 mA h) before the voltage hold to 70.4 mA h after the voltage hold, after which it then drops to 57.1 mA h and continues to fade slowly afterward. The decrease from 70.4 to 57.1 mA h is caused by the irreversible electrochemical oxidation of DHA2– or DHAL2–, the total decrease in capacity from 93.5 to 57.1 mA h corresponding to a capacity fade of 38.9%. Since the concentration decrease of DHAQ during the voltage hold is 40.0%, it is reasonable to conclude that the decomposition reaction of the DHAQ4– anions is the main cause for the capacity fade of the battery.
3. Conclusion
The use of in situ EPR and coupled online EPR/NMR metrologies to study flow batteries is demonstrated in this work. These methods allowed the formation of radical anions in anthraquinone-based flow batteries to be directly observed by EPR and the delocalization of the unpaired electron spin density over the DHAQ3–• and DBEAQ3–• radical anions to be determined. The concentrations of DHAQ3–• radical anions were quantified by spin counting from the EPR analysis and by measuring the bulk magnetization from the NMR analysis, and consistent results were obtained when using these two methods. The fraction of the radical anions was found to be concentration-dependent, the fraction decreasing as the total concentration of DHAQ increases. This suggests that, besides the comproportionation reaction between DHAQ2– and DHAQ4–, additional reaction equilibria exist. The most likely candidate for this involves a dimerization in which the two unpaired electrons of the DHAQ3–• radical anions are coupled, resulting in an EPR-silent anion (and a reduction in the susceptibility as measured by NMR). Further work will include exploring the concentration profiles of the various species with alternative methods. For example, complementary optical absorption spectroscopic measurements will be pursued to track dimer formation and loss of other species in the system. The role of dimer formation in the degradation mechanism remains another important question to be addressed.
EPR is more sensitive toward low concentrations of radical anions, enabling a reliable measurement of the concentration of the DHAQ3–• radical anions at high voltages. By simultaneously measuring the line-broadening of the 1H resonances of DHAQ4–, we were able to measure the rate constant for the intermolecular electron transfer from DHAQ4– to DHAQ3–• anion. The intermolecular electron transfer can be potentially used to relay electrons from the electrochemical cell to the electrolyte reservoir, thus reducing the pumping power and increasing the energy efficiency of the battery at the system level.
Decomposition of DHAQ4– to DHA2– and DHAL2– anions was observed for a 100 mM DHAQ solution, but not for a 10 mM DHAQ solution, suggesting that the decomposition reaction is concentration-dependent. Our NMR analysis reveals that DHA2– or DHAL2– anions undergo irreversible electrochemical oxidation reactions in the voltage window of 0.6–1.6 V. Furthermore, by correlating the concentration of DHAQ to the capacity of the battery quantitatively, we show that the decomposition of DHAQ is the main cause of the capacity fade of the battery. The elementary reaction steps that lead to the formation of DHA2– or DHAL2– and their further oxidation warrant systematic investigation. For example, the potential dependence of the decomposition could be investigated by varying the potentials of the anode or cathode side separately with the incorporation of a third reference electrode. This would allow the reaction kinetics and the elementary reaction steps to be studied.
Coupling the in situ NMR and EPR techniques, we demonstrate the possibility of multi-modal characterizations of redox flow batteries. Other characterization techniques such as optical spectroscopy or mass spectrometry could be incorporated into this concept. These characterization techniques could then be further extended to study other flow electrochemical systems, for example, to study electrochemical CO2 sequestration or water desalination.
4. Methods
Materials and Synthesis
2,6-Dihydroxyanthraquinone (2,6-DHAQ, A89502 technical grade, 90% purity), potassium hexacyanoferrate(II) trihydrate (P3289, ≥ 98.5% purity), potassium hexacyanoferrate(III) (1049730100, ≥ 99.0%), ≥ 99.0%), D2O (151882, 99.9 atom %), ethyl 4-bromobutyrate (167118, 95%), and 4-hydroxy-TEMPO (176164, 97%) were purchased from Sigma Aldrich Chemicals. Anhydrous N,N-dimethylformamide (43465, ≥99.9% purity), potassium ethoxide (14263.30, ≥ 95% purity), anhydrous potassium carbonate (A16625, ≥ 99.0% purity), isopropanol (20839.366, ≥ 99.0% purity), and glacial acetic acid (20103.364, 99–100%) were purchased from VWR. Synthesis of 4,4′-((9,10-anthraquinone-2,6-diyl)dioxy)dibutyric acid (2,6-DBEAQ) and 2,6-dihydroxyanthrone followed the previously reported methods.15,31 To synthesize a mixture of 2,6-dihydroxyanthrone and 2,6-dihydroxyanthranol, 2,6-DHAQ (2.40 g, 10.0 mmol) was mixed with tin dichloride dihydrate (18.32 g, 81.2 mmol, Aldrich 20,825-6, 98%) under an inert atmosphere. Hydrochloric acid (80 mL, degassed, Honeywell 07102-2.5L, 36.5–38%) was then added. The yellow-brown suspension was then taken to reflux (20.5 h) under a nitrogen atmosphere causing the suspension to become more yellow. After allowing the suspension to cool, the suspension was filtered under vacuum and washed with water (degassed) and dichloromethane (dry, degassed). The pale-yellow solid was then dried under vacuum. The solid was stored under an inert atmosphere in an opaque container before any further manipulation. The final product (4.567 g, 20 mmol) was adhesive and viscous in nature, suggesting that residual solvent or non-volatile trace byproducts remained despite the washings and extensive drying process.
Flow Battery Assembly
The assembly was described in detail in our previous work.7 Briefly, graphite flow plates with serpentine flow patterns were used for both electrodes. Each electrode comprised 4.6 mm carbon felt (SGL) with a 5 cm2 active area, which was used without further treatment. Nafion 212 was used as the ion transport membranes. Pretreatment of the Nafion 212 membranes was performed by first heating the membrane in 80 °C deionized water for 20 min and then soaking it in 5% hydrogen peroxide solution for 35 min.
Online EPR and NMR Setup
The setup consists of a flow battery (Scribner), two peristaltic pumps (MasterFlex L/S 07751-20, Cole-Parmer), an electrochemical cycler (SP-150, BioLogic SAS), a benchtop EPR (MS5000, Magnettech), and an NMR (300 MHz, Bruker) spectrometer (Figure S1). The battery and the EPR spectrometer are positioned outside the 5 G line of the NMR magnet. The electrolyte is pumped through the flow battery, then flowed through the EPR and NMR magnets, and finally back to the electrolyte reservoir. The direction of flow is from the bottom to the top of both magnets. PFA tubes (1/16 in.) are used to connect the electrolyte reservoir, the battery, and the EPR and NMR sampling tubes. The time it takes for a round-trip from and back to the electrolyte reservoir is 64 s at a flow rate of 13.6 cm3 min–1. The time it takes for the electrolyte solution to travel from the electrolyte reservoir to the battery is 3 s, from the battery to the EPR detection region is 3 s, from the EPR to the NMR detection region is 29 s, and from the NMR detection regions back to the electrolyte reservoir is 29 s. To minimize heating of the aqueous solution by microwave irradiation, a flat EPR cell (E4503, Magnettech) is used. A customized adaptor made of polyether ether ketone (PEEK) is used to connect the flat EPR cell to the 1/16 in. tube. The cell is orientated in the resonator such that the strength of the magnetic field is maximized and the strength of the electric field is minimized across the sample. The volume of the cell in the excitation region of the microwave is 0.03 cm3 (2.00 cm × 0.50 cm × 0.03 cm), giving a residence time of 0.13 s for the electrolyte solution at a flow rate of 13.6 cm3 min–1. Details of the NMR sampling tube are provided in our previous publication.7
For the coupled in situ EPR and NMR experiment, 27 mL of 10 mM DHAQ was used as the anolyte, and 60 mL of 15 mM K4[Fe(CN)6] and 3.75 mM K3[Fe(CN)6] was used as the catholyte. The solvent was D2O, with 1 M KOH dissolved in as the supporting electrolyte. The flow rate was 13.6 cm3 min–1. For the in situ NMR experiment, 20 mL of 100 mM DHAQ was used as the anolyte, and 40 mL of 150 mM K4[Fe(CN)6] and 50 mM K3[Fe(CN)6] was used as the catholyte. The solvent was D2O, with 1 M KOH. The flow rate was 33.3 cm3 min–1. For the in situ EPR experiments, 20 mL of 1 mM DHAQ or DBEAQ was used as the anolyte, and 40 mL of 20 mM K4[Fe(CN)6] and 10 mM K3[Fe(CN)6] was used as the catholyte. D2O and H2O with 1 M KOH were used as the solvents in two separated experiments. The flow rate was 13.6 cm3 min–1.
Spin Counting and EPR Parameters
The double integral, DI, of an EPR signal is proportional to the concentration of spins in the sample, CS, following32
| 6 |
where Cns is a constant including the normalized spectrometer settings, i.e., sweep time and number of accumulations, P is the microwave power, Bm is the modulation amplitude, Q is the resonator’s quality factor, nB is the Boltzmann factor for temperature dependence, S is the total electron spin, v is the volume of the sample, and f(B1,Bm) is the spatial distribution of microwave and modulation field in the sample. By measuring a standard sample of known CS, a linear relationship between DI and CS can be established. The concentration of a sample of interest can then be calculated from the DI of its EPR signal.
A set of 4-OH-TEMPO solutions of concentrations ranging from 0.1 mM to 125 mM was prepared. H2O was used as the solvent. 4-OH-TEMPO has a spin quantum number of a half, i.e., S = 1/2. EPR spectra were acquired using the flat cell under a flow rate of 13.6 cm3 min–1. This ensured that v and f(B1,Bm) remained the same throughout the measurements. The magnetic field was swept from 332.5 to 342.5 mT. The sweep time was 60 s for a single scan. P was 0.5 mW, and Bm was 0.001 mT. Q was recorded for each spectrum, in the range of 1670 ± 20. The first-derivative spectra (Figure S3a) were integrated to generate the absorption spectra (Figure S3b). Then the baselines of the absorption spectra were corrected by a fourth order polynomial fit after which the baseline-corrected spectra were integrated one more time to give the double integral, DI, which was fit linearly as a function of the concentrations of 4-OH-TEMPO (Figure S3c,d), and a slope of 4725.7 mM–1 was obtained.
For the in situ measurement of DHAQ3–• radical anions, the magnetic field was swept from 336.5 to 339 mT. The sweep time was 60 s per single scan. Bm was 0.001 mT, and Q was recorded for each spectrum. Q was 1306 ± 5. The temperature of the resonator was kept at 29 °C, and a time delay of 35 s was added between each scan. The in situ EPR spectra of the DHAQ3–• radical anions were doubly integrated, following the above procedure. Normalizing the EPR parameters following eq 6 and dividing the double integral by 4725.7 mM–1 gave the concentration of the DHAQ3–• radical anions.
The in situ measurement of DBEAQ was performed on an X-band spectrometer (EMX Micro, Bruker). The magnetic field was swept from 341.3 to 351.3 mT. The sweep time was 20 s per single scan. P was 2 mW, and Bm was 0.01 mT.
The measured g-factors of DHAQ3–• and DEBAQ3–• radical anions were corrected by using the g-factor of 4-OH-TEMPO as the reference. The g-factor of 4-OH-TEMPO measured on the Magnettech instrument is 2.0057, while the value reported in literature is 2.0059.33 A scaling factor of 1.0001 (2.0059/2.0057 = 1.0001) is therefore applied to the measured values using the Magnettech instrument. The corrected g-factor of DHAQ3–• is 2.0046 (2.0044 × 1.0001 = 2.0046). The g-factors of DHAQ3–• and DBEAQ3–• measured by the Bruker instrument are 2.0077 and 2.0080, respectively, which were corrected to 2.0046 and 2.0049.
NMR Parameters
Pseudo-2D NMR experiments were performed by direct excitation with a 90° radio-frequency pulse. Each NMR spectrum is acquired by collecting 16 free induction decays (FIDs) with a recycle delay of 5 s. The pulse width for a 90° pulse was 27 μs. All spectra were referenced to the water chemical shift at 4.79 ppm before battery cycling starts.
Bulk Magnetization and Radical Concentrations
Bulk magnetization of the electrolyte solution is linearly proportional to the concentration of DHAQ3–• radical anions. The derivation of the relationship between bulk magnetization, radical concentration (Crad), and the change in chemical shift of water (ΔδS) was provided in our previous work.7 The concentration of DHAQ3–• radical anions can be readily estimated by
| 7 |
Calculation of Rate Constant for the Electron Transfer Reaction
The result derived for the slow exchange regime was used for the calculations of the rate constants for the electron transfer between DHAQ3–• and DHAQ4– anion.7,34 When the concentration of radical anions is increased by Δ[P], the transverse nuclear relaxation of the nucleus is increased by ΔR2ex. Following eq 8, ΔR2ex is calculated by
| 8 |
where FWHM is the full width at the half maximum of the NMR signal, measured in Hz. FWHM0 is the full width at the half maximum of the NMR signal with a known concentration of radical anions, [P]0. The linewidth of the NMR signal during the voltage hold at 1.7 V for 10 h is chosen to be FWHM0, and it is equal to 26.7 Hz where [P]0 is 0.02 mM.
It takes 29 s for the flowing electrolyte to reach the NMR detection region from the EPR detection region. Within this time duration, some DHAQ4– anions are oxidized to the DHAQ3–• radical anions. Therefore, to obtain an accurate estimation of [P], we need to account for the increased concentration of DHAQ3–•, [P]1. During the voltage hold at 1.7 V, the current remains constant at 0.96 mA, corresponding to an electrochemical reduction rate of 9.95 × 10–4 mM s–1. The concentration of DHAQ3–• radical anions remains constant, suggesting that the system is in equilibrium and that the electrochemical reduction rate is equal to the oxidation rate. Therefore, during the time of flight of 29 s, 0.029 mM DHAQ3–• radical anions (9.95 × 10–4 mM s–1 × 29 s) have been produced, i.e., [P]1 = 0.029 mM. Δ[P] is calculated by
| 9 |
Rearranging eqs 4, 8, and 9, we obtain
| 10 |
Calculations of DHAQ, DHA, and DHAL Concentrations
The in situ NMR spectra were first baseline-corrected by a fourth-order polynomial function. The proton resonance at 7.48 ppm, which is well separated from the other peaks during the potential hold, was used for the calculations of DHA or DHAL concentrations. Note that we cannot differentiate the signals of DHA and DHAL, so the concentration calculated is for both of the molecules. The proton resonance at 7.96 ppm was used for the calculation of DHAQ4– concentration. This signal overlaps with two signals of DHA or DHAL at 7.88 and 8.01 ppm. To deduct the integrals of these two signals, a spectrum taken immediately after the voltage hold was stopped (at 27.93 h, see Figure 7a) was used to establish the ratios of the integrals of these two signals to that at 7.48 ppm. This spectrum was chosen because the signal A″ of DHAQ4– was broadened at this point to below the baseline due to the presence of approximately 1% DHAQ3–• radical anions and the corresponding intermolecular electron transfer between DHAQ3–• and DHAQ4–. As such, the signals of DHA or DHAL can be accurately integrated since they are unaffected by the radical-induced broadening. A ratio of 1.19:1.37:1.00 was obtained after the deconvolution of the three signals at 8.01, 7.88, and 7.48 ppm (Figure S6b). This ratio was used for the calculations of integrals at 8.01 and 7.88 ppm during the potential hold. The spectrum at the beginning of the potential hold (at 8.80 h, see Figure S6a) was used to calibrate the concentration since there are negligible decomposition products at this time. Signal A″ in this spectrum corresponds to 90 mM DHAQ4– (excluding the 10% impurity in the sample, as specified by the manufacturer).
Acknowledgments
E.W.Z. and C.P.G. acknowledge support from Centre of Advanced Materials for Integrated Energy Systems (CAM-IES), via EPSRC grant no. EP/P007767/1. E.W.Z., R.J., and C.P.G. acknowledge support from Shell. E.W.Z. acknowledges support from the Manifest exchange program via EPSRC grant no. EP/N032888/1. We thank the EPSRC EPR National Research Facility (NS/A000055/1) for access to their EPR spectrometers. We thank D. S. Wright from University of Cambridge for many fruitful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10650.
In situ NMR and EPR setup, additional 1H NMR spectra, and EPR spectra of DHAQ, DBEAQ, and 4-OH-TEMPO, calculations of reaction equilibria including Figures S1–S6 and Table S1 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ding Y.; Zhang C.; Zhang L.; Zhou Y.; Yu G. Molecular engineering of organic electroactive materials for redox flow batteries. Chem. Soc. Rev. 2018, 47 (1), 69–103. 10.1039/C7CS00569E. [DOI] [PubMed] [Google Scholar]
- Winsberg J.; Hagemann T.; Janoschka T.; Hager M. D.; Schubert U. S. Redox-flow batteries: from metals to organic redox-active materials. Angew. Chem., Int. Ed. 2017, 56 (3), 686–711. 10.1002/anie.201604925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung P.; Shah A. A.; Sanz L.; Flox C.; Morante J. R.; Xu Q.; Mohamed M. R.; Ponce de León C.; Walsh F. C. Recent developments in organic redox flow batteries: A critical review. J. Power Sources 2017, 360, 243–283. 10.1016/j.jpowsour.2017.05.057. [DOI] [Google Scholar]
- Dieterich V.; Milshtein J. D; Barton J. L; Carney T. J; Darling R. M; Brushett F. R Estimating the cost of organic battery active materials: a case study on anthraquinone disulfonic acid. Transl. Mater. Res. 2018, 5 (3), 034001. 10.1088/2053-1613/aacb0e. [DOI] [Google Scholar]
- Brushett F. R.; Aziz M. J.; Rodby K. E. On Lifetime and Cost of Redox-Active Organics for Aqueous Flow Batteries. ACS Energy Letters 2020, 5 (3), 879–884. 10.1021/acsenergylett.0c00140. [DOI] [Google Scholar]
- Kwabi D. G.; Ji Y.; Aziz M. J. Electrolyte Lifetime in Aqueous Organic Redox Flow Batteries: A Critical Review. Chem. Rev. 2020, 120 (14), 6467–6489. 10.1021/acs.chemrev.9b00599. [DOI] [PubMed] [Google Scholar]
- Zhao E. W.; Liu T.; Jónsson E.; Lee J.; Temprano I.; Jethwa R. B.; Wang A.; Smith H.; Carretero-González J.; Song Q.; Grey C. P. In situ NMR metrology reveals reaction mechanisms in redox flow batteries. Nature 2020, 579 (7798), 224–228. 10.1038/s41586-020-2081-7. [DOI] [PubMed] [Google Scholar]
- Maki A. H.; Geske D. H. Detection of Electrolytically Generated Transient Free Radicals by Electron Spin Resonance. J. Chem. Phys. 1959, 30 (5), 1356–1357. 10.1063/1.1730187. [DOI] [Google Scholar]
- Geske D. H.; Maki A. H. Electrochemical Generation of Free Radicals and Their Study by Electron Spin Resonance Spectroscopy; the Nitrobenzene Anion Radical. J. Am. Chem. Soc. 1960, 82 (11), 2671–2676. 10.1021/ja01496a004. [DOI] [Google Scholar]
- Goldberg I. B.; Bard A. J. Simultaneous electrochemical-electron spin resonance measurements. I. Cell design and preliminary results. J. Phys. Chem. 1971, 75 (21), 3281–3290. 10.1021/j100690a013. [DOI] [Google Scholar]
- Albery W. J.; Coles B. A.; Couper A. M. The tube electrode and e.s.r. J. Electroanal. Chem. 1975, 65 (2), 901–909. 10.1016/0368-1874(75)85165-3. [DOI] [Google Scholar]
- Albery W. J.; Compton R. G.; Chadwick A. T.; Coles B. A.; Lenkait J. A. Tube electrode and electron spin resonance. First-order kinetics. J. Chem. Soc., Faraday Trans. 1 1980, 76 (0), 1391–1401. 10.1039/f19807601391. [DOI] [Google Scholar]
- Lawton J. S.; Aaron D. S.; Tang Z.; Zawodzinski T. A. Electron spin resonance investigation of the effects of vanadium ions in ion exchange membranes for uses in vanadium redox flow batteries. ECS Trans. 2012, 41 (23), 53–56. 10.1149/1.3697454. [DOI] [Google Scholar]
- Lin K.; Chen Q.; Gerhardt M. R.; Tong L.; Kim S. B.; Eisenach L.; Valle A. W.; Hardee D.; Gordon R. G.; Aziz M. J.; Marshak M. P. Alkaline quinone flow battery. Science 2015, 349 (6255), 1529–1532. 10.1126/science.aab3033. [DOI] [PubMed] [Google Scholar]
- Kwabi D. G.; Lin K.; Ji Y.; Kerr E. F.; Goulet M.-A.; De Porcellinis D.; Tabor D. P.; Pollack D. A.; Aspuru-Guzik A.; Gordon R. G.; Aziz M. J. Alkaline quinone flow battery with long lifetime at pH 12. Joule 2018, 2, 1894–1906. 10.1016/j.joule.2018.07.005. [DOI] [Google Scholar]
- Ji Y.; Goulet M.-A.; Pollack D. A.; Kwabi D. G.; Jin S.; De Porcellinis D.; Kerr E. F.; Gordon R. G.; Aziz M. J. A phosphonate-functionalized quinone redox flow battery at near-neutral pH with record capacity retention rate. Adv. Energy Mater. 2019, 9, 1900039. 10.1002/aenm.201900039. [DOI] [Google Scholar]
- Wu M.; Jing Y.; Wong A. A.; Fell E. M.; Jin S.; Tang Z.; Gordon R. G.; Aziz M. J. Extremely Stable Anthraquinone Negolytes Synthesized from Common Precursors. Chem. 2020, 6 (6), 1432–1442. 10.1016/j.chempr.2020.03.021. [DOI] [Google Scholar]
- Eaton S. S.; Eaton G. R.. Chapter 2 - Relaxation Times of Organic Radicals and Transition Metal Ions. In Distance Measurements in Biological Systems by EPR; Berliner L. J., Eaton G. R., Eaton S. S., Eds.; Springer US: Boston, MA, 2000; pp 29–154. [Google Scholar]
- Weil J. A.; Bolton J. R.. Chapter 10 - Relaxation Times, Linewidths and Spin Kinetic Phenomena. In Electron Paramagnetic Resonance: Electron Paramagnetic Resonance: Elementary Theory and Practical Applications, 2nd ed.; John Wiley & Sons, Inc., Hoboken, NJ, 2006; pp 301–356. [Google Scholar]
- Ward R. L.; Weissman S. I. Electron Spin Resonance Study of the Electron Exchange between Naphthalene Negative Ion and Naphthalene. J. Am. Chem. Soc. 1957, 79 (9), 2086–2090. 10.1021/ja01566a017. [DOI] [Google Scholar]
- Carney T. J.; Collins S. J.; Moore J. S.; Brushett F. R. Concentration-dependent dimerization of anthraquinone disulfonic acid and its impact on charge storage. Chem. Mater. 2017, 29 (11), 4801–4810. 10.1021/acs.chemmater.7b00616. [DOI] [Google Scholar]
- Tong L.; Chen Q.; Wong A. A.; Gómez-Bombarelli R.; Aspuru-Guzik A.; Gordon R. G.; Aziz M. J. UV-Vis spectrophotometry of quinone flow battery electrolyte for in situ monitoring and improved electrochemical modeling of potential and quinhydrone formation. Phys. Chem. Chem. Phys. 2017, 19 (47), 31684–31691. 10.1039/C7CP05881K. [DOI] [PubMed] [Google Scholar]
- Wiberg C.; Carney T.; Brushett F.; Ahlberg E.; Wang E. Dimerization of 9,10-anthraquinone-2,7-Disulfonic acid (AQDS). Electrochim. Acta 2019, 317, 478. 10.1016/j.electacta.2019.05.134. [DOI] [Google Scholar]
- D’Souza F.; Deviprasad G. R. Studies on Porphyrin–Quinhydrone Complexes: Molecular Recognition of Quinone and Hydroquinone in Solution. J. Org. Chem. 2001, 66 (13), 4601–4609. 10.1021/jo0100547. [DOI] [PubMed] [Google Scholar]
- González Moa M. J.; Mandado M.; Mosquera R. A. A Computational Study on the Stacking Interaction in Quinhydrone. J. Phys. Chem. A 2007, 111 (10), 1998–2001. 10.1021/jp0661701. [DOI] [PubMed] [Google Scholar]
- Astudillo P. D.; Valencia D. P.; González-Fuentes M. A.; Díaz-Sánchez B. R.; Frontana C.; González F. J. Electrochemical and chemical formation of a low-barrier proton transfer complex between the quinone dianion and hydroquinone. Electrochim. Acta 2012, 81, 197–204. 10.1016/j.electacta.2012.07.078. [DOI] [Google Scholar]
- Michaelis L.; Boeker G. F.; Reber R. K. The Paramagnetism of the Semiquinone of Phenanthrenequinone-3-sulfonate. J. Am. Chem. Soc. 1938, 60 (1), 202–204. 10.1021/ja01268a058. [DOI] [Google Scholar]
- McConnell H. M. Indirect Hyperfine Interactions in the Paramagnetic Resonance Spectra of Aromatic Free Radicals. J. Chem. Phys. 1956, 24 (4), 764–766. 10.1063/1.1742605. [DOI] [Google Scholar]
- Comninellis C.; Plattner E. The electrochemical reduction of anthraquinone to anthrone in concentrated H2SO4. J. Appl. Electrochem. 1985, 15 (5), 771–773. 10.1007/BF00620574. [DOI] [Google Scholar]
- McCann G. M.; McDonnell C. M.; Magris L.; More O’Ferrall R. A. Enol–keto tautomerism of 9-anthrol and hydrolysis of its methyl ether. Journal of the Chemical Society, Perkin Transactions 2 2002, 4, 784–795. 10.1039/b109242c. [DOI] [Google Scholar]
- Goulet M.-A.; Tong L.; Pollack D. A.; Tabor D. P.; Odom S. A.; Aspuru-Guzik A.; Kwan E. E.; Gordon R. G.; Aziz M. J. Extending the lifetime of organic flow batteries via redox state management. J. Am. Chem. Soc. 2019, 141, 8014–8019. 10.1021/jacs.8b13295. [DOI] [PubMed] [Google Scholar]
- Manual and Documentation of Magnettech MiniScope MS 5000 and MS 5000X, Magnettech GmbH, Berlin, Germany. [Google Scholar]
- Ali M.; Zilbermann I.; Cohen H.; Shames A. I.; Meyerstein D. Properties of the Nickel(III) Complex with 1,4,8,11-Tetraazacyclotetradecane-1,4,8,11-tetraacetate in Aqueous Solution. Inorg. Chem. 1996, 35 (18), 5127–5131. 10.1021/ic950871n. [DOI] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G.; Ravera E.. Chapter 6 - Chemical exchange, chemical equilibria, and dynamics. NMR of Paramagnetic Molecules, 2nd ed.; Elsevier: Boston, 2017; pp 151–173. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.