
Figure 1. Structural Organization of SKIP.
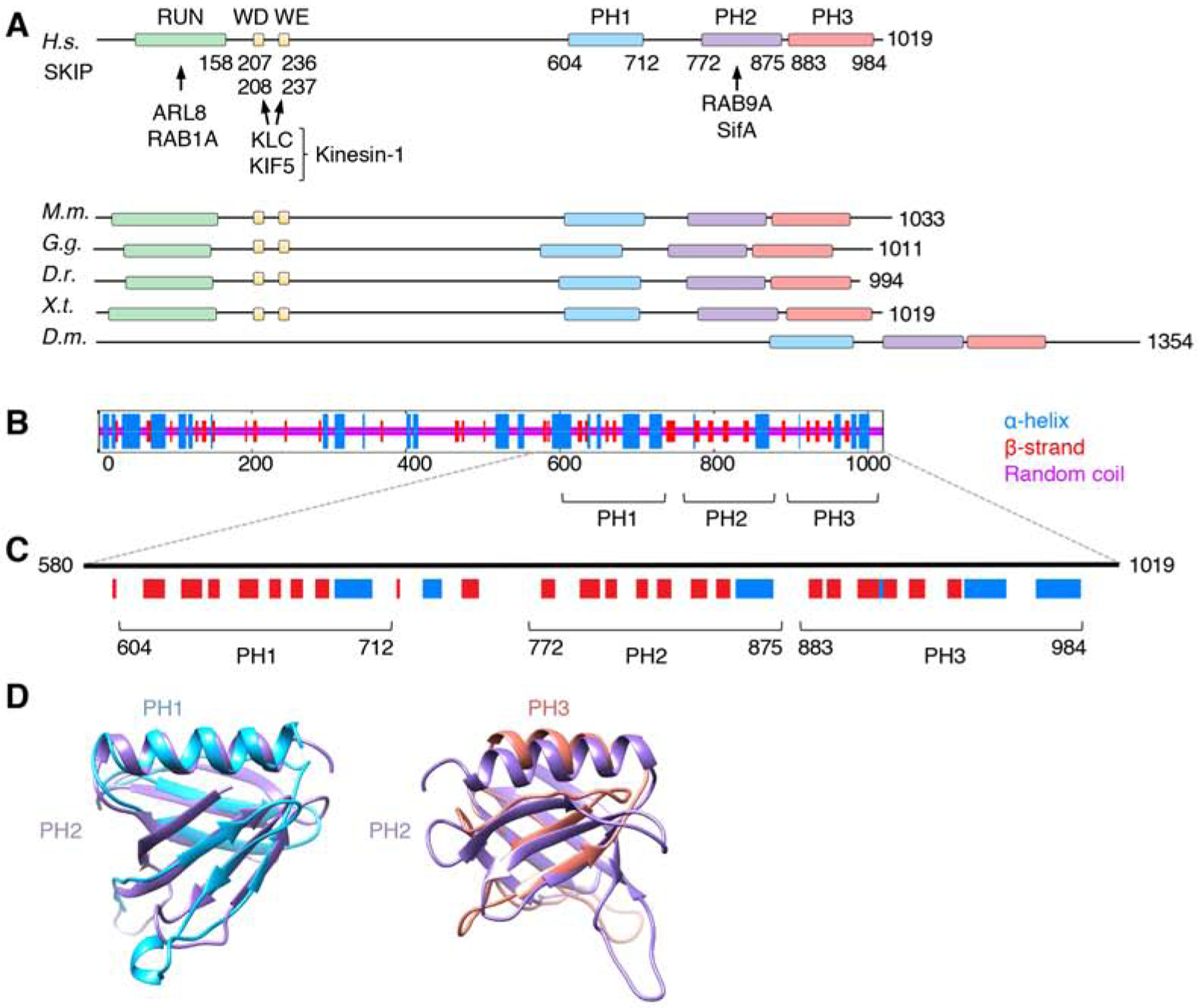
(A) Domain organization of SKIP from different animal species. Boxes indicate domains and motifs. RUN: RPIP8, UNC-14 and NESCA domain; WD: Trp-Asp motif; WE: Trp-Glu motif; PH: pleckstrin homology domain. Amino-acid numbers are indicated. Arrows indicate interactions with binding partners. H.s.: Homo sapiens, M.m.: Mus musculus, G.g.: Gallus gallus, D.r.: Danio rerio, X.t.: Xenopus tropicalis, D.m.: Drosophila melanogaster. Notice that Drosophila SKIP does not contain a RUN domain or WD/WE motifs, but its C-terminal domain is well conserved. The RUN domain mediates ARL8-dependent coupling to lysosomes [20] and RAB1A-dependent coupling to melanosomes [40]. Because melanosomes are not present in the cells used in this study, the role of RAB1A was not analyzed.
(B) Consensus secondary structure prediction of SKIP generated at the NPS-IBCP server (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_seccons.html).
(C) PH domain predictions by the PredictProtein server (https://www.predictprotein.org/). Notice the similarity in the secondary structure of the predicted PH1 and PH3 to the known PH2. HHpred (https://toolkit.tuebingen.mpg.de/tools/hhpred) also predicts PH1 and PH3 to be structurally similar to PH domains with high probabilities (95.2% for PH1 and 91.8% for PH3).
(D) Structural models of PH1 and PH3 according to the PHYRE2 Protein Fold Recognition Server superimposed on the known structure of PH2 (PDB ID: 3CXB). Confidence levels for PH1 and PH3 were 99.3% and 30.56%, respectively. Models were represented with UCSF Chimera.