

Abstract
Understanding the splicing code can be challenging as several splicing factors bind to many splicing-regulatory elements. The SMN silencer element ISS-N1 is the target of the antisense-oligonucleotide drug Spinraza, which is the treatment against Spinal Muscular Atrophy. However, limited knowledge about the nature of the splicing factors that bind to ISS-N1 and inhibit splicing exists. It is likely that the effect of Spinraza comes from blocking binding of these factors, but so far, an unbiased characterization has not been performed and only members of the hnRNP A1/A2 family have been identified by western blotting and NMR to bind to this silencer.
Employing an MS/MS based approach and SPRi, we show for the first time that splicing factor SRSF10 binds to ISS-N1. Furthermore, using splice-switching oligonucleotides we modulated the splicing of the SRSF10 isoforms generating either the long or the short protein isoform of SRSF10 to regulate endogenous SMN2 exon 7 inclusion. We demonstrate that the isoforms of SRSF10 regulate SMN splicing with different strength correlating with the length of their RS domain. Our results suggest that the ratio between the SRSF10 isoforms is important for splicing regulation.
Keywords: SRSF10, Spinal Muscular Atrophy, SMN2, antisense oligonucleotides (ASO), Alternative splicing
INTRODUCTION
Spinal Muscular Atrophy (SMA) is a neurodegenerative disease that affects motor neurons in spinal cord and lower brain stem. SMA is caused by homozygous loss of the SMN1 gene. Therefore, production of Survival of Motor Neuron (SMN) protein in patients depends solely on the diseasemodulating SMN2 gene (Clermont et al., 1995; Lefebvre et al., 1995). A silent c.840C>T (position +6 in exon 7) variation in SMN2 abolishes an SRSF1 binding exonic splicing enhancer (ESE) and creates an hnRNP A1 binding exonic splicing silencer (ESS) (Bruun et al., 2016; Cartegni & Krainer, 2002; Kashima & Manley, 2003). The SMN2 ESS acts in cooperation with other silencer motifs that bind hnRNP A1 (Bruun et al., 2016; Doktor et al., 2011; Singh, Singh, Androphy, & Singh, 2006). This cooperation between silencers inhibits the inclusion of exon 7, thereby leading to production of only a low amount of functional SMN protein from SMN2. A potent silencer in intron 7 (ISS-N1) is an important regulator of SMN splicing (Beusch, Barraud, Moursy, Clery, & Allain, 2017; Hua, Vickers, Okunola, Bennett, & Krainer, 2008; Singh et al., 2006). The ISS-N1 silencer comprises two hnRNP A1 motifs and binding of hnRNP A1 to ISS-N1 has been demonstrated by western blotting and NMR (Hua et al., 2008). Importantly, the recently approved and very successful antisense-oligonucleotide drug, Spinraza, is used as a treatment in SMA and binds to the ISS-N1 silencer. It is believed that Spinraza functions by blocking binding of hnRNP A1 to ISS-N1, thereby leading to increased inclusion of exon 7 from SMN2 and production of full length SMN protein (Chiriboga et al., 2016; Hua et al., 2008).
Ubiquitously expressed splicing factors like hnRNP A1 are important regulators of many splicing events both in constitutive and alternative splicing. HnRNP A1 functions as a global regulator and is known to regulate splicing across different tissues and cell types (Jean-Philippe, Paz, & Caputi, 2013; Kamma, Portman, & Dreyfuss, 1995). In contrast to hnRNP A1, the splicing factor SRSF10 (also known as neural salient serine/arginine-rich (NSSR) protein) exhibits a tissue-specific expression profile that better matches that of the affected tissues in SMA, with particularly high expression in motor neurons (K. J. Liu & Harland, 2005; L. Liu, Lin, Chen, Liu, & Xu, 2003). SRSF10 is a member of the serine- and arginine-rich (SR) family. The SRSF10 gene produces several mRNAs transcript as shown in fig 1A with the two main protein isoforms; a short isoform of 183 aa (ENST00000344989, NP_006616) and a long isoform of 262 aa (ENST00000492112, NP_473357) (Komatsu, Kominami, Arahata, & Tsukahara, 1999; Yang, Embree, Tsai, & Hickstein, 1998). SRSF10 is an unusual SR protein as it was initially characterized as a splicing repressor (Feng, Chen, & Manley, 2008; Shin & Manley, 2002). Furthermore, SRSF10 has been shown to act in a similar way as the hnRNP A/B family of splicing regulatory proteins and to antagonize the function of other SR proteins (Cowper, Caceres, Mayeda, & Screaton, 2001).
Figure 1. In vitro investigation of the ISS-N1 element by MS/MS analysis and RNA affinity pull down.
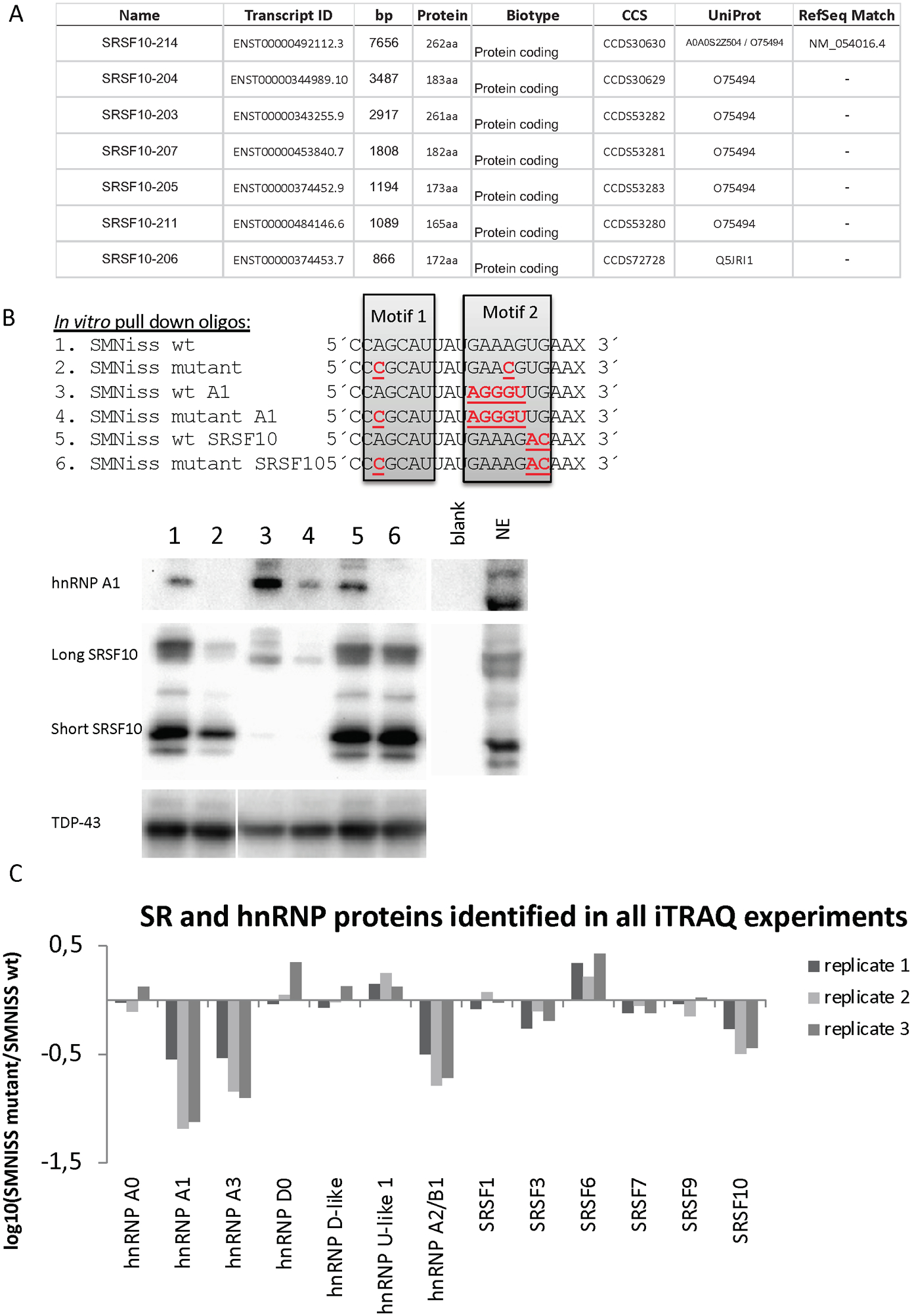
(A) List of SRSF10 transcripts with protein-coding products. (B) The RNA oligonucleotides used for the affinity pull down assay and western blot analysis. The nucleotides marked in red are mutated, compared to the wt sequence. Western blot analysis with antibodies against hnRNP A1 and SRSF10. The displayed blots are representative results from at least three pull down experiments. Western blotting for TDP-43 is included as a control. (C) Graphical representation of proteins identified in all three replicates. The Y-axis represents the ratio between the SMN ISS-N1 wt and SMN ISS-N1 mutant. Proteins binding significantly more to the wt sequence display a negative log-value on the y-axe and proteins found to bind more significantly to the mutant sequence display a positive log-value on the y-axe.
We noticed that the proposed SELEX sequence for SRSF10 matches a portion of the ISS-N1 silencer in the SMN genes and overlaps one of the hnRNP A1 motifs (Ray et al., 2009; Shin & Manley, 2002; Xiao et al., 2016). Therefore, we hypothesized that SRSF10 plays a role in the splicing of the SMN genes, and that SRSF10 can bind to a motif in the ISS-N1 silencer to suppress splicing of exon 7.
In this study, we used RNA affinity purification followed by global iTRAQ MS/MS analysis to identify splicing factors that bind to the ISS-N1 silencer. Next, we confirmed binding of SRSF10 and hnRNP A1 binding to different variant ISS-N1 RNA sequences by western blotting and analyzed binding kinetics by Surface Plasmon Resonance (SPRi). We investigated the functional effects of the two SRSF10 isoforms on SMN1/2 exon 7 splicing by overexpression and by employing MS2 coat fusion proteins. Finally, we show that we can efficiently re-direct the endogenous splicing of the two SRSF10 isoforms by employing splice-switching oligonucleotides (SSOs) and thereby affect SMN1/2 exon 7 splicing.
Our results demonstrate that the ISS-N1 silencer is a direct target for SRSF10 and that the long isoform of SRSF10 mediates the strongest splicing-regulatory effect.
RESULTS
SRSF10 binds to the SMNiss wt sequence
In order to identify proteins that bind to the ISS-N1 silencer in an unbiased manner, we performed RNA affinity purification from HeLa cell nuclear extracts employing biotin-labeled RNA oligonucleotides with the sequence of the wildtype (wt) or mutant (mut) ISS-N1 silencer. The SMN ISS-N1 mutant sequence has two A>C substitutions (A12C and A23C) in the hnRNP A1 motif 1 and motif 2 (Figure 1B). These substitutions were previously demonstrated to disrupt hnRNP A1 binding to a number of motifs (Bruun et al., 2016; Doktor et al., 2011; Hua et al., 2008; Nielsen et al., 2007; Olsen et al., 2014). The RNA-bound proteins were cleaved using trypsin and the resulting peptides were labeled with iTRAQ and processed for quantitative Liquid Chromatography (LC) tandem mass spectrometry (MS/MS). Only proteins that were identified to bind significantly more to either the wt or mutant sequences in all three replicates are displayed in Figure 1C. Criteria for protein identification were set for: 1) Minimum two unique identified peptides in each replicate and; 2) Protein identified in all three replicates. SRSF10 consistently showed more binding to the SMNiss wt sequence compared to the mutated sequence. As expected, our MS/MS analysis also confirmed binding of hnRNP A1, hnRNP A3 and hnRNP A2/B1 to the SMN ISS-N1 wt silencer (Hua et al., 2008). We confirmed binding of SRSF10 to the SMN ISS-N1 region by western blot analysis. As expected, this analysis revealed more hnRNP A1 and SRSF10 binding to the wt SMN ISS-N1 sequence compared to the mutant ISS-N1 sequence (Figure 1B). There are two major spliced isoforms of SRSF10, long and short, both of which interacted with SMN ISS-N1. We then replaced the original sequence of motif 2 of both the wt and mutated oligonucleotide with the hnRNP A1 consensus motif (Bruun et al., 2016; Burd & Dreyfuss, 1994) to investigate the relationship between the two hnRNP A1 motifs. This analysis showed that the largest amount of hnRNP A1 is bound when both motifs are functional. Because, the two hnRNP A1 motifs in ISS-N1 poorly match the hnRNP A1 consensus motif, it is likely that they both are needed to achieve efficient binding. Our results are consistent with results from Hua et al. 2008 and with the fact that the two RRM domains of hnRNP A1 have been demonstrated to bind each of the two motifs in ISS-N1 (Beusch et al., 2017). We next designed a set of RNA oligonucleotides in which the proposed SRSF10 SELEX sequence (AAAGACAA) (Shin & Manley, 2002; Xiao et al., 2016) is substituted into the motif 2 region. These RNA oligonucleotides showed binding of both isoforms of SRSF10 at a level comparable to the wt ISS-N1 despite the fact that, the wt ISS-N1 silencer has two mismatches to the published SELEX sequence (Figure 1B). Unlike hnRNP A1 binding, SRSF10 binding was not affected by disruption of motif 1, when motif 2 was substituted with the SRSF10 SELEX sequence, indicating that SRSF10 binds to the region overlapping motif 2. This result also demonstrates that SRSF10 binding is not mediated by hnRNP A1 binding to ISS-N1. Furthermore, substituting motif 2 in the wt ISS-N1 with the SRSF10 SELEX sequence did not affect hnRNPA1 binding, probably due to the fact that this motif retains an AG dinucleotide important for hnRNPA1 binding. Finally, the fact that substitution of motif 2 with the consensus hnRNP A1 motif caused a dramatic decrease in SRSF10 binding, also supports the conclusion that SRSF10 binding is not mediated through protein-protein interactions between hnRNP A1 and SRSF10. Taken together, these data indicate that SRSF10 binds in the distal part of the ISS-N1 motif 2, and that binding to this region may compete with hnRNP A1 binding. To obtain quantitative measurements of recombinant hnRNPA1 and SRSF10 protein binding, we performed Surface Plasmon Resonance imaging (SPRi) analysis with recombinant hnRNP A1 and recombinant SRSF10 protein (either the long or the short isoform) to the SMN oligo sequences used for iTRAQ and pulldown, using the IBIS MX 96 (Figure S1A, S1B and S1C). Consistent with the western blot analysis, we observed strong binding of hnRNP A1 to the SMN-ISS-N1 wt oligonucleotides compared to the mutated sequences. For the SRSF10 long isoform, strong binding was observed with SMN-ISS-N1 wt and with oligonucleotides containing the SELEX motif. The short SRSF10 isoform had the highest binding to the SRSF10 SELEX motif. As motif 2 and the presumed binding site for SRSF10 are located near the biotin-labeled end, we designed longer RNA oligonucleotides to test whether protein binding was affected by its position near the biotin-labeled end (Figure S2A). There was no significant difference in the pattern between the longer or shorter version of the two RNA oligonucleotides (Figure S2A and S2B). We also performed western blot analyses to validate the binding of other splicing factors identified by LC-MSMS to the ISS-N1 sequence (Figure S2C). Consistent with the MS/MS data, more SRSF3 bound to the ISS-N1 wt sequence compared to the SMN-ISS-N1 mutant sequence and to the sequence substituted with the SRSF10 SELEX motif. As expected, the hnRNP A2/B1 binding observed on the western blot resembled that observed for hnRNP A1.
The long isoform of SRSF10 increases skipping of exon 7
We next investigated the role of the long and short isoforms of SRSF10 in SMN1 and SMN2 splicing by overexpressing the two isoforms in HeLa cells together with SMN1 and SMN2 minigenes (Figure 2A). Interestingly, overexpression of the long isoform of SRSF10 caused a strong reduction of the inclusion of exon 7 from the SMN2 minigene, whereas overexpression of the short isoform of SRSF10 only had a very modest effect. As expected, we observed a significant decrease in the inclusion of exon 7 upon overexpression of hnRNP A1 (Figure 2B). Overexpression of hnRNP A1 caused significant exon 7 skipping from the SMN1 minigene, whereas overexpression of the long SRSF10 isoform had only a minor effect, and the short isoform no effect, on the SMN1 minigene. The more pronounced effect of hnRNP A1 could be due to competition with SRSF1 binding to the crucial splicing enhancer element within exon 7 in SMN1 (Cartegni & Krainer, 2002), whereas the SRSF10 isoforms does not have the same competition. We next investigated SMN minigenes with the two A>C mutations (A12C and A23C), which disrupt binding of hnRNP A1 and SRSF10 to ISS-N1. For the SMN1 ISS-mutant minigene, only low levels of exon 7 skipping were observed when overexpressing either the long isoform of SRSF10 or hnRNP A1, supporting the notion that disruption of hnRNP A1 and SRSF10 binding to ISS-N1 improves inclusion of exon 7 (Figure 2C). In the SMN2 ISS-mutant context, we observed that the long isoform of SRSF10 increased skipping of exon 7, indicating that overexpressed SRSF10 still inhibits splicing when the ISS-N1 motif is altered by the two A>C mutations (Figure 2D), despite the fact that binding to the mutated ISS-N1 motif is dramatically reduced in pull down experiments (Figure 1B). This could be explained by the fact that binding of hnRNP A1 to mutant ISS-N1 is completely abolished (Figure 1B), so the competition for binding is decreased, thereby allowing increased SRSF10 binding when it is overexpressed. Overexpression of hnRNP A1 with the SMN2 ISS-mutant minigenes also decreased the inclusion of exon 7, which can be explained by the fact that hnRNP A1 binds to several regulatory elements in the SMN2 gene (Bruun et al., 2016; Doktor et al., 2011), so the effect observed could well be a result of hnRNP A1 binding to other regulatory elements. When we used SMN minigenes with the proposed SRSF10 SELEX-based consensus motif inserted in ISS-N1 (Figure 2E and 2F), overexpression of both SRSF10 isoforms and hnRNP A1 showed the same tendency as with the normal SMN1 and SMN2 minigenes, indicating that the proposed SELEX motif (AAAGACAA) for SRSF10 and the wt ISS-N1 motif works equally well in binding SRSF10 and hnRNP A1, consistent with the pull-down experiments (Figure 1B). Finally, we substituted the hnRNP A1 consensus sequence into motif 2 of the ISS-N1 silencer and observed that overexpression of the long SRSF10 isoform still caused increased exon 7 skipping (Figure 2G and 2H). Because we observed only modest binding of SRSF10 to the RNA oligonucleotides in which motif 2 was substituted with the hnRNP A1 consensus motif, both by western blotting (Figure 1B) and SPRi (Figure S1B, we suspect that the effect observed by overexpressing the long isoform of SRSF10 could reflect binding to other motifs in SMN2. As expected, overexpression of hnRNP A1 caused almost complete skipping of exon 7 from the minigenes with the hnRNP A1 consensus motif (Figure 2G and 2H).
Figure 2. Splicing analysis of minigenes by overexpression of splicing factors in HeLa cells.
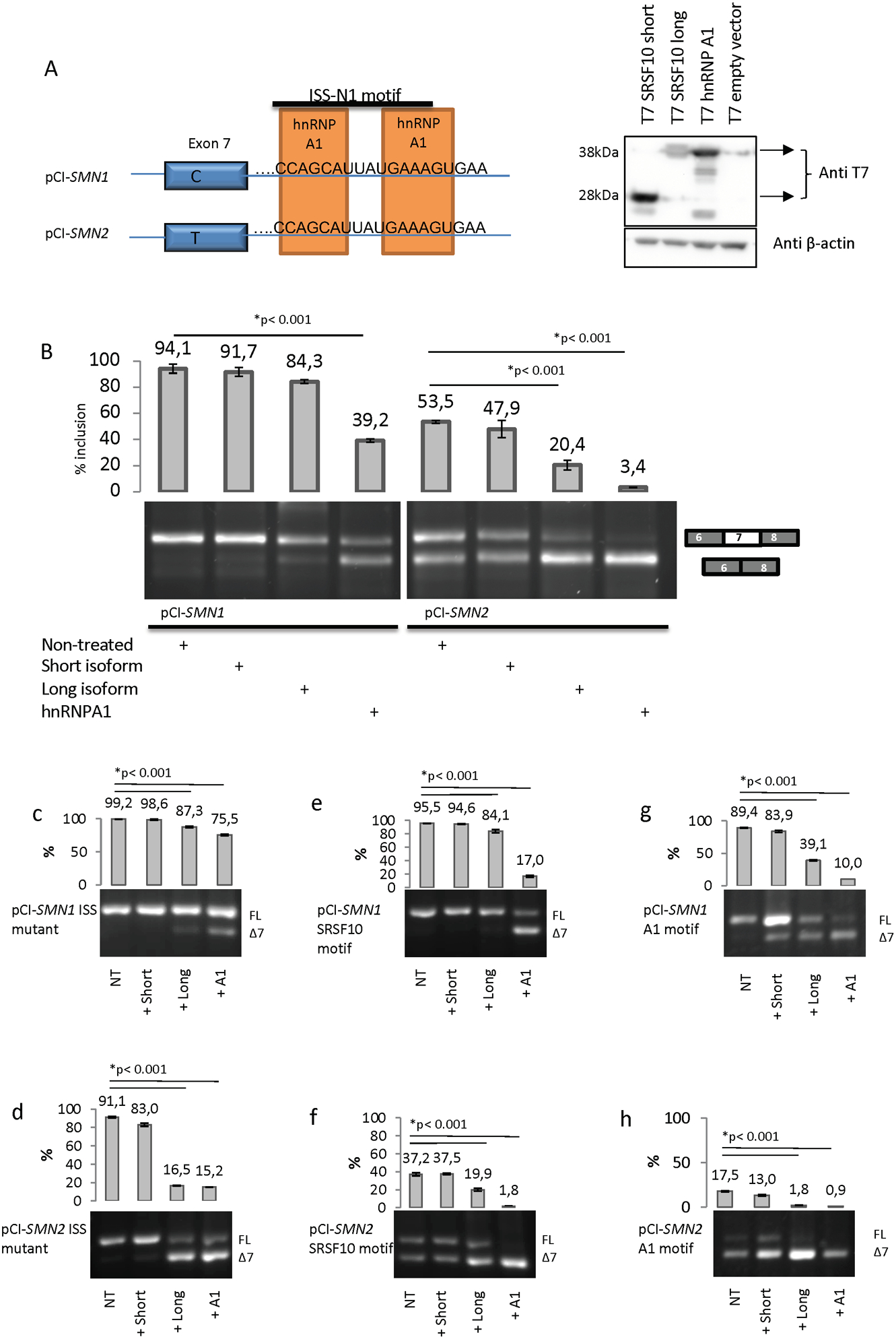
(A) Graphic overview of the pCI-SMN minigenes containing the ISS-N1 element, and validation of overexpression of the SRSF10 T7 expression vectors. (B) Overexpression of both short and long SRSF10 and hnRNP A1, together with pCI-SMN plasmids. The result is visualized on an agarose gel. The top band represents inclusion and the lower band represents skipping of exon 7. The inclusion of exon 7 was quantified using a Fragment Analyzer instrument. (*) indicates p<0.001 in a T test, compared to wt plasmid. Error bars are displayed as STD. N>3. The wt SMN1 and SMN2 minigenes were used to introduce mutations in the ISS-N1 element to investigate the function of the proposed binding motifs in ISS-N1. Six minigenes were made by mutagenesis to create SMN1 and SMN2 minigenes with a disrupted ISS-N1 element (C) and (D), SMN1 and SMN2 minigenes containing the SRSF10 SELEX motif (E) and (F) and SMN1 and SMN2 minigenes containing the consensus hnRNP A1 motif (G) and (H). HeLa cells were transfected with each minigene and the specific expression vector. After 48 h, the RNA was harvested and used for RT-PCR. The PCR products are visualized on an agarose gel. The top band represents the inclusion and the lower band represents the skipping of exon 7. The inclusion of exon 7 was quantified using a Fragment Analyzer instrument. Error bars are displayed as SD/sqrt(n). N>3. (*) indicates p<0.001 in a t test compared to wt.
SRSF10 affects endogenous SMN splicing
We next investigated the effect of the two SRSF10 isoforms on endogenous SMN splicing, using endoSMN PCR followed by digestion with DdeI to distinguish mRNA originating from either SMN1 or SMN2. DdeI only cuts SMN2 derived transcripts due to a SMN2 specific G>A variation (Gennarelli et al., 1995; Parsons et al., 1996). This analysis showed that both the long isoform of SRSF10 and hnRNP A1 mediate increased exon 7 skipping from endogenous SMN2 and SMN1 (Figure 3A and 3B), consistent with the effects observed when employing minigenes. Moreover, overexpression of the short SRSF10 isoform also increased skipping of exon 7 in the SMN2 context. These data demonstrate that both isoforms can repress exon 7 inclusion, but the long SRSF10 isoform is a more efficient repressor, as it is effective in both the SMN1 and SMN2 contexts.
Figure 3. Splicing analysis of overexpression of splicing factors in HeLa cells.
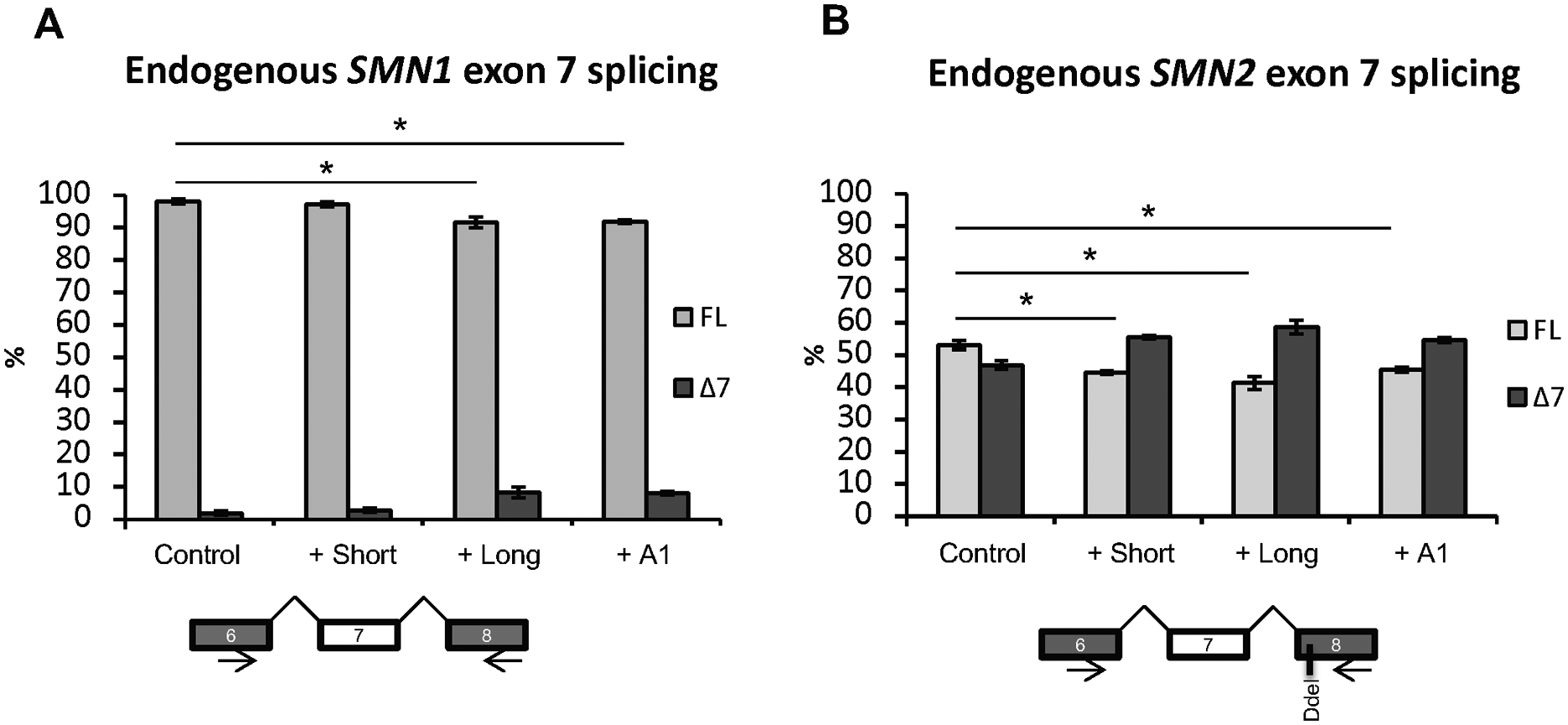
HeLa cells were transfected with expression vectors for SRSF10 long and short isoform or hnRNP A1. After 48 h the RNA was harvested and used in RT-PCR with primers located in exons 6 and 8. The PCR products were digested with DdeI at 37ᵒC to distinguish between SMN1 (A) and SMN2 (B) and quantified on an Agilent 2100 Bioanalyzer instrument. N=6 and (*) indicates statistical significance by T test, p<0.05. Error bars are displayed as Std/sqrt(n).
Investigation of SRSF10 binding using MS2 coat protein tethered binding assay
To further test the effect of the long and short isoforms of SRSF10, we performed a MS2 coat protein tethered binding assay employing SMN minigenes in which the ISS-N1 silencer was replaced by the MS2 hairpin sequence (Figure 4A). Overexpression of the SMN1-MS2 minigene with the MS2-tagged hnRNP A1 protein showed complete exon 7 skipping, whereas overexpression with the MS2-tagged long isoform of SRSF10 showed a very modest decrease in exon 7 inclusion (Figure 4B). For the SMN2-MS2 minigene, both MS2-tagged hnRNP A1 and the MS2-tagged SRSF10 isoforms showed severely decreased exon 7 inclusion, whereas the effect of the MS2-tagged short isoform of SRSF10 was slightly weaker than that of the MS2-tagged long isoform (Figure 4B).
Figure 4. Tethered MS2 coat protein binding assay.
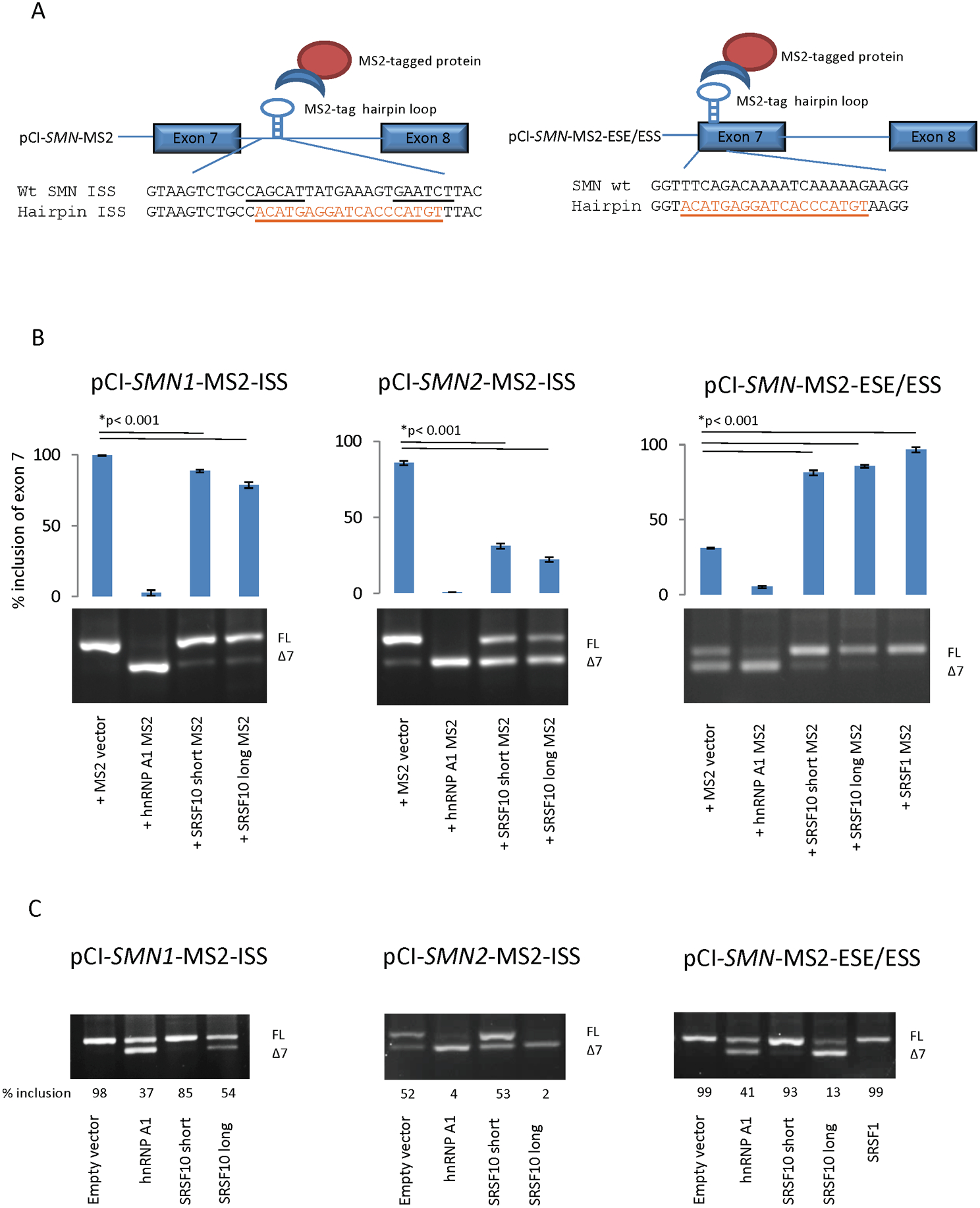
(A) Schematic representation of the MS2 coat protein system and sequences of the WT and MS2 hairpin inserted minigenes. The underlined motifs are the two hnRNP A1 motifs in the ISS. The underlined orange sequence is the MS2 hairpin. (B) Transfection of wt and MS2 minigenes with overexpression of MS2-tagged splicing factors. (C) Transfection of wt and MS2 minigenes with overexpression of untagged splicing factors. Hela cells were co-transfected for 48 h with either the pCI-SMN1 ISS MS2 minigene, the pCI-SMN2 ISS MS2 minigene or the pCI-SMN1-MS2-ESE/ESS minigene, together with MS2-tagged expression vectors for splicing-regulatory factors or untagged splicing-regulatory factors. The RNA was used for RT-PCR with plasmid-specific SMN primers and the products were separated by agarose gel electrophoresis. The inclusion of exon 7 was quantified using a Fragment analyzer instrument. Errors bars are displayed as SD/sqrt(n). N>3. (*) indicates p<0.001 in a t test compared to wt.
Because we noticed that the proposed SRSF10 SELEX motif (AAAAGACAAA) resembles the sequence of the SMN1 SRSF1-binding +6 ESE motif in exon 7 (TTTCAGACAAA), we tested whether SRSF10 could bind to this important regulatory region. In order to evaluate the effect of binding of the two SRSF10 isoforms, as well as SRSF1 and hnRNP A1, to the region of exon 7 harboring the +6 ESE in SMN1 and the +6 ESS in SMN2 we designed a SMN-ESE/ESS-MS2 minigene, in which the +6 ESE/ESS region of exon 7 was substituted with the MS2 hairpin sequence. Overexpression of MS2-tagged hnRNP A1 leads to complete exon 7 skipping, whereas overexpression of MS2-tagged SRSF1 caused complete inclusion of exon 7 (Figure 4B). This clearly corroborates the antagonistic effects of the two proteins (Mayeda, Helfman, & Krainer, 1993; Pollard, Krainer, Robson, & Europe-Finner, 2002), and it is consistent with previous studies showing that binding of SRSF1 to the +6 ESE stimulates exon 7 inclusion in SMN1 (Cartegni & Krainer, 2002) and that the +6 C>T variation in SMN2 disrupts the ESE and creates an ESS to which hnRNP A1 binds to repress SMN2 exon 7 inclusion (Cartegni, Hastings, Calarco, de Stanchina, & Krainer, 2006; Kashima & Manley, 2003). Overexpression of MS2 tag alone also caused some exon 7 skipping from the SMN-ESE/ESS-MS2 minigene, indicating that binding of even a small protein without functional domains at this position in exon 7 is disruptive. Interestingly, both MS2-tagged SRSF10 long and MS2-tagged SRSF10 short isoform increased inclusion, when bound at the +6 ESE/ESS region of exon 7, supporting the notion that the effect of SRSF10 binding is highly position-dependent (Wei et al., 2015). Quantitative LC-MSMS analysis of oligonucleotides harboring the +6 ESE/ESS region of exon 7 (Figure S3A–B) demonstrated that SRSF10 does not bind differential to the SMN1 and the SMN2 sequence, whereas, SRSF1 binds more to the ESE in exon 7 in SMN1 than to the disrupted ESE/ESS present in SMN2, in agreement with previous data (Cartegni & Krainer, 2002). We also observed more binding to the disrupted ESE region in SMN2 from members of the hnRNP A/B family, which is also consistent with previous studies, supporting the notion that the +6 ESE in SMN1 is converted to an ESS in SMN2 by the +6 C>T variation (Kashima & Manley, 2003). Additionally, we investigated the MS2-hairpin-containing minigenes with overexpression of the untagged proteins (Figure 4C). Interestingly, the long isoform of SRSF10 repressed exon 7 inclusion more strongly than hnRNP A1, when the +6 ESE/ESS sequence was replaced by the MS2-sequence, whereas repression of exon 7 inclusion by the long isoform of SRSF10 was weaker when the ISS-N1 sequence was replaced by the MS2 sequence (Figure 4C). This contrasts with the hnRNP A1-mediated repression of exon 7 inclusion, which was at comparable levels when the +6 ESE/ESS sequence or the ISS-N1 was replaced by the MS2-sequence.
Taken together, our data suggest that the inhibitory effect of hnRNP A1 is not only dependent on binding to ISS-N1, but is instead mediated by binding to multiple equally important sites, including ISS-N1. This is consistent with a proposed model according to which exon 7 repression is mediated by spreading of hnRNP A1 across multiple binding sites (Okunola & Krainer, 2009). In contrast, our data indicate that the repressive effect of SRSF10 on exon 7 inclusion is mediated to a large extent by binding to ISS-N1 (Figure 4C) and that binding to other, possibly intronic sites, like the ISS present at the +100 position in intron 8 of SMN2 (Kashima, Rao, & Manley, 2007), could also be important for SRSF10-mediated splicing repression. To validate this, we performed SPRi with oligonucleotides containing the SMN1 or SMN2 +100 sequence, with recombinant hnRNPA1, long SRSF10 and short SRSF10 protein (Figure S4). Both hnRNPA1 and SRSF10 bound to the +100 ISS of both SMN1 and SMN2.
Splice-switching oligonucleotides (SSOs) shift the ratio of the two SRSF10 isoforms by blocking and redirecting splicing
To investigate the effect of the two SRSF10 isoforms without using overexpression, we designed isoform-specific SSOs (Figure 5A). Transfection of HeLa cells with the anti-long SSO (Long→Short) showed a clear shift towards reduced amounts of the long SRSF10 isoform and increased amounts of the short SRSF10 isoform, whereas a reversed splicing pattern (i.e., reduced short SRSF10 isoform and increased long SRSF10 isoform) was observed from cells transfected with the anti-short SSO (S→L) (Figure 5B and 5C). Analysis of the expression of the long and short transcripts using qPCR confirmed that the SSOs do not merely block splicing of one isoform, but that splicing is redirected, resulting in elevated expression of the other isoform (Figure 5D). We went on to test if the shift of isoforms could affect endogenous SMN1 and SMN2 splicing. When cells were treated with the anti-short SSO (i.e., increased levels of long SRSF10 isoform and reduced levels of short SRSF10 isoform) we observed a significant increase in endogenous SMN2 exon 7 skipping and in SMN1 exon 7 skipping (Figure 5E–F). In contrast we did not observe a change in the splicing of endogenous SMN2 upon anti-long SSO treatment, meaning that when the short isoform predominates, no significant effect on splicing ensures.
Figure 5. Shifting the balance of SRSF10 transcripts by transfecting either anti-short (S→L) and anti-long (L→S) SSÓs into HeLa cells.
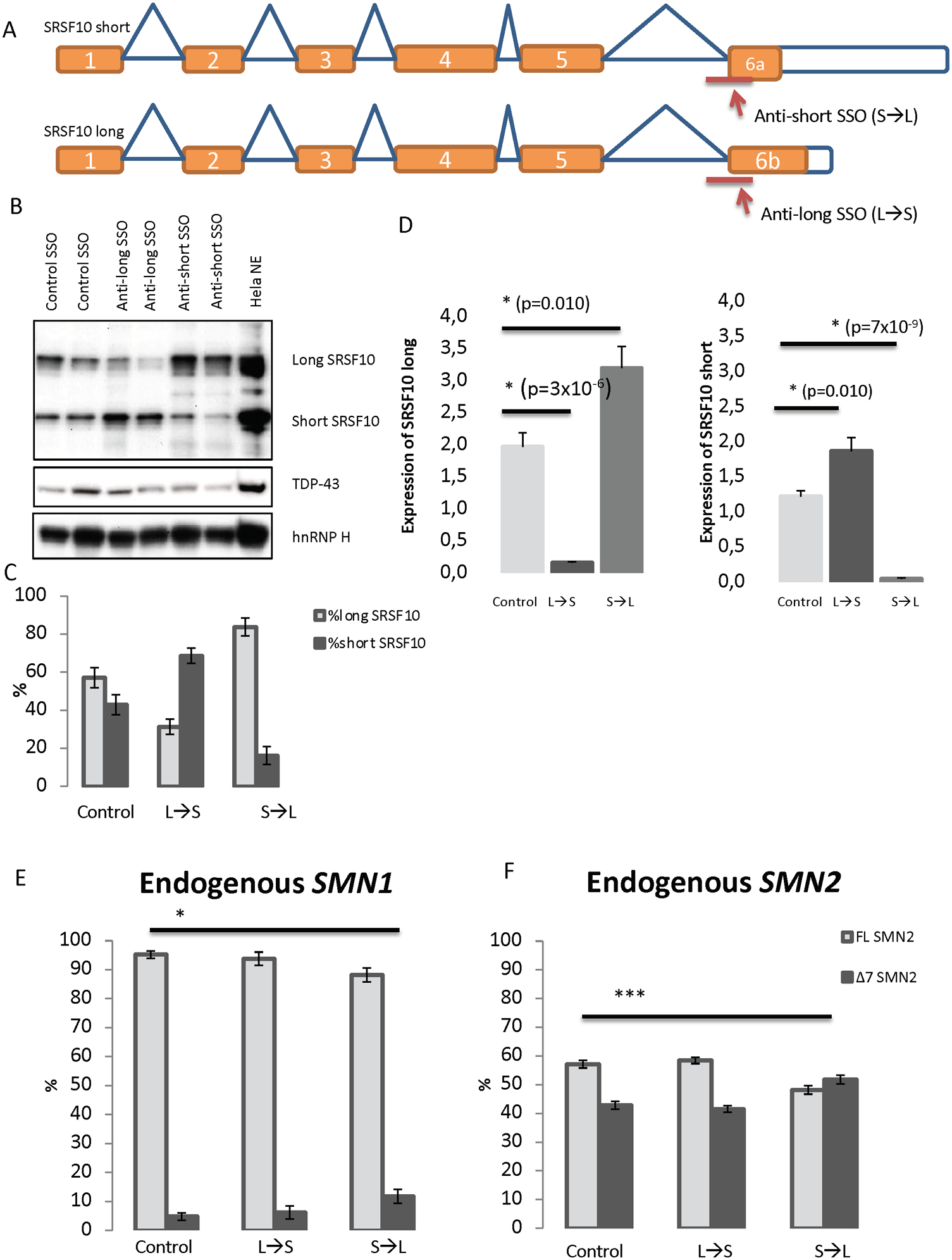
(A) SSOs were designed to target the 3śplice site of either exon 6a or exon 6b. This should alter the splicing of SRSF10 transcripts to either produce the long isoform (when treated with the anti-short SSO) or produce the short isoform (when treated with the anti-long SSO). (B) Validation of the shifted splicing pattern by Western blot analysis using antibody against SRSF10. Antibodies against TDP-43 and hnRNP H were used for controls. HeLa nuclear extract was included as a positive control. The blot displayed is representative of all replicates. (C) Densitometry quantification using ImageJ of replicates of Western blots. N=4. Error bars are displayed as SD/sqrt(n). (D) QPCR analysis with specific primers to detect the expression of SRSF10 long and SRSF10 short in SSO-treated samples. Expression was normalized to TBP. Errors bars are displayed as SD/sqrt(n). N>6. (E-F) HeLa cells were reverse-transfected with 40 nM anti-short, anti-long or control SSO for 48 h. The RNA was isolated and used for cDNA synthesis. PCR with primers in exons 6 and 8 of SMN was used. The PCR products were digested with DdeI to distinguish between PCR product originated from either SMN1 or SMN2. The digested products were quantified with an Agilent 2100 Bioanalyzer instrument. (*) indicates p<0.001 when performing a two-tailed Student T test. Error bars are displayed as STD. N=6.
These data show that the effect of SRSF10-specific SSOs can be observed on endogenous SMN, and that changing the ratio of the two SRSF10 isoforms, without increasing the overall SRSF10 expression, is sufficient to affect the SMN1/2 exon 7 splicing outcome.
MATERIALS AND METHODS
Affinity purification pulldown assay
The RNA affinity purification was performed as previously described (Nielsen et al., 2007). The following 3´-biotin labeled RNA oligonucleotides were used: SMNiss wt (5ĆCAGCAUUAUGAAAGUGAAX3´), SMNiss mutant (5ĆCCGCAUUAUGAACGUGAAX3´), SMNiss mutant Long (5ĆCCGCAUUAUGAACGUGAAUCUUX´3), SMNiss SRSF10 (5ĆCAGCAUUAUGAAAGACAAX´3), SMNiss SRSF10 Long (5ĆCAGCAUUAUGAAAGACAAUCUÚ3), SMNiss mutant SRSF10 (5´-CCCGCAUUAUGAAAGACAAX´3), SMNiss wt A1 (5´-CCAGCAUUAUAGGGUUGAAX´3), SMNiss mutant A1 (5´-CCCGCAUUAUAGGGUUGAAX´3), SMN1 (5´GGUUUCAGACAAAAUCAAX´3), SMN2 (5´GGUUUUAGACAAAAUCAAX´3) (DNA Technology, Denmark). 100 pmol RNA oligonucleotides were mixed with streptavidin-coupled magnetic beads (Invitrogen Co., Carlsbad, CA, US) and incubated with HeLa nuclear extract (Cilbiotech, S.A., Belgium). Eluted proteins were separated on a 4–12% Bis-Tris gel (Invitrogen) and the proteins were transferred on a PVDF membrane and probed with the following antibodies: anti-hnRNP A1 (sc-10029), anti-hnRNP K (sc-28380), anti-hnRNP H (sc-10042) and anti-hnRNP A2/B1 (sc-53531) all from Santa-cruz Biotechnology Inc., anti-SRSF10 (FUSIP1ab77209, Abcam), anti-SR-proteins (sc-53531, Invitrogen), anti-beta-actin (ab8229, Abcam), anti-HPRT (HPA006360, sigma - Aldrich), anti-TDP-43 (10782–2-AP, Proteintech group Inc.) and Anti-T7 (CSHL antibody facility #42 1–87).
Surface plasmon resonance imaging (SPRi)
Surface plasmon resonance imaging (SPRi) by IBIS MX-96 was used to measure the kinetics of recombinant hnRNP A1 (ab224866, Abcam), recombinant SRSF10 long (cat#OPCA02208, aviva system biology) and recombinant SRSF10 short (cat#TP321759, origene)binding to the immobilized RNA oligonucleotides also used for affinity purification pulldown assays, as well as +100 3’biotinylated oligonucleotides: SMN2+100G (5’-AUGUUAGAAAGUUGAAAGGUUAA’3) and SMN1+100A (5’-AUGUUAAAAAGUUGAAAGGUUAA’3). The oligonucleotides were immobilized on a Senseye G strep (SSENS) sensor chip in a 2×4×12 array by continuous flow in a CFM 2.0 printer (Wasatch microfluidics). The oligonucleotides were diluted in 1XTBS to a concentration of 1 μM and spotted for 20 min followed by 5-minute washing with TBS + 0.05% Tween-20. The sensor chip was inserted into the MX96 instrument (IBIS technologies A/S) and primed with SPR buffer (10 mM Hepes/KOH pH 7.9, 150 mM KCl, 10 mM MgCl2 and 0.075% Tween-80). Real-time binding was measured by following changes of the SPR angles at all printed positions of the array during 10-minute injections of hnRNP A1 protein over the entire surface, followed by 10-minute dissociation at 4μl/sec. Six injections of a 2-fold titration series from 6.25 to 200 nM hnRNP A1 were carried out (lowest to highest). Regeneration after each injection was carried out with 25 mM H3PO4, to remove any remaining protein before the next injection. Residual background binding was blocked by injecting 1 mg/ml BSA in SPR buffer onto the chip. To measure baseline and dissociation kinetics a continuous flow of SPR buffer flowed over the surface before, between and after the hnRNP A1 injections. Dissociation was measured for 5 min, by injecting SPR buffer over the chip at a rate of 8 μl/sec. Responses for a calibration curve were created after the concentration series by measuring SPR responses from defined dilutions of glycerol in running buffer (ranging from 5 to 0 % glycerol) and of pure water, as defined by the automated calibration routine of IBIS MX-96.
For data analysis, the SPRi data were imported into SPRINTX software (v. 2.1.1.0, IBIS technologies). The data were calibrated, reference subtracted, and the baselines of the responses before all hnRNP A1 injections were zeroed. The starting time point was aligned at the beginning of each new injection. Using Scrubber 2 (v. 2.1.1.0, Biologics Inc.) binding curves for all positions were fitted globally to the integrated rate equation that describes simple first order 1:1 binding kinetics to obtain kinetic association rate (ka), dissociation rate (kd) and equilibrium dissociation (KD = kd/ka) constants. To obtain a bimodal binding for 1:2 kinetics, the simulations were calculated using ClampXP (version 3.50, Biosensor Data Analysis) and using ggplot2 for plotting the resulting binding curves.
RNA affinity pulldown and iTRAQ labeling
The following 3´-biotin labeled RNA oligonucleotides were used: SMNiss wt (5´-CCAGCAUUAUGAAAGUGAAX3´), SMNiss mutant (5´-CCCGCAUUAUGAACGUGAAX3´), SMN1ese (5´GGUUUCAGACAAAAUCAAX3´), SMN2ese (5´-GGUUUUAGACAAAAUCAAX3´). An additional wash with 1xbinding bufferΔglycinΔKCL was performed to make the samples compatible with iTRAQ labeling. The samples for each run were mixed and an aliquot of 4μl was withdrawn, desalted on a R3 column and dried down before analysis by matrix-assisted laser desorption/ionization (MALDI) MS to check the labeling efficiency. The remaining sample was lyophilized and re-dissolved in 50 μl 0.1% trifluoroacetic acid (TFA) before being desalted on a C18 R3 microcolumn. The sample was eluted with 40 μl 60% acetonitrile/0.1% TFA and lyophilized prior to LC-MS/MS analysis.
Nano-liquid chromatography tandem mass spectrometry
The iTRAQ labelled sample was applied onto an EASY nano-LC system (Proxeon Biosystems, Odense, Denmark). The peptides were loaded directly onto a 20-cm 100-μm inner diameter, 360-μm outer diameter, ReproSil – Pur C18 AQ 3-μm (Dr. Maisch, Germany) homemade reversed phase capillary column. The peptides were eluted from the column into an LTQ-orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), using a gradient from 100% phase A (0.1 % formic acid) to 34 % phase B (0.1 % formic acid, 90%acetonitrile) in 120 min. at 250 nl min−1. The data-dependent analysis was performed using one MS full scan in the area 400 Da – 1800 Da performed in the Orbitrap with 30000 in resolution, followed by CID and HCD of the three most intense ions (Rewitz et al., 2009) The threshold for ion-selection was 10000 and the conditions for CID fragmentation were normalized collision energy 35, isolation width 2.0, activation time 30 ms, and for HCD fragmentation normalized collision energy 50, isolation width 2.0, activation time 1 ms.
Data processing – protein identification and quantitation
Raw files were analyzed using Proteome Discoverer v1.3 beta (Thermo Fisher Scientific, Bremen, Germany). MS/MS spectra were converted to .mgf files and searched against the Swissprot Human database using Mascot v2.3.02. The database search was performed with the following fixed parameters: precursor mass tolerance 8 ppm; MS/MS mass tolerance 0.8 Da (CID data) or 0.05 Da (HCD data); 2 possible missed cleavages, cysteine carbamidomethylation and full trypsin cleavage. Variable modifications included: N-terminal and lysine iTRAQ. iTRAQ quantification was performed using reporter ion area integration within a 50 ppm window. Ratios were normalized against the median peptide ratio.
Expression vector construct
All constructs were made using the pCI expression vector or the pCI SMN1 and pCI SMN2 wt minigenes. The pCI SMN mutant minigenes, the pCI SMN A1 motif minigenes and the pCI SMN SRSF10 motif minigenes were provided by GeneScript (GenScript, Piscataway, NJ, USA). The pCI vector was digested using Xbal and BamHI (New England Biolabs. Inc.) SRSF10 inserts were made using primer: SRSF10-F-NheI 5´-GGTGCTAGCATGTCCCGCTACCTGCGTCCC-3´, SRSF10v1-R-BamHI 5´- AGTGGATCCTCAGATCTTTCTTGAAGTGTAG-3´ and SRSF10v2-R-BamHI 5´- AGTGGATCCTCAGTGGCCACTGGACTTAGGACTAG-3´. All plasmids were sequenced to validate correct insertion.
Overexpression in HeLa cells
All transfections and co-transfections were performed using Xtremegene-9 (Roche) and HeLa cells were seeded in RPMI 1640+++10% (Lonza) (pen/strep, fetal calf serum and L-glutamine added) so they would be at 80–90% confluency on the day of transfection. RNA extraction was performed 48 h after transfection using Isol RNA lysis reagent (AH diagnostics) and cDNA synthesis was performed from 1 ug RNA using a High Capacity cDNA Reverse Transcription Kit (Invitrogen). Splicing products were analyzed using vector-specific pCIFwdB 5′-GACTCACTATAGGCTAGCCTCG-3′ and SMNtestex8as 5´-GTGGTGTCATTTAGTGCTGCTC-3´. The PCR was performed in 25 cycles (95°C in 15 min, 95°C in 30 sec, 54°C in 30 sec, 72°C in 30 sec, 72°C in 5 min) using TEMPase Hot start 2x master Mix C (Ampliqon). Endogenous SMN was amplified using SMN.endo.ex6s 5´-ATATGTCCAGATTCTCTTGATG-3´ and SMN.endo.ex8as 5´-ACCATTTGAAACATTTTAAGAC-3´. PCR product was digested with Ddel (R0175S, New England Biolabs) to distinguish their origin (SMN1 or SMN2). All PCR products were either quantified by Agilent 2100 Bioanalyzer or Advanced Analytical Technologies Inc., Fragment analyzer™.
SSO treatment of HeLa cells
Reverse transfection with SSÓs was performed using Lipofectamine RNAiMAX transfection reagent (Life Technologies) and 40 nM of each SSO. For co-transfection, cells were transfected with plasmids 24 h before using a forward transfection with SSOs and RNAiMAX. SSOs were phosphorothioate oligonucleotides with 2’-O-methyl modification on each sugar moiety (LGC Biosearch Technologies). The following SSOs were used: Control SSO: 5’GCUCAAUAUGCUACUGCCAUGCUUG3’, Anti-long SSO: 5ÚUUCGGUGUUUGAAUCUUUCAAAAC3´ and Anti-short: 5ÚCCAGCUGCAGUUUGGUCUUAAAUA-3´. After 48 h, protein and RNA were harvested for further analysis. Total protein was extracted using a 1 mM PMSF, 1xComplete ™ protease inhibitor (Roche) and M-PER lysis buffer (Thermo Fisher Scientific Inc.). After treatment with Benzonase (Merck Millipore, Darmstadt, Germany) proteins were loaded on a 4–12% Bis-Tris gel (Invitrogen) SDS-PAGE and Western blotted together with a positive control sample containing HeLa nuclear extract (Cilbiotech, S.A., Belgium). The following antibodies were used: anti-SRSF10 (FUSIP1ab77209, Abcam), anti-TDP-43 (10782–2-AP, Proteintech group Inc.) and anti-hnRNP H (sc-10042, Santa Cruz). RNA was used for cDNA synthesis and endogenous SMN was amplified and subsequently digested with DdeI as described above.
qPCR
1.0 μg of total RNA was reverse transcribed into cDNA using the Superscript VILO cDNA kit (Invitrogen, cat. 11754–050). For SRSF10 expression analysis in tissues, the Human Total RNA Master Panel II (Cat#636742/Lot#1202889A) was used. cDNA synthesis was performed according to manufactureŕs instructions, but with an extended synthesis at 42°C for 120 min. Subsequently, the cDNA was diluted 50-fold with MiliQ-water and cDNA concentration was measured by absorbance readings at 260, 280 and 230 nm (NanoDrop™1000 Spectrophotometer; Thermo Scientific, California, USA). Each cDNA (50 ng) was used in duplicate as template in a reaction volume of 8 μl containing 3.33 μl Fast Start Essential DNA Green Master (2x) (Roche Diagnostics, Hvidovre, Denmark), 0.33 μl primer premix (containing 10 pmol of each primer), and PCR-grade water. The qPCR was performed in a Light Cycler LC480 (Roche Diagnostics, Hvidovre, Denmark): 1 cycle at 95 °C/5 min followed by 45 cycles at 95 °C/10 sec, 57 to 65°C (primer-dependent)/10 sec, 72 °C/10 sec. Primers used for qPCR were: SRSF10ex4F 5ĆAGACGTTCTAGAAGCCG´3, SRSF10ex6R 5´GAGATGCAGACCTAGAACG´3, SRSF10ex7R 5´ GAAGTGTAGTAAGCAGAACTG´3, TBP-F 5´GTGACCCAGCATCACTGTTTĆ3, TBP-R 5´ GCAAACCAGAAACCCTTGCG´3. Threshold values were determined with Light Cycler software (LCS480 1.5.1.62 SP1) using Absolute Quantification Analysis/2nd derivative maximum. Each qPCR assay included: a standard curve of 7 serial dilution (2-fold) points of a cDNA mix of all the samples (250 to 3.9 ng), and a no-template control. PCR efficiency (=10(−1/slope) −1) were >70% and r2= 0.96 or higher. The specificity of each amplification was determined by melting curve analysis. Quantification cycle (Cq) was determined for each sample and the comparative method was used to detect a relative gene expression ratio (2−Cq) normalized to the reference gene TBP. The reference gene was chosen based on its observed stability across conditions. Significance was ascertained by the two-tailed Student’s t-test.
MS2 coat protein hairpin tethered function assay
To fuse the bacteriophage coat protein MS2 to both SRSF10 long and SRSF10 short, expression vectors were made by inserting the CDS into the pRBG4-MS2 vector using NheI-HF and HindIII restriction enzymes for cloning. The pRBG4-MS2 vector was generously provided by Akio Masuda and Kinji Ohno, Nagoya University.). The new expression vectors were transformed into One Shot™ TOP10 Chemically Competent E. coli (Invitrogen). Correct insertion was confirmed by sequencing of the whole insert. The pcDNA-MS2-SRSF1 (Rahman et al., 2015), pRBG4-MS2-hnRNP A1 and the pcDNA-MS2 (Rahman et al., 2013) were kind gifts from Akio Masuda and Kinji Ohno. The SMN minigenes harboring the MS2 coat protein hairpin sequence 5ÁCATGAGGATCACCCATGT´3 were made by Genscript (GenScript, Piscataway, NJ, USA). Western blots for validation were developed using anti-MS2 (ABE76-I, EMD Millipore).
DISCUSSION
Blocking of the ISS-N1 silencer in SMN2 by Spinraza is successful in treating patients with SMA. Despite this success, a comprehensive identification of the individual inhibitory splicing regulatory factors that bind to ISS-N1 and inhibit splicing has not yet been performed. Because a deeper understanding of SMA molecular pathology will require more comprehensive knowledge about the splicing factors involved in repressing SMN2 exon 7 splicing, we used an unbiased approach to identify factors that bind to ISS-N1 by employing RNA affinity purification and LC-MS/MS analysis. Not surprisingly, we observed significant binding of the ubiquitously expressed splicing repressor hnRNP A1, hnRNP A2/B1 (as well as other members of the hnRNP A family), which were previously demonstrated to bind and inhibit exon 7 splicing (Hua et al., 2008). However, because aberrant SMN2 splicing in spinal cord motor neurons causes the main disease symptoms in SMA, Spinraza is injected into the spinal fluid in SMA patients in order to block splicing repressors from binding to ISS-N1. Therefore, splicing regulatory factors that are highly expressed in neuronal tissues and able to bind to ISS-N1 to repress SMN2 exon 7 inclusion could play an especially important role in SMA pathogenesis. Interestingly, our unbiased pull-down analysis also identified SRSF10 as a new cis-acting factor that interacts with the ISS-N1 silencer, and we validated binding of both isoforms of SRSF10 to the silencer.
SRSF10 is highly expressed in neuronal tissues and was previously reported to be important in the context of cellular stress, the cell cycle, apoptosis, neurogenesis and oncogenesis (Feng et al., 2009; K. J. Liu & Harland, 2005; Qi et al., 2015; Shi, Nishida, Campigli Di Giammartino, & Manley, 2011; Shin, Feng, & Manley, 2004; Shin & Manley, 2002; Shkreta, Toutant, Durand, Manley, & Chabot, 2016; Yang, Chansky, & Hickstein, 2000; Yang et al., 1998). Due to its high expression in neuronal tissues, plus the fact that it can function as a splicing repressor and that its recognition motif resembles the ISS-N1 splicing silencer, we hypothesized that SRSF10 could be important in SMA pathogenesis by repressing SMN2 exon 7 inclusion.
Our results both from analyzing the effect on exon 7 splicing from SMN1/2 minigenes and endogenous SMN1/2 splicing clearly show that SRSF10 can repress splicing of SMN1/2 exon 7 and that this is mediated through binding to the ISS-N1 silencer. Interestingly, we also identified and validated binding of the splicing regulatory factor SRSF3 to the ISS-N1 silencer (Figure 1C and Figure S2C). This is consistent with a recent study that reported similarity between the binding motif of SRSF3 and SRSF10 (Xiao et al., 2016). SRSF3 was previously demonstrated to antagonize the effect of hnRNP A1 on exon 7 inclusion (Wang et al., 2012; Wee, Havens, Jodelka, & Hastings, 2014), indicating complex regulation at the ISS-N1 silencer, where the inhibitory factors hnRNP A1/A2 and SRSF10, and the stimulatory factor SRSF3, all compete for binding.
It is, however, clear from our data that SRSF10-mediated exon 7 repression also involves other splicing regulatory elements, because overexpressed SRSF10 also mediates exon 7 skipping from SMN2 minigenes in which ISS-N1 was mutated to sequences that do not bind SRSF10 in pull-down and SPRi analyses. In this context, it is noteworthy that the splicing silencer present at position +100 in intron 8 of SMN2, in addition to an hnRNP A1 motif (Kashima et al., 2007) also harbors a motif similar to that bound by SRSF10 in ISS-N1. The +100G, which is specific for SMN2, creates a GAAAGT motif identical to the SRSF10-binding motif in ISS-N1. Furthermore, downstream at position +107–112 is a second GAAAGG motif in both SMN1 and SMN2, which explains why we observe binding of SRSF10 to both oligonucleotides (Figure S4).
Recently, Cloutier et al reported that SRSF10, hnRNP A1 and Sam68 co-operate in regulating an alternative splicing response to DNA damage and stress (Cloutier et al., 2018). They suggested a direct protein-protein interaction between SRSF10 and the 14-3-3ε protein, which binds and protects the RS-domain of the long isoform from dephosphorylation and perhaps also mediates protein-protein interaction with hnRNP K in regulation of Bcl-X alternative splicing. This is coordinated with an interaction between hnRNP A1 and Sam68 in a very complex mechanism. Interestingly, Sam68 has also been demonstrated to bind next to the +6 ESS in SMN2 exon 7 to promote recruitment of hnRNP A1 to inhibit exon 7 inclusion (Pagliarini et al., 2015; Pedrotti et al., 2010). Knock down of Sam68 increased exon 7 inclusion and SMN expression in the cortex, cerebellum, spinal cord, and peripheral tissues of SMAΔ7 mice. Based on this, it could be speculated that SRSF10 plays a central role in a complex inhibition of SMN2 exon 7 inclusion, which involves both direct binding to splicing-regulatory elements, such as the ISS-N1 silencer, and protein-protein interactions with hnRNP K and other factors, and that this is co-regulated by hnRNP A1 and Sam68. Because both SRSF10 isoforms bind the ISS-N1 silencer, the difference in their inhibitory effect on SMN1/2 exon 7 splicing might be due to the different lengths of their RS domains. In a more complex scenario, the shorter RS domain of the short SRSF10 isoform might not be able to interact efficiently with the interacting partners (14-3-3ε protein etc.) thereby causing reduced inhibition. On top of this the two SRSF10 isoforms most likely also exhibit functional differences due to different phosphorylation of their RS-domains. This complex regulatory mechanism could be speculated also to be active in the cellular stress occurring in spinal cord in SMA (Doktor et al., 2017). Therefore, we speculate that SRSF10 plays a negative role in SMA pathogenesis.
Although the two isoforms of SRSF10 are present in comparable amounts in many tissues, only a few studies have investigated the effects of the two SRSF10 isoforms separately (Komatsu et al., 1999; Ling & Estus, 2010; Matsushita et al., 2007; Shin & Manley, 2002). According to the Human Protein Atlas database (Figure S3C) SRSF10 protein is highly expressed in several tissues, including high expression of SRSF10 protein in the brain, heart, skeletal muscle and testis in accordance with literature (Thul et al., 2017; Uhlen et al., 2015). Because our results show that the repressive effect of the two SRSF10 isoforms is different, we investigated the relative expression of the two SRSF10 isoforms in a panel of RNA from 20 different human tissues (Figure S3D). This analysis showed that splicing is directed towards the long SRSF10 isoform in brain tissues whereas the short isoform is more predominant in tissues like adrenal gland and liver (Fushimi, Osumi, & Tsukahara, 2005; L. Liu et al., 2003; Qi et al., 2015). This result is consistent with fact that SMN2 exon 7 skipping and consequently disease symptoms are more pronounced in neuronal tissues, where expression of the long isoform of SRSF10 is highest. Based on our data, in particular the observed effect of the splice-switching SSO towards the long isoform, it is clear that the preferential expression of the long SRSF10 isoform in neuronal tissues will result in a more pronounced inhibitory effect on SMN1/2 exon 7 splicing in these tissues.
An earlier study of siRNA-mediated SRSF10 knockdown did not report an effect on SMN exon 7 splicing (Wee et al., 2014), but previous studies did not consider the different effects of the two isoforms. Other studies hypothesized that the long isoform is the “active” isoform and that the short isoform is a “passive and regulatory” isoform, which mainly functions by binding and blocking access for the functional long isoform (Fushimi et al., 2005; Shin, Kleiman, & Manley, 2005). It is likely that for many binding sites, the combined effect of knocking down both isoforms are neutral. Therefore, the repressive effect on SMN1/2 exon 7 splicing that we observe in this study, either by overexpression or using splice-switching SSOs, is an effect of increasing the long SRSF10 isoform, rather than depleting cells of both SRSF10 isoforms.
Our data emphasize the importance of a better understanding of the separate functions of the two SRSF10 isoforms. The repressing role of SRSF10 on SMN2 exon 7 splicing may be critical in SMA patients, as they have low amounts of SMN protein and high levels of neuronal stress. Our data suggest that Spinraza functions not only by blocking hnRNP A1 from binding to the ISS-N1 silencer, but that blocking binding of the long SRSF10 isoform to ISS-N1 may also be important.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mette Larsen and Kasper W. Stonor for technical assistance. We are grateful to Kinji Ohno and Akio Masuda for the generous gift of expression vectors for MS2 tagged hnRNP A1 and SRSF1 fusion proteins.
Funding
This work was supported by a grant from the Lundbeck foundation (R54-2010-5706 to BSA and R231-2016-2823 to TKD), The Danish Medical Research Council (FSS no. 11-107174 to BSA), Natur og Univers, Det Frie Forskningsråd (4181-00515 to BSA) and Muskelsvindfonden (to TKD). Y.H. and A.R.K acknowledge support from NIH grant R37-GM42699. The funders had no role in the study design, collection of data or manuscript preparation. The Villum Center for Bioanalytical Sciences at SDU is acknowledged for access to state-of-the-art mass spectrometric instrumentation.
Footnotes
Publisher's Disclaimer: aThis is the accepted version of the following article: Frederiksen SB, Holm LL, Larsen MR, et al. Identification of SRSF10 as a regulator of SMN2 ISS-N1. Human Mutation. 2020;1–15, which has been published in final form at [https://doi.org/10.1002/humu.24149]. This article may be used for non- commercial purposes in accordance with the Wiley Self-Archiving Policy [http://www.wileyauthors.com/self- archiving].
The authors have no conflict of interest to declare.
REFERENCES
- Beusch I, Barraud P, Moursy A, Clery A, & Allain FH (2017). Tandem hnRNP A1 RNA recognition motifs act in concert to repress the splicing of survival motor neuron exon 7. Elife, 6. doi: 10.7554/eLife.25736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruun GH, Doktor TK, Borch-Jensen J, Masuda A, Krainer AR, Ohno K, & Andresen BS (2016). Global identification of hnRNP A1 binding sites for SSO-based splicing modulation. BMC Biol, 14, 54. doi: 10.1186/s12915-016-0279-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CG, & Dreyfuss G (1994). RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. Embo j, 13(5), 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Hastings ML, Calarco JA, de Stanchina E, & Krainer AR (2006). Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet, 78(1), 63–77. doi: 10.1086/498853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, & Krainer AR (2002). Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet, 30(4), 377–384. doi: 10.1038/ng854 [DOI] [PubMed] [Google Scholar]
- Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, … Bishop KM (2016). Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology, 86(10), 890–897. doi: 10.1212/wnl.0000000000002445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clermont O, Burlet P, Lefebvre S, Burglen L, Munnich A, & Melki J (1995). SMN gene deletions in adult-onset spinal muscular atrophy. Lancet, 346(8991–2), 1712–1713. doi: 10.1016/s0140-6736(95)92881-2 [DOI] [PubMed] [Google Scholar]
- Cloutier A, Shkreta L, Toutant J, Durand M, Thibault P, & Chabot B (2018). hnRNP A1/A2 and Sam68 collaborate with SRSF10 to control the alternative splicing response to oxaliplatin-mediated DNA damage. Sci Rep, 8(1), 2206. doi: 10.1038/s41598-018-20360-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowper AE, Caceres JF, Mayeda A, & Screaton GR (2001). Serine-arginine (SR) protein-like factors that antagonize authentic SR proteins and regulate alternative splicing. J Biol Chem, 276(52), 48908–48914. doi: 10.1074/jbc.M103967200 [DOI] [PubMed] [Google Scholar]
- Doktor TK, Hua Y, Andersen HS, Broner S, Liu YH, Wieckowska A, … Andresen BS (2017). RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res, 45(1), 395–416. doi: 10.1093/nar/gkw731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doktor TK, Schroeder LD, Vested A, Palmfeldt J, Andersen HS, Gregersen N, & Andresen BS (2011). SMN2 Exon 7 Splicing is Inhibited by Binding of hnRNP A1 to a Common ESS Motif That Spans the 3 ‘ Splice Site. Human Mutation, 32(2), 220–230. doi: 10.1002/humu.21419 [DOI] [PubMed] [Google Scholar]
- Feng Y, Chen M, & Manley JL (2008). Phosphorylation switches the general splicing repressor SRp38 to a sequence-specific activator. Nat Struct Mol Biol, 15(10), 1040–1048. doi: 10.1038/nsmb.1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Valley MT, Lazar J, Yang AL, Bronson RT, Firestein S, … Manley JL (2009). SRp38 regulates alternative splicing and is required for Ca(2+) handling in the embryonic heart. Dev Cell, 16(4), 528–538. doi: 10.1016/j.devcel.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushimi K, Osumi N, & Tsukahara T (2005). NSSRs/TASRs/SRp38s function as splicing modulators via binding to pre-mRNAs. Genes Cells, 10(6), 531–541. doi: 10.1111/j.1365-2443.2005.00855.x [DOI] [PubMed] [Google Scholar]
- Gennarelli M, Lucarelli M, Capon F, Pizzuti A, Merlini L, Angelini C, … Dallapiccola B (1995). Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem Biophys Res Commun, 213(1), 342–348. doi: 10.1006/bbrc.1995.2135 [DOI] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Okunola HL, Bennett CF, & Krainer AR (2008). Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet, 82(4), 834–848. doi: 10.1016/j.ajhg.2008.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Philippe J, Paz S, & Caputi M (2013). hnRNP A1: the Swiss army knife of gene expression. Int J Mol Sci, 14(9), 18999–19024. doi: 10.3390/ijms140918999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamma H, Portman DS, & Dreyfuss G (1995). Cell type-specific expression of hnRNP proteins. Exp Cell Res, 221(1), 187–196. doi: 10.1006/excr.1995.1366 [DOI] [PubMed] [Google Scholar]
- Kashima T, & Manley JL (2003). A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet, 34(4), 460–463. doi: 10.1038/ng1207 [DOI] [PubMed] [Google Scholar]
- Kashima T, Rao N, & Manley JL (2007). An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci U S A, 104(9), 3426–3431. doi: 10.1073/pnas.0700343104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kominami E, Arahata K, & Tsukahara T (1999). Cloning and characterization of two neural-salient serine/arginine-rich (NSSR) proteins involved in the regulation of alternative splicing in neurones. Genes Cells, 4(10), 593–606. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, … et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 80(1), 155–165. [DOI] [PubMed] [Google Scholar]
- Ling IF, & Estus S (2010). Role of SFRS13A in low-density lipoprotein receptor splicing. Hum Mutat, 31(6), 702–709. doi: 10.1002/humu.21244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KJ, & Harland RM (2005). Inhibition of neurogenesis by SRp38, a neuroD-regulated RNA-binding protein. Development, 132(7), 1511–1523. doi: 10.1242/dev.01703 [DOI] [PubMed] [Google Scholar]
- Liu L, Lin JJ, Chen X, Liu X, & Xu P (2003). Neural expression and regulation of NSSR1 proteins. Neuroreport, 14(14), 1847–1850. doi: 10.1097/01.wnr.0000091134.75061.e3 [DOI] [PubMed] [Google Scholar]
- Matsushita H, Blackburn ML, Klineberg E, Zielinska-Kwiatkowska A, Bolander ME, Sarkar G, … Yang L (2007). TASR-1 regulates alternative splicing of collagen genes in chondrogenic cells. Biochem Biophys Res Commun, 356(2), 411–417. doi: 10.1016/j.bbrc.2007.02.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeda A, Helfman DM, & Krainer AR (1993). Modulation of exon skipping and inclusion by heterogeneous nuclear ribonucleoprotein A1 and pre-mRNA splicing factor SF2/ASF. Mol Cell Biol, 13(5), 2993–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen KB, Sørensen S, Cartegni L, Corydon TJ, Doktor TK, Schroeder LD, … Gregersen N (2007). Seemingly neutral polymorphic variants may confer immunity to splicing-inactivating mutations: a synonymous SNP in exon 5 of MCAD protects from deleterious mutations in a flanking exonic splicing enhancer. The American Journal of Human Genetics, 80(3), 416–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okunola HL, & Krainer AR (2009). Cooperative-binding and splicing-repressive properties of hnRNP A1. Mol Cell Biol, 29(20), 5620–5631. doi: 10.1128/mcb.01678-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RK, Broner S, Sabaratnam R, Doktor TK, Andersen HS, Bruun GH, … Andresen BS (2014). The ETFDH c.158A>G variation disrupts the balanced interplay of ESE- and ESS-binding proteins thereby causing missplicing and multiple Acyl-CoA dehydrogenation deficiency. Hum Mutat, 35(1), 86–95. doi: 10.1002/humu.22455 [DOI] [PubMed] [Google Scholar]
- Pagliarini V, Pelosi L, Bustamante MB, Nobili A, Berardinelli MG, D’Amelio M, … Sette C (2015). SAM68 is a physiological regulator of SMN2 splicing in spinal muscular atrophy. J Cell Biol, 211(1), 77–90. doi: 10.1083/jcb.201502059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, McAndrew PE, Monani UR, Mendell JR, Burghes AH, & Prior TW (1996). An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: further evidence for SMN as the primary SMA-determining gene. Hum Mol Genet, 5(11), 1727–1732. [DOI] [PubMed] [Google Scholar]
- Pedrotti S, Bielli P, Paronetto MP, Ciccosanti F, Fimia GM, Stamm S, … Sette C (2010). The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. Embo j, 29(7), 1235–1247. doi: 10.1038/emboj.2010.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard AJ, Krainer AR, Robson SC, & Europe-Finner GN (2002). Alternative splicing of the adenylyl cyclase stimulatory G-protein G alpha(s) is regulated by SF2/ASF and heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) and involves the use of an unusual TG 3’-splice Site. J Biol Chem, 277(18), 15241–15251. doi: 10.1074/jbc.M109046200 [DOI] [PubMed] [Google Scholar]
- Qi Y, Li Y, Cui SC, Zhao JJ, Liu XY, Ji CX, … Chen XH (2015). Splicing factor NSSR1 reduces neuronal injury after mouse transient global cerebral ischemia. Glia, 63(5), 826–845. doi: 10.1002/glia.22787 [DOI] [PubMed] [Google Scholar]
- Rahman MA, Azuma Y, Nasrin F, Takeda J, Nazim M, Bin Ahsan K, … Ohno K (2015). SRSF1 and hnRNP H antagonistically regulate splicing of COLQ exon 16 in a congenital myasthenic syndrome. Sci Rep, 5, 13208. doi: 10.1038/srep13208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MA, Masuda A, Ohe K, Ito M, Hutchinson DO, Mayeda A, … Ohno K (2013). HnRNP L and hnRNP LL antagonistically modulate PTB-mediated splicing suppression of CHRNA1 pre-mRNA. Sci Rep, 3, 2931. doi: 10.1038/srep02931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Chan ET, Pena Castillo L, Chaudhry S, Talukder S, … Hughes TR (2009). Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat Biotechnol, 27(7), 667–670. doi: 10.1038/nbt.1550 [DOI] [PubMed] [Google Scholar]
- Rewitz KF, Larsen MR, Lobner-Olesen A, Rybczynski R, O’Connor MB, & Gilbert LI (2009). A phosphoproteomics approach to elucidate neuropeptide signal transduction controlling insect metamorphosis. Insect Biochemistry and Molecular Biology, 39(7), 475–483. doi: 10.1016/j.ibmb.2009.04.005 [DOI] [PubMed] [Google Scholar]
- Shi Y, Nishida K, Campigli Di Giammartino D, & Manley JL (2011). Heat shock-induced SRSF10 dephosphorylation displays thermotolerance mediated by Hsp27. Mol Cell Biol, 31(3), 458–465. doi: 10.1128/mcb.01123-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Feng Y, & Manley JL (2004). Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature, 427(6974), 553–558. doi: 10.1038/nature02288 [DOI] [PubMed] [Google Scholar]
- Shin C, Kleiman FE, & Manley JL (2005). Multiple properties of the splicing repressor SRp38 distinguish it from typical SR proteins. Mol Cell Biol, 25(18), 8334–8343. doi: 10.1128/mcb.25.18.8334-8343.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, & Manley JL (2002). The SR protein SRp38 represses splicing in M phase cells. Cell, 111(3), 407–417. [DOI] [PubMed] [Google Scholar]
- Shkreta L, Toutant J, Durand M, Manley JL, & Chabot B (2016). SRSF10 Connects DNA Damage to the Alternative Splicing of Transcripts Encoding Apoptosis, Cell-Cycle Control, and DNA Repair Factors. Cell Rep, 17(8), 1990–2003. doi: 10.1016/j.celrep.2016.10.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NK, Singh NN, Androphy EJ, & Singh RN (2006). Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Molecular and Cellular Biology, 26(4), 1333–1346. doi: 10.1128/mcb.26.4.1333-1346.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, … Lundberg E (2017). A subcellular map of the human proteome. Science, 356(6340). doi: 10.1126/science.aal3321 [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, … Ponten F (2015). Proteomics. Tissue-based map of the human proteome. Science, 347(6220), 1260419. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- Wang Z, Chatterjee D, Jeon HY, Akerman M, Vander Heiden MG, Cantley LC, & Krainer AR (2012). Exon-centric regulation of pyruvate kinase M alternative splicing via mutually exclusive exons. J Mol Cell Biol, 4(2), 79–87. doi: 10.1093/jmcb/mjr030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee CD, Havens MA, Jodelka FM, & Hastings ML (2014). Targeting SR proteins improves SMN expression in spinal muscular atrophy cells. PLoS One, 9(12), e115205. doi: 10.1371/journal.pone.0115205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei N, Cheng Y, Wang Z, Liu Y, Luo C, Liu L, … Feng Y (2015). SRSF10 Plays a Role in Myoblast Differentiation and Glucose Production via Regulation of Alternative Splicing. Cell Rep, 13(8), 1647–1657. doi: 10.1016/j.celrep.2015.10.038 [DOI] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, … Yang YG (2016). Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell, 61(4), 507–519. doi: 10.1016/j.molcel.2016.01.012 [DOI] [PubMed] [Google Scholar]
- Yang L, Chansky HA, & Hickstein DD (2000). EWS.Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J Biol Chem, 275(48), 37612–37618. doi: 10.1074/jbc.M005739200 [DOI] [PubMed] [Google Scholar]
- Yang L, Embree LJ, Tsai S, & Hickstein DD (1998). Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem, 273(43), 27761–27764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.