

Abstract
Protein translation is essential for cell physiology, and dysregulation of this process has been linked to aging-related diseases such as type 2 diabetes. Reduced protein level of a requisite scaffolding protein of the initiation complex, eIF4G1, downstream of nutrients and insulin signaling is associated with diabetes in humans and mice. In the current study, we tested the hypothesis that eIF4G1 is critical for β-cell function and glucose homeostasis by genetically ablating eIF4G1 specifically in β-cells in vivo (βeIF4G1 knockout [KO]). Adult male and female βeIF4G1KO mice displayed glucose intolerance but normal insulin sensitivity. β-Cell mass was normal under steady state and under metabolic stress by diet-induced obesity, but we observed increases in proliferation and apoptosis in β-cells of βeIF4G1KO. We uncovered deficits in insulin secretion, partly due to reduced mitochondrial oxygen consumption rate, glucose-stimulated Ca2+ flux, and reduced insulin content associated with loss of eIF4E, the mRNA 5′ cap-binding protein of the initiation complex and binding partner of eIF4G1. Genetic reconstitution of eIF4E in single β-cells or intact islets of βeIF4G1KO mice recovers insulin content, implicating an unexplored role for eIF4G1/eIF4E in insulin biosynthesis. Altogether these data demonstrate an essential role for the translational factor eIF4G1 on glucose homeostasis and β-cell function.
Introduction
Protein translation is a fundamental cellular process and has emerged as an important checkpoint in the regulation of protein expression during organ development and cell function for both normal and in disease, for various cell types, including the pancreatic insulin-producing β-cell. The β-cell functions as a highly specialized metabolic factory with unique ability to continually sense nutrient and metabolic signals and respond with appropriate levels of insulin synthesis and secretion. In response to elevated glucose concentrations, β-cells preferentially release newly synthesized insulin granules (1,2). To match the demand for insulin, β-cells synthesize large quantities of insulin by recruiting preproinsulin mRNA to the endoplasmic reticulum (ER), thereby enhancing its translational efficiency (3–5).
To synthesize and process insulin, the β-cells possess a highly developed ER, which plays a central role in protein biosynthesis (6). Translation of the insulin precursor protein, preproinsulin, occurs on ribosomes associated with the ER while nascent proinsulin undergoes final folding within the lumen of the ER. Proinsulin is then trafficked through the Golgi complex and into exocytotic granules where prohormone convertase (PC) 2, PC1/3, and carboxypeptidase E (CPE) work together to convert proinsulin to mature insulin and C-peptide (7).
The first and rate-limiting step of the translation initiation is the binding of eukaryotic translation initiation factor 4 F (eIF4F) to the 5′ cap (m7GpppN). The eIF4F complex consists of the major m7G cap-binding protein eIF4E, the RNA helicase eIF4A, and a large protein scaffold that interacts with several components of the translation initiation machinery eIF4G. The eIF4F-eIF4G1 complex is required for 5′ cap scanning-dependent translation (8). The formation of eIF4F-eIF4G1 complex recruits ribosomes to mRNA, allowing scanning-dependent translation to proceed.
eIF4G1 is an integral component of the initiation complex, acting as scaffolding protein to recruit its binding proteins (i.e., eIF4E, eIF4A). The role of eIF4G1 in mRNA translation is well studied in cancer models, where it plays a critical role in cell growth, proliferation and differentiation, and dysregulation of the eIF4F-eIF4G1 complex, or overexpression of eIF4G1 promotes tumorigenesis in various cell types (9–11). In pancreatic β-cells, overexpression of eIF4G1 has been implicated in the translational regulation of CPE and the manifestation of a prediabetic hyperproinsulinemic phenotype in mice (12–14). Furthermore, islets isolated from patients with type 2 diabetes (T2D) and the ob/ob mouse model of T2D show decreased levels of eIF4G1 protein, suggesting that loss of this translation factor may contribute to the pathogenesis of T2D (12). However, the role of eIF4G1 in glucose homeostasis and β-cell function in vivo has not been specifically tested.
Currently, we have used genetic manipulation to generate a mouse model of β-cell–specific deletion of eIF4G1 (βeIF4G1 knockout [KO]) and uncovered a novel role for eIF4G1 in glucose metabolism. βeIF4G1KO mice are glucose intolerant in part by multiple defects at the β-cell level contributed by reduced islet insulin content and secretion associated with altered Ca2+ signaling and mitochondrial dysfunction. βeIF4G1KO mice display normal β-cell mass, with high turnover marked by elevated apoptosis and proliferation rates. Altogether, these data underscore the novel role of eIF4G1 in β-cell function.
Research Design and Methods
Animal Models and In Vivo Mouse Procedures
The following breeders were used in the study: C57Bl/6J, eIF4G1 flox/flox (The Jackson Laboratory), and mice harboring one allele of Cre-recombinase under the rat insulin 2 promoter (Rip-Cre; provided by Dr. Pedro Herrera, University of Geneva) or under the glucagon promoter with tdTomato Cre-reporter (Gcg-Cre;Tomato+/−; provided by Dr. George Gittes, University of Pittsburg). Rip-Cre used in Fig. 1 are the littermate control to eIF4G1 flox and Rip-Cre;eIF4G1 floxed mice. Wild-type (WT) are referred to as Rip-Cre positive or negative animals without any floxed gene. All mice were generated on a C57Bl/6J background and group housed on a 14:10 light-dark cycle. eIF4G1 floxed contains eIF4G1 with point mutation of R1207H and is treated as an independent biological variable. A high-fat diet (HFD) (60% kcal of fat, D12492) was purchased from Research Diets. Glucose and insulin tolerance tests and in vivo glucose-stimulated insulin secretion assays were performed as previously described in age-matched or littermates mice (15). Random-fed serums were collected via facial vein. For BrdU proliferation assay, cohorts of mice were injected with BrdU (100 mg/kg) every 4 days for total span of 12 days. All procedures were performed in accordance with the University of Minnesota Animal Studies Committee (Institutional Animal Care and Use Committee [ACUC] #1806A36072).
Figure 1.
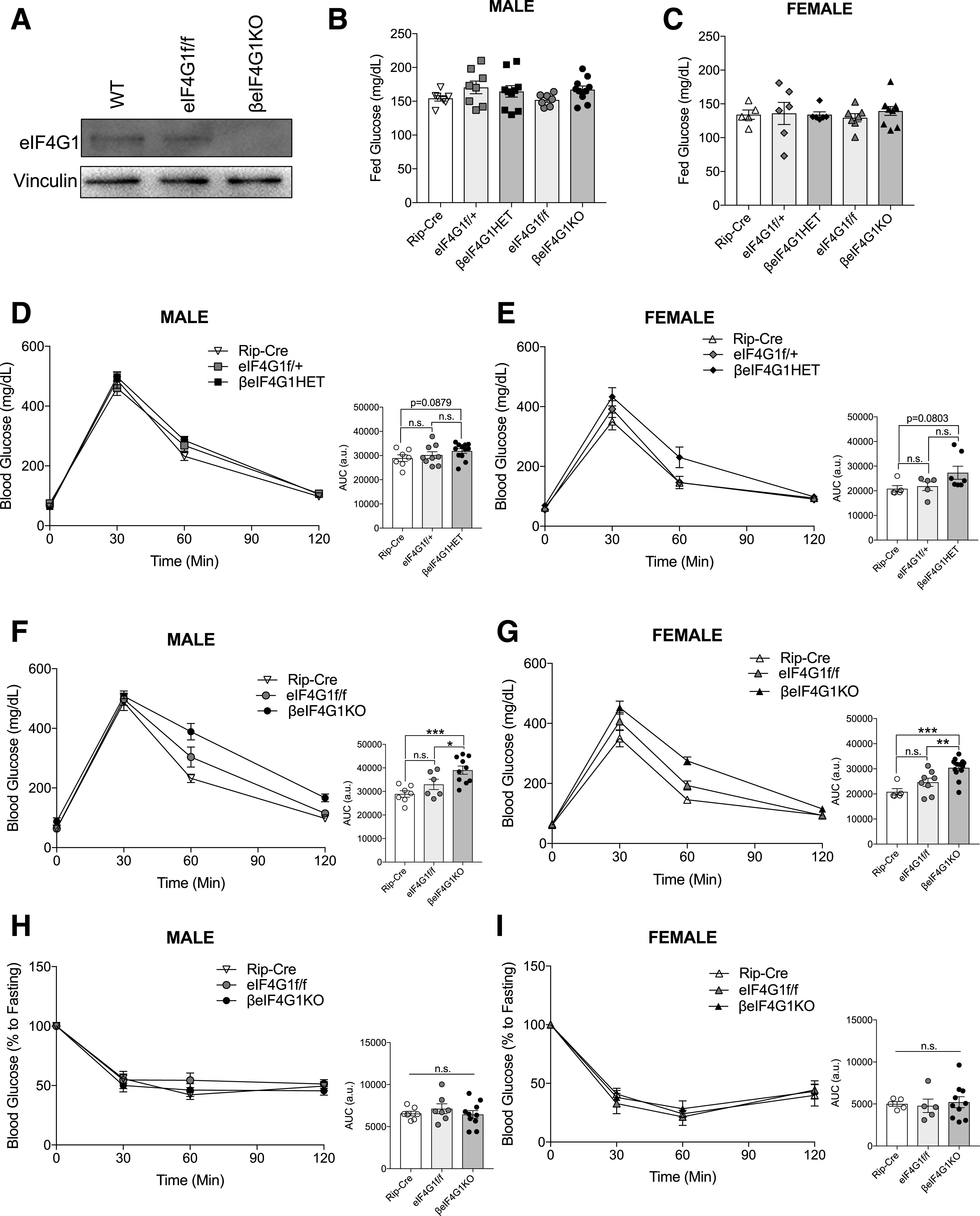
βeIF4G1KO mice exhibit glucose intolerance and normal insulin sensitivity. A: Representative islet level of eIF4G1 protein in WT, eIF4G1f/f, and βeIF4G1KO (n = 2). Random-fed blood glucose level of 2–3 months of age for male (B) and female (C) Rip-Cre, eIF4G1f/+, βeIF4G1HET, eIF4G1f/f, and βeIF4G1KO mice (n = 8). In vivo glucose tolerance tests (2 g/kg glucose, i.p.) were performed on 2-month-old littermate Rip-Cre WT, eIF4G1f/+, and βeIF4G1HET (n = 7, 9, 12 males; n = 5, 5, 7 females) (D and E) or eIF4G1f/f and βeIF4G1KO (n = 7, 6, 10 males; n = 5, 8, 13 females) (F and G). Insulin sensitivity (0.75 units/kg insulin, i.p.) at 8–10 weeks of age for Rip-Cre WT, eIF4G1f/f, and βeIF4G1KO (n = 7, 7, 10 males; n = 5, 5, 10 females) (H and I). Area under curve (AUC) of the blood glucose curves are presented in arbitrary units (a.u.) for each figure. Statistical analyses were conducted using unpaired, two-way Student t test with significance: *P < 0.05, **P < 0.01, ***P < 0.001.
Islet Isolation, Insulin Secretion Assay, Western Blot, Electron Microscope
We have previously described our islet isolation, insulin secretion assay technique (15,16), Western blot, and electron microscope (EM) imaging (14). See Supplementary Material for more details.
Insulin and Proinsulin ELISA
Insulin and proinsulin levels from random-fed serum, lysed isolated islets or INS-1 cells, and islet secretion solutions were measured using Mouse Ultrasensitive Insulin and Proinsulin ELISA (ALPCO), according to kit instruction. Content data were normalized to DNA, as determined by Quant-iT Pico Green dsDNA Assay (Molecular Probes) or by total protein, as determined by bicinchoninic acid assay.
Cell Culture Plasmid Transfection and Adenoviral Infection
INS-1 cells (gift from Dr. Peter Arvan, University of Michigan) were maintained in 11 mmol/L glucose RPMI 1640 media (14). pT7-EGFP-HseIF4E was purchased from Addgene (#79437; generated by Dr. Elisa Izaurralde, Max Planck Institute for Developmental Biology). Transfections of the plasmids were performed using Lipofectamine 2000 (for single islet cells) or Lonza Nucleofector Kit V (for INS-1 cells), following manufacturer’s instructions. eIF4E adenovirus and siRNA lentivirus (scrambled siCtrl and siRNA against eIF4G1) were purchased from Applied Biological Materials. Then, 10–20 islets dispersed by gentle pipetting with 0.05% Trypsin-EDTA or INS-1 cells were plated on tissue culture six-well plates before infection at 10–15 multiplicity of infection. The cells were collected 48-h after transfection or infection for lysis/analysis or further testing (cycloheximide [CHX] 50 μg/mL for up to 12 h).
Immunofluorescence/TUNEL Imaging
Sections were incubated in primary antibodies, followed by secondary antibodies conjugated to fluorophores (Supplementary Table 1), and DAPI solution (Thermo Fisher Scientific). Stained slides were imaged on a motorized microscope (ECLIPSE NI-E; Nikon). β-Cell mass was assessed as described previously (15). β-Cell size was determined by β-catenin or e-cadherin and insulin-positive area/number of nuclei, analyzing 60 islets per mouse in sections imaged at magnification ×20. TUNEL staining was performed as previously described (17). For proliferation analysis, isolated islets from BrdU-injected mice were isolated and processed for sectioning as described in the published protocol (18). Then, sectioned islets were stained against Ki67 and BrdU for imaging and counting. A minimum of 7,000 β-cells were analyzed per mouse for the quantifications for proliferation and apoptosis.
Nonradioactive Pulse Label Experiment
INS-1 cells were starved with methionine-free media for 45 min before incubation with Click-IT AHA (l-azidohomoalanine) (Invitrogen) in methionine-free media for 30 min (Pulse) before harvest. The cells were lysed, and the lysates were subjected to Click-IT reaction with biotin-conjugate alkyne, according to the manufacturer’s protocol. The biotin-labeled lysates and immunoprecipitates were resolved by Western blot and detected by horseradish peroxidase-conjugated streptavidin (STV).
Single Islet Cell Insulin Content Assay and Calcium Measurement
Approximately 100–125 isolated islets dispersed in 0.25% trypsin (Gibco) were plated onto coverslips before transfection with green fluorescent protein (GFP) or GFP-tagged eIF4E plasmids. Media was replaced after 24 h. At 48 h after transfection, individual GFP-positive islet cells were handpicked using a fluorescence microscope (BX51; Olympus) under magnification ×60, lysed in 20 μL of RIPA buffer (plus inhibitors), and assayed for insulin content. Insulin values from tdTomato-positive α-cells, from Gcg-Cre;Tomato+/− mice, were used to determine a minimum inclusionary threshold for insulin value (0.01 ng/mL insulin). Intracellular calcium was measured as previously described (16). The average Fura-2 signal (Fig. 7M) was calculated by averaging all Fura-2 signal points in the given glucose concentration.
Figure 7.
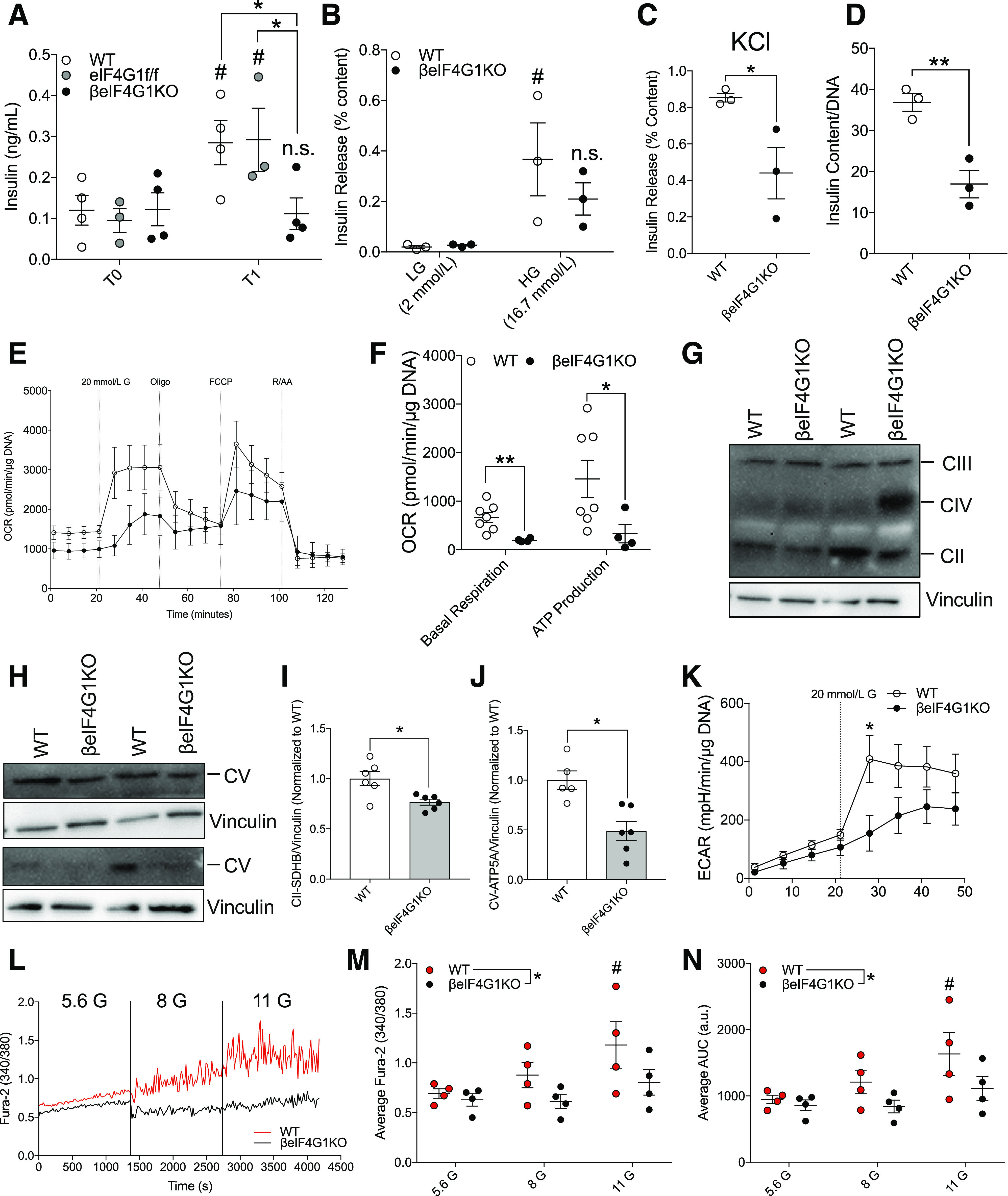
Defective mitochondrial function and Ca2+ influx contributes to reduced insulin secretory capacity in βeIF4G1KO mice. (A) In vivo glucose-stimulated insulin secretion assays were performed on 6-month-old male WT, eIF4G1f/f, and βeIF4G1KO mice (n = 3–5). WT and βeIF4G1KO islet secretory characteristics showing glucose-stimulated secretion (low glucose [LG] and high glucose [HG]; percentage insulin content) (B), KCl-stimulated secretion (30 mmol/L, percentage insulin content) (C), and total islet insulin content (normalized to DNA) (n = 3 (D). E: Representative OCR from isolated islets (pmol/min/normalized to µg DNA content). FCCP, carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone; R/AA, rotenone and antimycin A. F: Quantification of basal mitochondrial respiration and glucose-stimulated ATP production. G–J: Immunoblot of electron transport chain components (complex [C]III: UQCRC2, CIV: MTCO1, CII: SDHB, and CV: ATP5A) levels (relative to vinculin, normalized to WT) from isolated islets. K: Extracellular acidification rate (ECAR; mpH/min/normalized to μg DNA content) (n = 4–7). G, glucose. A representative trace (L), average Fura-2 AM 340-to-380 ratio (M), and average trace area under the curve (AUC) in arbitrary units (a.u.) (N) for calcium measurements from groups of 100 WT and βeIF4G1KO islets, in response to increasing concentrations of glucose (5.6, 8 and 11 mmol/L) (n = 4). WT are a mix of Rip-Cre (+) and Rip-Cre (−) mice with no floxed genes. Statistical analyses were conducted using unpaired, two-way Student t test or two-way ANOVA (A and B; *P = genotype, #P = LG vs. HG) and two-way ANOVA (M and N; *P = genotype, #P = 5.6 vs. 11 G) with significance: *P < 0.05, **P < 0.01; #P < 0.05.
Cell Mitochondrial Stress Test Using Seahorse Analyzer
Approximately 70–100 islets were seeded onto wells precoated with Cell-Tak in 30 μL serum-free RPMI media/well and incubated at 37°C for 1 h. Then, 70 μL of complete islet media were added per well and incubated overnight. Islets were washed with freshly prepared Seahorse media, following the manufacturer’s recommendations and incubated in the same media, 175 μL/well. Cell Mitochondrial Stress Test (Agilent) was performed following the protocol provided by the manufacturer with the addition of a step including a 20 mmol/L glucose injection before the oligomycin injection. After the test, islets were collected in RIPA lysis buffer and sonicated for DNA quantification as described. INS-1 cells were tested in a similar manner with few differences: no Cell-Tak precoating before plating and no 20 mmol/L glucose injection.
RT-Quantitative PCR
Quantitative (q)PCR was performed as described previously (19). Relative gene expression was calculated with ΔΔcycle threshold normalized to β-Actin. Primer sequences are listed in Supplementary Table 2.
Statistical Analysis
Data are presented as mean ± SEM and were analyzed using the two-tailed unpaired Student t test. Multiple outcome data were assessed using repeated measures two-way ANOVA. Statistical analyses were performed in GraphPad Prism version7 with a significance threshold of P < 0.05.
Data and Resource Availability
The reagents generated during the current studies are available from the corresponding author upon reasonable request.
Results
β-Cell–Specific Deletion of eIF4G1 Leads to Glucose Intolerance in Male and Female Mice
To investigate the significance of eIF4G1 in regulating β-cell mass and function in vivo, we generated a β-cell–specific loss of eIF4G1 model (Rip-Cre;eIF4G1 flox/+ or Rip-Cre;eIF4G1 flox/flox, herein referred to as βeIF4G1HET and βeIF4G1KO, respectively). They are generated in the global eIF4G1 R1207H mutant background, a mutation associated with increased susceptibility to Parkinson disease (20). While the locomotive and metabolic phenotypes of these mutant mice have not been described, we found that by 2 months of age, WT and mutant mice were indistinguishable based on physical appearance, home cage activity, or response to handling. At 5 months of age, male motor coordination was normal as assessed by an accelerating rotarod test (Supplementary Fig. 1A), suggesting the R1207H mutation does not present gross locomotive dysfunction up to this age in mice. For appropriate controls in the initial experiments and in vivo assessments, we included both Rip-Cre–positive and Rip-Cre–negative floxed (eIF4G1f/+ or eIF4G1f/f) mice. Moreover, we recently validated the fidelity of the Rip-CreHerrera for β-cell–specific expression (19) and demonstrated no Cre-dependent effects, as has been seen in other Rip-Cre models (21).
To validate the model, we confirmed that eIF4G1 protein was completely absent in islets isolated from βeIF4G1KO compared with WT and eIF4G1f/f islets (Fig. 1A). Next, we assessed the effect of eIF4G1 on glucose homeostasis. At 2 months old, male and female mice from all genotypes displayed normal random blood glucose levels (Fig. 1B and C) and body weight compared with littermate controls (Supplementary Fig. 1B and C). Male and female mice with partial loss of eIF4G1 (βeIF4G1HET) demonstrated a trend in mild glucose intolerance compared with WT littermates (Fig. 1D and E). A full deletion of β-cell eIF4G1 resulted in glucose intolerance compared with both controls (Fig. 1F and G), which persisted in a cohort of 5-month-old βeIF4G1KO mice (Supplementary Fig. 1D), emphasizing the importance of eIF4G1 in glucose homeostasis. A nonsignificant trend in glucose intolerance was found between eIF4G1f/f and Rip-Cre, which may suggest a possible effect of the mutation in nonpancreas tissues. Comparable insulin sensitivity was observed across all genotypes in both sexes (Fig. 1H and I), pointing toward a defect at the pancreatic β-cell level.
Reduced Insulin Levels but Normal Insulin Processing in βeIF4G1KO Islets
To investigate β-cell mechanisms contributing to the glucose intolerance, first we examined the impact of eIF4G1 on the proinsulin-to-insulin ratio, because a decrease in eIF4G1 level has been associated with hyperproinsulinemia through dysregulation of the insulin processing enzyme, CPE, in vitro (12,14,22). We measured nonfasted serum insulin and proinsulin levels and found no differences between βeIF4G1KO and controls in vivo (Fig. 2A–C). However, when we examined isolated islets, in the absence of external hormonal and neural stimuli, we observed decreases in both insulin and proinsulin content in the βeIF4G1KO (Fig. 2D and E). The proinsulin-to-insulin ratio, a key indicator of the hyperproinsulinemic phenotype, showed no alterations (Fig. 2F). Likewise, immunofluorescence staining showed dimmer insulin and proinsulin staining in βeIF4G1KO islets versus WT (Fig. 2G). Furthermore, a reduction of total insulin granules per β-cell, both insulin-rich dark core and immature light core granules, in EM images was observed in βeIF4G1KO islets versus WT (23,24) (Fig. 2H and I). The lack of proinsulin-to-insulin ratio defect in βeIF4G1KO mice was supported by normal islet CPE protein levels, as detected by immunofluorescence staining or Western blot (Fig. 3A and B). Despite a normal level of CPE, we observed a sixfold increase in CPE transcript levels in βeIF4G1KO islets (Fig. 3C), a possible compensation for a constitutive loss of eIF4G1 in vivo, which will be further explored in a later section. When INS-1 cells were used, acute eIF4G1 knockdown in vitro showed a mild but significant loss of CPE protein with a trend toward increased in CPE mRNA (P = 0.0674) (Fig. 3D and E and Supplementary Fig. 1E). The reduction in CPE protein in eIF4G1 knockdown appears to be independent of protein stability (Fig. 3F and G). Congruent with previous studies, CPE was regulated by eIF4G1 at the translational level, as we observed reduced pulse-labeled biosynthesis of CPE in eIF4G1 knockdown cells (Fig. 3H–J). Together, these data demonstrate that eIF4G1 is essential for CPE translation.
Figure 2.
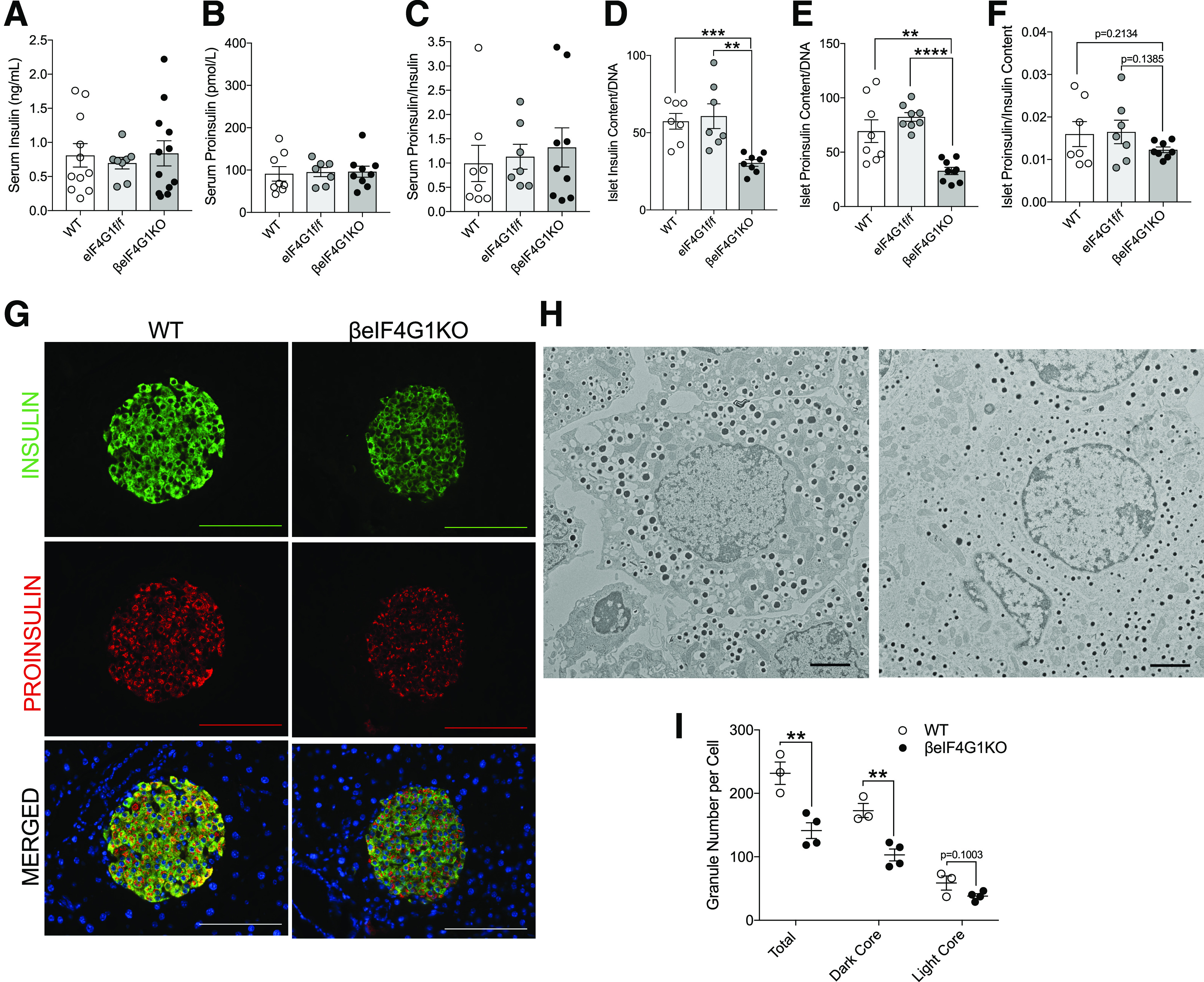
Normal insulin processing persists in β-cell–specific eIF4G1KO mice. Phenotype of 15-week-old WT, eIF4G1f/f, and βeIF4G1KO mice showing insulin, proinsulin, and proinsulin-to-insulin ratio values from in vivo random-fed serum (n = 8, 7, 9, respectively) (A–C), and isolated islets (n = 7, 7, 8, respectively) (D–F). G: Immunofluorescent images of pancreatic sectioned islets from 20-week-old animals showing insulin (green), proinsulin (red), and DAPI (blue). Immunofluorescent image scale bars, 100 μm. EM image of β-cells between WT and βeIF4G1KO islets (H), with quantification of insulin granules: total number, dark core, and light core (I) (n = 3 to 4 animals; 5–15 β-cells counted per animal). EM images scale bars, 2 μm. Representative of three to four animals per group for imaging data. WT are mix of Rip-Cre(+) and Rip-Cre(−) mice with no floxed genes. Statistical analyses were conducted using unpaired, two-way Student t test with significance: **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure 3.
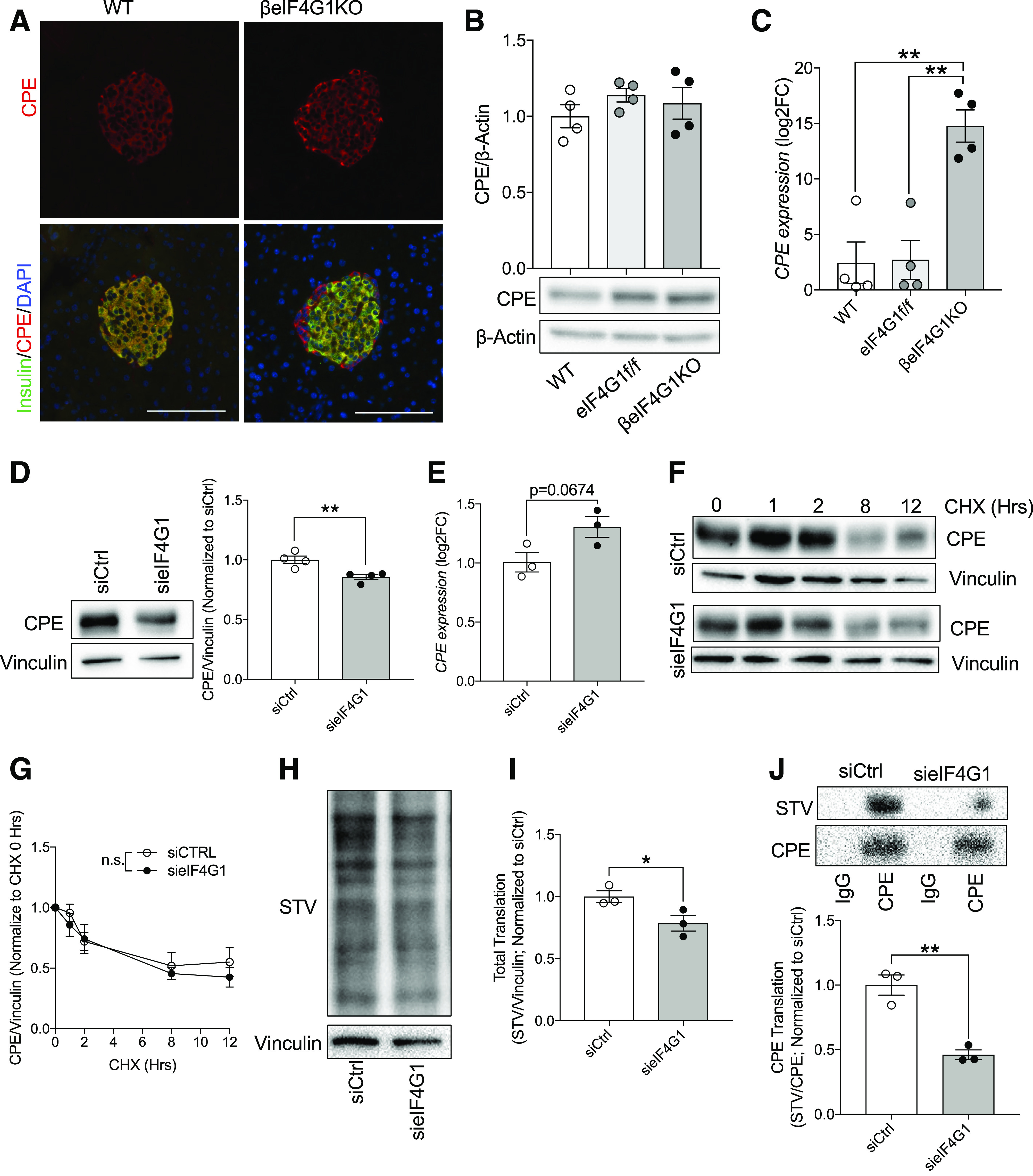
eIF4G1 regulates CPE translation in β-cells. Immunofluorescent images of CPE (red) + insulin (green)/DAPI (blue) (A), islet CPE levels (relative to β-actin, normalized to WT) by immunoblot (B), and qPCR gene expression (relative to β-Actin, log2 fold-change [FC] to WT) (C) (n = 4). Scale bars, 100 μm for immunofluorescent images. Representative of three to four animals per group for imaging data. sieIF4G1 knockdown INS-1 CPE protein level (relative to vinculin, normalized to siCtrl) (D), qPCR gene expression (relative to β-Actin, log2FC to siCtrl) (E), and protein stability (relative to vinculin, normalized to CHX 0 h) (F and G) (n = 3–4). eIF4G1 knockdown NS1 pulse labeled with modified methionine, followed by biotin/STV detection: total translate (STV/vinculin; normalized to siCtrl) (H and I) and CPE translation (STV/CPE; normalized to siCtrl) (J) (n = 3). WT are mix of Rip-Cre (+) and Rip-Cre (−) mice with no floxed genes. Statistical analyses were conducted using unpaired, two-way Student t test and two-way ANOVA with significance: *P < 0.05, **P < 0.01.
Altered β-Cell Turnover, Without Changes to the Overall β-Cell Mass in βeIF4G1KO Mice
Because we observed glucose intolerance in βeIF4G1KO mice, without any deficit in the proinsulin-to-insulin ratio, we evaluated β-cell mass and function. Normal pancreas weight and β-cell mass was observed in βeIF4G1KO mice (Fig. 4A and B). The βeIF4G1KO and WT mice both displayed comparable distribution of islet sizes or total islet count across the pancreas (Fig. 4C). Nevertheless, the total number of β-cells per islet was increased in a quantification of imaged islets (Fig. 4D). The increase in total cell number appeared to be compensated, in terms of total mass, by a decrease in average β-cell size (Fig. 4E and F and Supplementary Fig. 2A). This finding was corroborated by islet EM images that revealed a clear decrease in β-cell density within a field of view in islets of βeIF4G1KO compared with WT (Fig. 4G and H). Furthermore, the reduced β-cell size was associated with a reduction in the mTORC1 downstream target S6, a known regulator of β-cell growth and size (Supplementary Fig. 2D–F).
Figure 4.
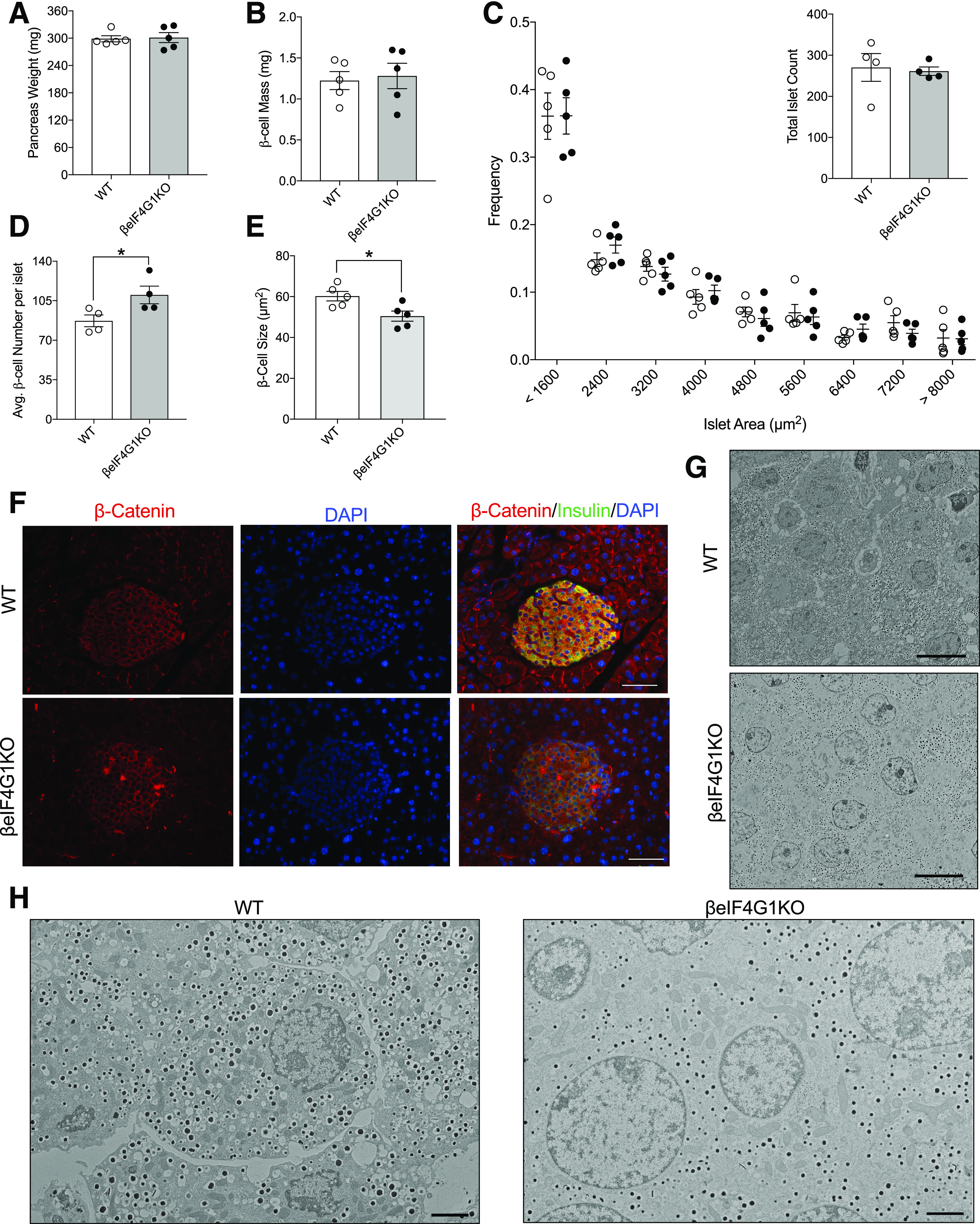
βeIF4G1KO exhibit normal β-cell mass, but with altered β-cell size and number. Pancreas weight (mg) (A) and β-cell mass (mg) (B) of 20-week-old WT and βeIF4G1KO (n = 5). C: Islets imaged at magnification ×4 were binned according to the area, from 800 μm2 (small) to 8,000 μm2, and the number of islets from each mouse was counted from five representative sections of the pancreas (n = 5). Average β-cell number per islet (D) and average β-cell size (E) was determined from immunofluorescence staining of pancreatic sections of islets with β-catenin (red), insulin (green), and DAPI (blue) (F) (scale bars, 100 μm) (n = 4–5). Islet EM image between WT and βeIF4G1KO at magnification ×600 (scale bars, 10 μm) (G) and at magnification ×2,500 (scale bars, 2 μm) (H). WT are mix of Rip-Cre (+) and Rip-Cre (−) mice with no floxed genes. Statistical analyses were conducted using unpaired, two-way Student t test with significance: *P < 0.05.
To further dissect the mechanisms underlying the increased number in the βeIF4G1KO, we assessed apoptosis and proliferation rates in β-cells. Unexpectedly, we observed greater β-cell turnover, marked by both an increase in apoptosis, measured by TUNEL staining, (Fig. 5A and B) and a higher rate of proliferation, measured by Ki67 expression and BrdU incorporation (Fig. 5C and E) in the islets of βeIF4G1KO versus WT. Consistent with our observation, we found increased expressions of both tumor suppressive (p53 and p19Arf) and proproliferative (Mdm2 and Cyclin D2) genes in βeIF4G1KO islets (Fig. 5F–I). ER stress levels have been correlated to the proliferative capacity of the β-cells (25). In the βeIF4G1KO model, reduction in ER stress was evident via assessment of reduced Bip and phosphorylated PERK protein levels (Fig. 5J–L), as well as reduced mRNA expressions of IRE1a, ATF6, XBP splicing, and Chop, compared with the control (Fig. 5M and N and Supplementary Fig. 2B and C). In sum, while gross β-cell mass remained normal, loss of eIF4G1 led to an increased in β-cell turnover.
Figure 5.
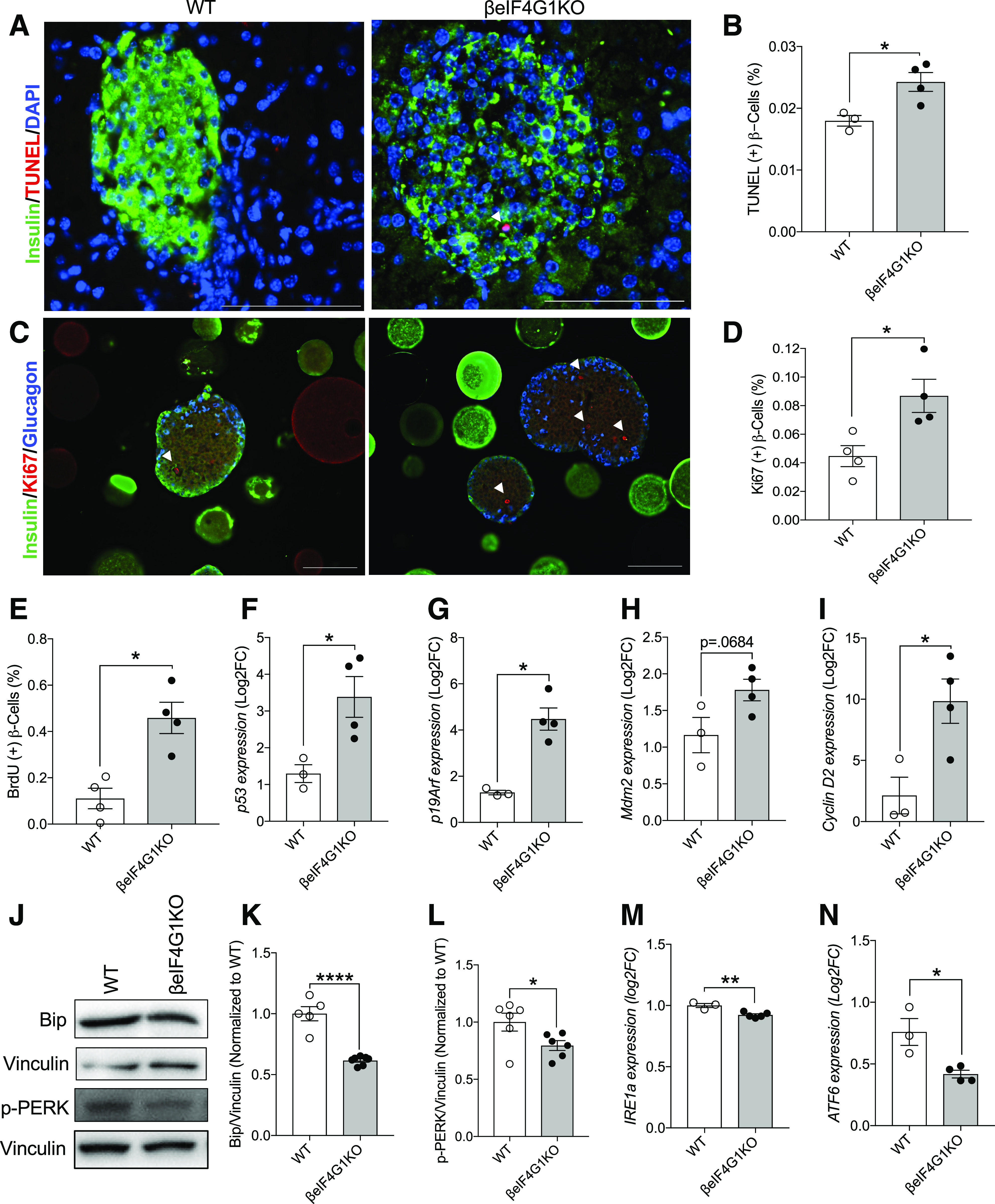
Enhanced β-cell turnover in βeIF4G1KO mice. A and B: Apoptosis of β-cells was measured through TUNEL immunofluorescence staining in WT and βeIF4G1KO pancreas (TUNEL+ β-cells percentage to total β-cells counted) (n = 3–4). β-Cell replication was measured through Ki67 (C and D) and BrdU incorporation (E) in WT and βeIF4G1KO islet sections (Ki67+ or BrdU+ β-cells percentage to total β-cells counted) (n = 4). White arrows in images indicate TUNEL+ and Ki67+ β-cells. Scale bar, 100 μm. F–I: qPCR gene expressions of p53, p19Arf, Mdm2, Cyclin D2 from isolated islets. Bip and phosphorylated (p)-PERK levels (relative to vinculin, normalized to WT) by immunoblot (J–L) (n = 5–7) and mRNA expressions of IRE1a (M) and ATF6 (N) relative to β-Actin, log2 fold-change (FC) to WT (n = 3–5) from isolated islets. B, D, and E: Minimum of 7,000 β-cells were analyzed per mouse for β-cell count quantifications. WT are a mix of Rip-Cre (+) and Rip-Cre (−) mice with no floxed genes. Statistical analyses were conducted using unpaired, two-way Student t test with significance: *P < 0.05, **P < 0.01, ****P < 0.0001.
βeIF4G1KO Mice Fail to Develop Hyperinsulinemia, Despite Normal β-Cell Mass Expansion, Under HFD-Induced Obesity
To test their response to metabolic stress and to assess the ability of the β-cells to expand under hypernutrient conditions, 11-week-old βeIF4G1KO and control mice were fed a 60% kcal HFD for 10 weeks. During the course of HFD feeding, the βeIF4G1KO mice exhibited lower body weight gain compared with WT mice (Fig. 6A and B). Despite lack of obesity, worsening of glucose intolerance was observed at 5 weeks post-HFD (Fig. 6C), without alterations in insulin sensitivity tested at 7 weeks post-HFD in βeIF4G1KO compared with the control mice (Fig. 6D). βeIF4G1KO mice also displayed hyperglycemia by 10 weeks post-HFD, consistent with their failure to develop compensatory hyperinsulinemia in that time frame (Fig. 6E and F). Despite irregularities in the proliferation and apoptotic rate we previously documented under the standard diet (SD), βeIF4G1KO achieved a similar level of β-cell mass by 10 weeks of the HFD as the control (Fig. 6G), both significantly higher than the SD (data produced from Fig. 4B for comparison), indicating that the βeIF4G1KO β-cells were able to adapt and increase β-cell mass in response to metabolic stress. We determined higher proliferation and apoptosis as well as reduction in cell size in the βeIF4G1KO β-cells compared with control in HFD (Supplementary Fig. 3A–F), overall contributing to relative normal β-cell mass expansion, which is insufficient to normalize glucose intolerance in βeIF4G1KO mice. Together, these data suggest that the glucose intolerance observed under both SD and HFD conditions in the βeIF4G1KO mice arises from defects in β-cell function rather than mass.
Figure 6.
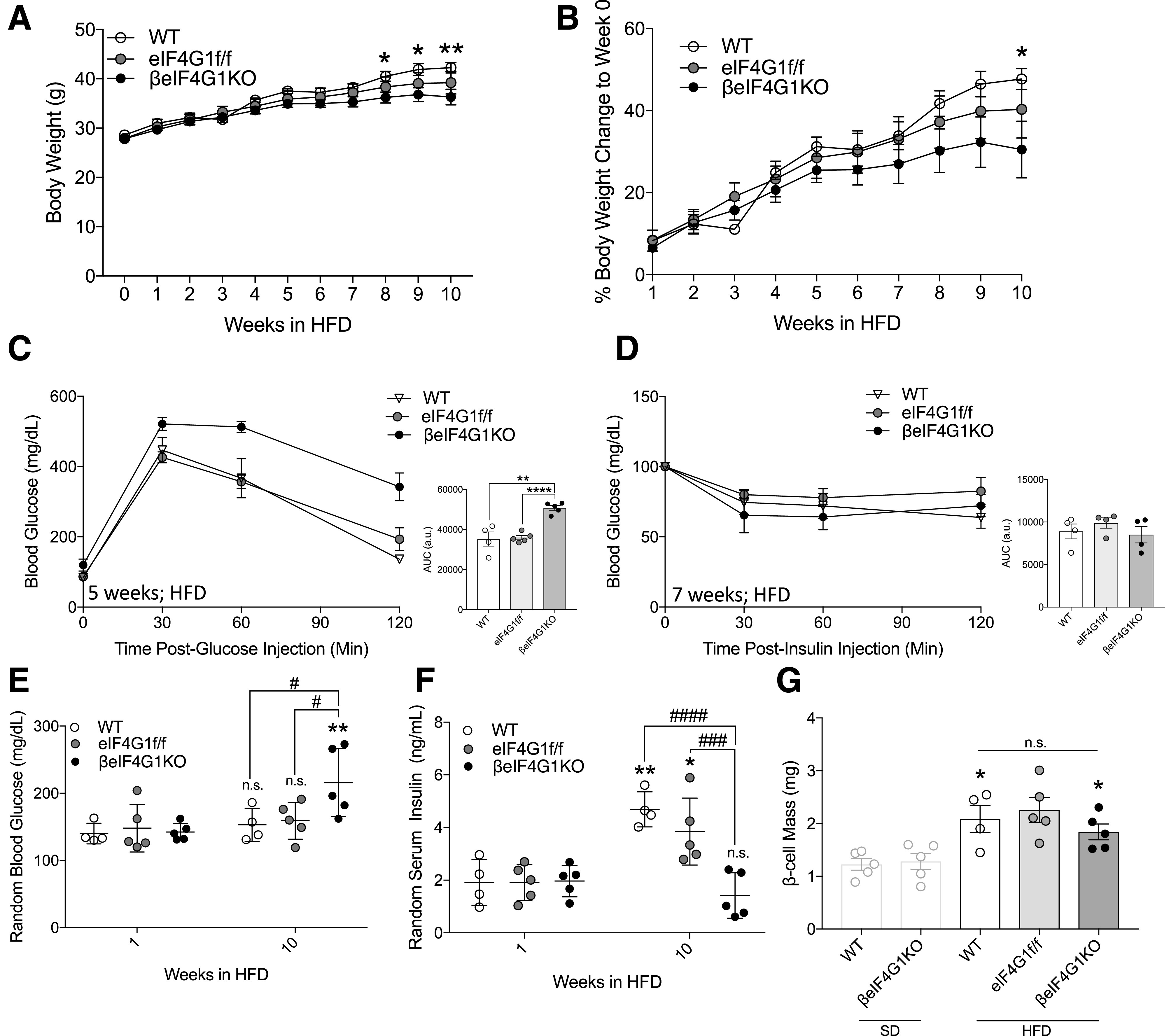
βeIF4G1KO mice fail to develop hyperinsulinemia under HFD milieu. Body weight (g) (A) and body weight gain (percentage body weight a day before HFD feeding) (B) over 10 weeks of the HFD. C: In vivo glucose tolerance (2 g/kg glucose, i.p.) was assessed 5 weeks post-HFD in WT, eIF4G1f/f, and βeIF4G1KO. The area under the curve (AUC) is shown in arbitrary units (a.u.). Insulin sensitivity was performed 7 weeks post-HFD (0.75 units/kg insulin, i.p.) in WT, eIF4G1f/f, and βeIF4G1KO. Blood glucose (E) and serum insulin (F) at 1 and 10 weeks of the HFD in WT, eIF4G1f/f, and βeIF4G1KO. G: β-Cell mass (mg) after 10 weeks of the HFD in WT, eIF4G1f/f, and βeIF4G1KO mice. (*P = 1 week vs. 10 weeks; #P = genotypes). The light-gray bar represents β-cell mass of the SD-fed mice shown in Fig. 4B (n = 4 WT, n = 5 eIF4G1f/f, and n = 5 βeIF4G1KO). WT are mix of Rip-Cre (+) and Rip-Cre (−) mice with no floxed genes. Statistical analyses were conducted using the unpaired, two-way Student t test and two-way ANOVA with significance: *P < 0.05; #P < 0.05, ###P < 0.001, ####P < 0.0001.
Impaired Insulin Secretion Associated With Reduced Mitochondrial Function and Calcium Signaling in βeIF4G1KO Mice
Subsequently, we assessed β-cell secretory function, uncovering severely diminished insulin response to stimulatory glucose in multiple cohorts in vivo (Fig. 7A and Supplementary Fig. 4A) and validated at the islet level in vitro (Fig. 7B). Islet insulin release was also significantly lower from βeIF4G1KO islets in response to the membrane-depolarizing agent potassium chloride (KCl) (Fig. 7C). Reiterating our previous findings, average βeIF4G1KO islet insulin content, assessed after the secretion assay, was less than half of the level observed in WT islets (Fig. 7D), which likely contributed to the hyposecretory defect.
As mitochondrial function, particularly nutrient-driven ATP production, is critical for insulin secretion, we assessed overall oxygen consumption rate (OCR) of isolated islets using the Seahorse Mitochondrial Stress Assay (Fig. 7E). We observed a significant reduction in both basal and glucose-stimulated ATP-linked respiration in βeIF4G1KO (Fig. 7F). There were no significant changes in the maximum respiration, proton leak, spare capacity, nonmitochondrial, or glucose-stimulated OCR (Supplementary Fig. 4B, and calculations were done based on schematic shown in Supplementary Fig. 4C), although the limited sample size may underrepresent potential alterations of these different mitochondrial fitness parameters. We observed reductions in complex II and V subunits of the electron transport chain without significant changes in complex III and IV in islets from the βeIF4G1KO mice (Fig. 7G–J and Supplementary Fig. 4D). We recapitulated the defect in mitochondrial fitness in acute eIF4G1 knockdown INS-1 with reduced basal and ATP-linked respiration as well decreased expressions of complex I, II, III, and V (Supplementary Fig. 4E–G). Additionally, we observed a trend toward decreased glycolysis, measured by the extracellular acidification rate, in response to glucose in βeIF4G1KO islets, hinting at a defect upstream of mitochondrial glucose metabolism that warrants future investigation (Fig. 7K).
We assessed Ca2+ signaling by Fura-2 fluorescence and found a blunted response to increasing glucose levels in βeIF4G1KO islets compared with WT (Fig. 7L–N and Supplementary Fig. 4H). As an important Ca2+ transporter for defining β-cell stimulus-secretion coupling, we checked the SERCA2 protein levels but found no change (Supplementary Fig. 5A and B). We also detected no change in Kir6.2, a subunit of the K+/ATP channel upstream of Ca2+ influx (Supplementary Fig. 5C and D), suggesting that the upstream stimulus signal (i.e., ATP production) may indeed contribute to the Ca2+ flux defect. Altogether, our data show defects in mitochondrial function, Ca2+ signaling, and islet insulin content, which all may contribute to the insulin secretion dysfunction in βeIF4G1KO mice.
Restoration of eIF4E Protein Loss Rescues Insulin Content in βeIF4G1KO β-Cells and Islets
A defect in insulin production is major contributing factor to insulin secretion deficit, especially when insulin secretion is reduced in response to the KCl depolarizing agent (Fig. 7C). Therefore, we sought to identify the mechanisms behind the reduction of insulin content in βeIF4G1KO mice. Despite the decrease in insulin content in βeIF4G1KO islets, we found no changes in Ins2 transcript expression but increased in mRNA transcripts of Ins1 and NeuroD1, a critical insulin transcription factor (Fig. 7A and B and Supplementary Fig. 6A). No change was detected in the protein expression of PDX-1, an important insulin transcription factor and regulator of β-cell function (26,27) (Supplementary Fig. 6B). A previous report suggested that glucose-responsive insulin translation in humans is mostly dependent on 5′ cap-dependent translation (28), so we assessed the mRNA and protein levels of important translation initiation factors in βeIF4G1KO islets by qPCR and Western blot, respectively: eIF4G2, eIF4G3, eIF4A, and eIF4E (Fig. 8C). Among the eIF4G homologs tested across genotypes, we found only a difference in eIF4G2 mRNA between WT and βeIF4G1KO islets (Supplementary Fig. 6C–F). The significant increase in eIF4G2 mRNA detected in the βeIF4G1KO islets was associated with trend toward increased eIF4G2 protein (Supplementary Fig. 6D). No change was detected in mRNA or protein levels of eIF4A, a binding partner of eIF4G1 (Supplementary Fig. 6G and H). We observed a significant reduction (∼60%) of eIF4E, an important 5′ cap-binding protein that complexes with eIF4G1, at both protein and mRNA level (Fig. 8D and E). Acute eIF4G1 knockdown in INS-1 cells in vitro also demonstrated reduced protein and mRNA of eIF4E (Fig. 8F–H), independent of its protein stability (Fig. 8I and J). Reduction in insulin content was apparent as well in these knockdown cells (Fig. 8K), prompting us to further investigate the contribution of eIF4E on regulating insulin content in the eIF4G1 loss model.
Figure 8.
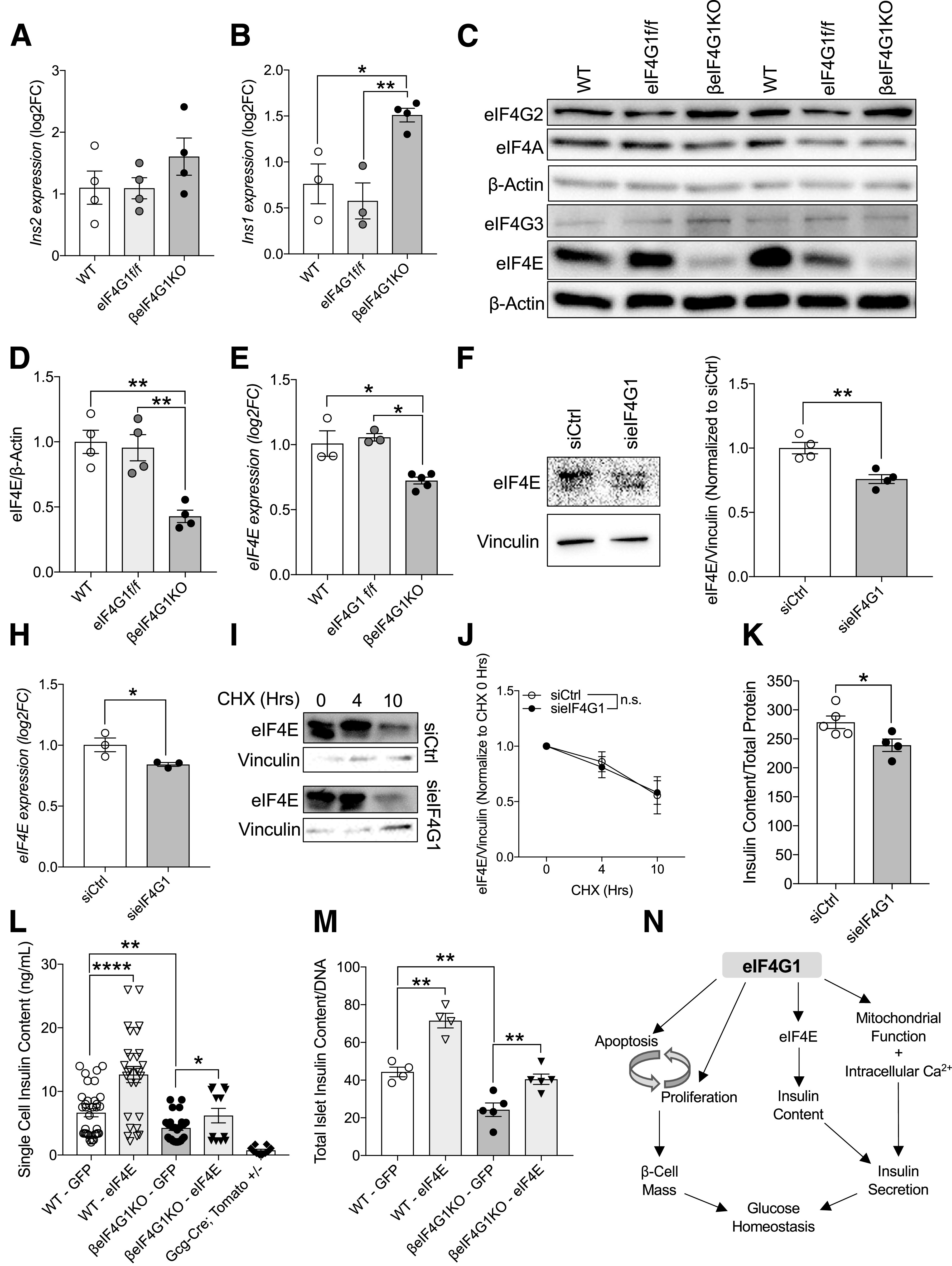
eIF4E-eIF4G1 interaction is important in regulating insulin content in β-cells. qPCR measuring Ins2 (A) and Ins1 (B) transcripts from isolated islets (relative to β-Actin, log2 fold-change [FC] to WT). Representative immunoblot (C) and quantitation of protein levels in WT, eIF4G1f/f, and βeIF4G1KO islets (relative to β-actin, normalized to WT) (n = 4) for eIF4E (D) (n = 4). E: mRNA level of eIF4E (relative to β-Actin, log2FC to WT) from isolated islets (n = 3–5). eIF4E protein (F and G), mRNA (H), and stability (I and J) from sieIF4G1 knockdown INS-1 cells (n = 3). K: INS-1 cell insulin content from siCtrl and sieIF4G1 knockdown (n = 4–5). L: Insulin content from handpicked GFP plus dispersed individual islet cells from WT and βeIF4G1KO (n = 3 experiments per panel) using a microscope with magnification ×60. Td-Tomato plus cells from Gcg-cre;Tomato+/− islets were included in (L) as an insulin-negative control threshold. M: Insulin content from ∼10 to 25 whole dispersed islets infected with adenoviral GFP or eIF4E (n = 4–5). WT are a mix of Rip-Cre (+) and Rip-Cre (−) mice with no floxed genes. Statistical analyses were conducted using the unpaired, two-way Student t test with significance at P < 0.05. N: Schematic diagram depicts the role of eIF4G1 expression regulating 1) β-cell turnover in modulation of both proliferative and apoptotic signals, 2) insulin content through its regulation of 5′ cap-binding protein, eIF4E, and 3) insulin secretory capacity contributed by mitochondrial/intracellular Ca2+ signals. *P < 0.05; **P < 0.01, ****P < 0.001.
An initial experiment found a trend toward increased insulin content (P = 0.08) in the eIF4E-transfected and FACS-sorted transformed INS-1 β-cell line (Supplementary Fig. 6I), implicating eIF4E availability as a potential regulator of insulin content downstream of eIF4G1 loss in our hypoinsulinemic mouse model. To test the hypothesis that eIF4E is required for insulin content, we transiently overexpressed GFP-tagged eIF4E in dispersed islet cells isolated from WT or βeIF4G1KO mice and assessed the insulin content of individual β-cells 48 h after transfection (see diagram in Supplementary Fig. 6J). We set the minimum threshold for the “insulin” signal at 2 ng/mL to subtract background noise as determined experimentally from a parallel collection of glucagon-driven red fluorescing cells (tdTomato-positive Gcg-Cre;tdTomato+/−). As in our earlier experiments, dispersed β-cells from βeIF4G1KO mice were hypoinsulinemic relative to WT cells, while GFP-eIF4E expressing WT β-cells demonstrated a higher insulin content than GFP control-transfected cells (Fig. 8L). Furthermore, eIF4E overexpression in βeIF4G1KO cells resulted in insulin levels comparable to WT controls (Fig. 8L). We achieved a similar outcome by infecting whole intact islets with adenovirus overexpressing eIF4E, which was sufficient to fully rescue the insulin content deficit of the βeIF4G1KO islets (Fig. 8M). These results strongly point to the role of eIF4E in defining insulin content and highlight a previously unreported role for eIF4E/eIF4G1 as an important regulator of insulin biosynthesis.
Discussion
The effect of eIF4G1 on glucose homeostasis was independent of sex and emerged only in full deletion of β-cell eIF4G1. We observed normal glucose tolerance and insulin sensitivity in eIF4G1f/f mice, suggesting that the mutation of eIF4G1 on R1207H does not impact the function of eIF4G1 on glucose homeostasis or β-cell function. The homologous mutation of R1207H in human eIF4G1 is R1205H, and this mutation site is controversially associated with autosomal dominant familial Parkinson disease (29,30). The functional consequences of the homologous eIF4G1-R1207H mutation are unclear, but in Drosophila, result in ∼30% downregulation of 5′ cap-dependent translation (31). On the basis of our data, R1207H mutation was insufficient to impact insulin content, glucose-stimulated insulin secretion, and β-cell mass, although further studies in other metabolic tissues may be needed to fully understand the consequence of the mutant. However, only full deletion of eIF4G1 protein in β-cells was sufficient to induce glucose intolerance, highlighting the importance of its expression level and role as a scaffolding protein in β-cell function.
In the current study, constitutive genetic deletion of eIF4G1 precipitated lack of defect in the proinsulin-to-insulin ratio in vivo in part due to normal CPE protein levels. However, a dramatic increase in CPE transcription (sixfold relative to control) likely played a compensatory mechanism that led to a normal CPE protein, and thus, a normal proinsulin-to-insulin ratio. It is important to recognize that in the O-linked β-N-acetylglucosamine transferase (OGT) KO or insulin receptor (IR) KO background, CPE loss was associated with no change or mildly increased CPE mRNA, respectively (12,14). Thus, it is possible that lack of transcriptional upregulation in CPE contributed to protein reduction. Indeed, acute eIF4G1 knockdown in vitro caused a decrease in CPE protein and a trend toward an increase in CPE mRNA. The reduction in CPE protein was due to translational defect but not stability. Future studies will aim to identify other possible compensatory mechanisms in the constitutive βeIF4G1KO and to assess mechanisms of gene transcription in the constitutive deletion and not in the acute knockdown of eIF4G1.
β-Cell mass was comparable between βeIF4G1KO and littermate control mice, which could account for normal insulin serum levels in these mice. Although no change in β-cell mass was observed, we noted a reduction in average β-cell size, that was accompanied by an increased β-cell number per islet in the βeIF4G1KO mice. The prominent reduction in average β-cell size was associated with reduced phosphorylated mTORC1/S6 (Ser240), a well-known regulator of β-cell size (32,33). eIF4G1 loss may also be associated with increased autophagy, downstream of mTORC1 signaling, which has contributed to smaller cell size in other models (34,35). In addition to changes in cell morphology, we observed higher β-cell turnover in the βeIF4G1KO islets under the SD or HFD diets. In β-cells, overexpression of S6 kinase levels is associated with a similar phenotype in terms of high cell turnover from enhanced apoptosis and proliferation (32), although in the current study, only minor changes in S6K activity were observed. During mitosis, repression of 5′ cap-dependent translation allows a cis-regulatory element, such as an internal ribosome entry site (IRES), to recruit ribosome to the mRNA through IRES–trans-acting factors to promote translation related to cell progression and apoptosis, which includes Bcl-2, CDK1, and p53 (36,37). In the islets of constitutive eIF4G1KO, eIF4G2 (DAP5) transcript was increased and protein was trending toward increased, which may contribute to translation of cap-independent mechanisms by the IRES element (38). Additionally, we found reduced ER stress status (measured by Bip and phosphorylated PERK) in the βeIF4G1KO model, which is also known to have a role in regulating the cell division cycle in β-cells (25,39). Loss of eIF4G1 may potentially result in β-cell immaturity, which is marked with lower insulin biosynthesis associated with high proliferation rates (40). The molecular mechanisms behind increased proliferation and apoptosis in βeIF4G1KO mice warrant further investigation, which include a combination of proteomics studies in addition to polyribosomal profiling to identify eIF4G1-sensitive transcripts that may contribute to the observed phenotype.
Despite normal β-cell mass, βeIF4G1KO mice displayed a deficit in glucose-stimulated insulin secretion in vivo. Underlying the insulin secretion dysfunction in βeIF4G1KO mice, we uncovered several cellular dysregulations that may involve Ca2+ signaling and mitochondrial function. While there are no reports of eIF4G1 directly regulating Ca2+ homeostasis, we showed the failure of the βeIF4G1KO islets to upregulate Ca2+ influx and enhance the mitochondrial OCR. Several electron transport chain subunits were downregulated in eIF4G1-deficient cells, which may contribute to the reduced basal mitochondrial and ATP-linked oxygen consumption. In addition to changes in total insulin content, failure to generate a sufficient amount of ATP in response to glucose may impact glucose-stimulus coupling. Future studies can be directed to assess the role of eIF4G1 in the glucose-sensing pathways, mitochondrial function, and calcium handling.
βeIF4G1KO islets showed severe deficits in insulin secretion imparted by reduced total islet insulin content independent of Ins1/Ins2 transcription. Interestingly, the loss in total insulin content in islets was not observed in the serum (at the time we measured it), and this is likely due to sampling time or that the releasable pool of insulin was not altered. The reduction in total insulin content was caused in part by a reduction in the 5′ cap-binding protein eIF4E, an important component to insulin translation (28,41,42). Mechanistically, eIF4E protein loss was associated with reduction in its transcript in βeIF4G1KO islets. HuR, a critical mRNA stabilizer of eIF4E mRNA, was shown to be reduced in another eIF4G1 knockdown model, pointing to mRNA stability as a possible node of eIF4E regulation (43). By genetic manipulation, eIF4E overexpression in β-cells increased insulin content, consistent with previous reports suggesting a relationship between 5′ cap/untranslated region–dependent translation and insulin production (28,41,42). The direct reconstitution of eIF4E in the βeIF4G1KO rescues insulin content, which suggests that eIF4E regulates insulin translation. The role of eIF4E on insulin content has not been explored previously. Genetic ablation of 4E-BP2, which releases eIF4E and favors the interaction with eIF4G1, has been reported to increase β-cell mass and not insulin secretion at the islet level (44). However, deletion of 4E-BP2 rescues decreased insulin serum levels in mice lacking β-cell mTORC1 (13), supporting the idea that eIF4E can regulate circulating insulin levels.
In summary, we identified the roles for the translation initiation factor eIF4G1 in regulating glucose homeostasis through pancreatic β-cell function (Fig. 8N), where it regulates both β-cell morphology/turnover and insulin secretory pathways. The current study highlights translation initiation complex proteins, eIF4E and eIF4G1, as regulators of insulin biosynthesis in the pancreatic β-cells and provides the knowledge behind the functional consequences of the observed eIF4G1 dysregulation in islets of murine T2D models and individuals with diabetes.
Article Information
Acknowledgments. The authors thank David Bernlohr (University of Minnesota), Peter Arvan (University of Michigan), and Ernesto Bernal-Mizrachi (University of Miami) for discussion. The authors acknowledge Briana Clifton, Alicia Wong, and Ingrid Bender (University of Minnesota) for technical support. The authors thank Dr. Thomas Pengo (University of Minnesota Imaging Center) for his assistance with the in Fiji analysis and the University of Minnesota Imaging Center for technical support. The authors thank Dr. Michael Benneyworth, of the University of Minnesota Mouse Behavior Core, for his consultancy and assistance in acquiring the rotarod test equipment. The authors thank Dr. Gail Celio, of the University of Minnesota Imaging Center, for assistance in EM imaging.
Funding. This work was supported by National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases grant (1F31DK113694 to A.L. and R21DK112144 and R01DK115720 to E.U.A.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. S.J. designed experiments, generated and analyzed data, interpreted the data, and wrote and edited the final manuscript. A.L., R.M., N.E., R.S., N.P., A.E., and E.G. designed experiments, generated and analyzed data, assisted with manuscript preparation, and approved the final version. E.U.A. conceived the study, designed experiments, interpreted the data, and wrote and edited the manuscript. E.U.A. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 78th Scientific Sessions of the American Diabetes Association, Orlando, FL, 22–26 June 2018.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.13110335.
References
- 1.Halban PA Differential rates of release of newly synthesized and of stored insulin from pancreatic islets. Endocrinology 1982;110:1183–1188 [DOI] [PubMed] [Google Scholar]
- 2.Ivanova A, Kalaidzidis Y, Dirkx R, et al. . Age-dependent labeling and imaging of insulin secretory granules. Diabetes 2013;62:3687–3696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenman IC, Gomez E, Moore CE, Herbert TP. The selective recruitment of mRNA to the ER and an increase in initiation are important for glucose-stimulated proinsulin synthesis in pancreatic beta-cells. Biochem J 2005;391:291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Itoh N, Okamoto H. Translational control of proinsulin synthesis by glucose. Nature 1980;283:100–102 [DOI] [PubMed] [Google Scholar]
- 5.Wicksteed B, Alarcon C, Briaud I, Lingohr MK, Rhodes CJ. Glucose-induced translational control of proinsulin biosynthesis is proportional to preproinsulin mRNA levels in islet beta-cells but not regulated via a positive feedback of secreted insulin. J Biol Chem 2003;278:42080–42090 [DOI] [PubMed] [Google Scholar]
- 6.Evans-Molina C, Hatanaka M, Mirmira RG. Lost in translation: endoplasmic reticulum stress and the decline of β-cell health in diabetes mellitus. Diabetes Obes Metab 2013;15(Suppl. 3):159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cawley NX, Rodriguez YM, Maldonado A, Loh YP. Trafficking of mutant carboxypeptidase E to secretory granules in a beta-cell line derived from Cpe(fat)/Cpe(fat) mice. Endocrinology 2003;144:292–298 [DOI] [PubMed] [Google Scholar]
- 8.Haimov O, Sehrawat U, Tamarkin-Ben Harush A, et al. . Dynamic interaction of eukaryotic initiation factor 4G1 (eIF4G1) with eIF4E and eIF1 underlies scanning-dependent and -independent translation. Mol Cell Biol 2018;38:e00139-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaiswal PK, Koul S, Palanisamy N, Koul HK. Eukaryotic translation initiation factor 4 gamma 1 (EIF4G1): a target for cancer therapeutic intervention? Cancer Cell Int 2019;19:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao Y, Wei M, Li B, et al. . Functional role of eukaryotic translation initiation factor 4 gamma 1 (EIF4G1) in NSCLC. Oncotarget 2016;7:24242–24251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Badura M, Braunstein S, Zavadil J, Schneider RJ. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc Natl Acad Sci U S A 2012;109:18767–18772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liew CW, Assmann A, Templin AT, et al. . Insulin regulates carboxypeptidase E by modulating translation initiation scaffolding protein eIF4G1 in pancreatic β cells. Proc Natl Acad Sci U S A 2014;111:E2319–E2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, et al. . Loss of mTORC1 signalling impairs β-cell homeostasis and insulin processing. Nat Commun 2017;8:16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jo S, Lockridge A, Alejandro EU. eIF4G1 and carboxypeptidase E axis dysregulation in O-GlcNAc transferase-deficient pancreatic β-cells contributes to hyperproinsulinemia in mice. J Biol Chem 2019;294:13040–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lockridge AD, Baumann DC, Akhaphong B, Abrenica A, Miller RF, Alejandro EU. Serine racemase is expressed in islets and contributes to the regulation of glucose homeostasis. Islets 2016;8:195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lockridge A, Jo S, Gustafson E, et al. . Islet O-GlcNAcylation is required for lipid potentiation of insulin secretion through SERCA2. Cell Rep 2020;31:107609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baumann D, Wong A, Akhaphong B, et al. . Role of nutrient-driven O-GlcNAc-post-translational modification in pancreatic exocrine and endocrine islet development. Development 2020;147:dev186643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong Y, Ebrahimpour P, Liu Y, Yang C, Alonso LC. Pancreatic islet embedding for paraffin sections. J Vis Exp 2018:57931. [DOI] [PMC free article] [PubMed]
- 19.Alejandro EU, Bozadjieva N, Kumusoglu D, et al. . Disruption of O-linked N-acetylglucosamine signaling induces ER stress and β cell failure. Cell Rep 2015;13:2527–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulte EC, Mollenhauer B, Zimprich A, et al. . Variants in eukaryotic translation initiation factor 4G1 in sporadic Parkinson’s disease. Neurogenetics 2012;13:281–285 [DOI] [PubMed] [Google Scholar]
- 21.Lee JY, Ristow M, Lin X, White MF, Magnuson MA, Hennighausen L. RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J Biol Chem 2006;281:2649–2653 [DOI] [PubMed] [Google Scholar]
- 22.Naggert JK, Fricker LD, Varlamov O, et al. . Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nat Genet 1995;10:135–142 [DOI] [PubMed] [Google Scholar]
- 23.Wijesekara N, Dai FF, Hardy AB, et al. . Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 2010;53:1656–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alarcon C, Boland BB, Uchizono Y, et al. . Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes 2016;65:438–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szabat M, Page MM, Panzhinskiy E, et al. . Reduced insulin production relieves endoplasmic reticulum stress and induces β cell proliferation. Cell Metab 2016;23:179–193 [DOI] [PubMed] [Google Scholar]
- 26.Fujimoto K, Polonsky KS. Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes Metab 2009;11(Suppl. 4):30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gauthier BR, Wiederkehr A, Baquié M, et al. . PDX1 deficiency causes mitochondrial dysfunction and defective insulin secretion through TFAM suppression. Cell Metab 2009;10:110–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fred RG, Sandberg M, Pelletier J, Welsh N. The human insulin mRNA is partly translated via a cap- and eIF4A-independent mechanism. Biochem Biophys Res Commun 2011;412:693–698 [DOI] [PubMed] [Google Scholar]
- 29.Chartier-Harlin MC, Dachsel JC, Vilariño-Güell C, et al. . Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet 2011;89:398–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nichols N, Bras JM, Hernandez DG, et al. . EIF4G1 mutations do not cause Parkinson's disease. Neurobiol Aging 2015;36:2444.e1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jia H The Role of Parkinson’s Disease Associated Mutations in eIF4G1 on Protein Translation and Neurodegeneration Baltimore, MD, Johns Hopkins University Library, 2017 [Google Scholar]
- 32.Elghazi L, Balcazar N, Blandino-Rosano M, et al. . Decreased IRS signaling impairs beta-cell cycle progression and survival in transgenic mice overexpressing S6K in beta-cells. Diabetes 2010;59:2390–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruvinsky I, Sharon N, Lerer T, et al. . Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 2005;19:2199–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramírez-Valle F, Braunstein S, Zavadil J, Formenti SC, Schneider RJ. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J Cell Biol 2008;181:293–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang RC, Levine B. Autophagy in cellular growth control. FEBS Lett 2010;584:1417–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pyronnet S, Pradayrol L, Sonenberg N. A cell cycle-dependent internal ribosome entry site. Mol Cell 2000;5:607–616 [DOI] [PubMed] [Google Scholar]
- 37.Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D. The translational landscape of the mammalian cell cycle. Mol Cell 2013;52:574–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de la Parra C, Ernlund A, Alard A, Ruggles K, Ueberheide B, Schneider RJ. A widespread alternate form of cap-dependent mRNA translation initiation. Nat Commun 2018;9:3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma RB, O’Donnell AC, Stamateris RE, et al. . Insulin demand regulates β cell number via the unfolded protein response. J Clin Invest 2015;125:3831–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puri S, Roy N, Russ HA, et al. . Replication confers β cell immaturity. Nat Commun 2018;9:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wicksteed B, Herbert TP, Alarcon C, Lingohr MK, Moss LG, Rhodes CJ. Cooperativity between the preproinsulin mRNA untranslated regions is necessary for glucose-stimulated translation. J Biol Chem 2001;276:22553–22558 [DOI] [PubMed] [Google Scholar]
- 42.Wicksteed B, Uchizono Y, Alarcon C, McCuaig JF, Shalev A, Rhodes CJ. A cis-element in the 5′ untranslated region of the preproinsulin mRNA (ppIGE) is required for glucose regulation of proinsulin translation. Cell Metab 2007;5:221–227 [DOI] [PubMed] [Google Scholar]
- 43.Vosler PS, Gao Y, Brennan CS, et al. . Ischemia-induced calpain activation causes eukaryotic (translation) initiation factor 4G1 (eIF4GI) degradation, protein synthesis inhibition, and neuronal death. Proc Natl Acad Sci U S A 2011;108:18102–18107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blandino-Rosano M, Scheys JO, Jimenez-Palomares M, et al. . 4E-BP2/SH2B1/IRS2 are part of a novel feedback loop that controls β-cell mass. Diabetes 2016;65:2235–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]