

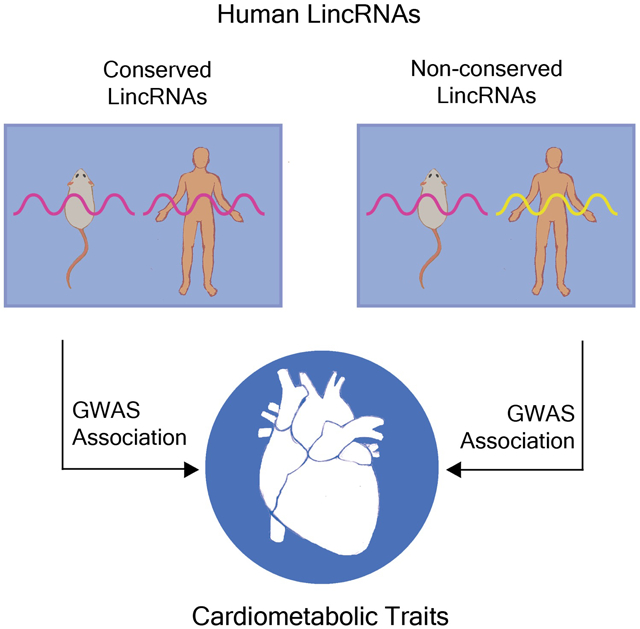
Abstract
Objective:
Transcriptome profiling of human tissues has revealed thousands of long intergenic non-coding RNAs (lincRNAs) at loci identified through large-scale genome wide studies for complex cardiometabolic traits. This raises the question of whether genetic variation at non-conserved lincRNAs has any systematic association with complex disease, and if so, how different this pattern is from conserved lincRNAs. We evaluated whether the associations between non-conserved lincRNAs and eight complex cardiometabolic traits resemble or differ from the pattern of association for conserved lincRNAs.
Approach and Results:
Our investigation of over 7,000 lincRNA annotations from Gencode Release 33 – GRCh38.p13 for complex trait genetic-associations leveraged several large, established meta-analysis genome-wide association study (GWAS) summary data resources – including GIANT, UK Biobank, GLGC, Cardiogram and DIAGRAM/DIAMANTE. These analyses revealed that: (1) non-conserved lincRNAs associate with a range of cardiometabolic traits at a rate that is generally consistent with conserved lincRNAs; (2) these finding persist across different definitions of conservation; and (3) overall across all cardiometabolic traits approximately one third of GWAS-associated lincRNAs are non-conserved and this increases to about two thirds using a more stringent definition of conservation.
Conclusions:
These findings suggest that the traditional notion of conservation driving prioritization for functional and translational follow-up of complex cardiometabolic genomic discoveries may need to be revised in the context of the abundance of non-conserved lncRNAs in the human genome and their apparent predilection to associate with complex cardiometabolic traits.
Keywords: Cardiometabolic traits, conservation, expression, GWAS, long intergenic non-coding RNAs (lincRNAs), synteny
Graphical Abstract
Introduction
Most loci identified through large-scale genome wide studies for complex cardiometabolic traits fall in intergenic regions and many of these overlap genomic features that confer cell-specific regulatory functions. Indeed, transcriptome profiling of human tissues has revealed thousands of long intergenic non-coding RNAs (lincRNAs), representing the majority of all long non-coding RNAs (lncRNAs), transcribed in a cell- and tissue-specific manner at many of these loci raising the question as to whether these lincRNAs could be causal elements for cardiometabolic trait associations at these intergenic loci 1. Convention in the field suggests that genetic elements that are conserved across many species are more likely to be functional and, if disrupted by mutations or common variation, contribute to rare diseases and complex traits respectively. Evolutionary profiling show that the majority of human lincRNAs mapped by RNA sequencing (RNA-seq) is not conserved outside of primate species and it has been suggested that some proportion of these may not be true functional lncRNAs but rather by-products of pervasive transcription 2-4. Recent work, however, is revealing many examples of non-conserved human lincRNAs that are functional and biologically important including a subset that may be the causal element at loci for human cardiometabolic and other diseases 1, 4-15.
These perspectives raise important questions as to whether genetic variation at non-conserved lincRNAs has any systematic association with complex cardiometabolic diseases, and if so, how different is this pattern from conserved lincRNAs. This is an important question in determining which human lincRNAs should be prioritized for functional and translational study. If non-conserved lincRNAs warrant systematic interrogation, this requires a shift in mind-set and application of innovative in vivo humanized models to address the physiological roles and disease impact of non-conserved lincRNAs. More broadly, because human genomes contain mostly non-conserved lincRNAs, the traditional notion of conservation driving functional prioritization for mechanistic studies in cardiometabolic model systems may need revision in context of our expanding knowledge of diverse, non-conserved, functional regulatory features.
In the current work, we evaluated the likelihood that non-conserved lincRNAs have association with a complex cardiometabolic trait, and whether this resembles or differs from the pattern of association for conserved lincRNAs. This included comprehensive consideration of summary data from multiple large meta-analysis genome-wide association study (GWAS) for eight cardiometabolic disease related traits: waist to hip ratio adjusted for body mass index (WHRadjBMI); body mass index (BMI) 16-20; height 21; high-density lipoprotein cholesterol (HDL-C); low-density lipoprotein cholesterol (LDL-C); triglycerides (TGs) 22; coronary artery disease (CAD) 23 and type-2 diabetes (T2D) 24. For lincRNA interrogation, we utilized a well-defined and comprehensive set of over 7,000 multi-exon lincRNAs that have been rigorously annotated (GENCODE Release 33 – GRCh38.p13) 25. Conservation was defined using multiple distinct strategies, primarily based on the broad perspective of synteny, or positional genomic conservation 2, 3, 26, 27, with secondary incorporation of additional information on expression in mouse tissues 25, and an exploratory consideration of conservation based on base-pair sequence scoring 28. Our analyses revealed that: (1) non-conserved lincRNAs associate with cardiometabolic traits at a rate that is consistent with conserved lincRNAs; (2) these finding persist across different definitions of conservation; and (3) overall across all traits approximately one third of GWAS-associated lincRNAs are non-conserved and this increases to about two thirds using a more stringent definition of conservation.
Materials and Methods
Anonymized data and materials are collected from existing publicly available repositories as indicated below.
Supplement Figure I provides a schematic illustration of how synteny is defined and summarizes our analytic pipeline. Key aspects of data preparation and analysis are described here. Additional methods description including gene set enrichment analyses are described in the Supplemental Material and Methods.
Conservation determination
Conservation based on synteny.
LincRNAs with expression in human tissues were identified using the Human Gencode v33 gene annotation build hg38 25 and were designated as intergenic if no protein-coding gene (PCG) start or stop locations were contained within the lincRNA start and stop locations. For primary analysis, a lincRNA was conserved if it as syntenic and non-conserved if it was not syntenic. LincRNAs were defined as syntenic if: 1) the nearest upstream and downstream neighboring PCGs in humans had one-to-one mouse homologs based on the Ensembl genome database release 47; and 2) the homologs were on the same chromosome with consistent relative orientation based on Mouse Gencode v24 25. Neighbors were defined as within 900Kb of the start and end position of the lincRNA as described previously 2.
A subset (18.5%) of lincRNAs without two neighboring PCGs within this region were designated as absent neighbors and were not classified as either syntenic or non-syntenic. Furthermore, for a small subset of lincRNAs (1.2%) that had upstream and downstream one-to-one mouse homologs but with inconsistent PCG orientation between human and mouse homologs, the lincRNA was designated as inconsistent orientation and not included in subsequent modeling. The relative orientation of mouse homologs for two lincRNAs could not be determined as they were not present in Mouse Gencode v24 and therefore, these lincRNAs were designated as having absent neighbors. This classification approach is summarized in the top panel of Supplement Figure I.
Conservation based on synteny and expression.
For secondary analysis, alternative definitions of lincRNA conservation were applied. A lincRNA was considered conserved if it was both syntenic at the genomic level in mouse and was also expressed in mouse tissues as defined by Mouse Gencode v24. A lincRNA was non-conserved if it was syntenic but not expressed in mouse tissues or not syntenic in mouse. Syntenic lincRNAs were defined as expressed if a lincRNA was present in mouse between the two identified PCG homologs based on Mouse Gencode v24. Syntenic lincRNAs with overlapping mouse homologs were unclassified under this definition of conservation.
Conservation based on sequence scoring.
Finally, as an exploratory analysis using a sequence-level approach to determining conservation, we calculated 7-way phastCons scores 28 for the 200bp region of each lincRNA transcript’s transcription start site (TSS). The average across the TSS region was calculated and the maximum across all transcripts of a lincRNAs was used as an alternative measure of conservation.
Merging of lincRNAs
For focused interrogation of lincRNAs with GWAS summary data, lincRNA boundaries were extended by 5Kb, in order to include single nucleotide polymorphisms (SNP) in the canonical 5’ promoter and 3’ UTR regulatory regions of lincRNAs. Resultant overlapping lincRNAs on the same strand were consolidated and treated as a single lincRNA. In the case that overlapping lincRNAs were on opposite strands, the lincRNA on the positive (+) strand was retained and the lincRNA on the negative (−) strand was removed. In merging lincRNAs, the following decision rules were applied: 1) if any of the merged lincRNAs were syntenic, the new merged lincRNA was classified as syntenic; 2) if none of the lincRNAs were syntenic but at least one was non-syntenic, the new merged lincRNA was classified as non-syntenic; 3) if none were syntenic or non-syntenic but at least one had inconsistent orientation, the new merged lincRNA was classified as inconsistent orientation; and 4) in all remaining cases, new merged lincRNAs were classified as absent neighbors. For the secondary definition of conservation, a similar approach was applied where syntenic is replaced with syntenic and expressed and a final category is included based on overlapping homologs. Merged lincRNAs are removed from the exploratory analysis based on phastCons. The approach to merging lincRNAs is presented in the bottom left panel of Supplement Figure I.
GWAS data selection and signal determination
Cardiometabolic trait GWAS summary datasets with large participant numbers were selected in order to provide statistical power to evaluate the disease-association of conserved versus non-conserved lincRNAs. Height was included because it is a defining complex genetic trait, has very large GWAS sample sizes and in recent years height has been shown to share causal pathways with those for atherosclerotic cardiovascular disease 29. SNPs were mapped from hg19 to hg38 using LiftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver). If the minimum SNP level p-value within a lincRNA (+/− 5Kb as outlined above) was less than the corresponding threshold (provided in Table 1) the lincRNA was classified as having a GWAS signal. For WHRadjBMI and BMI, the minimum p-value was determined across meta-analyses of men, women and men and women combined. In all other cases, results were based on meta-analysis results for men and women combined. GC content was calculated using sequence data from Human Gencode v33. For each lincRNA, exons were identified and merged if overlapping and GC content was defined as the proportion of G’s and C’s in the exon sequences. Transposable element (TE) coverage was defined as the proportion of the lincRNA exon sequences that overlap with TEs. For this calculation, the positions of TE types "LINE", "SINE", "LTR", and "DNA" were identified using UCSC Genome Browser RepeatMasker 30.
Table 1.
Summary of GWAS data resources
| Number of SNPs |
Sample size |
Coverage* | # LincRNAs mapped to SNPs |
Signal threshold† |
Source | |
|---|---|---|---|---|---|---|
| WHRadjBMI | 27,364,379 | 694,649 | 183 (123.5, 388) | 7011 | 5 x 10−8 | GIANT/UKBb 18 |
| BMI | 27,369,701 | 806,834 | 183 (124, 388.5) | 7011 | 5 x 10−8 | GIANT/UKBb 18 |
| Height | 2,332,944 | ~700,000 | 16 (8, 37) | 6611 | 5 x 10−8 | GIANT/UKBb 21 |
| HDL | 2,445,954 | 188,577 | 17(8, 39) | 6704 | 5 x 10−6 | GLGC 22 |
| LDL | 2,437,751 | 188,577 | 17 (8, 39) | 6698 | 5 x 10−6 | GLGC 22 |
| TGs | 2,439,264 | 188,577 | 17 (8, 39) | 6698 | 5 x 10−6 | GLGC 22 |
| CAD | 9,455,778 | 184,305 | 67 (40, 142) | 6859 | 5 x 10−6 | Cardiogram 23 |
| T2D | 21,635,866 | 898,130 | 146 (98, 309) | 6977 | 5 x 10−8 | DIAGRAM 24 |
Median number of SNPs per lincRNA and interquartile range (IQR) (25th, 75th)
Signal threshold was set to 5 x 10−8 for analysis of GIANT/UKBb and DIAGRAM data to correct for multiple comparisons. A less stringent but still suggestive threshold of 5 x 10−6 was used for the analysis of GLGC and Cardiogram data as the sample sizes and therefore power for detecting association are lower in these settings.
Statistical Analysis
The primary outcome was GWAS signal defined as an indicator that the minimum SNP level p-value within the lincRNA (+/− 5Kb as outlined above) was less than a pre-defined threshold (Table 1). Conservation was defined based on synteny for primary analysis and based on synteny and expression for secondary analysis. The proportions of conserved (p1) and non-conserved (p2) lincRNAs respectively with GWAS signal are reported. A non-inferiority test given by H0: p1 − p2 > δ versus HA: p1 − p2 < δ is applied for each trait with δ = 0.01. A corresponding p-value less than 0.05 was considered statistically meaningful and suggested that the proportion of non-conserved lincRNAs with GWAS signal was not significantly less than the proportion of conserved lincRNAs with GWAS signal in unadjusted analysis.
Additionally, multivariable logistic regression models were fitted separately for each trait and adjusted for number of SNPs (natural log transformed), GC content (natural log transformed) and TE coverage. LincRNAs were treated as the unit of analysis and data were limited to lincRNAs that were classified as conserved or not conserved. Wald tests of a difference in the probability of GWAS signal between conserved and non-conserved lincRNAs based on adjusted models are reported. Odds ratios (ORs) and corresponding 95% confidence intervals corresponding to the odds of GWAS signal for conserved lincRNAs compared to the odds of GWAS signal for non-conserved lincRNAs are also provided. The estimated probabilities of GWAS signal for conserved and non-conserved lincRNAs and the corresponding prediction interval were determined based on the multivariable fitted logistic model. A summary of the statistical analysis approach is provided in the righthand panel of Supplement Figure I.
Results
Descriptive characteristics of lincRNAs
Publicly available GWAS summary data used in the analysis are summarized in Table 1 and included: WHRadjBMI and BMI 18; height 21; HDL-C, LDL-C and TGs 22; CAD 23; and T2D 24. Table 2 illustrates the distributions of transcript length, GC content, exon count and TE coverage for lincRNAs that are classified as conserved or non-conserved (defined based on synteny as described in Methods). Summary data are reported as medians and interquartile ranges (IQR) as these measures are robust to skewness in the data. Information on lincRNAs unclassified due to absence of a PCG upstream or downstream (or both) or with inconsistent PCG relative orientation is provided in Supplement Table I.
Table 2.
Characteristics of conserved and non-conserved lincRNAs.
| Characteristic* | Conservation based on synteny | Conservation based on synteny and expression |
Total (n=7089) |
||
|---|---|---|---|---|---|
| Conserved (n=4243) |
Non-conserved (n=1445) |
Conserved (n=2262) |
Non-conserved (n=3398) |
||
| Length | 15960 (11918, 32571) |
15851 (11984, 29670) |
19120 (12510, 44262) |
14721 (11702, 26402) |
17130 (12093, 36922) |
| GC content | 0.458 (0.416, 0.506) |
0.460 (0.415, 0.504) |
0.452 (0.413, 0.498) |
0.463 (0.418, 0.509) |
0.450 (0.407, 0.499) |
| Exon Count | 3 (2, 4) |
3 (2, 4) |
3 (2, 5) |
2 (2, 4) |
3 (2, 5) |
| TE coverage | 0.346 (0.154, 0.543) |
0.329 (0.130, 0.541) |
0.334 (0.149, 0.518) |
0.347 (0.147, 0.558) |
0.346 (0.155, 0.545) |
| # SNPs† | 178 (123, 354) |
161 (108, 293) |
214 (136, 484) |
157 (111, 272) |
183 (123.5, 388) |
| # SNPs/length† | 0.010 (0.009, 0.012) |
0.010 (0.008, 0.012) |
0.011 (0.009, 0.012) |
0.010 (0.009, 0.012) |
0.010 (0.009, 0.012) |
Median and interquartile range (IQR) (25th, 75th) across lincRNAs within corresponding category.
Summary results for number of SNPs per lincRNA and number of SNPs divided by lincRNA length are based on subset of n=7011 lincRNAs and GWAS SNPs for WHRadjBMI (see Table 1).
Conserved lincRNAs tend to be longer than non-conserved lincRNAs and this difference is more pronounced when expression in mouse is considered [median length: syntenic lincRNAs = 15960 bps; non-syntenic lincRNAs = 15851 bps; syntenic and expressed lincRNAs = 19120 bps; syntenic and not expressed or non-syntenic lincRNAs = 14721 bps]. Moreover, unclassified lincRNAs tend to be significantly longer with a lower GC content and higher TE coverage (Supplement Table I). The number of SNPs per lincRNA (based on WHRadjBMI data) tracks with the length of the lincRNA, so that the distribution of number of SNPs divided by lincRNA length is approximately the same in all categories. Overall these findings support the use of multivariable adjusted analyses including these variables as potential confounders in characterizing the relationship between lincRNA conservation and GWAS signal.
Analysis using primary definition of conservation based on synteny
The counts and percentages of lincRNAs by conservation and GWAS signal are provided in Table 3. In this unadjusted analysis based on the primary definition of conservation, the estimated proportion of lincRNAs with GWAS signal for non-conserved lincRNAs is less than the corresponding proportion for conserved lincRNAs for BMI (6.2% vs 6.9%, non-inferiority p > 0.05) and height (16.8% vs. 18.8%, non-inferiority p > 0.05) while this estimated proportion is greater in non-conserved compared to conserved lincRNAs for WHRadjBMI (5.7% vs. 5.0%, non-inferiority p < 0.01), HDL-C (1.0% vs. 0.7%, non-inferiority p < 0.001), LDL-C (1.2% vs 0.6%, non-inferiority p < 0.001), TGs (1.1% vs. 0.7%, non-inferiority p < 0.001), CAD (0.6 vs 0.4%, non-inferiority p < 0.001) and T2D (1.6% vs. 1.1%, non-inferiority p < 0.001).
Table 3.
GWAS signal counts by trait and conservation (unadjusted analysis)
| Conservation defined based on synteny: | ||||||
|---|---|---|---|---|---|---|
| No signal |
Signal (col %) | Total | % Signal | Test of non- inferiority* |
||
| WHRadjBMI (n=5635) |
Non-conserved | 1315 | 80 (27.6%) | 1395 | 5.7% | 0.00796 |
| Conserved | 4030 | 210 | 4240 | 5.0% | ||
| BMI (n=5635) |
Non-conserved | 1308 | 87 (23.0%) | 1395 | 6.2% | 0.345 |
| Conserved | 3949 | 291 | 4240 | 6.9% | ||
| Height (n=5319) |
Non-conserved | 968 | 195 (20.0%) | 1163 | 16.8% | 0.212 |
| Conserved | 3375 | 781 | 4156 | 18.8% | ||
| HDL (n=5395) |
Non-conserved | 1208 | 12 (29.3%) | 1220 | 1.0% | <0.001 |
| Conserved | 4146 | 29 | 4175 | 0.7% | ||
| LDL (n=5389) |
Non-conserved | 1203 | 14 (36.8%) | 1217 | 1.2% | <0.001 |
| Conserved | 4148 | 24 | 4172 | 0.6% | ||
| TGs (n=5389) |
Non-conserved | 1204 | 13 (29.5%) | 1217 | 1.1% | <0.001 |
| Conserved | 4141 | 31 | 4172 | 0.7% | ||
| CAD (n=5534) |
Non-conserved | 1301 | 8 (33.3%) | 1309 | 0.6% | <0.001 |
| Conserved | 4209 | 16 | 4225 | 0.4% | ||
| T2D (n=5616) |
Non-conserved | 1354 | 22 (31.4%) | 1376 | 1.6% | <0.001 |
| Conserved | 4192 | 48 | 4240 | 1.1% | ||
| Conservation defined based on synteny and expression: | ||||||
| No signal |
Signal (col %) | Total | % Signal | Test of non- inferiority* |
||
| WHRadjBMI (n=5607) |
Non-conserved | 3173 | 173 (59.9%) | 3346 | 5.2% | 0.0336 |
| Conserved | 2145 | 116 | 2261 | 5.1% | ||
| BMI (n=5607) |
Non-conserved | 3115 | 231 (61.3%) | 3346 | 6.9% | 0.0196 |
| Conserved | 2115 | 146 | 2261 | 6.5% | ||
| Height (n=5292) |
Non-conserved | 2481 | 579 (59.6%) | 3060 | 18.9% | 0.0160 |
| Conserved | 1840 | 392 | 2232 | 17.6% | ||
| HDL (n=5368) |
Non-conserved | 3100 | 28 (68.3%) | 3128 | 0.9% | <0.001 |
| Conserved | 2227 | 13 | 2240 | 0.6% | ||
| LDL (n=5362) |
Non-conserved | 3097 | 26 (68.4%) | 3123 | 0.8% | <0.001 |
| Conserved | 2227 | 12 | 2239 | 0.5% | ||
| TGs (n=5362) |
Non-conserved | 3094 | 29 (65.9%) | 3123 | 0.9% | <0.001 |
| Conserved | 2224 | 15 | 2239 | 0.7% | ||
| CAD (n=5506) |
Non-conserved | 3233 | 17 (70.8%) | 3250 | 0.5% | <0.001 |
| Conserved | 2249 | 7 | 2256 | 0.3% | ||
| T2D (n=5588) |
Non-conserved | 3280 | 47 (68.1%) | 3327 | 1.4% | <0.001 |
| Conserved | 2239 | 22 | 2261 | 1.0% | ||
Test of non-inferiority is based on delta=0.01.
Overall, these findings suggest that it is as likely for non-conserved lincRNAs as for conserved lincRNAs to include a GWAS-association SNP. In addition, a substantial number of GWAS associated lincRNAs are not conserved, as indicated by the column percentages in Supplement Table II. For example, 80/290 = 27.6% of lincRNAs with a GWAS signal for WHRadjBMI are non-conserved. The percentage of GWAS-associated lincRNAs that are not conserved ranges from 20% (for height) to 36.8% (for LDL-C).
Multivariable models
The results of multivariable modeling (Table 4, Figure 1, Supplement Figure II, Supplement Figure III) are consistent with findings of unadjusted analyses with the exception that the predicted probability of GWAS signal for BMI is now very slightly higher for non-conserved compared to conserved lincRNAs. The corresponding adjusted estimated odds ratio (OR) of conserved, relative to non-conserved, lincRNA association with traits is less than one for all traits except height (p < 0.05 for LDL-C; p > 0.05 for all other traits) and ranges from 0.451 [95% CI = (0.231, 0.878)] for LDL to 1.126 [95% CI = (0.947, 1.338)] for height.
Table 4.
Multivariable adjusted model estimates for effect of conservation on GWAS signal by trait.
| Conservation defined based on synteny: | |||||
|---|---|---|---|---|---|
| Estimate for Syntenic* |
Std. Error | z value | Pr(>∣z∣) | OR (95% CI) | |
| WHRadjBMI | −0.220 | 0.136 | −1.613 | 0.107 | 0.803 (0.614, 1.048) |
| BMI | −0.005 | 0.391 | −0.040 | 0.968 | 0.995 (0.775, 1.277) |
| Height | 0.118 | 0.088 | 1.343 | 0.179 | 1.126 (0.947, 1.338) |
| HDL | −0.412 | 0.346 | −1.189 | 0.235 | 0.663 (0.336, 1.306) |
| LDL | −0.796 | 0.340 | −2.341 | 0.019 | 0.451 (0.231, 0.878) |
| TG | −0.442 | 0.334 | −1.325 | 0.185 | 0.643 (0.334, 1.236) |
| CAD | −0.546 | 0.436 | −1.253 | 0.210 | 0.579 (0.247, 1.361) |
| T2D | −0.415 | 0.262 | −1.589 | 0.112 | 0.660 (0.395, 1.102) |
| Conservation defined based on synteny and expression: | |||||
| Est. for Syntenic & Expressed* |
Std. Error | z value | Pr(>∣z∣) | OR (95% CI) | |
| WHRadjBMI | −0.085 | 0.127 | −0.667 | 0.505 | 0.919 (0.716, 1.179) |
| BMI | −0.276 | 0.114 | −2.419 | 0.016 | 0.759 (0.607, 0.949) |
| Height | −0.155 | 0.075 | −2.077 | 0.038 | 0.856 (0.740, 0.991) |
| HDL | −0.634 | 0.348 | −1.820 | 0.069 | 0.530 (0.268, 1.050) |
| LDL | −0.745 | 0.364 | −2.047 | 0.041 | 0.475 (0.233, 0.969) |
| TG | −0.551 | 0.331 | −1.664 | 0.096 | 0.576 (0.301, 1.103) |
| CAD | −0.761 | 0.462 | −1.648 | 0.099 | 0.467 (0.189, 1.155) |
| T2D | −0.488 | 0.267 | −1.825 | 0.068 | 0.614 (0.363, 1.037) |
Separate multivariable models are fitted for each trait. Models are adjusted for number of SNPs (natural log transformed), GC content (natural log transformed) and TE coverage. In the model for the WHRadjBMI signal with conservation defined based on synteny, the OR corresponding to a one unit change in natural log GC content is 3.20 [95% CI = (1.36, 7.48), p=0.007] and the OR for one unit change in TE coverage is 0.837 [95% CI = (0.522, 1.35), p=0.459. This suggests that GC content is significantly associated with the probability of a GWAS signal for WHRadjBMI. Adjustment for these additional covariates supports the unadjusted finding that the likelihood for a non-conserved lincRNAs to include a GWAS signal SNP is similar to that of a conserved lincRNA.
Figure 1.

Predicted probabilities of GWAS signal for conserved and non-conserved lincRNAs.
Predicted probabilities and corresponding 95% prediction intervals are calculated based on multivariable models using average of observed median values for GC content and TE coverage and observed trait-specific median number of SNPs. The predicted probability of GWAS signal is greater for non-conserved lincRNAs than conserved lincRNAs for all traits considered except height based on the primary definition of conservation. The results based on the secondary definition of conservation are consistent though in this case, the predicted probably of GWAS signal is greater for non-conserved lincRNAs than conserved lincRNAs for all traits including height (results not shown). The consistently overlapping confidence intervals suggest that the likelihood of GWAS association for conserved and non-conserved lincRNAs is comparable and, therefore, the traditional metrics of conservation for prioritizing lncRNAs for functional studies needs to be reconsidered.
Illustrative examples
As illustrative examples, Supplement Figure IV presents locus plots for several examples of genetic loci containing non-conserved as well as conserved lincRNAs that are associated with CAD and WHRadjBMI, two well-studied and clinically important cardiometabolic traits.
Secondary analysis using alternative definitions of conservation
Using the secondary definition of conservation that requires lincRNA expression in mouse and human as well as synteny, the predicted probability of GWAS signal is higher in non-conserved lincRNAs compared to conserved lincRNAs for all traits (p < 0.05 for BMI, height and LDL-C; p > 0.05 for all other traits, Table 4, Supplement Figure III). Notably, for both definitions of conservation, the point estimate for the probability of GWAS signal is consistently greater in non-conserved lincRNAs compared to conserved lincRNAs. Although this difference is not statistically significant for most traits considered individually, the overall trend suggests that the notion that GWAS signal would be lower in non-conserved regions needs to be reconsidered. Similar to the first definition of conservation, a substantial number of GWAS associated lincRNAs are not conserved based on the secondary definition (Supplement Table II). In this case, 173/289 = 59.9% of GWAS-associated lincRNAs for WHRadjBMI are non-conserved. This percentage of GWAS-associated lincRNAs that are not conserved, based on the secondary definition, ranges from 59.6% (for height) to 70.8% (for CAD).
Additional analyses
In order to compare the strength of lincRNA GWAS signals, we plotted the density of the maximum within lincRNA SNP-level z-score among trait-associated lincRNAs for conserved and non-conserved lincRNAs using our primary syntenic definition of conservation (Supplement Figure V). No apparent trend is observed to suggest that the magnitude of the association signal in conserved lincRNAs is greater than non-conserved lincRNAs.
To probe features of lincRNAs that were unclassified in our primary syntenic definition of conservation (i.e., the 18.5% lincRNAs that lack two neighboring PCGs within 900Kb of their start and end positions), counts and associated models comparing the set of unclassified lincRNAs to lincRNAs that are classified as either conserved or non-conserved are provided in Supplement Tables III and IV. These results generally suggest a lower probability of GWAS signal in more isolated genomic regions within which the majority of unclassified lincRNAs is found.
In exploratory analysis of sequence-level conservation, the distribution of lincRNA level phastCons scores by GWAS-association for WHRadjBMI and CAD are provided in Supplement Figure VI. For lincRNAs associated with compared to lincRNAs not associated with these traits, the median phastCons score is higher in lincRNAs associated with WHRadjBMI (Wilxocon rank sum test p-value<0.001, left hand panel) but not lincRNAs associated with CAD (Wilxocon rank sum test p-value=0.310, right hand panel). Although there is a statistically significant difference in the median phastCons score for WHRadBMI, the distribution of phastCons for WHRadjBMI associated lincRNAs ranges from 0 to 1 with a large proportion of relatively low scores and a low average phastCons score for WHRadjBMI- as well as for CAD- associated lincRNAs.
To explore lincRNA regulatory and functional features, we examined whether neighboring PCGs of conserved and non-conserved disease-associated lincRNAs were enriched in different pathways that might hint at differences in their regulatory functions in cardiometabolic traits. Using WHRadjBMI as an example, we performed pathway-based analysis using Database for Annotation, Visualization and Integrated Discovery (DAVID) (DAVID; https://david.ncifcrf.gov/) 31, 32 based on neighboring PCGs of trait-associated conserved and non-conserved lincRNAs. Each interrogation of DAVID categories showed similar findings so we present the results from UniProt Keyword (UP_Keyword) annotations in Supplement Table V. For WHRadjBMI-associated lincRNAs, biological processes were quite different for PGCs at conserved versus those at non-conserved lincRNAs - PCG neighbors of conserved lincRNA are significantly enriched in transcriptional regulation and DNA-binding whereas PCG neighbors of non-conserved lincRNA enrich for major histocompatibility complex I, immunity and cell division.
Discussion
A large portion of human lncRNAs lack conservation; yet, emerging evidence suggest non-conserved lncRNAs are functional 1, 4-15, 26, 33, 34. Motivated by this, we evaluated the likelihood that non-conserved lincRNA loci have genetic association with complex human cardiometabolic traits and compared this to the pattern of association for conserved lincRNAs. Focusing on eight established cardiometabolic disease-related traits 35, 36, we found that non-conserved lincRNAs have a similar likelihood of associating with cardiometabolic traits as conserved lincRNAs and that this association was broadly consistent across different definitions of conservation and different cardiometabolic traits. Moreover, approximately one third of trait-associated lincRNAs loci were non-conserved based on a syntenic definition of conservation and closer to two thirds were not conserved based on a more rigorous definition that included both synteny and expression in mouse. These findings suggest that the traditional notion of conservation driving prioritization for functional and translational follow-up of human cardiometabolic genomic discoveries may need to be revised in the context of the abundance of non-conserved lincRNAs in the human genome and their apparent predilection to associate with complex disease traits.
Species conservation, at DNA and protein sequence levels, has been considered an important feature, and often used for primary triage, when determining whether a PCG is likely to be functional. This perspective is reinforced by decades of using model organisms particularly mouse genetic models, relative to human or primate studies, to study in vivo function. However, a primary focus on conservation and use of mouse models may be de-prioritize important genetic signals for human diseases when considering genomic and regulatory features, including alternative splicing, tissue-specific enhancers and lincRNAs, that are prominent features of primate evolution 37. Although the protein coding genome is largely conserved between primates and non-primates, many cell-specific regulatory features are not conserved outside primates. This should not be altogether surprising because the specialized cell and organ functions that have emerged with primate evolution cannot be explained by changes in numbers of PCGs. This lack of conservation is particularly marked for lincRNAs and our work 26, 27 and that of others 2, 3 suggests that the majority of human lincRNAs is not conserved in mice.
An alternative measure of conservation that is applied to PCGs is base pair sequence homology 4, 38. However, human lincRNAs that are syntenic, expressed in mouse tissues and functionally conserved often have very limited nucleotide sequence homology across species 2-4. For this reason, we focus in this work on genomic synteny between human and mouse as a primary measure of conservation. In our exploratory analysis of sequence conservation, while the central tendency of phastCons scores is higher in WHRadjBMI associated lincRNAs compared to non-associated lincRNAs, the low average phastCons score for WHRadjBMI- and CAD-associated lincRNAs, relative to PCGs, confirms a low sequence-level conservation for trait-associated lincRNAs. This suggests poor utility of sequence level conservation scores in discriminating disease-associated from non-disease-associated lincRNAs.
While it has been proposed that many non-conserved lncRNA molecules that are identified through RNA-seq technologies may be non-functional, several lines of evidence suggest that this is not the case. Genomic markers of function including tissue-enrichment, binding of tissue-specific transcription factors at lncRNA enhancers and promoters, and regulation in response to physiological stressors, do not differ significantly between conserved and non-conserved myeloid and other tissue lincRNAs 26, 27, 39, 40. Several groups have also published genomic criteria, not dependent simply on conservation, and experimental methods, including CRISPR screens, to predict lncRNA functionality and prioritize candidates 1, 4, 5, 9. Multiple examples have emerged of lincRNAs that overlap loci for human cardiometabolic traits 1, 12, 15, including ANRIL, H19, MALAT1, MEXIS, LOC157273, and LASER 6-8, 10, 11, 13, 14. Of these, there are several examples of conserved (syntenic) lincRNAs including MALAT1 and LOC157273 (RP11-10A14.4). There are also examples of functionally characterized non-conserved lincRNAs at loci for cardiometabolic disease traits despite limited functional studies including H19 which also has been shown to have higher plasma levels of H19 in patients with CAD 34.
In a recent pre-publication, the GTEx consortium performed “colocalization” analysis connecting genetic variation, gene expression and traits for a set of 690 human lncRNAs by integrating results from GWAS for 48 traits and expression quantitative trait loci (eQTL) for 48 tissues in the latest GTEx v8 data 1. Of 4,694 significant eQTL-GWAS SNP colocalization events for these lncRNAs and traits, a striking 80% lacked any colocalization with protein-coding genes 1. Although the GTEx work did not focus on measures of lncRNA conservation, our current findings suggest that a large proportion of lncRNAs that colocalize at loci for complex cardiometabolic traits lacks conservation in mice. Further, many primate-specific lincRNAs, not found in rodents or other model organisms, have emerged as important regulators in cellular processes, such as pluripotency and differentiation, and as noted above several have been implicated in human cardiometabolic disorders 5, 26, 41-44. These data and our exploratory finding of differences in gene-pathway enrichment for neighboring PCGs suggest there may be utility in considering regulatory and functional features as well as disease association, rather than an initial triage using conservation, to identify and prioritize human lincRNAs for translational study.
A reluctance to study non-conserved lncRNAs also may hamper the development of rigorous and reproducible model systems to address pathophysiological functions of non-conserved lncRNAs and other genomic elements. Recent advances in tissue engineering have established stem cell-based organoids as “near-physiological” systems to study human physiology and diseases 45, 46. Modulation of PCGs and microRNAs by RNAi or transgene have been used in non-human primates in translational or pre-clinical studies. However, non-human primates are scarce and costly, limiting feasibility. Much work on functional models is needed including remains to be done transgene approaches that can express primate-specific lincRNAs in non-primate animal models – indeed, a few studies show that protein or RNA partners of such lincRNAs are conserved and can interact with primate-specific lincRNAs in non-primate models e.g., 41. Bacterial artificial chromosome (BAC) transgene mouse models can include the gene body and large fragments of genomic regulatory DNA of non-conserved lincRNA loci to drive human lincRNA expression in mouse models in vivo 47. An additional in vivo approach is to engraft human cells expressing primate-specific lincRNAs in rodent models with immune deficiency as has been used to study the roles of human lincRNA in tumor development and metastasis 48, 49.
In our analyses, a substantial subset of lincRNAs (18.5%) were characterized as “unclassified” in terms of synteny because they lacked PCG within the published range of 900kb 2 that we applied to examine PCGs upstream or downstream of a given lincRNA. These unclassified lincRNAs tend to be longer with a lower GC content and higher TE coverage relative to classified lincRNAs (Supplement Table I). Using an established minimum range cut-point for “gene deserts” of absence of a PCG within 250Kb upstream and 250Kb downstream 50-52, 55.1% of unclassified compared to 7.3% of classified lincRNAs reside within gene deserts. Gene deserts, and lincRNAs within such regions, are enriched in ancient duplications, have lower GC content and lower conservation than other parts of the human genome, and may have specific long-distance cis- and trans- regulatory functions related to their unique evolutionary and genomic characteristics 50-52. Although unrelated to our primary focus on the role of lincRNA conservation in human complex diseases, further study of these unique unclassified lincRNAs in gene deserts is of interest to the field. Indeed, there are well recognized loci in gene desert that associate with complex traits at GWAS including the 9p21 locus with CAD and T2D 53 and the 8q24 locus with several cancers 54. Our analyses, however, suggest a lower probability of GWAS signal for unclassified lincRNAs that lie in more isolated genomic regions and gene deserts compared to classified lincRNAs (Supplement Tables III and IV).
Our study has several limitations. For example, the are no established standards in the field regarding the definition of lincRNA conservation and therefore we chose somewhat arbitrary, although previously published 2, 3, 26, 27, definitions of synteny. For example, we excluded certain lincRNAs that lacked PCGs within 900Kb of lincRNAs. We also merged overlapping lincRNAs and this may not accurately reflect the precise lincRNA and isoform expression in individual tissues or across tissues. Although GENCODE as a resource for lincRNAs is widely used and well cross-validated, it may lack sensitivity to many lncRNAs as expression of some functional lincRNAs can be highly context specific and found at low levels and therefore missed in the GENCODE resource. Indeed, our group 26, 27 and others 55 have published such findings in several prior papers. Although our trait selection is comprehensive, we did not interrogate an all-encompassing set of cardiometabolic traits. Rather, we focused primarily on traits with adequately powered GWAS datasets that provided sufficient numbers of trait-associated SNPs in both conserved and non-conserved lincRNAs. In addition, our use of large SNP-based GWAS datasets rather than whole genome data did not permit interrogation or rare functional variation and lincRNA exonic regions and did not provide the level of coverage required for a fine-mapping subset analysis focused on SNPs within exons and introns. As larger whole genome datasets emerge, there will be opportunities to focus on rare functional variations in lincRNAs as well as analysis that can weight for enriched signals in 5’, 3’, exonic and intronic SNPs and regions of lincRNAs.
In conclusion, we found that non-conserved lincRNAs have a non-trivial and consistent likelihood of association with a broad array of complex cardiometabolic traits. Indeed, we found that non-conserved lincRNAs associate with cardiometabolic traits at a rate that is consistent with conserved lincRNAs, that these finding are robust across different definitions of conservation, and strikingly that across all traits as much as two thirds of GWAS-associated lincRNAs may be non-conserved depending on the definition applied. Given these findings, computational, high-throughput functional and human pathophysiological approaches 1, 4, 5, 9, rather than traditional metrics of conservation, should be applied to prioritize lncRNAs for functional studies. Expansion of research strategies using non-traditional model systems is urgently required to address physiological and pathophysiological functions of non-conserved lncRNAs and other genomic elements in human cardiometabolic disorders.
Data and code availability
All data used in the analyses contributing to this manuscript are publicly available at the sites indicated. Code is available upon request to the corresponding author.
Supplementary Material
Highlights.
Of 1000s of long intergenic non-coding RNAs (lincRNAs) in the human genome, non-conserved lincRNAs associate with cardiometabolic traits at a rate that is similar to that for conserved lincRNAs.
These findings are consistent across multiple cardiometabolic traits and persist using different definitions of conservation.
For all cardiometabolic traits, more than one third of GWAS-associated lincRNAs are non-conserved based on syntenic positional conservation and this increases to as much as two thirds using a more stringent definition of conservation.
Acknowledgements.
We remember Xuan Zhang Ph.D., a friend and outstanding scientist, who tragically and suddenly passed away while contributing to an early draft of this manuscript.
Sources of Funding.
Support for this research was provided by grants from the National Institutes of Health (NIH) R01 GM127862 and R15 GM126485 to ASF, and R01 HL132561, R01 HL113147 and K24 HL107643 to MPR.
Abbreviations
- BMI
Body mass index
- CAD
Coronary artery disease
- GWAS
Genome-wide association study
- HDL-C
High-density lipoprotein cholesterol
- LDL-C
Low-density lipoprotein cholesterol
- LincRNA
Long intergenic non-coding RNA
- OR
Odds ratio
- PCG
Protein-coding gene
- T2D
Type-2 diabetes
- TGs
Triglycerides
- WHRadjBMI
Waist to hip ratio adjusted for body mass index
Footnotes
Disclosures.
All authors declare no competing interests
References
- 1.de Goede OM, Ferraro NM, Nachun DC, Rao AS, Aguet F, Barbeira AN, et al. Long non-coding rna gene regulation and trait associations across human tissues. bioRxiv. 2019 [Google Scholar]
- 2.Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I. Principles of long noncoding rna evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015;11:1110–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, et al. The evolution of lncrna repertoires and expression patterns in tetrapods. Nature. 2014;505:635–640 [DOI] [PubMed] [Google Scholar]
- 4.Gil N, Ulitsky I. Regulation of gene expression by cis-acting long non-coding rnas. Nat Rev Genet. 2019 [DOI] [PubMed] [Google Scholar]
- 5.Zhang X, Li DY, Reilly MP. Long intergenic noncoding rnas in cardiovascular diseases: Challenges and strategies for physiological studies and translation. Atherosclerosis. 2019;281:180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shan K, Jiang Q, Wang XQ, Wang YN, Yang H, Yao MD, et al. Role of long non-coding rna-rncr3 in atherosclerosis-related vascular dysfunction. Cell death & disease. 2016;7:e2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sallam T, Jones M, Thomas BJ, Wu X, Gilliland T, Qian K, et al. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding rna. Nat Med. 2018;24:304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, et al. Long noncoding rna malat1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389–1397 [DOI] [PubMed] [Google Scholar]
- 9.Mattioli K, Volders PJ, Gerhardinger C, Lee JC, Maass PG, Mele M, et al. High-throughput functional analysis of lncrna core promoters elucidates rules governing tissue specificity. Genome Res. 2019;29:344–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C, Hu Z, Zhang W, Yu J, Yang Y, Xu Z, et al. Regulation of cholesterol homeostasis by a novel long non-coding rna laser. Scientific reports. 2019;9:7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inouye M, Ripatti S, Kettunen J, Lyytikainen LP, Oksala N, Laurila PP, et al. Novel loci for metabolic networks and multi-tissue expression studies reveal genes for atherosclerosis. PLoS Genet. 2012;8:e1002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holdt LM, Teupser D. Long noncoding rna anril: Lnc-ing genetic variation at the chromosome 9p21 locus to molecular mechanisms of atherosclerosis. Front Cardiovasc Med. 2018;5:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao W, Zhu M, Wang H, Zhao S, Zhao D, Yang Y, et al. Association of polymorphisms in long non-coding rna h19 with coronary artery disease risk in a chinese population. Mutation research. 2015;772:15–22 [DOI] [PubMed] [Google Scholar]
- 14.Congrains A, Kamide K, Oguro R, Yasuda O, Miyata K, Yamamoto E, et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of anril and cdkn2a/b. Atherosclerosis. 2012;220:449–455 [DOI] [PubMed] [Google Scholar]
- 15.Ballantyne RL, Zhang X, Nunez S, Xue C, Zhao W, Reed E, et al. Genome-wide interrogation reveals hundreds of long intergenic noncoding rnas that associate with cardiometabolic traits. Hum Mol Genet. 2016;25:3125–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winkler TW, Justice AE, Graff M, Barata L, Feitosa MF, Chu S, et al. Correction: The influence of age and sex on genetic associations with adult body size and shape: A large-scale genome-wide interaction study. PLoS Genet. 2016;12:e1006166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Magi R, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of european ancestry. Hum Mol Genet. 2019;28:166–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rask-Andersen M, Karlsson T, Ek WE, Johansson A. Genome-wide association study of body fat distribution identifies adiposity loci and sex-specific genetic effects. Nat Commun. 2019;10:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lumish HS, O'Reilly M, Reilly MP. Sex differences in genomic drivers of adipose distribution and related cardiometabolic disorders: Opportunities for precision medicine. Arterioscler Thromb Vasc Biol. 2019:ATVBAHA119313154 [DOI] [PubMed] [Google Scholar]
- 21.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50:1505–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, et al. Gencode reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Xue C, Lin J, Ferguson JF, Weiner A, Liu W, et al. Interrogation of nonconserved human adipose lincrnas identifies a regulatory role of linc-adal in adipocyte metabolism. Sci Transl Med. 2018;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Xue C, Wang Y, Shi J, Zhang X, Li W, et al. Deep rna sequencing uncovers a repertoire of human macrophage long intergenic noncoding rnas modulated by macrophage activation and associated with cardiometabolic diseases. J Am Heart Assoc. 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson CP, Hamby SE, Saleheen D, Hopewell JC, Zeng L, Assimes TL, et al. Genetically determined height and coronary artery disease. N Engl J Med. 2015;372:1608–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smit AFA, Hubley R, Green P. Repeatmasker open-3.0. http://www.repeatmasker.org 1996-2010 [Google Scholar]
- 31.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nature protocols. 2009;4:44–57 [DOI] [PubMed] [Google Scholar]
- 32.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stender S, Smagris E, Lauridsen BK, Kofoed KF, Nordestgaard BG, Tybjaerg-Hansen A, et al. Relationship between genetic variation at ppp1r3b and levels of liver glycogen and triglyceride. Hepatology. 2018;67:2182–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z, Gao W, Long QQ, Zhang J, Li YF, Liu DC, et al. Increased plasma levels of lncrna h19 and lipcar are associated with increased risk of coronary artery disease in a chinese population. Scientific reports. 2017;7:7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karlsson T, Rask-Andersen M, Pan G, Hoglund J, Wadelius C, Ek WE, et al. Contribution of genetics to visceral adiposity and its relation to cardiovascular and metabolic disease. Nat Med. 2019;25:1390–1395 [DOI] [PubMed] [Google Scholar]
- 36.Emdin CA, Khera AV, Natarajan P, Klarin D, Zekavat SM, Hsiao AJ, et al. Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. JAMA. 2017;317:626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ule J, Blencowe BJ. Alternative splicing regulatory networks: Functions, mechanisms, and evolution. Mol Cell. 2019;76:329–345 [DOI] [PubMed] [Google Scholar]
- 38.Ulitsky I Evolution to the rescue: Using comparative genomics to understand long non-coding rnas. Nat Rev Genet. 2016;17:601–614 [DOI] [PubMed] [Google Scholar]
- 39.Ferguson JF, Xue C, Gao Y, Tian T, Shi J, Zhang X, et al. Tissue-specific differential expression of novel genes and long intergenic noncoding rnas in humans with extreme response to evoked endotoxemia. Circ Genom Precis Med. 2018;11:e001907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Shi J, Hachet MA, Xue C, Bauer RC, Jiang H, et al. Crispr/cas9-mediated gene editing in human ipsc-derived macrophage reveals lysosomal acid lipase function in human macrophages-brief report. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:2156–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rani N, Nowakowski TJ, Zhou H, Godshalk SE, Lisi V, Kriegstein AR, et al. A primate lncrna mediates notch signaling during neuronal development by sequestering mirna. Neuron. 2016;90:1174–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Durruthy-Durruthy J, Sebastiano V, Wossidlo M, Cepeda D, Cui J, Grow EJ, et al. The primate-specific noncoding rna hpat5 regulates pluripotency during human preimplantation development and nuclear reprogramming. Nat Genet. 2016;48:44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao T, Liu L, Li H, Sun Y, Luo H, Li T, et al. Long noncoding rna adinr regulates adipogenesis by transcriptionally activating c/ebpalpha. Stem Cell Reports. 2015;5:856–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rigoutsos I, Lee SK, Nam SY, Anfossi S, Pasculli B, Pichler M, et al. N-blr, a primate-specific non-coding transcript leads to colorectal cancer invasion and migration. Genome Biol. 2017;18:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin X, Mead BE, Safaee H, Langer R, Karp JM, Levy O. Engineering stem cell organoids. Cell Stem Cell. 2016;18:25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease. Nat Cell Biol. 2016;18:246–254 [DOI] [PubMed] [Google Scholar]
- 47.Van Keuren ML, Gavrilina GB, Filipiak WE, Zeidler MG, Saunders TL. Generating transgenic mice from bacterial artificial chromosomes: Transgenesis efficiency, integration and expression outcomes. Transgenic Res. 2009;18:769–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Pitchiaya S, Cieslik M, Niknafs YS, Tien JC, Hosono Y, et al. Analysis of the androgen receptor-regulated lncrna landscape identifies a role for arlnc1 in prostate cancer progression. Nat Genet. 2018;50:814–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Zeng X, Wang N, Zhao W, Zhang X, Teng S, et al. Long noncoding rna dancr, working as a competitive endogenous rna, promotes rock1-mediated proliferation and metastasis via decoying of mir-335-5p and mir-1972 in osteosarcoma. Mol Cancer. 2018;17:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor J Clues to function in gene deserts. Trends Biotechnol. 2005;23:269–271 [DOI] [PubMed] [Google Scholar]
- 51.Ovcharenko I, Loots GG, Nobrega MA, Hardison RC, Miller W, Stubbs L. Evolution and functional classification of vertebrate gene deserts. Genome Res. 2005;15:137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302:413. [DOI] [PubMed] [Google Scholar]
- 53.Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huppi K, Pitt JJ, Wahlberg BM, Caplen NJ. The 8q24 gene desert: An oasis of non-coding transcriptional activity. Front Genet. 2012;3:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Volders PJ, Anckaert J, Verheggen K, Nuytens J, Martens L, Mestdagh P, et al. Lncipedia 5: Towards a reference set of human long non-coding rnas. Nucleic Acids Res. 2019;47:D135–D139 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in the analyses contributing to this manuscript are publicly available at the sites indicated. Code is available upon request to the corresponding author.