

Abstract
To shed light on the impact of systemic physiology on the pathology of traumatic brain injury (TBI), we examine the effects of TBI (concussive injury) and dietary fructose on critical aspects of lipid homeostasis in the brain and liver of young-adult rats. Lipids are integral components of brain structure and function, and the liver has a role on the synthesis and metabolism of lipids. Fructose is mainly metabolized in the liver with potential implications for brain function. Lipidomic analysis accompanied by unbiased sparse partial least squares discriminant analysis (sPLS-DA) identified lysophosphatidylcholine (LPC) and cholesterol ester (CE) as the top lipid families impacted by TBI and fructose in the hippocampus, and only LPC (16:0) was associated with hippocampal-dependent memory performance. Fructose and TBI elevated liver pro-inflammatory markers, interleukin-1α (IL-1α), Interferon-γ (IFN-γ) that correlated with hippocampal-dependent memory dysfunction, and monocyte chemoattractant protein-1 (MCP-1) positively correlated with LPC levels in the hippocampus. The effects of fructose were more pronounced in the liver, in agreement with the role of liver on fructose metabolism and suggest that fructose could exacerbate liver inflammation caused by TBI. The overall results indicate that TBI and fructose interact to influence systemic and central inflammation by engaging liver lipids. The impact of TBI and fructose diet on the periphery provides a therapeutic target to counteract the TBI pathogenesis.
Keywords: Lysophosphatidylcholine, cPLA2, liver, fructose, traumatic brain injury, cognition
Introduction
Traumatic brain injury (TBI) is a health burden in civilian and military populations in which severe cognitive and emotional deficits can evolve to serious neuropsychiatric disorders. Brain contains the second highest concentration of lipids in the human body, in which lipids are main components of plasma membranes, myelin sheaths, and provide signaling infrastructure to events underlying higher order functions such as learning and memory [1, 2]. Alterations in brain lipid and their downstream products is an important burden in the TBI pathology [3], and this is an understudied area of research. An additional complication is that dietary factors affect lipid homeostasis and function acting at various levels in the brain and body. The liver plays a critical role on lipid metabolism with the potential to influence brain function [4, 5], and the extent of the TBI pathology [6]. New evidence suggests that the liver plays a role in the acute inflammatory response to TBI [7–9]. In turn, recent studies indicate that concussive brain injury in rodents causes disruptions in lipid homeostasis in the liver, with subsequent effects on inflammatory events across body and brain [9, 10]. The double effects of TBI on brain and liver has prompted us to evaluate how TBI coordinate its effects in the regulation of lipid homeostasis in brain and liver, and how this interplay could be influenced by fructose.
Fructose is widely consumed in the Western society but has only recently been recognized as a powerful contributor to the current epidemic of metabolic disorders [11]. A fructose diet affects lipid peroxidation in the brain [12] and the liver [10], and neuronal plasticity and signaling are compromised in the brain [13]. The fact that fructose is metabolized in the liver makes of fructose a critical instigator of lipid disorders as observed on non-alcoholic fatty liver disease (NAFLD) and systemic inflammation [14, 15] and the devastating neurological sequel of hepatic neuropathy [16, 17]. Proper liver function is crucial for brain plasticity since several brain lipids are metabolized or processed in the liver [18]. The liver is the primary organ for converting excess of carbohydrates into fatty acids and triglycerides [19], which are exported to other tissues such as brain [20]. The current studies provide information about the action of fructose in the TBI pathology acting on the brain and liver, with translational relevance for the design of dietary programs to reduce the burden of TBI.
The metabolism of brain lipids is tightly regulated through a liver-brain interaction in which the autonomic nervous system plays a crucial role. Liver dysfunction can be aggravated by consumption of fructose as fructose is mainly metabolized in the liver where is converted into fatty acids that can reach the brain and expand the inflammatory reaction started in the periphery [21, 22]. Lysophosphatidylcholine (LPC) is a major component of oxidized low-density lipoprotein, and is produced by hydrolysis of Phosphatidylcholine (PC) under the action of phospholipase A (cPLA2). LPC has a pro-inflammatory action, and it has been associated with atherosclerosis [23, 24], diabetes [25] and liver disease [26], and its levels are increased in plasma of Alzheimer’s disease (AD) patients [27] and in the brain of an AD mice model [28]. In turn, cholesterol ester (CE) has important actions on brain cellular signaling, metabolic disease, and inflammation especially in the context of neurodegeneration [29, 30].
2. Methods
2.1. Animals
Male Sprague-Dawley rats (Charles River Laboratories) were housed in polyacrylic cages and maintained under standard housing conditions, with 12 h light/dark cycle. All experiments were performed in accordance with the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals. The animal studies and experimental procedures were approved by the University of California at Los Angeles Chancellor’s Animal Research Committee (ARC). Rats were randomly assigned to either regular or fructose (15 % v/v) drinking water for 3 weeks before they received FPI.
2.2. Fluid Percussion Injury (FPI)
In brief, animals were anesthetized by using a Laboratory Animal Anesthesia System (VetEquip Inc., CA, USA) that provides a mixture of isoflurane and oxygen. Animals were maintained in a deep anesthetic state during surgery with 2–5% isoflurane mixed with 100% O2 at a flow rate of 0.4 l/min via nose cone.3.0 mm-diameter craniotomy was made over the left parietal cortex, 3.0mm posterior to bregma, and 6.0mm lateral (left) to the midline with a high-speed drill (Dremel, WI, USA). A plastic injury cap was placed over the craniotomy with dental cement. When the dental cement hardened, the cap was filled with 0.9% saline solution. Anesthesia was discontinued and the injury cap was attached to the fluid percussion device. At the first sign of hind-limb withdrawal to a paw pinch, a moderate fluid percussion pulse (2.7 atm) was administered to the epidural space. Immediately upon responding to a paw pinch, anesthesia was restored, and the skull was sutured. Neomycin was applied on the suture and the rats were placed in a heated recovery chamber before being returned to their cages. Sham animals underwent an identical preparation with the exception of the lesion. Moderate FPI was used based on its capacity to model concussive brain injury involving minimal neuronal death and bleeding while resulting in reproducible cognitive impairment [31, 32].
2.3. Barnes Maze test
Animals were trained with two trials per day for five consecutive days to locate a dark escape chamber hidden underneath a hole positioned around the perimeter of a disk on the Barnes maze (BM). A trial was started by placing the animal at the center of the maze covered under a cylindrical start chamber; after a 10s delay, the start chamber was raised. The disk was brightly illuminated by four overhead halogen lamps to provide aversive stimulus. A training session ended after the animal had entered the escape chamber or when a pre-determined time (5 min) had elapsed, whichever came first. Rats were then randomly assigned to fructose feeding or water for 3 weeks. FPI was then performed in half of the animals and memory retention (two trials) was evaluated 10 days later. We measured the latency to the first approach to the target hole to evaluate memory performance. The maze was manufactured from acrylic plastic to form a disk 1.5 cm thick and 115 cm in diameter, with 18 evenly spaced 7 cm holes at its edges. All trials were recorded simultaneously by a video camera installed directly overhead at the center of the maze.
2.4. Tissue collection
10 days after FPI the animals were killed immediately by decapitation and the fresh brains and livers were dissected out and stored at −70°C until use.
2.5. Lipidomic analysis
50-100 mg of tissue were collected in tube pre-loaded with 2.8mm ceramic beads (Omni #19-628). Tissue is homogenized with PBS in the Omni Bead Ruptor Elite (3 cycles of 10 seconds at 5 m/s with a 10 second dwell time), and 3-6mg of homogenate were used for extraction (Bligh and Dyer extraction (Bligh, 1959)). Prior to biphasic extraction, a 13-lipid class Lipidyzer Internal Standard Mix is added to each sample (AB Sciex, 5040156). Following two successive extractions, pooled organic layers were dried in a Genevac EZ-2 Elite. Lipid samples were resuspended in 1:1 methanol/dichloromethane with 10mM Ammonium Acetate and transferred to robovials (Thermo 10800107) for analysis. Samples were analyzed on the Sciex Lipidyzer Platform for targeted quantitative measurement of 1100 lipid species across 13 classes. Differential Mobility Device on Lipidyzer is tuned with SelexION tuning kit (Sciex 5040141). Data analysis performed on Lipidyzer software and quantitative values were normalized to mg amounts used.
2.6. Western-Blot analysis
The tissues were homogenized in lysis buffer containing 137 mM NaCl, 20 mM Tris–HCl pH 8.0, 1%NP40, 10% glycerol, 1 mM phenylmethylsulfonylfluoride (PMSF), 10 μg/ml aprotinin, 0.1 mM benzethonium chloride, 0.5 mM sodium vanadate. The homogenates were then centrifuged at 12000g (4C) for 30 min; the supernatants were collected, and total protein concentration was determined according to MicroBCA procedure (Pierce, IL, USA), using bovine serum albumin (BSA) as standard. Briefly, 20 μg of total hippocampus protein or 25 μg of total liver protein was loaded in each well and separated by electrophoresis either on a 10% polyacrylamide gel or 4-15 % gel for ApoB (Biorad #4561086). Proteins were then transferred to a PVDF membrane (Millipore, MA, USA). Non-specific binding sites were blocked in Tris-buffered saline (TBS), pH 7.6, containing 5% non-fat dry milk. Membranes were rinsed in buffer (0.05% Tween-20 in TBS) and then incubated with one of the following specific primary antibodies anti-P-cPLA2 (SCT, 2831), anti-cPLA2 (SCT, 2832) anti-FAS (sc-48357), anti-ABCA1 (Novus NB 400-105), anti-ApoB (NB, 200-527) and anti-β-actin (sc-1616) or followed by anti-rabbit or anti-goat or anti-mouse IgG horseradish peroxidase-conjugate (1:10,000; Santa Cruz Biotechnology, CA, USA). After rinsing with buffer, the protein bands were detected using a chemiluminescent HRP substrate kit (WBKLS0500, Millipore). The membranes were scanned (Image Lab Software, Version 3.0, Bio Rad). The relative density of each band of interest was measured with the software Image J v 1.51. The intensity of the interest band was normalized to β-actin.
2.7. Rat cytokines detection
Cytokines in the liver were detected using a rat cytokine antibody array-membrane kit ab 133991 (abcam, MA), according to manufacturer protocol. The commercial Rat Cytokine Antibody Array detects both the precursor (uncleaved) and the cleaved mature form of IL-1α as the immunogen used to produce the antibodies is located within amino acids 115-270. For MCP-1 the full-length protein was used as immunogen to generate the antibody and the supplier cannot ascertain whether it is the cleaved or uncleaved form or both that is/are detected with ab133991. Briefly, liver tissue was homogenized in cell lysis buffer, 250 μg of total liver proteins was used, and membranes were scanned using chemiluminescence detection (Biorad). We measure the density of each spot in the array using Image J (v 1.51) and calculated the abundance of each cytokine normalized to the positive control.
2.8. Statistical Analysis
All groups were compared by multivariate analysis (sparse partial least squares discriminant analysis, sPLS-DA) to determine the lipid profile of each group. The abundance of each lipid, behavioral changes, and protein levels in the hippocampus and the liver were analyzed using a two-way ANOVA followed by Fisher LSD post-hoc (p≤0.05). Pearson correlation analysis was performed to assess associated among variables such as latency, lipid levels and cytokines levels. The data are presented as mean ± EMS, n =5-6 per group. The lipidomic data was analyzed in R 3.6.1 using MixOmics package and Graph Pad Prism 7.
3. Results
3.1. TBI and fructose consumption affect lipid species in the brain
Sparse partial least squares discriminant analysis (sPLS-DA) discriminated lipid profile separation on conglomerates of lipid species in response to the combined effects of TBI and fructose as is shown in the score plot (Fig. 1 A). The same analysis identified 60 over a total of 261 lipid species in the hippocampus. In particular, lipid species from the combination of both TBI and fructose (TF) showed a distinct lipid profile relative to the SW group (Fig. 1A). Specific lipid species such as LPC (16:0), CE (16:0), CE (20:4), CE (18:1) showed a higher contribution to the separation between the groups as evidenced in the top 30 lipid species according to comp2 values of sPLS-DA (Fig. 1B), in which, LPC appeared as the top contributor to the separation.
Figure 1. Lipidomic analysis showing the effects of TBI and fructose consumption in the hippocampus.
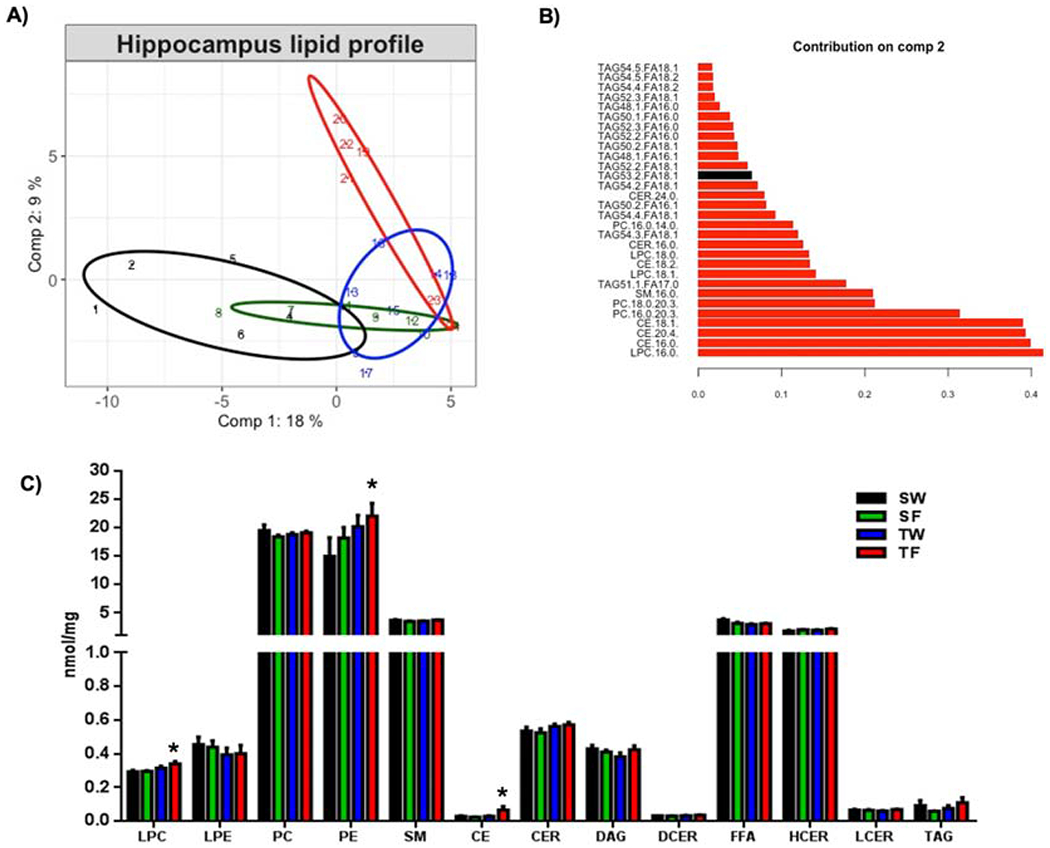
(A) Score plot of the sparse partial least squares analysis (sPLS-DA) showing the lipid profile of each experimental group. (B) Top 30 lipid species according to loading 2 values of the sPLS-DA. (C) shows concentrations of each lipid class in the different experimental groups. 2-way ANOVA with Fisher’s LSD post-test (*p<0.05 vs SW-group), Abbreviations: LPC, Lysophosphatidylcholine, LPE, Lysophosphatidylethanolamine, PC, Phosphatidylcholine, PE, Phosphatidylethanolamine, SM, Sphingomyelin, CE, Cholesterol ester, CER, Ceramides, DAG, Diacylglycerol, DCER, Dihydroceramides,FFA, Free fatty acids, HCER, Hexosyl ceramides, LCER, Lactosyl ceramides, TAG, Triglyceride. Animal groups: SW, Sham-Water; SF, Sham-Fructose; TW, TBI-Water; TF, TBI-Fructose.
Subsequent quantitative lipidomic analysis identified numerous lipid classes in the hippocampus (Fig. 1C), including Lysophosphatidylcholine (LPC), Lysophosphatidylethanolamine (LPE), Phosphatidylcholine (PC), Phosphatidylethanolamine (PE), Sphingomyelin (SM), Cholesterol ester (CE), Ceramides (CER), Diacylglycerol (DAG), Dihydroceramides (DCER), Free fatty acids (FFA), Hexosyl ceramides (HCER), Lactosyl ceramides (LCER) and Triglyceride (TAG). The actions of TBI and fructose diet were evidenced by increased levels of LPC, PE and CE lipid classes in response to TBI and fructose diet (TF) compared with sham/water group (SW) (Fig. 1C, *p < 0.05).
3.2. Effects of fructose and TBI on levels of LPC and cholesterol ester (CE) species in the hippocampus
The hippocampus is the main locus for the processing of spatial memory in the brain, and specific lipids may provide structural and signaling support. We used quantitative lipidomic to assess abundance of specific lipid species belonging to LPC and CE main lipid classes that contributed to the lipid profile separation. We assessed seven LPC species according to their fatty acyl composition, including saturated, monounsatured and polyunsaturated species (Fig. 2A). As predicted by the sPLS-DA profile analysis, quantitative lipidomic revealed a significant increase in LPC (16:0) and LPC (18:1) (Fig. 2A) contents in the hippocampus of the TF group as compared to SW and sham/fructose (SF) (*p<0.05) groups. In addition, we found that three species of CE (Fig. 2B): CE (16:0), CE (18:1), CE (22:6) were more abundant in the TF group as compared to SW or sham/fructose (SF) groups (*p<0.05; Fig. 2B) and compared to TBI/water group (TW) (#p<0.05; Fig. 2B). The analysis did not yield significant effects of TBI and fructose on any given specific PE species (data not shown) besides the effects on the total PE class.
Figure 2. Altered concentrations of Lysophosphatidylcholine (LPC, A) and Cholesteryl esters (CE, B) species in the hippocampus in response to TBI and fructose.
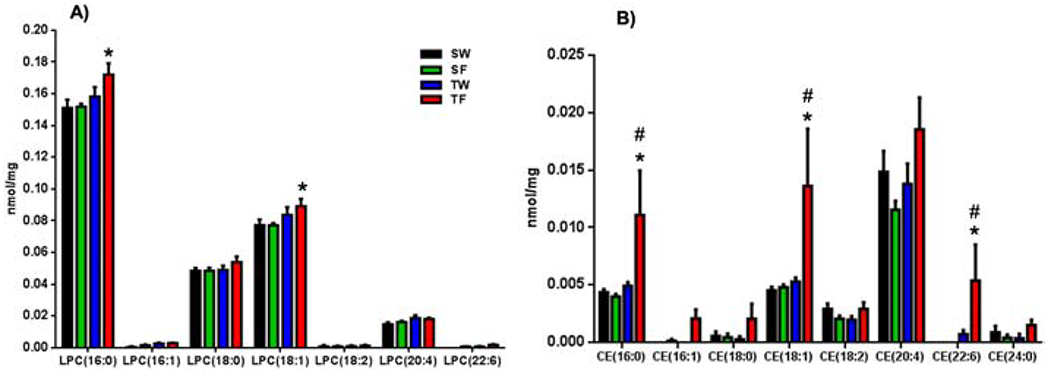
(A) Concentrations of LPC (A) and CE (B) species according to fatty acyl composition. Statistical significance was evaluated by 2-way ANOVA with Fisher’s LSD post-test (*p<0.05 vs SW group and SF group, # p<0.05 vs TW group).
3.3. Hippocampal LPC (16:0) levels changed in proportion to BM performance
The latency to find the escape hole in the BM serves as an indicator of memory performance, and trended to be higher in the TW group compared to the SW (†p = 0.09), while the TF-group showed a latency significantly higher than SW-group (*p≤0.05, Fig. 3 A). Next, we sought to determine a possible association between the two main lipid classes (LPC, CE) identified with the sPLS-DA vs behavior. Correlation analysis showed that escape latency changed in proportion with LPC 16:0 concentration (R=0.44, p=0.03; Fig. 3 B) but not with regards to CE 20:4 concentration (R=0.22, p=0.32; Fig. 3C). These results suggest that the proinflammatory action of LPC 16:0 could interfere with memory function. Given that LPC was the only lipid specie associated with behavior, the subsequent analysis was devoted to determine main aspects of the possible regulation of LPC along our paradigm.
Figure 3. LPC (16:0) levels change in proportion to BM performance.
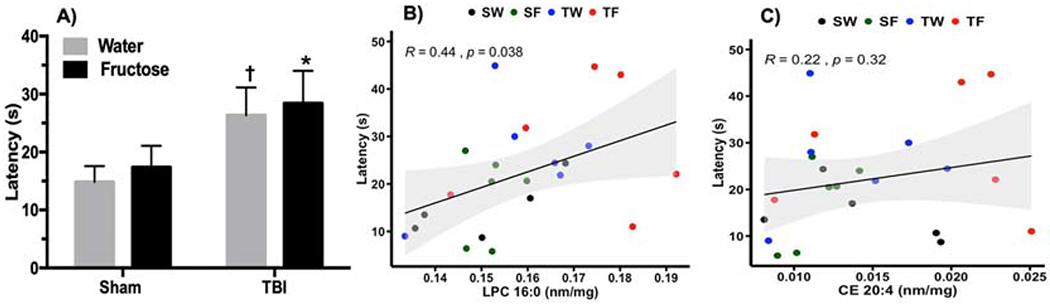
(A) Latency to find the escape hole during memory test in BM. 2-way ANOVA with Fisher’s LSD posttest (*p≤0.05 vs SW) († p = 0.09 vs SW). (B) Correlation analysis using the latency as a variable versus the concentration of LPC (16:0) (Pearson correlation, R=0.44, p=0.03). (C) Correlation analysis using the latency as a variable versus the concentration of CE (20:4) (Pearson correlation, R=0.22, p=0.32)
3.4. Action of fructose and TBI on select pro-inflammatory markers in liver and possible association with brain function
Given that fructose is largely metabolized in the liver, we wanted to examine possible effects of the interventions on lipid inflammation that could influence the brain. We evaluated the effects of the interventions on cytokine levels including interferon-γ (IFN-γ), interleukin 1-α (IL-1α), tumoral necrosis factor- α (TNF-α), and monocyte chemoattractant protein-1 (MCP-1) in the liver. We found that the combination of TBI and fructose (TF-group) elevated IFN-γ, IL-1α and MCP-1 levels in comparison to SW-group (*p≤0.05, Fig. 4 A). We used regression analysis to evaluate a possible association between liver inflammation and latency to find the escape hole in the BM test. Results showed that the latency in the BM changed in proportion to levels of IL-1α, (R=0.51, p=0.015; Fig. 4B) and TNF-α, R=0.40, p=0.06; Fig.4C). In addition, we searched for possible associations between the pro-inflammatory markers and levels of hippocampal LPC (16:0) and CE (16:0, 18:1, 20:0). We found a positive correlation between levels of liver MCP-1 and hippocampal LPC (R=0.48, p= 0.024; Figure 4D) but not CE.
Figure 4. Effects of TBI and fructose on pro-inflammatory cytokines (IFN-γ, IL-1α, MCP-1, TNF-α) in the liver.
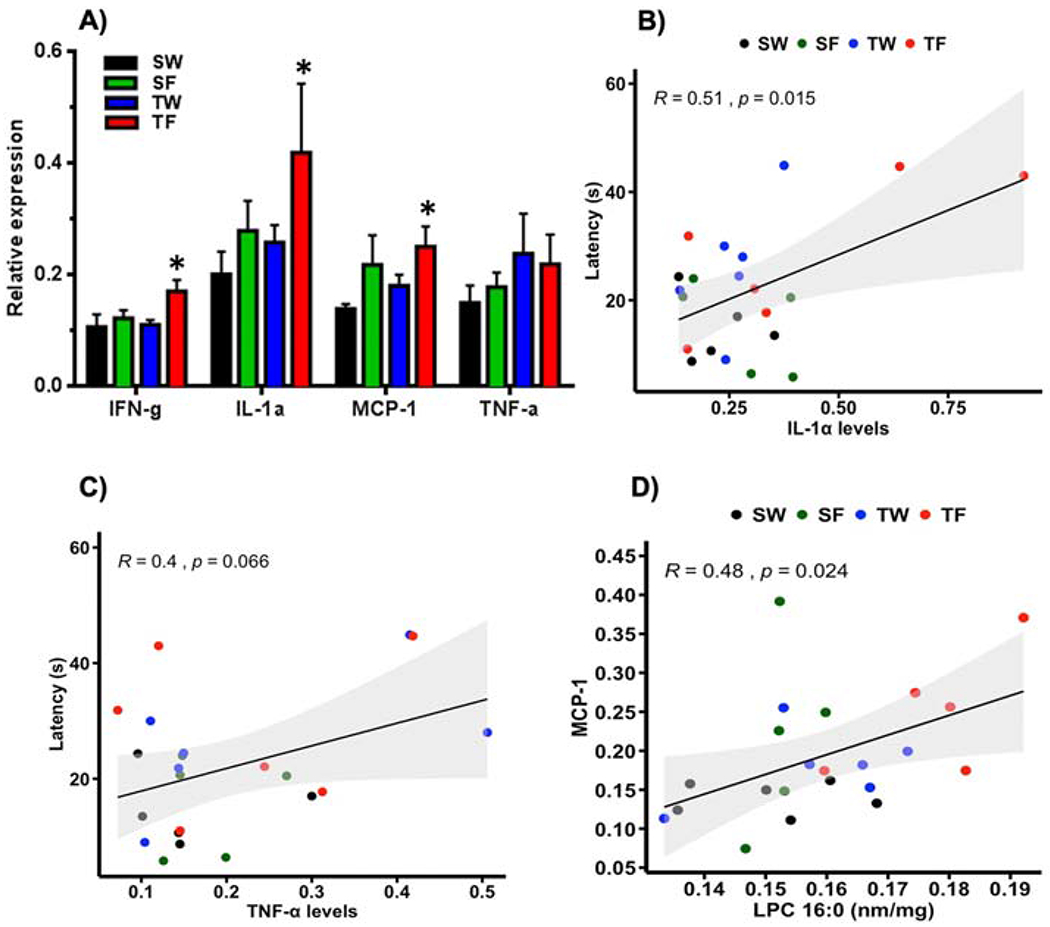
(A); 2-way ANOVA with Fisher’s LSD post-test (*p<0.05 vs SW group). (B) Correlation analysis using the latency as a variable shows association between IL-1α (R=0.51, p=0.015) and TNF-α (C, R=0.40, p=0.06) levels with memory performance. (D) Correlation analysis shows association between MCP-1 and LPC (16:0) levels (R= 0.48, p=0.024). Pearson correlation, n=5-6 per group.
3.5. Differential effects of fructose/TBI on cPLA2 in the hippocampus vs liver
LPC is mainly generated by the hydrolysis of Phosphatidylcholine (PC) involving the action of phospholipase cPLA2, acting on the brain and liver. We sought to determine possible differential effects of TBI and fructose on brain vs. liver. Liver is a major site for the synthesis of phospholipids and has the potential to act as a gate for the action of fructose on the brain. We assessed total levels of cPLA2 and its phosphorylated form (P-cPLA2), and the ratio (P-cPLA2/cPLA2) to get insight about the actions of TBI and fructose on cPLA2 activation. Levels of P-cPLA2 were significantly increased only under the action of TBI (TW-group) compared to SW-group (*p<0.05; Fig. 5A) in the hippocampus. In turn, in the liver, the ratio P-cPLA2/cPLA2 was increased in the TW-group and TF-group compared to SW-group (***p<0.001, **p<0.01, Fig. 5B). These results seem to indicate that the liver cPLA2 is more vulnerable to the action of fructose, in agreement with a mayor role of liver on fructose metabolism, with implications for brain function.
Figure 5. Altered activation of cPLA2 in the hippocampus and liver in response to TBI and fructose.
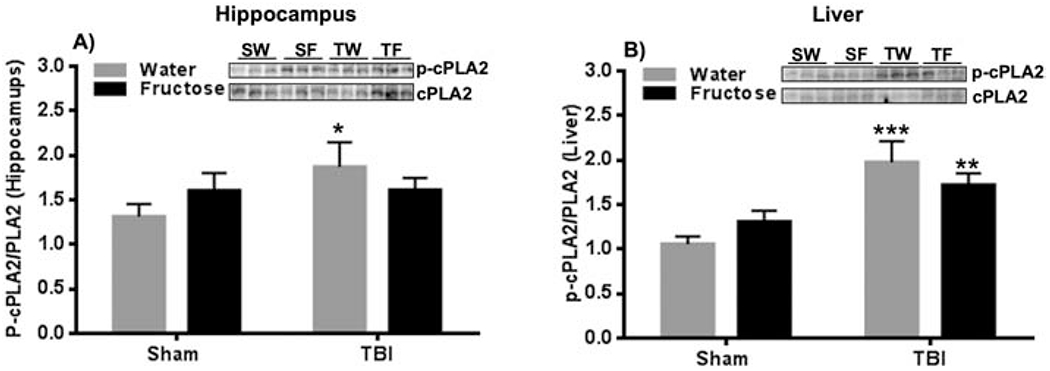
(A) Quantification of ratio from p-cPLA2/cPLA2 in the hippocampus for each experimental group. (B) Quantification of ratio P-cPLA2/cPLA2 in the liver for each group. n=6 per group. 2-way ANOVA with Fisher LSD posttest, *p<0.05, **p<0.01, ***p<0.001 vs SW-group. The uncropped western blots are shown in the supplementary information (Supplementary Figure 1).
3.6. Fructose and TBI affect lipid transport systems in the liver
Given that the liver is an important station for the metabolism of fructose, we sought to shed light on how fructose and TBI affects systems involved with lipid export from liver. Levels of the lipogenic enzyme fatty acid synthase (FAS) were increased by fructose consumption in both SF and TF groups as compared to their control groups (*p<0.05 vs SW group and #p<0.05 vs TW group, Fig. 6A). Liver lipids are packaged into VLDLs particles that are released into the blood stream such that assessment of VLDLs particles provide an estimate of lipid export from the liver. ApoB100 is a main lipoprotein present in VLDL which levels can be used to estimate changes in VLDL and its transport from liver [33], and we used to have an indication of the impact of TBI and fructose on VLDL. Results showed that fructose was vital to elevate the synthesis of ApoB100 (**p<0.01 vs SW group, # p<0.05 vs TW group; Fig. 6B) and ApoB48 (**p<0.01 vs SW group, # p<0.05 vs TW group; Fig. 6C) in both SF and TF-groups compared to their respective control groups. ATP-binding cassette transporter A1 (ABCA1) is engaged in the transport of lipids such as cholesterol and phospholipid across the plasma membrane. There was a statistical interaction (F (1,20) = 24.99, p<0.0001) between the effects of diet and injury (Fig. 5D), and ABCA1 levels were elevated in the TW-group as compared with the SW-group (*p<0.05, Fig. 6D). In addition, fructose potentiated the effects of TBI on ABCA1 levels in the TF group (### p<0.001 vs TW-group, ***p<0.001 vs SW-group, Fig. 6D). Interestingly, the liver levels of ABCA1 positively correlated with hippocampal levels of LPC (16:0) (R =0.48, p=0.017, Fig. 6E), which could indicate that brain LPC benefits from transport of phospholipids across liver plasma membranes.
Figure 6. Lipid metabolism in the liver in response to TBI and fructose.
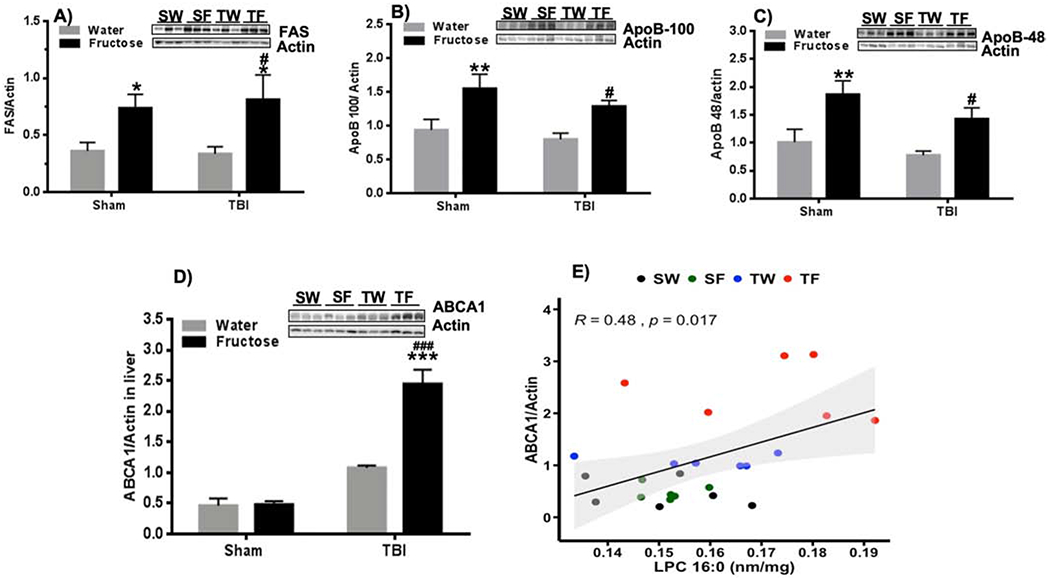
(A) Quantification of Fatty acid synthase (FAS; A), ApoB-100 (B), (C) ApoB-48 (C), and ABCA1 (D) in the liver. n=6 per group. 2-way ANOVA with Fisher’s LSD posttest, (*p<0.05, **p<0.01, *** p<0.001 vs SW-group, #p<0.05, ### p<0.001 vs TW-group). The uncropped western blots are shown in the supplementary information (Supplementary Figure 2). (E) Correlation analysis shows association between ratio ABCA1/actin and LPC 16:0 levels (R= 0.48, p=0.017).
Discussion
Lipids are major structural components of brain, crucial for cell communication, and involved in higher order functions during homeostatic and disease conditions. Emerging evidence indicates that a disruption in lipid homeostasis poses a heavy toll for brain function and can contribute to the pathogenesis of TBI [3, 34]. In addition, the fact that lipid homeostasis in the liver is under the range of action of dietary sugars, makes it crucial to better understand how diet interacts with the TBI pathology. Our results show that TBI and fructose have critical actions on lipid homeostasis in the brain and liver. Based on lipidomic analysis accompanied by unbiased sPLS-DA, we found that LPC and CE were the top lipid classes impacted by TBI and fructose in the hippocampus; however, LPC was the only lipid species associated with memory performance. The effects of fructose were more pronounced in the liver, in agreement with the role of liver on fructose metabolism. Correlation analysis showed that levels of select pro-inflammatory markers in the liver increased in proportion to hippocampal-dependent memory dysfunction. We found that fructose collaborates with TBI to influence systems that transport lipids from liver to bloodstream and could make lipids more accessible to brain. This interaction is particularly important for disease stages involving a breakdown of the blood brain barrier (BBB) such as the case of TBI [35]. Moderate FPI was chosen to assay our paradigm based on its capacity to model concussive head injury involving minimal neuronal death while resulting in reproducible neurological symptoms, including cognitive impairment [31, 32]. We used the Barnes maze to assess hippocampal-dependent function by testing learning and memory[36]. The overall results suggest that peripheral physiology is a confluent target for the effects of TBI and fructose diet, and that systemic dysfunction could, in turn, influence brain pathogenesis.
Lipids and cognitive dysfunction.
Brain lipids provide support to several molecular processes underlying higher order functions such as learning and memory [1, 2]. Our results showed that LPC levels changed in proportion to memory performance in the hippocampus (a region deeply involved in learning and memory), in which higher levels of LPC (16:0) were associated with worse performance. The detrimental impact of LPC in the pathogenesis of TBI was exacerbated by consumption of the fructose diet. These results are in general agreement with recent studies showing that LPC injection into the brain of rodents caused cognitive deficit [37, 38]. Our findings have enormous implications for the control of TBI pathogenesis as LPC has a pro-inflammatory action in the brain and the liver, and because of the fact that inflammation is an underlying mechanism for disruption of cell function and plasticity.
LPC is produced by the hydrolysis of phosphatidylcholine (PC) under the action of phospholipase A2 (PLA2) [39]. Therefore, elevated activation of cPLA2 in response to TBI in the hippocampus could have been operational to increase the levels of LPC and inflammation. A recent study shows that cPLA2 in membranes of lysosomes increases LPC (16:0) and promotes neurodegeneration through inhibition of autophagy in a brain injury model [40]. LPC has been implicated in other neurological disorders including multiple sclerosis, stroke and Alzheimer’s disease [41–43]. In addition, LPC fosters demyelination in spinal cord [44], and microglial cells are main conveyor of an inflammatory environment [42]. The differential actions of LPC on tissues depend primarily on the degree of saturation of the LPC acyl chain. The saturated acyl LPC form has been shown to promote inflammation [45], while the polyunsaturated acyl LPC reverses the saturated acyl LPC-induced inflammation [46]. In the present study, TBI and fructose diet primarily increased the levels of saturated acyl LPC (16:0) in the hippocampus, which is pro-inflammatory.
Effects of TBI and fructose on liver
Our results show that levels of phosphorylated cPLA2 were also elevated in the liver of rodents exposed to TBI and that fructose exacerbated the action of TBI. The fact that fructose had a larger effect on the liver is in general agreement with abundant evidence that dietary fructose is largely metabolized in the liver (see FGP, Royes, BBA 2020, in press). In addition, the action of cPLA2 in the liver has been deemed important for secretion of liver lipids, particularly LPC. PC is required for hepatic very low-density lipoprotein (VLDL) assembly and secretion [47]. LPC is derived from the turnover of PC by involving the enzymatic action of cPLA2, and promotes hepatic VLDL production and its subsequent liver secretion [48, 49]. The overall evidence suggests that LPC synthesis and metabolism could be co-regulated in the brain and liver in the pathogenesis of TBI, and that fructose can have a preponderant action on the liver.
We assessed levels of various molecules with recognized role on lipid liver homeostasis to gain insight into the role of liver on the TBI pathogenesis. Our results showed that fructose diet elevates synthesis of the lipogenic enzyme fatty acid synthase (FAS), and ApoB-100 that is required for the synthesis and secretion of VLDL from liver to blood. Transport of lipids across plasma membranes is crucial for lipid function, and a highly regulated process. We found an increment of ABCA1 levels in the liver of the TW-group and the effect was exacerbated by fructose. ABCA1 is a member of the ABC transporter family that enables lipids such as cholesterol and phospholipids across the membrane [50]. ABCA1 transporter is involved in the regulation of cholesterol and phospholipids efflux from hepatocytes to plasma [51]. Serum amyloid A (SAA) is a liver protein that is elevated during the acute phase of TBI, and is involved in systemic and central inflammation [7, 8]. Recent studies show that SAA promotes cholesterol and phospholipid efflux involving the action of ABCA1 [52, 53]. Our results additionally show that levels of liver ABCA1 changed in proportion to hippocampal LPC levels and suggest the possibility that TBI and fructose may influence lipid mobilization and lipoprotein metabolism in the liver with implications for the central inflammatory response. There is evidence of communication between the brain with the liver in a bidirectional manner to regulate hepatic lipid metabolism and lipoprotein production [54].
Effects of fructose and TBI on pro-inflammatory systems in liver
Cytokines are critical components of the acute inflammatory response in the liver [55], and our results show that TBI and fructose elevate pro-inflammatory cytokines such as IFN-γ, IL-1α and the chemokine MCP-1. The systemic production of cytokines and chemokines by the liver is an essential aspect of the inflammatory secondary response to injury with subsequent effects on brain [56]. LPC (16:0) has been shown to induce the synthesis and secretion of pro-inflammatory cytokines in macrophages [57, 58]. Our results showing that memory deficit correlated with IL1-α and TNF-α levels suggest that systemic inflammation may contribute to disrupt cognitive function, and these results shed light to observations that systemic inflammation is commonly associated with loss of neurological function in TBI patients [59, 60]. In addition, results showing that levels of liver MCP-1 changed in proportion to levels of hippocampal LPC (16:0) suggest a link between liver inflammation and brain LPC. Increases in BBB permeability after TBI [61, 62] result in influx of immune cells, and serum proteins from blood, and provide a route for liver-brain communication with inflammatory consequences [56]. LPC levels have been found elevated in the liver of a non-alcoholic steatohepatitis (NASH) mice model [26], and in plasma of AD patients [27].
An increasing line of clinical and experimental evidence indicates that high fructose consumption correlates with rising rates of neurologic disorders [63]. Research so far indicates that fructose can directly affect the brain [64]; however, the fact that most fructose is metabolized in the liver makes of liver as a crucial mediator for the action of fructose on brain [65]. The liver plays a critical role on lipid metabolism with the potential to influence brain function [4, 5], and the extent of the TBI pathology [6] (see Figure 7). Given the essential role of lipids for neuronal signalling and overall brain function [66], lipid dysfunction can have devastating consequences for the outcome of TBI pathology. The results of this study are critical for prevention and managing of TBI pathogeneis.
Figure 7. Proposed mechanisms by which brain interacts with liver in response to the actions of TBI and fructose.
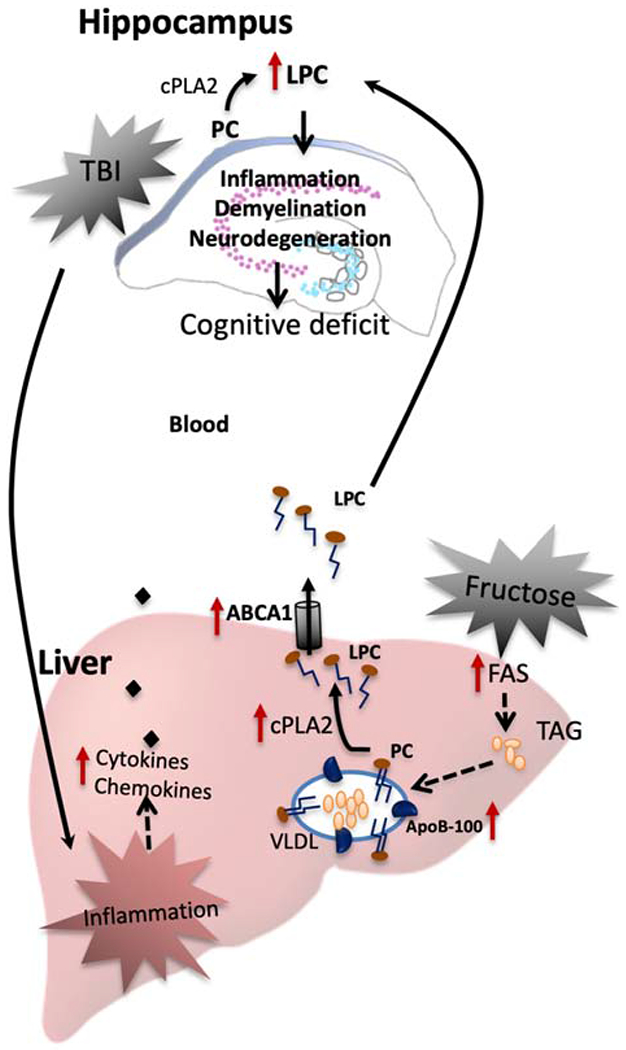
The effects of TBI on brain function and plasticity involve the action of liver on lipid metabolism. Lysophosphatidylcholine (LPC) is a lipid inflammatory mediator that it is derived from the turnover of phosphatidylcholine (PC) involving cPLA2 and secreted as component of very low-density lipoprotein (VLDL). TBI in combination with fructose diet alter lipid species in the brain such as LPC (16:00) that uses precursor molecules synthesized in the liver. The liver is a main target for the action of fructose and then fructose byproducts can influence TBI pathogenesis. Fructose increases the secretion of VLDL to the blood and could promote the conversion of PC to LPC by cPLA2. The hepatic ABCA1 transporter exports cholesterol and phospholipids such as LPC from liver to the bloodstream. Thereby, fructose can help LPC to reach the brain and to amplify brain degeneration started by TBI with subsequent effects on cognitive performance. TBI and fructose induce systemic inflammation that can affects the brain, i.e., TBI stimulates the release of pro-inflammatory cytokines from liver which are transported into brain, taking advantage of a breakdown of the blood brain barrier (BBB). Brain inflammation can enhance degenerative events resulting in cognitive dysfunction, demyelination, and cell degeneration.
Conclusions
We sought to shed light how TBI pathogenesis can involve the brain and systemic physiology. Our results show that TBI in lipid metabolism in the brain and liver, and that a fructose diet exacerbate the action of TBI, particularly in the liver. The action of liver on lipid metabolism seems crucial for control of brain function after TBI. LPC (16:0) appears as a lipid species highly susceptible to the action of TBI and fructose, and results suggest loss of lipid homeostasis as a possible route by which periphery can harm the brain. Results show that LPC brain levels are associated with cognitive dysfunction, and that liver inflammation may contribute to brain dysfunction. Fructose appears as an important modulator of brain function and plasticity, which action on liver can amplify TBI pathogenesis. These results suggest a series of future studies to mechanistically show how liver byproducts of lipid metabolism could influence the brain in the TBI pathogenesis. These results are important for building prevention programs based on diet to improve outcome in TBI patients.
Supplementary Material
Highlights.
TBI affects select lipid species in the brain
Levels of hippocampal lysophosphatidylcholine are proportional to memory deficit
Fructose exacerbates the effects of TBI on the lipid metabolic machinery in liver
Select hepatic pro-inflammatory cytokines levels are proportional to memory defict
Lysophosphatidylcholine appears as mediator between liver and brain in response to TBI and fructose
Acknowledgments
This research was supported by NIH R01NS50465 (F.G.P), R01NS111378 (F.G.P)
Dr. Victoria Palafox-Sanchez was supported by a fellowship from UC MEXUS-CONACYT Program 2019-2020.
Abbreviations:
- ABCA1
ATP-binding cassette transporter A1
- AD
Alzheimer’s Disease
- ApoB
Apolipoprotein- B
- BBB
Blood brain barrier
- BM
Barnes Maze
- BSA
Bovine Serum Albumin
- CE
Cholesterol ester
- CER
Ceramides
- cPLA2
cytosolic phospholipase 2
- DAG
Diacylglycerol
- DCER
Dihydroceramides
- FAS
Fatty Acid Synthase
- FFA
Free fatty acids
- HCER
Hexosyl ceramides
- IFN- γ
Interferon – γ
- IL-1α
Interleukin-1α
- LCER
Lactosyl ceramides
- LPC
Lysophosphatidylcholine
- LPE
Lysophosphatidylethanolamine
- MCP-1
Monocyte chemoattractant protein-1
- NAFLD
Nonalcoholic Fatty Liver Disease
- P-cPLA2
phospho-cytoplasmic phospholipase A 2
- PC
Phosphatidylcholine
- PE
Phosphatidylethanolamine
- PVDF
Polyvinylidene difluoride
- SM
Sphingomyelin
- sPLS-DA
sparse partial least squares discriminant analysis
- TAG
Triglyceride
- TBI
traumatic brain injury
- TNF- α
Tumoral necrosis factor- α
- VLDL
very low-density lipoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests Statement
The authors declare no competing interests.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Bazan NG, Lipid signaling in neural plasticity, brain repair, and neuroprotection. Mol Neurobiol, 2005. 32(1): p. 89–103. [DOI] [PubMed] [Google Scholar]
- [2].Cermenati G, et al. , Lipids in the nervous system: from biochemistry and molecular biology to patho-physiology. Biochim Biophys Acta, 2015. 1851(1): p. 51–60. [DOI] [PubMed] [Google Scholar]
- [3].Lamade AM, et al. , Mitochondrial damage & lipid signaling in traumatic brain injury. Exp Neurol, 2020. 329: p. 113307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Filipović B, Marković O, and Ðurić V, Cognitive Changes and Brain Volume Reduction in Patients with Nonalcoholic Fatty Liver Disease. Can J Gastroenterol Hepatol, 2018. 2018: p. 9638797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Seo SW, et al. , Nonalcoholic fatty liver disease is associated with cognitive function in adults. Neurology, 2016. 86(12): p. 1136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].McDonald SJ, et al. , Beyond the Brain: Peripheral Interactions after Traumatic Brain Injury. J Neurotrauma, 2020. 37(5): p. 770–781. [DOI] [PubMed] [Google Scholar]
- [7].Villapol S, et al. , Hepatic expression of serum amyloid A1 is induced by traumatic brain injury and modulated by telmisartan. Am J Pathol, 2015. 185(10): p. 2641–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wicker E, et al. , Serum Amyloid A Protein as a Potential Biomarker for Severity and Acute Outcome in Traumatic Brain Injury. Biomed Res Int, 2019. 2019: p. 5967816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nizamutdinov D, et al. , Hepatic alterations are accompanied by changes to bile acid transporter-expressing neurons in the hypothalamus after traumatic brain injury. Sci Rep, 2017. 7: p. 40112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rege SD, et al. , Brain Trauma Disrupts Hepatic Lipid Metabolism: Blame It on Fructose? Mol Nutr Food Res, 2019. 63(15): p. e1801054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cowie CC, et al. , Full accounting of diabetes and pre-diabetes in the U.S. population in 1988–1994 and 2005–2006. Diabetes Care, 2009. 32(2): p. 287–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Agrawal R, et al. , Dietary fructose aggravates the pathobiology of traumatic brain injury by influencing energy homeostasis and plasticity. J Cereb Blood Flow Metab, 2016. 36(5): p. 941–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Agrawal R and Gomez-Pinilla F, ‘Metabolic syndrome’ in the brain: deficiency in omega-3 fatty acid exacerbates dysfunctions in insulin receptor signalling and cognition. J Physiol, 2012. 590(Pt 10): p. 2485–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Stanhope KL, et al. , Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest, 2009. 119(5): p. 1322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Abdelmalek MF, et al. , Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology, 2010. 51(6): p. 1961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pflugrad H, et al. , Cerebral white matter lesions in patients with cirrhosis - causative for hepatic encephalopathy or bystanders? Liver Int, 2015. 35(7): p. 1816–23. [DOI] [PubMed] [Google Scholar]
- [17].Petta S, et al. , The Presence of White Matter Lesions Is Associated With the Fibrosis Severity of Nonalcoholic Fatty Liver Disease. Medicine (Baltimore), 2016. 95(16): p. e3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pinçon A, et al. , Non-Alcoholic Fatty Liver Disease, and the Underlying Altered Fatty Acid Metabolism, Reveals Brain Hypoperfusion and Contributes to the Cognitive Decline in APP/PS1 Mice. Metabolites, 2019. 9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Faeh D, et al. , Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes, 2005. 54(7): p. 1907–13. [DOI] [PubMed] [Google Scholar]
- [20].Banks WA, et al. , Triglycerides cross the blood-brain barrier and induce central leptin and insulin receptor resistance. Int J Obes (Lond), 2018. 42(3): p. 391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lanaspa MA, et al. , Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun, 2013. 4: p. 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ishimoto T, et al. , High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology, 2013. 58(5): p. 1632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou Z, et al. , Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab, 2011. 13(5): p. 592–600. [DOI] [PubMed] [Google Scholar]
- [24].Zhou X, et al. , Identification of Lysophosphatidylcholines and Sphingolipids as Potential Biomarkers for Acute Aortic Dissection via Serum Metabolomics. Eur J Vasc Endovasc Surg, 2019. 57(3): p. 434–441. [DOI] [PubMed] [Google Scholar]
- [25].Drzazga A, et al. , Lysophosphatidylcholine and its phosphorothioate analogues potentiate insulin secretion via GPR40 (FFAR1), GPR55 and GPR119 receptors in a different manner. Mol Cell Endocrinol, 2018. 472: p. 117–125. [DOI] [PubMed] [Google Scholar]
- [26].Kakisaka K, et al. , Caspase-independent hepatocyte death: A result of the decrease of lysophosphatidylcholine acyltransferase 3 in non-alcoholic steatohepatitis. J Gastroenterol Hepatol, 2019. 34(7): p. 1256–1262. [DOI] [PubMed] [Google Scholar]
- [27].Dorninger F, et al. , Alterations in the Plasma Levels of Specific Choline Phospholipids in Alzheimer’s Disease Mimic Accelerated Aging. J Alzheimers Dis, 2018. 62(2): p. 841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Grimm MO, et al. , From brain to food: analysis of phosphatidylcholins, lyso-phosphatidylcholins and phosphatidylcholin-plasmalogens derivates in Alzheimer’s disease human post mortem brains and mice model via mass spectrometry. J Chromatogr A, 2011. 1218(42): p. 7713–22. [DOI] [PubMed] [Google Scholar]
- [29].Liu L, et al. , Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell, 2015. 160(1–2): p. 177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu L, et al. , The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab, 2017. 26(5): p. 719–737.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dixon CE, et al. , A fluid percussion model of experimental brain injury in the rat. J Neurosurg, 1987. 67(1): p. 110–9. [DOI] [PubMed] [Google Scholar]
- [32].Lyeth BG, et al. , Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res, 1990. 526(2): p. 249–58. [DOI] [PubMed] [Google Scholar]
- [33].Schonfeld G, et al. , Fatty liver in familial hypobetalipoproteinemia: triglyceride assembly into VLDL particles is affected by the extent of hepatic steatosis. J Lipid Res, 2003. 44(3): p. 470–8. [DOI] [PubMed] [Google Scholar]
- [34].Anthonymuthu TS, et al. , Oxidized phospholipid signaling in traumatic brain injury. Free Radic Biol Med, 2018. 124: p. 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hu Y, et al. , Longitudinal Characterization of Blood-Brain Barrier Permeability after Experimental Traumatic Brain Injury by. J Neurotrauma, 2020. [DOI] [PubMed] [Google Scholar]
- [36].Krishna G, et al. , 7,8-Dihydroxyflavone facilitates the action exercise to restore plasticity and functionality: Implications for early brain trauma recovery. Biochim Biophys Acta Mol Basis Dis, 2017. 1863(6): p. 1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang G, et al. , Neuroprotective effect of l-serine against white matter demyelination by harnessing and modulating inflammation in mice. Neuropharmacology, 2019. 146: p. 39–49. [DOI] [PubMed] [Google Scholar]
- [38].Mousavi Majd A, et al. , Inhibition of GABA A receptor improved spatial memory impairment in the local model of demyelination in rat hippocampus. Behav Brain Res, 2017. 336: p. 111–121. [DOI] [PubMed] [Google Scholar]
- [39].Kudo I and Murakami M, Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat, 2002. 68-69: p. 3–58. [DOI] [PubMed] [Google Scholar]
- [40].Sarkar C, et al. , PLA2G4A/cPLA2-mediated lysosomal membrane damage leads to inhibition of autophagy and neurodegeneration after brain trauma. Autophagy, 2020. 16(3): p. 466–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Freeman L, et al. , NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med, 2017. 214(5): p. 1351–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Inose Y, et al. , Activated microglia in ischemic stroke penumbra upregulate MCP-1 and CCR2 expression in response to lysophosphatidylcholine derived from adjacent neurons and astrocytes. Neuropathology, 2015. 35(3): p. 209–23. [DOI] [PubMed] [Google Scholar]
- [43].Kim H, et al. , Nicotinamide attenuates the decrease in dendritic spine density in hippocampal primary neurons from 5xFAD mice, an Alzheimer’s disease animal model. Mol Brain, 2020. 13(1): p. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lu C, et al. , G-Protein-Coupled Receptor Gprl7 Regulates Oligodendrocyte Differentiation in Response to Lysolecithin-Induced Demyelination. Sci Rep, 2018. 8(1): p. 4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aiyar N, et al. , Lysophosphatidylcholine induces inflammatory activation of human coronary artery smooth muscle cells. Mol Cell Biochem, 2007. 295(1-2): p. 113–20. [DOI] [PubMed] [Google Scholar]
- [46].Hung ND, Sok DE, and Kim MR, Prevention of 1-palmitoyl lysophosphatidylcholine-induced inflammation by polyunsaturated acyl lysophosphatidylcholine. Inflamm Res, 2012. 61(5): p. 473–83. [DOI] [PubMed] [Google Scholar]
- [47].Rinella ME, et al. , Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res, 2008. 49(5): p. 1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hollie NI and Hui DY, Group 1B phospholipase A2 deficiency protects against diet-induced hyperlipidemia in mice. J Lipid Res, 2011. 52(11): p. 2005–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li Z, et al. , Lysophosphatidylcholine acyltransferase 3 knockdown-mediated liver lysophosphatidylcholine accumulation promotes very low density lipoprotein production by enhancing microsomal triglyceride transfer protein expression. J Biol Chem, 2012. 287(24): p. 20122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Borst P, Zelcer N, and van Helvoort A, ABC transporters in lipid transport. Biochim Biophys Acta, 2000. 1486(1): p. 128–44. [DOI] [PubMed] [Google Scholar]
- [51].Timmins JM, et al. , Targeted inactivation of hepatic Abcal causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest, 2005. 115(5): p. 1333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Stonik JA, et al. , Serum amyloid A promotes ABCA1-dependent and ABCA1-independent lipid efflux from cells. Biochem Biophys Res Commun, 2004. 321(4): p. 936–41. [DOI] [PubMed] [Google Scholar]
- [53].Abe-Dohmae S, et al. , Serum amyloid A generates high density lipoprotein with cellular lipid in an ABCAl- or ABCA7-dependent manner. J Lipid Res, 2006. 47(7): p. 1542–50. [DOI] [PubMed] [Google Scholar]
- [54].Taher J, Farr S, and Adeli K, Central nervous system regulation of hepatic lipid and lipoprotein metabolism. Curr Opin Lipidol, 2017. 28(1): p. 32–38. [DOI] [PubMed] [Google Scholar]
- [55].Woodcock T and Morganti-Kossmann MC, The role of markers of inflammation in traumatic brain injury. Front Neurol, 2013. 4: p. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Glushakova OY, Johnson D, and Hayes RL, Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J Neurotrauma, 2014. 31(13): p. 1180–93. [DOI] [PubMed] [Google Scholar]
- [57].Zhang SY, et al. , Adipocyte-derived Lysophosphatidylcholine Activates Adipocyte and Adipose Tissue Macrophage Nod-Like Receptor Protein 3 Inflammasomes Mediating Homocysteine-Induced Insulin Resistance. EBioMedicine, 2018. 31: p. 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Corrêa R, et al. , Lysophosphatidylcholine Induces NLRP3 Inflammasome-Mediated Foam Cell Formation and Pyroptosis in Human Monocytes and Endothelial Cells. Front Immunol, 2019. 10: p. 2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chiaretti A, et al. , Interleukin 1beta and interleukin 6 relationship with paediatric head trauma severity and outcome. Childs Nerv Syst, 2005. 21(3): p. 185–93; discussion 194. [DOI] [PubMed] [Google Scholar]
- [60].Sun Y, et al. , Elevated Serum Levels of Inflammation-Related Cytokines in Mild Traumatic Brain Injury Are Associated With Cognitive Performance. Front Neurol, 2019. 10: p. 1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tagge CA, et al. , Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain, 2018. 141(2): p. 422–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Doherty CP, et al. , Blood-Brain Barrier Dysfunction as a Hallmark Pathology in Chronic Traumatic Encephalopathy. J Neuropathol Exp Neurol, 2016. 75(7): p. 656–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang DM, Jiao RQ, and Kong LD, High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions. Nutrients, 2017. 9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Johnson RJ, et al. , Cerebral Fructose Metabolism as a Potential Mechanism Driving Alzheimer’s Disease. Frontiers in Aging Neuroscience, 2020. 12(299). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lanaspa MA, et al. , Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun, 2013. 4: p. 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bruce KD, Zsombok A, and Eckel RH, Lipid Processing in the Brain: A Key Regulator of Systemic Metabolism. Front Endocrinol (Lausanne), 2017. 8: p. 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Parcianello Cipolat R, Freire Royes L Dietary Fructose as a model to explore the influence of peripheral metabolism on brain function and plasticity, BBA in press [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.