

Abstract
Autoimmune regulator+ (Aire) medullary thymic epithelial cells (mTECs) play a critical role in tolerance induction. Several studies demonstrated that Aire+mTECs differentiate further into Post‐Aire cells. Yet, the identification of terminal stages of mTEC maturation depends on unique fate‐mapping mouse models. Herein, we resolve this limitation by segmenting the mTEChi(MHCIIhiCD80hi) compartment into mTECA/hi (CD24−Sca1−), mTECB/hi (CD24+Sca1−), and mTECC/hi (CD24+Sca1+). While mTECA/hi included mostly Aire‐expressing cells, mTECB/hi contained Aire+ and Aire− cells and mTECC/hi were mainly composed of cells lacking Aire. The differential expression pattern of Aire led us to investigate the precursor‐product relationship between these subsets. Strikingly, transcriptomic analysis of mTECA/hi, mTECB/hi, and mTECC/hi sequentially mirrored the specific genetic program of Early‐, Late‐ and Post‐Aire mTECs. Corroborating their Post‐Aire nature, mTECC/hi downregulated the expression of tissue‐restricted antigens, acquired traits of differentiated keratinocytes, and were absent in Aire‐deficient mice. Collectively, our findings reveal a new and simple blueprint to survey late stages of mTEC differentiation.
Keywords: autoimmune regulator, differentiation, medullary thymic epithelial cell, thymus, tolerance induction
Although the expression of Aire was initially considered to mark a terminal stage of mTEC maturation, recent studies have demonstrated that mTECs extend their differentiation beyond Aire. Integrating flow cytometry, functional, and transcriptomic analyses, Ferreirinha et al. provide an effective phenotypic‐based method to resolve Early‐, Late‐ and Post‐Aire mTECs.
Introduction
Within the thymus, cortical (c) and medullary (m) thymic epithelial cells (TECs) provide dedicated microenvironments for the development and selection of T cells that are simultaneously reactive to foreign antigens and tolerant to the body's own constituents [1, 2]. While cTECs direct T cell commitment and positive selection, mTECs regulate negative selection of autoreactive thymocytes or their deviation into the T regulatory cell lineage [2]. The critical role of mTECs in tolerance induction depends on their capacity to express virtually all tissue‐restricted antigens (TRAs), which are otherwise confined to peripheral tissues [3]. This process is regulated, in part, by the Autoimmune regulator (Aire) protein, which is encoded by the gene mutated in human autoimmune disorder APECED. Deficiency in Aire alters TRA expression in mTECs, causes failures in negative selection and T regulatory cell differentiation, leading to an increased susceptibility to autoimmunity [4].
Since the discovery of Aire, there have been considerable advances in the understanding of the development of Aire+mTECs, which represent a fraction of mature mTEChi (MHCIIhiCD80hi) and derive from precursors within mTEClo (MHCIIloCD80lo) [2]. Although the expression of Aire was initially considered to identify a terminal step of mTEC maturation [5], subsequent studies demonstrated that Aire+mTECs can further differentiate into a Post‐Aire stage [6, 7, 8, 9, 10]. Post‐Aire mTECs downregulate the expression of Aire, certain TRAs, CD80, and MHCII, while acquiring traits of terminally differentiated keratinocytes, including keratin 10 (K10) and involucrin (Ivl) [6, 7, 8, 10]. Still, the identification of later stages of Aire+mTEC maturation remains dependent on the use of unique fate‐mapping mouse models and advanced high‐content single‐cell RNA sequencing, the latter being incompatible with functional studies. In this study, we provide an efficient method to prospectively identify Aire+mTECs and their Post‐Aire descendants, which will prove useful to examine the murine medullary microenvironment, and ultimately, study its role in tolerance induction.
Results and discussion
CD24 and Sca1 expression identifies new subsets of mTEChi with distinct levels of Aire
Compelling evidence indicates that the cartography of mature mTEC differentiation is more complex than initially recognized [4]. To resolve the heterogeneity of mTECs (defined as indicated in Supporting Information Fig. 1A), we integrated in their conventional phenotypic profiling (CD45−EpCAM+UEA+MHCIIlo/hiCD80lo/hi) the analysis of two markers, CD24 and Sca1, which are routinely used to mark progenitor cells in a variety of hematopoietic, mammary, and intestinal epithelial lineages [11, 12]. Examination at defined stages of postnatal life revealed the existence of three prime novel mTEC subsets: CD24−Sca1− (referred hereafter as mTECA), CD24+Sca1− (mTECB), and CD24+Sca1+ (mTECC) (Fig. 1A). The representation of these subsets was largely maintained over time, and their numbers increase with the expansion of the mTEC compartment during early adulthood (Fig. 1A). Next, we assessed whether the mTECA‐C subtypes specifically segregated into conventional mTEClo or mTEChi (defined as in Supporting Information Fig. 1B). We found that mTEClo contained comparable proportions of mTECA (mTECA/lo), mTECB (mTECB/lo) and mTECC (mTECC/lo) during postnatal life, with a reduction in mTECA/lo and an accumulation of mTECC/lo unfolding at 6 weeks (Fig. 1B and Supporting Information Fig. 1C). In contrast, mTEChi were continuously enriched in mTECA (mTECA/hi) (Fig. 1C). We also found rare cells with a CD24−Sca1+ phenotype, which appeared as an extension of mTECC/lo (Fig. 1B) and mTECA/hi (Fig. 1C) instead of defined populations. Thus, we focused our study on mTECA‐C. Since Aire+mTECs represent a specialized functional subset within mature mTEChi [4], we determined their relationship to mTECA‐C/hi subtypes. Notably, mTECA/hi were mainly Aire+, mTECB/hi included a combination of Aire+/− cells, and mTECC/hi largely lacked Aire expression (Fig. 1C). Complementary analysis confirmed that the majority of Aire+ cells were confined to the mTECA/hi subtype (Fig. 1D). The sequential changes in the pattern of Aire expression within mTECA‐C/hi was intriguing and led us to further study whether these subsets identified discrete stages of Aire+mTEC differentiation. Since mTECC/hi and mTECB/hi largely lack or express intermediate levels of Aire, we hypothesized that they could either represent immediate precursors of Aire+mTECs (Pre‐Aire) or cells completing their final stages of maturation (Post‐Aire). Alternatively, mTECC/hi could represent a subset with an independent lineage relationship to Aire+mTECs. The subsequent experiments were designed to address these possibilities.
Figure 1.
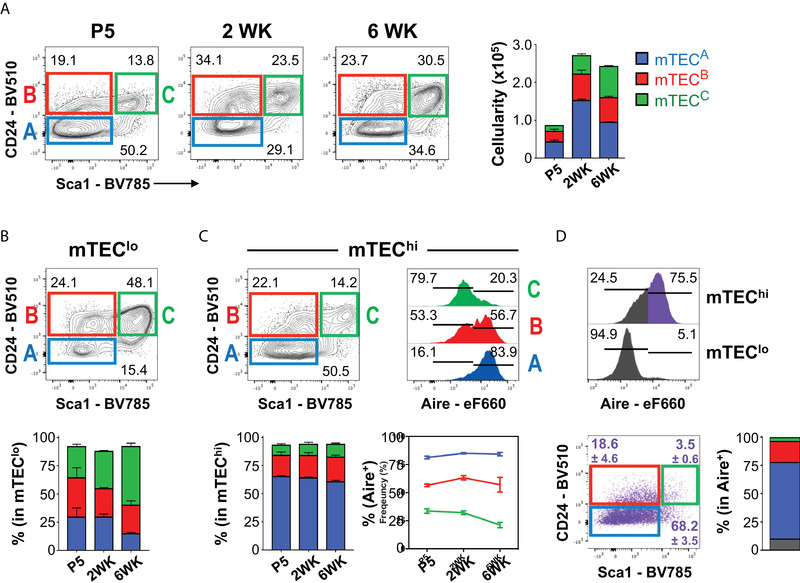
The combined analysis of CD24 and Sca1 defines new mature mTEC subsets with distinct patterns of Aire expression. (A) Thymi from C57BL/6 mice were isolated at the indicated time points and cells were stained with PerCP‐Cy5‐conjugated anti‐CD45.2, PE‐conjugated anti‐Ly51, APC‐eFlour 780‐conjugated anti‐MHCII (I‐A/I‐E), eFlour 660‐cojugated anti‐AIRE, BV421‐conjugated anti‐EpCAM, BV650‐conjugated anti‐CD80, BV510‐conjugated anti‐CD24, BV786‐conjugated anti‐Sca1; Biotinylated UEA: BV711‐conjugated streptavidin. Total mTECs (CD45−EpCAM+UEA+) were subdivided into three subpopulations based on the expression pattern of CD24 and Sca1: mTECA (CD24−Sca1−, blue), mTECB (CD24+Sca1−, red), and mTECC (CD24+Sca1+, green). Bar graph displays the number of mTEC subsets at different time points. Representation of mTECA, mTECB, and mTECC within mTEClo (B) and mTEChi (C) at different time points. (A–C) Data are representative of at least three experiments per time point with 2–4 mice per experiment. Numbers in plots indicate the frequency of cells found within each gate. Bar graph displays the frequency of mTECA, mTECB and mTECC within mTEClo and mTEChi, as mean plus SD. P – Postnatal day; WK – Postnatal weeks. Histograms in (C) show Aire expression within mTECA‐C/hi subsets. Numbers in plots indicate the frequency of cells found within each gate. Bar graph below displays the frequency of Aire+ cells within mTECA‐C/hi subsets across time. (D) Aire expression in mTEChi (top) and mTEClo (bottom). The expression of CD24 and Sca1 was analysed on Aire+mTEChi (pseudo‐coloured purple). Total mTECs were plotted in gray. Column chart illustrates the relative composition of mTECA/hi (blue), mTECB/hi (red) and mTECC/hi (green) subsets within the total Aire+mTEChi. Data are representative of at least three experiments per time point with 2–4 mice per experiment.
The genetic program mTECA‐C/hi maps successive stages of Aire+mTEC differentiation
To determine whether mTECB‐C/hi defined early or later stages of Aire+ mTEC differentiation, we performed RNA sequencing analysis of mTECA‐C/hi, including mTECA‐C/lo as additional reference populations. The overall snap‐shot of their transcriptome revealed that the biological replicates of each subset clustered together, emphasizing that these populations had stable genetic programs and low intrapopulation variability (Fig. 2A). Second, mTECA‐C/hi primarily segregated from their mTEClo counterparts and presented the highest number of detected genes, indicating a presumable increased promiscuous gene expression capacity. Interestingly, mTECA/hi and mTECB/hi were more closely related to each other compared to mTECC/hi (Fig. 2A). To further study the functional significance of mTECA‐C/hi, we extracted a publicly available list of Aire‐dependent and Aire‐independent TRAs [13] and determined their expression pattern in the analyzed subsets. We found that Aire‐dependent and Aire‐independent TRA genes were predominantly upregulated in mTECA/hi and mTECB/hi, with Aire‐dependent targets displaying the most marked augment in these subsets (Fig. 2B and Supporting Information Table 1). These observations are in line with the expression pattern of Aire in mTECA‐B/hi subsets (Fig. 1D) and the TRA expression capacity of mature Aire+mTECs [13].
Figure 2.
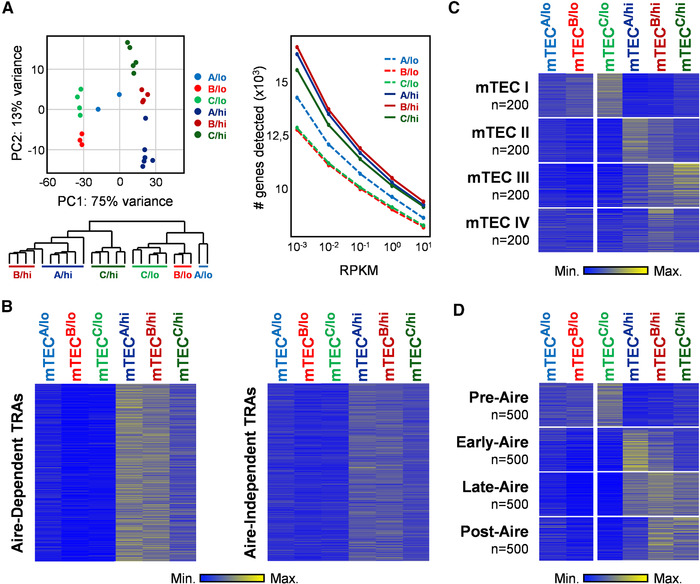
Transcriptomic analysis of mTECA‐C with mTEClo and mTEChi, and their relationship to the genetic profile of distinct stages of the Aire+mTEC differentiation. RNA‐Seq analysis was performed on independently sorted biological replicates of mTECA/lo (n = 2), mTECB/lo (n = 3), mTECC/lo (n = 5), mTECA/hi (n = 6), mTECB/hi (n = 4), and mTECC/hi (n = 5). (A) Principal component analysis (PCA) and dendrogram shows the hierarchical clustering between the distinct biological samples; graph shows the total amount of genes detected in each subset. (B) Heat maps representing the deviation to the average expression of Aire‐dependent (left) and Aire‐independent (right) TRAs in mTECA‐C/lo‐hi. (C) Heat maps represent the deviation to the average expression of the top 200 specifically upregulated genes of the mTEC I‐IV subsets in mTECA‐C/lo‐hi. (D) Heat maps represent the deviation to the average expression of the top 500 specifically upregulated genes of Pre‐, Early‐, Late‐, and Post‐Aire subsets in mTECA‐C/lo‐hi.
Recent single‐cell RNA‐sequencing studies have reclassified mTECs into four subtypes, termed mTEC I‐IV [14]. Using their available transcriptome and in silico analysis, we defined the top 200 genes specifically upregulated in mTEC I‐IV (details in section Material and Methods) and cross‐examined how their expression oscillated in mTECA‐C/lo‐hi subtypes. We found that the genetic signatures of mTEC I, mTEC II, mTEC III, and mTEC IV were respectively augmented in mTECC/lo, mTECA/hi, mTECC/hi, and mTECB/hi. In contrast, mTECA/lo and mTECB/lo did not reveal a particular link to any of the mTEC I‐IV subtypes (Fig. 2C; Supporting Information Table 2). While the molecular similarities between mTEC I:mTECC/lo and mTEC II:mTECA/hi pairs were in line with their mTEClo/hi nature and the expression of Aire, the association between mTEC III:mTECC/hi and mTECIV:mTECB/hi was intriguing. Since mTEC III and mTEC IV contained cells that expressed molecular features linked to Post‐Aire cells [14], we examined the possibility that mTECB‐C/hi defined later stages of Aire+mTEC differentiation. To do so, we curated recently published transcriptional profiles of Pre‐, Early‐, Late‐, and Post‐Aire mTECs, which were originally obtained from the analysis of an Aire‐Cre fate‐mapping mouse model [15]. Following a similar strategy to the aforementioned, we deconvoluted the top 500 genes specifically enriched in Pre‐, Early‐, Late‐, and Post‐Aire mTECs and surveyed their expression pattern in mTECA‐C/lo‐hi subtypes (Fig. 2D; Supporting Information Table 3). As above, mTECA‐B/lo did not reveal a particular association to the analyzed genetic profiles. We found that gene sets specifically associated to Pre‐ and Early‐Aire mTECs were respectively upregulated in mTECC/lo and mTECA/hi subtypes. The genetic program of Late‐Aire cells was detected across mTECA‐C/hi, being mostly marked in mTECB/hi. Strikingly, specific genetic features of Post‐Aire cells were upregulated in mTECB/hi and mTECC/hi. Supportive of their Post‐Aire nature, the expression of genes associated to Post‐Aire cells, and shared by terminally differentiated keratinocytes: Krt10 (keratin‐10), Ivl (Involucrin), Dsg3 (Desmoglein‐3), Dsc1 (Desmocollin‐1), Asprv1 (Aspartic Peptidase Retroviral Like 1), and Krt6a and Krt6b [6, 7, 8, 10, 15], was progressively augmented in mTECB‐C/hi. Contrarily, the expression of Cd80, Cd86 and Aire was gradually downregulated from mTECA/hi to mTECB‐C/hi, replicating the dynamics observed in the transition from Early‐ to Late and Post‐Aire stages (Supporting Information Fig. 2) [7, 10, 14, 15, 16]. Together, our genome‐wide transcriptional analysis suggested the following developmental trajectory: mTECA/hi (Aire+; i.e. Early‐Aire) turn on CD24 and Sca1 expression during their transition to mTECB‐C/hi (Aire+/− to Aire−; i.e., Late to Post‐Aire stages).
mTECB/hi and mTECC/hi are absent in Aire‐deficient mice
The findings described above implicated that mTECB‐C/hi derived from Aire‐expressing mTECA/hi. To study the precursor‐product relationship, we purified (cell sorting) mTECA‐C/hi and determined their developmental potential using reaggregate thymus organ cultures (RTOCs). Sorted C57BL/6‐derived mTEC subsets were mixed with BALB/c‐ derived embryonic thymic cells (carriers), and differences in MHCI expression (H2‐Kb for C57BL/6 and H2‐Kd for BALB/c) were used to trace their progeny within hybrid RTOCs (Supporting Information Fig. 3A and B). Since mTEClo contained precursors of Aire+mTEChi [2] and mTECC/lo presented transcriptomic traits coupled to the pre‐Aire stage (Fig. 2D), we first evaluated the capacity of mTECA‐C/lo, which lacked Aire, to contribute to the Aire lineage. Interestingly, both mTECB/lo and mTECC/lo subsets contained cells that were able to differentiate into mature mTEChi, including Aire+mTEChi. While some cells maintained their original mTECB‐C/lo phenotype, Aire+ cells that developed under these conditions resided within the mTECA/hi subset as their in vivo counterparts (Supporting Information Fig. 3C). We failed to detect the progeny of mTECA/lo (data not shown). Further studies are required to deconstruct whether mTECB‐C/lo represent developmentally unrelated precursors or transitional stages of the same differentiation pathway, as well as their relationship to recently defined embryonic (e.g., Cld3/4+SSEA−RANK) and post‐natal (Podoplanin (PDPN)+) mTEC‐restricted precursors [17]. Still, our results are in line with the observations that Aire+mTEC precursors (e.g., mTECB‐C/lo) downregulate CD24 expression during their differentiation into Aire+ cells [18].
Relatively to the developmental potential of mTECA‐C/hi subsets, we found that a large fraction of mTECA/hi remained mTEChi and either lacked or maintained Aire expression. While Aire+ generally maintained their mTECA/hi phenotype, emerging Aire− contained cells that acquired mTECB‐C/hi traits (Supporting Information Fig. 3D). Yet, we failed to detect the progeny of mTECB‐C/hi (data not shown), suggesting that these ex vivo isolated subtypes had a poor survival in RTOCs. We infer that the reduced ability to engraft and thrive in RTOCs may be related to their terminal maturation stage. Albeit our data suggest a linear relationship between mTECA/hi and mTECB‐C/hi, hybrid RTOCs experiments have limitations related with the integration and sensitivity of detection of “spiked” mature mTECs and their competition with embryonic‐derived carrier cells. Thus, we undertook a complementary in vivo genetic approach to provide further mechanistic evidence that mTECB‐C/hi represent end‐stages of Aire+mTEC maturation.
Apart from its role in regulating the expression of TRAs, Aire expression regulates mTEC differentiation [9, 19, 20, 21]. This notion is supported by the skewed mTEClo/mTEChi ratio, in favor of mTEChi, and the absence of Post‐Aire cells in Aire−/− mice [9, 14, 15, 20, 22]. To study whether the homeostasis of mTECB‐C/hi was Aire‐dependent, we compared their frequency in control and Aire−/− mice. As expected, Aire−/− mice presented an accumulation of mTEChi. Interestingly, the mTEChi compartment of Aire‐deficient mice was mostly composed of mTECA/hi and virtually lacked mTECB/hi and mTECC/hi subsets (Fig. 3A). We consider that the absence of mTECB‐C/hi does not result from a defective expression of CD24 and Sca1 in Aire‐deficient mice. CD24 and Sca1 have been reported as Aire‐independent genes [13] and CD24 and Sca1 were expressed in mTECB‐C/lo. Instead, we favor that the accumulation of mTECA/hi in Aire‐deficient mice define mature cells, which immediately preceded Aire expression but ceased their differentiation at this stage as a result of the lack of Aire protein. Lastly, the absence of mTECB‐C/hi (Post‐Aire) under Aire‐deficient conditions further supports the notion that Aire controls mTEC differentiation.
Figure 3.
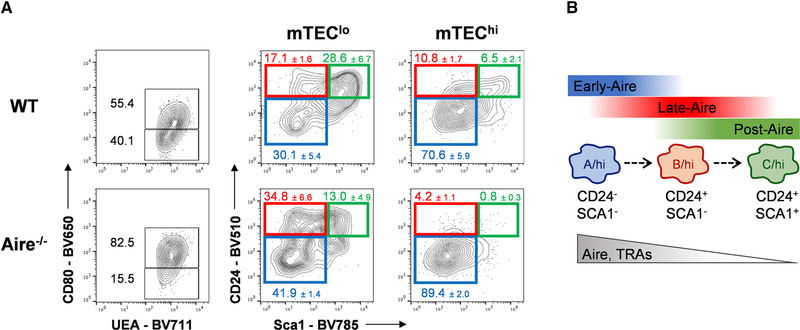
Altered mTECA‐C/hi composition in Aire‐deficient mice. (A) Thymic cells were stained with PerCP‐Cy5‐conjugated anti‐CD45.2, PE‐conjugated anti‐Ly51, APC‐eFlour 780‐conjugated anti‐MHCII (I‐A/I‐E), BV421‐conjugated anti‐EpCAM, BV650‐conjugated anti‐CD80, BV510‐conjugated anti‐CD24, BV786‐conjugated anti‐Sca1; Biotinylated UEA: BV711‐conjugated streptavidin. Total mTECs (CD45−EpCAM+UEA+) from 6‐week‐old Aire−/− and control (WT) C57BL/6 mouse thymi were analyzed for the expression of CD80, and mTECA‐C subsets were respectively identified with mTEClo and mTEChi. Plots shown are of a representative analysis out of three independent experiments with 1–2 mice of each genotype per experiment. Frequencies for mTECA‐C populations are shown as a mean plus SD. (B) Proposed model for the phenotypic discrimination of Early‐, Late, and Post‐Aire stages of mTEC differentiation.
Concluding remarks
Our understanding of the role of Aire+mTECs has considerably improved in the past years. Albeit the expression of Aire was initially considered to mark a terminal stage of mTEC maturation, several studies have shown that mTECs extend their differentiation beyond Aire (Post‐Aire) [19]. The most recent evidence of the complexity of mTEC cartography is the identification of tuft‐like mTECs, some of which progress through an Aire‐expressing stage before terminating Aire expression [14, 15]. The isolation of Post‐Aire cells has been hampered by the paucity of a reliable set of cell surface markers to define them. To date, the identification of these dedicated mTEC populations depends on lineage‐tracing systems and complex single‐cell RNA sequencing approaches, which are neither always easily accessible nor compatible with functional analysis. Our study solves this conundrum, offering an efficient and novel phenotypic chart for the direct analysis of Early‐, Late‐, and Post‐Aire stages of mTEC maturation. To our knowledge, the simultaneous analysis of CD24 and Sca1 expression within mTECs provides the first mean to prospectively segregate later stages of Aire+mTEC differentiation. First, our observation that mTECC/hi mirror cells in a Post‐Aire stage are consistent with the genome‐wide molecular signature of Post‐Aire mTECs obtained from the analysis of fate‐mapping mouse models [6, 8, 9]. Second, the expression of Aire‐dependent TRAs was downregulated in mTECC/hi compared to mTECA‐B/hi, an observation that matches the pattern of Aire expression in mTECC/hi (Aire−). Lastly, mTECC/hi express markers linked to terminally differentiated keratinocytes and depend on Aire. Although Post‐Aire cells terminate their differentiation process as mTEClo, our populational RNAseq analysis was not able to dissect if those cells are diluted into a specific mTECA‐C/lo subset. Still, as the majority of cells that lost Aire expression downregulate MHCII and CD80 but remain as mTEChi [8] or reside at the mTEChi/lo boundary [6, 9], we reason that our phenotypic definition (CD24+Sca1+MHCII+CD80+) enables the prospective isolation of a large fraction of Post‐Aire cells. Hence, we propose that mTECA/hi‐mTECB/hi‐mTECC/hi sequentially mark transitional steps of Aire+mTEC differentiation into a Post‐Aire stage (Fig. 3B). Our study provides an integrative road map that will prove of general interest to survey the dynamic of the medullary microenvironment in several experimental and autoimmune conditions.
Material and methods
Mice
C57BL/6 mice were housed under specific pathogen‐free conditions at I3S’ animal facility. Aire−/− mice were described previously [22] and housed at the animal facility of Tokushima University. Experiments were performed under the European guidelines for animal experimentation.
Isolation and flow cytometry analysis of TECs
Thymi were dissected at the indicated time points and TEC isolation and staining was performed as previously described [23]. The following antibodies were used for flow cytometry analysis of isolated cells: PerCP‐Cy5‐conjugated anti‐CD45.2 (clone 104), PE‐conjugated anti‐Ly51 (clone 6C3), APC‐eFlour 780‐conjugated anti‐I‐A/I‐E (clone M5/114‐15‐2), Alexa eFluor 647‐conjugated anti‐EpCAM (clone G8.8), PE‐conjugated anti‐CD80 (clone 16‐10A1) and eFlour 660‐cojugated anti‐AIRE (all from eBioscience); BV421‐conjugated anti‐EpCAM (clone G8.8), BV650‐conjugated anti‐CD80 (clone 16‐10A1), BV510‐conjugated anti‐CD24 (clone M1/69), BV786‐conjugated anti‐Sca1 (clone D7), APC‐Fire‐conjugated anti‐CD24 (clone M1/69), and Alexa 488‐conjugated anti‐Sca1 (clone D7) (all from Biolegend); Biotinylated UEA (vectorshield) was revealed with BV711‐conjugated (Biolegend), or PE‐Cy7‐conjugated streptavidin (eBioscience). Flow cytometry analyses were performed on an LSRFortessa and cells sorted on a FACS ARIA II (both from BD Bioscience) with purities above 95%. Data were analyzed on FlowJo software (Tree Star Inc). Our study was conducted according to the guidelines for the use of flow cytometry and cell sorting in immunological studies [24].
RNA sequencing
Total RNA library preparation and high‐throughput sequencing of sorted postnatal (P5) mTECA‐C subsets were performed at Gene Core facility (EMBL, Germany), as previously described [23]. Thirteen sequencing libraries (three for mTECA/lo, five for mTECB/lo, two for mTECC/lo, four for mTECA/hi, five for mTECB/hi, and six for mTECC/hi) were prepared using NEB Next RNA ultraprotocol (#E7530 NEB). Obtained libraries were quantified fluorimetrically, pooled in equimolar amounts and sequenced on the Illumina NextSeq sequencer (NextSeqHi‐75) in single‐end mode (75 bases), following manufacturer's instructions (Illumina). Sequencing reads were submitted to ENA (http://www.ebi.ac.uk/ena) and are accessible under the accession number PRJEB32793. The reads were mapped to the mouse genome (mm10) using STAR (version 2.4.2a) with mm10 GTF annotation. The number of reads per gene was generated during alignment step (quantMode GeneCounts) and gene counts were then analyzed with DESeq2 package [25]. Reads per kb of genes per million mapped reads (RPKM) values were computed from normalized read counts. Gene ontology (GO) enrichment analysis was done using model‐based gene set analysis (MGSA) [26].
In silico comparative transcriptomic analyses
To obtain the deviation to average expression in mTEC I, mTEC II, mTEC III, and mTEC IV, we calculated the mean of the deviation to the average expression in clusters X3‐X18, X19‐X28, X29‐33, and X34‐36 [14]. To determine the gene sets specifically upregulated in mTEC I‐IV, we further identified the top 200 most upregulated genes from each population considering a twofold increase relatively to the remaining three. To acquire the gene sets specifically upregulated in Early‐, Pre‐, Late‐, and Post‐Aire mTECs [15], we calculated the deviation of expression of each gene in each subset (mean of the four biological replicates) relatively to the average expression across all samples. To identify the top 500 most upregulated genes in each subpopulation, we considered genes with an expression >10 rpkm and genes in which the upregulation in the selected population is specifically (at least) twofold higher relatively to any of the other three.
Reaggregate thymus organ culture (RTOC)
Reaggregate thymus organ cultures (RTOCs) were established as previously described [27] by combining 7 × 105 total thymic cells from E14.5 BALB/c thymus and 2–3 × 104 sorted mTECs (EpCAM+CD45−UEA+) of mTECA‐C/lo‐hi subsets obtained from newborn C57BL/6 thymic lobes. After 7 days in culture, RTOCs were dissociated and analyzed by flow cytometry.
Author Contributions
N.L.A. conceived and performed experiments, wrote the manuscript, and secured funding, P.F and C.R. conceived and performed experiments and wrote the manuscript, J.M. J.J.M.L., C.M., and L.A. performed experiments, and M.M., V.B., and M.M. provided scientific expertise and feedback.
Conflict of interest
The authors have no commercial or financial conflict of interests.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202048764.
Supporting information
Supporting information
Acknowledgments
This work was supported by a starting grant from the European Research Council (ERC) under the project 637843 and by FEDER ‐ Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020 ‐ Operacional Programme for Competitiveness and Internationalisation (POCI), Portugal 2020, and by Portuguese funds through FCT ‐ Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Ensino Superior in the framework of the project POCI‐01‐0145‐FEDER‐029129 (PTDC/MED‐IMU/29129/2017). N.L.A is supported by the FCT program “Scientific Employment Stimulus”. M.M. is supported by the Japanese Agency for Medical Research and Development‐Core Research for Evolutional Science and Technology (JSPS KAKENHI Grant Numbers JP16K21731, JP16H06496 and JP16H05342). We thank Drs. Sofia Lamas and the caretakers from the animal facility for technical assistance. We thank Dr. Pedro Rodrigues for discussions and critical reading.
References
- 1. Alves, N. L. , Takahama, Y. , Ohigashi, I. , Ribeiro, A. R. , Baik, S. , Anderson, G. and Jenkinson, W. E. , Serial progression of cortical and medullary thymic epithelial microenvironments. Eur. J. Immunol. 2014. 44: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson, G. and Takahama, Y. , Thymic epithelial cells: working class heroes for T cell development and repertoire selection. Trends Immunol. 2012. 33: 256–263. [DOI] [PubMed] [Google Scholar]
- 3. Klein, L. , Kyewski, B. , Allen, P. M. and Hogquist, K. A. , Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat. Rev. Immunol. 2014. 14: 377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abramson, J. and Anderson, G. , Thymic Epithelial Cells. Annu. Rev. Immunol. 2017. 35: 85–118. [DOI] [PubMed] [Google Scholar]
- 5. Gray, D. , Abramson, J. , Benoist, C. and Mathis, D. , Proliferative arrest and rapid turnover of thymic epithelial cells expressing Aire. J. Exp. Med. 2007. 204: 2521–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nishikawa, Y. , Hirota, F. , Yano, M. , Kitajima, H. , Miyazaki, J. , Kawamoto, H. , Mouri, Y. and Matsumoto, M. , Biphasic Aire expression in early embryos and in medullary thymic epithelial cells before end‐stage terminal differentiation. J. Exp. Med. 2010. 207: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. White, A. J. , Nakamura, K. , Jenkinson, W. E. , Saini, M. , Sinclair, C. , Seddon, B. , Narendran, P. et al., Lymphotoxin signals from positively selected thymocytes regulate the terminal differentiation of medullary thymic epithelial cells. J. Immunol. 2010. 185: 4769–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Metzger, T. C. , Khan, I. S. , Gardner, J. M. , Mouchess, M. L. , Johannes, K. P. , Krawisz, A. K. , Skrzypczynska, K. M. and Anderson, M. S. , Lineage tracing and cell ablation identify a post‐aire‐expressing thymic epithelial cell population. Cell Rep. 2013. 5: 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nishikawa, Y. , Nishijima, H. , Matsumoto, M. , Morimoto, J. , Hirota, F. , Takahashi, S. , Luche, H. et al., Temporal lineage tracing of Aire‐expressing cells reveals a requirement for Aire in their maturation program. J. Immunol. 2014. 192: 2585–2592. [DOI] [PubMed] [Google Scholar]
- 10. Wang, X. , Laan, M. , Bichele, R. , Kisand, K. , Scott, H. S. and Peterson, P. , Post‐Aire maturation of thymic medullary epithelial cells involves selective expression of keratinocyte‐specific autoantigens. Front. Immunol. 2012. 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shackleton, M. , Vaillant, F. , Simpson, K. J. , Stingl, J. , Smyth, G. K. , Asselin‐Labat, M. L. , Wu, L. et al., Generation of a functional mammary gland from a single stem cell. Nature. 2006. 439: 84–88. [DOI] [PubMed] [Google Scholar]
- 12. Holmes, C. and Stanford, W. L. , Concise review: stem cell antigen‐1: expression, function, and enigma. Stem. Cells. 2007. 25: 1339–1347. [DOI] [PubMed] [Google Scholar]
- 13. Sansom, S. N. , Shikama‐Dorn, N. , Zhanybekova, S. , Nusspaumer, G. , Macaulay, I. C. , Deadman, M. E. , Heger, A. et al., Population and single‐cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self‐antigen expression in thymic epithelia. Genome. Res. 2014. 24: 1918–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bornstein, C. , Nevo, S. , Giladi, A. , Kadouri, N. , Pouzolles, M. , Gerbe, F. , David, E. et al., Single‐cell mapping of the thymic stroma identifies IL‐25‐producing tuft epithelial cells. Nature 2018. 559: 622–626. [DOI] [PubMed] [Google Scholar]
- 15. Miller, C. N. , Proekt, I. , von Moltke, J. , Wells, K. L. , Rajpurkar, A. R. , Wang, H. , Rattay, K. et al., Thymic tuft cells promote an IL‐4‐enriched medulla and shape thymocyte development. Nature 2018. 559: 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang, J. , Sekai, M. , Matsui, T. , Fujii, Y. , Matsumoto, M. , Takeuchi, O. , Minato, N. and Hamazaki, Y. , Hassall's corpuscles with cellular‐senescence features maintain IFNalpha production through neutrophils and pDC activation in the thymus. Int. Immunol. 2019. 31: 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alves, N. L. and Ribeiro, A. R. , Thymus medulla under construction: Time and space oddities. Eur. J. Immunol. 2016. 46: 829–833. [DOI] [PubMed] [Google Scholar]
- 18. Akiyama, N. , Takizawa, N. , Miyauchi, M. , Yanai, H. , Tateishi, R. , Shinzawa, M. , Yoshinaga, R. et al., Identification of embryonic precursor cells that differentiate into thymic epithelial cells expressing autoimmune regulator. J. Exp. Med. 2016. 213: 1441–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matsumoto, M. , Contrasting models for the roles of Aire in the differentiation program of epithelial cells in the thymic medulla. Eur. J. Immunol. 2011. 41: 12–17. [DOI] [PubMed] [Google Scholar]
- 20. Yano, M. , Kuroda, N. , Han, H. , Meguro‐Horike, M. , Nishikawa, Y. , Kiyonari, H. , Maemura, K. et al., Aire controls the differentiation program of thymic epithelial cells in the medulla for the establishment of self‐tolerance. J. Exp. Med. 2008. 205: 2827–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gillard, G. O. , Dooley, J. , Erickson, M. , Peltonen, L. and Farr, A. G. , Aire‐dependent alterations in medullary thymic epithelium indicate a role for Aire in thymic epithelial differentiation. J. Immunol. 2007. 178: 3007–3015. [DOI] [PubMed] [Google Scholar]
- 22. Kawano, H. , Nishijima, H. , Morimoto, J. , Hirota, F. , Morita, R. , Mouri, Y. , Nishioka, Y. and Matsumoto, M. , Aire expression is inherent to most medullary thymic epithelial cells during their differentiation program. J. Immunol. 2015. 195: 5149–5158. [DOI] [PubMed] [Google Scholar]
- 23. Rodrigues, P. M. , Ribeiro, A. R. , Perrod, C. , Landry, J. J. M. , Araujo, L. , Pereira‐Castro, I. , Benes, V. et al., Thymic epithelial cells require p53 to support their long‐term function in thymopoiesis in mice. Blood 2017. 130: 478–488. [DOI] [PubMed] [Google Scholar]
- 24. Cossarizza, A. , Chang, H.D. , Radbruch, A. , Acs, A. , Adam, D. , Adam‐Klages, S. , Agace, W. W. et al., Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol. 2019. 49: 1457–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Love, M. I. , Huber, W. and Anders, S. , Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome. Biol. 2014. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bauer, S. , Gagneur, J. and Robinson, P. N. , GOing Bayesian: model‐based gene set analysis of genome‐scale data. Nucleic. Acids. Res. 2010. 38: 3523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ribeiro, A. R. , Rodrigues, P. M. , Meireles, C. , Di Santo, J. P. and Alves, N. L. , Thymocyte selection regulates the homeostasis of IL‐7‐expressing thymic cortical epithelial cells in vivo. J. Immunol. 2013. 191: 1200–1209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information