

Graphical abstract
Keywords: SARS-CoV-2 virus, Spike-protein, Structure refinement, Electronic structure, Interatomic bonding, Density functional calculation
Abstract
The COVID-19 pandemic poses a severe threat to human health with an unprecedented social and economic disruption. Spike (S) glycoprotein of the SARS-CoV-2 virus is pivotal in understanding the virus anatomy, since it initiates the first contact with the ACE2 receptor in the human cell. We report results of ab initio computation of the spike protein, the largest ab initio quantum chemical computation to date on any bio-molecular system, using a divide and conquer strategy by focusing on individual structural domains. In this approach we divided the S-protein into seven structural domains: N-terminal domain (NTD), receptor binding domain (RBD), subdomain 1 (SD1), subdomain 2 (SD2), fusion peptide (FP), heptad repeat 1 with central helix (HR1-CH) and connector domain (CD). The entire Chain A has 14,488 atoms including the hydrogen atoms but excluding the amino acids with missing coordinates based on the PDB data (ID: 6VSB). The results include structural refinement, ab initio calculation of intra-molecular bonding mechanism, 3- dimensional non-local inter-amino acid interaction with implications for the inter-domain interaction. Details of the electronic structure, interatomic bonding, partial charge distribution and the role played by hydrogen bond network are discussed. In the interaction among structural domains, we present new insights for crucial hinge-like movement and fusion process. Extension of such calculation to the interface between the S-protein binding domain and ACE2 receptor can provide a pathway for computational understanding of mutations and the design of therapeutic drugs to combat the COVID-19 pandemic.
1. Introduction
The COVID-19 pandemic has been with us for nearly 12 months now, with no clear end in sight, and with unabashed destruction of human life and mounting danger to the world economy [1], [2]. Intensive research on all aspects of combating this disease is pervasive, ranging from strategies to prevent spreading, role played by the physical distance, nature of virus infection process, vaccine development, drug discovery and post-infection recovery to the long-term psychological damage and more [3], [4], [5]. From the perspective of fundamental science related to COVID-19, it is fair to say that this is one of the most outstanding scientific challenges of this century that has incited many scientists in different disciplines towards timely contributions. The causative agent of COVID-19 is the virus dubbed the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the World health organization (WHO) [6]. SARS-CoV-2 is a single positive strand RNA virus composed of four structural proteins: the spike (S), the envelope (E), the membrane (M), and the nucleocapsid (N) proteins. Of the four, the S-protein is the most important since it is facing the external bathing solution and hence controls the infectivity and transmissibility [7], [8]. The other two proteins, E and M, are located between the spikes, while the N-protein encloses a long ss-RNA genome with 29,900 nucleotides [9].
The structure of S-protein was released in late February 2020 using Cryo-electron microscopy with a resolution of 3.5 Å [10] (PDB: ID 6VSB). Similar experiments were conducted immediately by other teams on S-protein itself, as well as related structures [11], [12], [13], [14]. This is the first step for a detailed analysis of the structure, properties and functionality of the S-protein, enabling computational studies of S-protein using a variety of methods and approaches at different levels of complexity [15], [16], [17], [18], [19]. Here, we focus on the ultra-large-scale ab initio quantum chemical computation of the SARS-CoV-2 S-protein consisting of three similar chains A, B, C (shown in Fig. 1). Of these three, Chain A in the up conformation, is the most critical one since it is receptor accessible [10]. Each chain in the S-protein has two main subunits, the receptor binding subunit S1 and the membrane fusing subunit S2. S1 consists of a signal sequence (SS), N-terminal domain (NTD), receptor binding domain (RBD), subdomain 1 (SD1) and subdomain 2 (SD2). S2 consists of fusion peptide (FP), heptad repeat 1 (HR1), central helix (CH), connector domain (CD), heptad repeat 2 (HR2), transmembrane domain (TM), and cytoplasmic tail (CT). We performed quantum chemical calculations on all domains except SS, HR2, TM and CT, since their position coordinates are missing in 6VSB. The entire Chain A of 6VSB has a total of 959 amino acids (AA) and 14,488 atoms, including the hydrogen atoms but excluding the amino acids with missing coordinates in 6VSB.
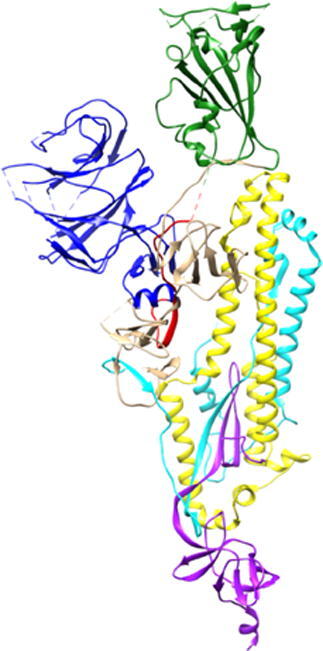
Fig. 1.

Structure of the trimeric S-protein of SARS-CoV-2 in the prefusion conformation. Chain A is shown in ribbon representation with their seven structural domains colored by blue for NTD, red for SD1, green for RBD, tan for SD2, cyan for FP, yellow for HR1-CH and violet for CD. The RBD of Chain A is in the up conformation whereas Chain B and C are in down conformation. Chain B and C are shown in surface representations with powder-blue and light-yellow colors respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Such ultra-large-scale ab initio calculations on a complex biomolecular system are obviously impossible at present. We thus devised a divide and conquer strategy to tackle this monumental challenge. Based on the available position coordinates of 6VSB, we divided the S-protein into seven structural domains: NTD, RBD, SD1, SD2, FP, heptad repeat 1 with central helix (HR1-CH), and CD (shown in Fig. 2). FP, HR1-CH and CD in S2 are divided in such a way that they include all amino acids available in Wrapp et al. [10]. We performed the calculations with these seven structural domains individually and connected the results in an insightful way for the entire S-protein. The largest domain is NTD with 226 AAs and the smallest domain is SD1 with 24 AAs. For each of the seven structural domains, we first refine their structures to high accuracy in order to perform the density functional theory (DFT) calculations on each of them separately. This enables us to investigate the intra-molecular bonding mechanism, with the implications for the inter-domain interaction. The results will include electronic structure, interatomic bonding, partial charge distribution, the hydrogen bonding network and the non-local AA-AA interaction. More importantly, our calculation and methodology demonstrates that it is possible to perform similar atomic-scale calculations also with the S-protein and the ACE2 receptor binding domain that can provide a pathway to computational understanding of the effects of amino acid mutation as well as enable and guide the design of therapeutic drugs. What we accomplished is probably the largest, unprecedented quantum chemical calculation based on DFT [20], [21]. To the best of our knowledge, the current and the most rigorous quantum chemical calculations using the Gaussian package [22] are generally limited to just to a few hundred atoms at most.
Fig. 2.

The schematic representation of S-protein sequence in SARS-CoV-2 (ID: 6VSB). S-protein is composed of two subunits S1 (left side of the horizontal bar with domains NTD, SD1, RBD and SD2) and S2 (right side of the horizontal bar with domains FP, HR1-CH and CD). The domains with their information are shown in similar color. The number of amino acids (AA), atoms (including hydrogen atoms), and their sequence number range are marked in the upper part of the horizontal bar. The numbers in the bottom part of the horizontal bar indicates the sequence number for domains. The missing position coordinates are shown in the vertical white column with their sequence numbers shown in pink boxes marked by dashed arrows. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
In what follows, we first briefly describe the methods used. This is followed by the description of the structural relaxation for each structural domain (NTD, SD1, RBD, SD2 in S1 and FP, HR1-CH, CD in S2). This part consumes the most computational resources and is the most demanding part of our research. The results of the DFT calculation for each of the seven structural domains are presented in Section 3 and discussed in Section 4 with the overall goal of connecting them to the properties and implications of the entire S-protein. We end up with a brief conclusion and our vision of the large-scale computational modeling for complex biomolecular systems in general.
2. Methods
2.1. Structural reconstruction and relaxation.
The structure of the S-protein in SARS-CoV-2 is obtained from PDB (ID: 6VSB) [10], where many of the amino acid positions have missing atomic coordinates due to experimental difficulties. To proceed with our calculations, we first eliminated these amino acids without complete atomic coordinates in the seven structural domains. This created some gaps in the remaining sequences. More importantly, the deposited PDB data does not include the H atoms and they have to be added using the standard software (Chimera) [23]. This initial structure is then fully relaxed by using Vienna ab initio simulation package (VASP) [24] known for its efficiency in structure optimization. We used the projector augmented wave (PAW) method with Perdew-Burke-Ernzerhof (PBE) exchange correlation functional [25] within the generalized gradient approximation (GGA). While there exist other more elaborate potentials within DFT they come at the expense of prohibitive computational cost for large complex biomolecular systems such as the S-protein.
Our past experience and detailed tests suggest the use of following input parameters: energy cut-off 500 eV, electronic convergence of 10−4 eV; force convergence criteria for ionic steps at −10−2 eV/Å and a single k-point sampling. All VASP structure relaxations were carried out at the National Energy Research Scientific Computing (NERSC) facility at the Lawrence Berkeley Laboratory and also at the Research Computing Support Services (RCSS) of the University of Missouri System. The structures of each of the seven structural domains in the spike protein are fully relaxed with accuracy in atomic positions estimated to be about 0.01 Å. The total energies of the initial unrelaxed and the fully relaxed structural domains from VASP are listed in Table 1. For the smallest subdomain SD1 with only 24 amino acids and 391 atoms, the calculated total energy decreases from −2370.90 eV to −2379.21 eV or only 0.02 eV (2.05 kJ/mol) per atom. The VASP-relaxed structure is used as the input for the electronic structure and interatomic bonding calculations described below.
Table 1.
Calculated total energy with reduction in the energy per atom in the seven structural domains.
| Domain | Cell dimen: a × b × c (Å) | # of atoms | Initial energy (eV) | Final energy (eV) | ΔE/atom (eV/atom) | ΔE/atom (kJ/mol) | ΔE/atom (kcal/mol) |
|---|---|---|---|---|---|---|---|
| NTD | 64.428 × 51.855 × 73.509 | 3459 | −21120.58 | −21224.03 | 0.0299 | 2.8857 | 0.6897 |
| SD1 | 38.487 × 61.424 × 61.096 | 391 | −2370.90 | −2379.21 | 0.0212 | 2.0502 | 0.4900 |
| RBD | 48.453 × 48.825 × 63.166 | 2100 | −12890.48 | −12944.23 | 0.0256 | 2.4696 | 0.5902 |
| SD2 | 63.635 × 69.596 × 82.795 | 1912 | −11631.10 | −11692.36 | 0.0320 | 3.0913 | 0.7388 |
| FP | 73.587 × 76.165 × 96.654 | 2130 | −12913.56 | −12964.66 | 0.0240 | 2.3149 | 0.5533 |
| HR1-CH | 53.277 × 47.476 × 109.584 | 2786 | −16854.37 | −16923.84 | 0.0249 | 2.4060 | 0.5751 |
| CD | 48.347 × 64.427 × 90.932 | 1710 | −10497.60 | −10548.65 | 0.0299 | 2.8810 | 0.6886 |
2.2. Electronic structure and interatomic bonding
For the electronic structure and interatomic interactions in the S-protein we use a very different DFT method, the all-electron orthogonalized linear combination of atomic orbitals (OLCAO) method [26], developed in-house. The efficacy of using the combination of these two different DFT codes is well documented [27], [28], [29], [30] and it is especially effective for large complex biomolecular systems such as the SARS-CoV-2 virus. The key feature of the OLCAO method is the provision for the effective charge () on each atom and the bond order (BO) values ραβ between any pairs of atoms. They are obtained from the ab initio wave functions with atomic basis expansion calculated quantum mechanically:
| (1) |
| (2) |
In the above equations, are the overlap integrals between the orbital in atom and the orbital in the atom. are the eigenvector coefficients of the occupied molecular orbital. The partial charge (PC) or () is the deviation of the effective charge from the neutral atomic charge on the same atom. The BO quantifies the strength of the bond between two atoms and usually scales with the bond length (BL), being also influenced by the surrounding atoms. The calculation of PC and BO are based on the Mulliken scheme [31], [32], hence are basis-dependent. Comparisons of BO values using different basis or different methods should be treated with caution.
The atomic-scale interactions based on DFT calculations are critical for providing the accurate information necessary for their fundamental understanding and are rarely done for large proteins. In the present case, the largest domain NTD consists of a total of 3459 atoms. It is obviously challenging to obtain the accurate atomic partial charges for each atom and the bond order values between all pairs of atoms. More details on the OLCAO method can be found in Refs. [33], [26].
2.3. Extension to amino acid interactions in proteins
The bond order values in Eq. (2) above can be calculated for every pair of atoms since their positions are precisely defined after optimization. In biological systems the focus is mostly on whole amino acids whose exact positions are ill-defined. Amino acids or residues are essentially biomolecules containing different atoms with different configurations and orientations. Strictly speaking, assigning a distance of separation between AAs in a protein in order to describe their interactions, is a vague and arbitrary parameter. However, with the quantum mechanically based OLCAO method and with the interatomic interaction between all atoms available, we can define the bonding between two AAs u and v with no ambiguity, which we dub as amino acid bond pair (AABP) [34]:
| (3) |
where the summations are over atoms in and atoms in . This is a far more rigorously defined quantity and can be further extended to different units, such as all seven structural domains in the spike protein, if necessary. The merit of the above scheme is that AABP for selected groups of AAs can be obtained by adding their BOs for relative comparisons. AABP includes all possible bonding between two amino acids such as covalent and hydrogen bonding. This single quantitative parameter reflects the internal bonding strength among amino acids. It can be further resolved into nearest neighbor (NN) and non-local bonding and it ideal to understand inter amino acid bonding.
3. Results
3.1. Structural refinement
The S-protein of SARS-CoV-2 virus consists of three chains A, B, and C, as shown in Fig. 1. These three chains are similar but not the same and contain the same subunits and domains. The Chain A is considered to be the most important one since its RBD is in the receptor accessible up conformation. We chose the orientation of Chain A to fix the Cartesian coordinate (x, y, z) for all the structures in the S-protein. Fig. 2 delineates with specific details their structural arrangement including the location of missing amino acids groups or gaps of Chain A in the present study. There are two main subunit S1 on the left and S2 on the right. Based on the position coordinates available, S1 is divided into NTD, SD1, RBD and SD2. S2 is divided into FP, HR1-CH, and CD. The entire Chain A has 14,488 atoms including the hydrogen atoms but excluding the amino acids with missing coordinates [10]. In the divide and conquer strategy designed for this study, the seven structural domains are separately relaxed to high accuracy using supercomputer facility. This is the most resources demanding part of the whole computation, because accurate final atomic scale structures are pivotal to the reliability of the results reported in the following section. Fig. 3 shows the ribbon structures of Chain A and its seven structural domains in the S-protein. Fig. 4 display the ball and stick figures of the seven structural domains of Fig. 3(b) after full optimization that are used as the input for the electronic structure calculation using the OLCAO method. The position coordinates of optimized seven structural domains are provided in PDB format in the supplementary data.
Fig. 3.

Ribbon structure of (a) the Chain A of S-protein SARS-CoV-2 with their (b) seven structural domains NTD, SD1, RBD, SD2, FP, HR1-CH, and CD colored corresponding to Fig. 1.
Fig. 4.

Ball and stick figure of seven structural domains after full optimization. (a) NTD, (b) SD1, (c) RBD, (d) SD2, (e) FP, (f) HR1-CH, (g) CD. The degree of accuracy on their optimization in terms of energy is listed in Table 1.
Fig. 5 shows the frequency distributions of all the 959 AAs in the S-protein among the 20 known amino acids, and the components of each type of residue in the seven structural domains. It can be seen:
Fig. 5.

Frequency distribution of 959 AAs in Chain A of the S-protein over 20 canonical amino acids.
(1) Leu has the largest count of 82 followed by Thr and Val (78 counts each).
(2) Trp has the lowest count 6 with presence only in NTD, RBD, HR1-CH and CD. This is followed by Met with 8 counts with presence only in FP, HR1-CH and CD.
(3) It is observed that residues Ala, Asp, Cys, His, Leu, Met, Trp were not occurring in SD1 since it is the smallest domain. NTD and RBD contains all residues except Met. SD2 contains all residues except Met and Trp. FP contains all residues except His and Trp. HR1-CH contains all residues except His and CD contains all 20 types of residues.
(4) The most important domain RBD contains the largest number of Tyr closely followed by NTD.
In Table 1 we list the total energies of the initial and final structures and the reduction in the total energy for each of these seven structural domains to demonstrate the accuracy of optimization.
As can be seen, the decrease in energy/per atom ranges from 2.05 kJ/mol in SD1 and then all the way to 3.09 kJ/mol in SD2, with 2.47 kJ/mol for RBD somewhere in the middle of the range. Such reasonable variations depend on several factors such as the size (number of atoms in the structural domains), the internal structures of each structural domain, and the availability of computing cycles over the time of roughly four months. We estimate that the accuracy reached is about 0.01 Å in atomic positions since this is the key parameter for the accuracy in the DFT calculations, not the total energy.
To demonstrate the importance of the accuracy in atomic positions, we recently carried out a test calculation in another model of the S-protein RBD bound to the ACE2 receptor (PDB ID: 6M0J) shown in Fig. 6 (a), that has claimed a resolution of 2.45 Å [8]. Our test model (Fig. 6 (b)) includes only the key interacting residues at the interface between the receptor binding motif (RBM) of RBD and the ACE2 receptors α1, α2, β1, β2 (PDB ID 6M0J). This interface model contains the residues from Ser438 to Tyr508 of the SARS-CoV-2 RBM (71 AAs) and residues from Ser19 to Ile88 of the receptor binding motifs α1 and α2 plus residues from Gly319 to Thr365 of motifs β3 and β4 and some other residues of the ACE2 (117 AAs). This interface model has a total of 2924 atoms after addition of hydrogen atoms and does not have any missing AAs.
Fig. 6.

(a) The sketch of the interface model of the SARS-CoV-2 RBD bound to the ACE2 receptor. (b) The interface model between the RBM and α1, α2, β1 and β2 used in VASP relaxation. (c) Strong HBs likely to form between the RBM and α1 shown as green dots. Key residues are labeled and shown in sticks. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The interatomic separations between atoms forming possible HBs from experimentally reported crystal data [8] are listed in Table 2. After the addition of H atom, the potential HBs are compared with the interatomic separations of potential HBs between the initial unoptimized structure, the partially optimized structure, and the fully optimized structure using VASP relaxation. The comparation shows that the unoptimized structure has mostly larger HB separation distances compared to the optimized structures. Moreover, the experimental X-ray crystal structure itself is not sufficiently accurate. For instance, the values colored in red in Table 2 are not predicted in our HBs analysis after optimization and those may not be actual HBs. These data clearly indicate that accurate structural optimization is extremely important for interatomic interaction. Our analysis reveals the presence of strong HBs between RBM and α1 of the ACE2 from the fully optimized structure with interatomic separations of less than 2.0 Å (see Fig. 6(c)). These stronger HBs could explain the high binding affinity between the S-protein and the ACE2 receptor. We used the BIOVIA Discovery Studio Visualizer [35] and Chimera for these HBs analysis. The details of this work are still in progress and will be reported elsewhere.
Table 2.
Hydrogen bond distance at the RBM–ACE2 interface from X-ray crystal structure and partial and fully optimized structures.
| SARS-CoV2 RBM | ACE2 | 6M0J# (Å) | SARS-CoV2 RBM including H atoms | ACE2 including H atoms | Distance (Å) |
||
|---|---|---|---|---|---|---|---|
| Initial* | Partial† | Full† | |||||
| Asn487(ND2) | Gln24(OE1) | 2.6 | Asn487(HD21) | Gln24(OE1) | 1.89 | 1.88 | 1.89 |
| Gln493(NE2) | Glu35(OE2) | 2.8 | Gln493(HE22) | Glu35(OE2) | 2.46 | 1.87 | 1.87 |
| Tyr505(OH) | Glu37(OE2) | 3.2 | Tyr505(HH) | Glu37(OE2) | 4.0 | 5.0 | 5.1 |
| Tyr449(OH) | Asp38(OD2) | 2.7 | Tyr449(HH) | Asp38(OD2) | 1.82 | 1.54 | 1.54 |
| Thr500(OG1) | Tyr41(OH) | 2.6 | Thr500(HG1) | Tyr41(HH) | 2.87 | 1.77 | 1.77 |
| Asn501(O) | Tyr41(OH) | 3.7 | Asn501(OD1) | Tyr41(HH) | 3.7 | 4.36 | 4.43 |
| Gly446(O) | Gln42(NE2) | 3.3 | Gly446(O) | Gln42(HE21) | 2.35 | 2.02 | 1.99 |
| Tyr449(OH) | Gln42(NE2) | 3.0 | Tyr449(OH) | Gln42(HE22) | 2.02 | 2.05 | 2.02 |
| Tyr489(OH) | Tyr83(OH) | 3.5 | Tyr489(HH) | Tyr83(HH) | 2.80 | 2.95 | 3.01 |
| Asn487(OD1) | Tyr83(OH) | 2.7 | Asn487(OD1) | Tyr83(HH) | 3.4 | 5.34 | 5.38 |
| Gly502(N) | Lys353(O) | 2.8 | Gly502(H) | Lys353(O) | 1.79 | 1.86 | 1.85 |
# The separation distances from Ref. [8], * unoptimized structure, † optimized structure.
The above example clearly shows the importance of accurate atomic positions is pivotal in analyzing the electronic interaction and nature of HBs in complex biomolecules. Pure experimentally measured data, no matter how advanced still has its limitations. Very recently, there have been reports of revolutionary development of cryo electron microcopy (cryo-EM) techniques, both in instrumentation and software development that could increase the atomic resolution down to 2.5 Å or lower [36], [37]. These are wonderful news. Nevertheless, our current work clearly demonstrates the important role played by high accuracy computational optimization based on the most powerful supercomputers. They complement each other in advancing future research direction for biomolecular systems, not just limited to COVID-19 virus. Another computational approach that is generally used is classical molecular dynamics (MD). We nevertheless need to point out that the set of assumptions of the two methodologies, classical MD and ab initio DFT are fundamentally different, and they have not been in general reconciled, especially not for the size of the computational effort described in our MS.
3.2. Electronic structure
In the calculation for small biomolecules, the electronic structure is usually displayed in the form of molecular energy levels separated by the HOMO (highest occupied molecular orbitals) and LUMO (lowest unoccupied molecular orbitals) gap. In large complex biomolecules such as proteins this is unpractical and we present them in the form of density of states (DOS) and partial density of states (PDOS), commonly used in materials science and condensed matter physics. Fig. S1 shows the calculated TDOS and atom-resolved PDOS for the seven structural domains of the S-protein. It can be seen that grosso modo their features are very similar, as they should be, but there are minor differences which can be succinctly summarized as follows:
-
(1)
The overall features of the TDOS for the seven structural domains are very similar since they all consist of AAs with similar atomic components. The occupied valence band (VB) is separated from the unoccupied conduction band (CB) by a sizable HOMO-LUMO gap ranging from 1.31 eV in HR1-CH to 2.45 eV in RBD.
-
(2)
The atom-resolved PDOS shows some differences between the different units. They all contain the S atom in the Cys and some from Met residue except for the smallest structural domain, the SD1. The PDOS of S consists of several very sharp peaks in the VB and CB since S is the only atom that contains the 3 s and 3p atomic orbitals. The LUMO state originates from the S atoms.
-
(3)
The PDOS of the H atoms is very different from that of O, C, and N. It has more states in the CB region than in the VB region, reflecting the CB states from the anti-bonding states of the O–H, C–H and N–H pairs.
-
(4)
The difference in the PDOS of O, C, and N are mainly in the top portion of the VB and also in the CB. They can all be attributed to the electronic configuration of 2s22p4, 2s22p2, and 2s22p3 of O, C and N respectively.
In principle, the OLCAO method can resolve the PDOS into individual AA as demonstrated by us in the past investigation unrelated to COVID-19 [27], [28], [29]. Such detailed analysis in the present case is neither practical nor useful. However, it may provide additional insights on specific AA under mutation such as in the case of D614G mutation that will be discussed in Section 4.4.
3.3. Partial charge distribution
From the electronic structure the effective charge Q* and the corresponding partial charge (PC) on every atom can be calculated (see Eq. (1) in Method Section) as demonstrated in Ref. [33] for RBD and SD1-SD2 subunits, assuming that each domain is charge neutral from computational point of view. We have listed all the calculated PC for the seven structural domains in the tables from Table S1 to Table S7. We have shown the PC distribution on the protein surface (defined as the solvent accessible surface (SAS)) of each structural domain in Fig. 7 and Fig. S2, and marked those amino acids on this surface with extraordinarily large positive or negative PC. The size and the orientation of these structural domains are close to those depicted in Fig. 1 for Chain A. Fig. S2 shows the same PC distributions as in Fig. 7 for the same seven structural domains but with orientation of 90° and 180° about the vertical axis. This enables us to delineate the potential electrostatic interactions between these structural domains and the implication for the functionality of the S-protein, which is the ‘conquer’ part of our strategy.
Fig. 7.

Partial charge distributions for the seven structural domains (a) NTD, (b) RBD, (c) SD2, (d) SD1, (e) FP, (f) HR1-CH, (g) CD on the solvent accessible surface. The AAs with large PC are marked.
The PC values for the largest positively and negatively charged AAs on the SAS shown in Fig. 7 are listed in Table S1 to Table S7 for all seven structural domains. They are Tyr695 (2.049 e−) in FP followed by Tyr612 (2.021 e−) in SD2, Arg328 (2.018 e−) in SD1, Asp1146 (1.963 e−) in CD, Arg246 (1.890 e−) in NTD and relatively lower PC in Pro521 (1.170 e−) of RBD and Lys947 (1.048 e−) in HR1-CH. The largest negatively charged amino acids on the SAS in all seven structural domains are Asp985 (−1.125 e−) in HR1-CH followed by Arg237 (−1.124 e−) in NTD, Ser305 (−1.082 e−) in SD1, Asp663 (−1.033 e−) in SD2, Val687 (−1.014 e−) in FP and relatively lower PC in Arg357 (−1.007 e−) in RBD and Glu1092 (−0.993 e−) in CD. The absolute values for negative PC are generally less than the positive PC values. These ab initio computed PC values can be compared with the canonical charges of different AAs at neutral pH [38] bearing in mind of course that the canonical values refer to fully hydrated deprotonated negatively charged AAs (Asp, Glu, Tyr, and Cys) and protonated positively charged AAs (Arg, Lys, and His). In what follows we will delimit ourselves only to the most charged AAs.
In the NTD the positively charged Arg246 and Lys304 are consistent with the canonical assignment of charge, with Gly142 and Leu176 being anomalous, while the negatively charged Phe79 and Arg237 are both anomalous. In the RBD only the positively charged Arg454 is canonical and Pro521 is not, while of the negatively charged AAs only the Tyr449 is canonically charged with Leu335, Val503, Pro491 and Arg357 being anomalous. In the SD2 the positively charged Tyr612 is anomalous while negatively charged Asp663 is canonical. In the SD1 the positively charged Arg328 is canonical, while the negatively charged Arg319 and Ser305 are not. In the CD the positively charged Asp1146 is anomalous. In the FP the positively charged Lys811 is canonical and Tyr695 is anomalous, while negatively charged Val687 is anomalous. In HR1-CH only the negatively charged Asp985 is canonical. In summary, the positively charged Arg246 and Lys304 in NTD, Arg454 in RBD, Arg328 in SD1 and Lys811 in FP are all canonically charged, while the only canonically negatively charged AAs are Asp663 in SD2 and Asp985 in HR1-CH. The PC of the positively charged AAs thus seems to be more conserved, irrespective of the local solvent environment and could signify some structural charge stability that is not partitioned equally among the AAs of the SAS.
Electrostatic interaction is an important component of the protein binding energetics and charge patterning of different subunits of the S-protein can affect the binding properties with other proteins, certainly playing an important role in the interaction with the ACE2 and its recombinant varieties. One could thus expect that mutations that are not disruptive to the charge patterning would be preferred in the course of virus mutational trajectory.
3.4. Interatomic bonding
One of the great strengths of the OLCAO method is that it allows for the quantification of the strength of interatomic bonding by the provision of the BO values between every pair of atoms in the system under study. In Fig. S3, we display the BO vs. BL distributions for the seven structural domains in the spike protein. On the first glance, all seven displays look similar except for the number of data points which depends on the size of the unit. The atomic pairs with short BL close to 1.0 to 1.2 Å originate from the strong O–H, N–H and C–H covalent bonds of varying BO values within different residues. The next group of atomic pairs are between 1.3 Å and 1.6 Å originating from the much stronger C–O, N–C, C–C. The C–C bonds can be roughly separated in two groups, one with higher BO from around 0.50 e− to 0.65 e− and the other with lower BO. These higher BO pairs are from double bonds and those with lower BO are from single bonds. It is clear that the BO values of these group varies substantially. It is also noted that some of the N–H bonds occur at the pair separation larger than 1.5 Å. These bonds occur between atoms from same AAs in the same structural domain.
An interesting observation is that the C-S and S-S bond pairs at roughly1.82 Å and 2.03 Å apart respectively, display fairly large BO values. They originate from the S atoms in the Cys residue. The BO values decrease rapidly for BL greater than 2.1 Å because of the larger separation between atoms of different AAs. Notable is the presence of many HBs (O∙∙∙H, N∙∙∙H) at BL from 1.5 Å to larger than 2.0 Å, and some of these HBs have sufficiently large BO values that are greater than 0.1 e. These will be discussed separately in the next section. The plethora of different BO distributions in these complex biomolecular units and the ability to analyze them in details is quite impressive.
3.5. Hydrogen bonding network
This section emphasizes the importance of hydrogen bonding in biomolecular systems. HB is a much weaker bond than covalent or ionic bonding but they are ubiquitous. The sheer number of possible HBs plays a decisive role in many of their properties especially those involving the aqueous solvent. As can been seen from Fig. S3 in the BO vs. BL plots they usually range from 1.5 Å upward and the BO values can be as high as 0.1 e− in special cases. In Fig. 8 (a), we replot the combined HBs in these seven structural domains in a different scale. We have identified strong HBs with BO larger than 0.1 e− for all seven structural domains in Table 3. The strongest HB among these structural domains is in NTD between Asp53-Lys195 with BO of 0.123 e− which is followed by Ile410-Lys378 in RBD with BO of 0.122 e−. It is noted that these strong HBs are all O⋯H type and none are N⋯H type.
Fig. 8.

(a) The intramolecular and intermolecular hydrogen bonds for S-protein. (a). Bond order vs bond length showing the intramolecular hydrogen bonding for the seven structural domains. (b) Potential intermolecular HBs (red dots) between domains. (c) 90° orientation of (b). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 3.
Stronger HBs in structural domains.
| BL(Å) | BO (e−) | AA1 | AA2 | |
|---|---|---|---|---|
| NTD | ||||
| O⋯H | 1.566 | 0.123 | Asp53: OD1 | Lys195: HZ3 |
| RBD | ||||
| O⋯H | 1.505 | 0.109 | Val503: O | Tyr508: HH |
| O⋯H | 1.567 | 0.122 | Ile410: O | Lys378: HZ2 |
| O⋯H | 1.594 | 0.106 | Glu340: O | Lys356: HZ3 |
| SD2 | ||||
| O⋯H | 1.539 | 0.108 | Asp614: OD2 | Arg646: HH11 |
| O⋯H | 1.582 | 0.113 | Glu619: OE2 | Ser591: HG |
| FP | ||||
| O⋯H | 1.539 | 0.117 | Glu819: OE2 | Ser816: HG |
| HR1-CH | ||||
| O⋯H | 1.549 | 0.119 | Asp979: OD2 | Ser974: HG |
| O⋯H | 1.559 | 0.105 | Glu1017: OE1 | Ser1021: HG |
| O⋯H | 1.562 | 0.116 | Glu868: OE1 | Thr866: HG1 |
| O⋯H | 1.583 | 0.103 | Glu918: OE2 | Asn914: HD21 |
Before analyzing the intermolecular HBs, we aligned each domain with respect to the initial structure of Chain A of S-protein (6VSB). The clash and contact have been checked using standard parameters of UCSF Chimera and followed by minimization steps to remove them. Finally, the root-mean-square-deviation (RMSD) of heavy atoms of relaxed structure is computed in reference to initial structure of Chain A of S-protein (6VSB). The RMSD value between these two structures is 0.85 Å, which indicates that the relaxed structures are highly conserved and less deviated. The Ramachandran plot for the initial and relaxed structure of S-protein are shown in Fig. S4 (a) and (b) respectively.
In Fig. 8 (b) and Fig. 8 (c), we plot the possible HBs between different domains as red dots connecting them to show the potential HB network in spike protein. We omit those within each domain since there will be many of them as to be impractical. This is a very busy and complex figure since we attempt to plot the HB bonding network in a 2-dimensional projection of a 3-dimensional distribution. As can be seen, an HB network between different domains does exist and its role in SARS-CoV-2 virus has not been explored so far. There are 103 inter-domain HBs as listed in Table S8. An interesting observation is the lack of HBs between RBD and the NTD. Based on the number of inter-domain HBs one can hypothesize the strength of the interaction between them, based on this criterion the SD1 – SD2 is strongest, followed by FP – HR1-CH, HR1-CH – CD, FP – CD, FP – SD2, RBD – SD2. Of course, this sequence is only conditional as the water mediated HB network is missing. To further exemplify the inter-domain HB network, we separate display in Fig. 9 the enlarged version on 4 out of the 7 possible inter-domain HBs between specific amino acids in different domains listed in Table S8 as dotted lines. They are: (a) SD2 – RBD. (b) SD1 – NTD, (c) SD2 – SD1 and (d) FP – CD.
Fig. 9.

Examples of some intermolecular HBs between different domains: (a) SD2 – RBD. (b) SD1 – NTD, (c) SD2 – SD1 and (d) FP – CD. For simplicity, only the atoms forming HBs are represented in stick and specific AAs involved are marked.
3.6. Three-dimensional AA-AA interaction
Recently, we have demonstrated AABP analysis of two specific structural domains RBD and SD1 [34]. This is the first time that the non-local 3-dimensional (3D) AA interactions were carefully demonstrated and analyzed beyond the traditional approach based only on the amino acid primary sequence or their nearest neighbors in the sequences based on which the conventional sequence conservation is characterized. It is now possible to calculate and analyze the 3D AA interactions in all of the seven structural domains of the S-protein, giving a broader picture of the AA interaction in 3D network of the Spike protein. Such endeavors involve exceptionally large and resource demanding efforts. Nevertheless, we take on this challenge and calculated and analyzed the AABP for all the remaining 5 structural domains. The results for the 7 structural domains are presented in Table S9 to Table S15 and Fig. S5. They further reveal some unexpected observations and helpful to understand the overall mechanism of possible interaction between the inter-domain interactions in S-protein. More detailed breakdown of different types of interatomic bonding in each structural domain are listed in Table S16 to Table S22.
In the following, we describe the results for the seven structural domains from the AABP calculations individually as illustrated in Fig. S5 (a)-(g). All the figures for the 7 domains are presented in the same format and style to minimize repetition. The figures are in the form of bar graph distribution following the amino acid sequence in the domain from left to right using single letter designation for each amino acid with the first and the last amino acid marked. These two (first and last) AAs have only one NN in the primary sequence similar to those AAs with gaps in the sequence (marked with vertical dashed lines in each figure). The y-axis indicates the calculated AABP values. Each bar consists of three segments, those from 2 NN AAs in the primary sequence (yellow) or only 1 NN (light blue), and those from off-diagonal AAs in 3D-space (grey). In each domain, some specific AAs are highlighted with a vertical colored arrow and with their AA name and sequence number indicated. The red and blue arrow corresponds to AAs with negative and positive PCs lower than −0.4 e− and higher than 0.4 e− respectively. There are two black arrows, one in NTD and the other in RBD which are amino acids with exceptional large AABP values. In the following we describe the distribution of AABP in the seven structure domains separately.
(1) NTD
NTD is the largest domain in S-protein with 226 AAs, so Fig. S5 (a) has two rows. Detailed observation indicates that majority of AAs (176 AA, 77.9%) have the off-diagonal contributions. It is also noted the Ser297 has the largest total AABP value of 1.449 e− in NTD with 0.542 e− from the off-diagonal component. AAs with large contribution in off-diagonal AABP are important since they denote either large number of non-local bonding or stronger non-local bonding. It demonstrates that the non-local interaction is indeed included in the twist and turn complexity of the 3D structure of AAs. The other AAs with large off-diagonal AABP ranging from 0.160 e− to higher values include Tyr37, Asp40, Lys41, Arg44, Asp111, Lys113, Glu132, Glu191, Lys195, Tyr204, Lys206, Glu224, Asp228, Lys278, Asn280, Asp287, Cys291, Asp294, Ser297, Lys300, and Cys301. They are dominated by the amino acids Asp, Glu and Lys. Among these 21 AAs having large off-diagonal AABP 15 of them coincide with those with large positive PC (blue arrow) or large negative PC (red arrow) shown in Fig. S5 (a).
(2) SD1
In contrast to NTD, SD1 is the smallest domain with only 24 AAs and with a long-elongated shape (Fig. 4 (b)). As a result, it has only one amino acid Glu309 with 2 NNs, a modest non-local AABP contribution and a large negative PC (red arrow) shown in Fig. S5 (b). Val327 has largest AABP value of 1.001 e− in SD1. The amino acids in the two ends (Ser305 and Arg328) have large negative PC and large positive PC respectively. There are other AAs with 2 NN also have large PC. Lys310 has positive PC, Arg319 and Glu334 have negative PC.
(3) RBD
The calculation and analysis of AABP in RBD has been reported in ref. [34] in excruciating detail. Here we recapture most of them as part of the presentation for the seven structural domains in S-protein. Fig. S5 (c) shows the bar graph plot of the AABP for RBD. Among 144 AA, 103 AAs (71. 5%) have off-diagonal AABP. The most distinguished feature is that Gly504 has the largest total AABP value of 1.513 e− and with 1.442 e− from the 2 NNs in the primary sequence and only a modest contribution of 0.070 e− from the off-diagonal AAs. The AAs with substantial off-diagonal AABP ranging from 0.160 e− or higher include Cys336, Lys356, Cys361, Lys378, Cys379, Lys386, Asp389, Cys432, Asp442, Thr500, Tyr505, and Arg509. The dominant AA in this list is Cys containing Sulphur S followed by Lys. Among these 12 AAs, 7AAs coincides with AAs with large positive or large negative PC as indicted by blue or red arrows in Fig. S5 (c).
It is possible to provide more detailed inter-amino acids interactions at atomic level as shown in Fig. 6 of ref. [34] for RBD including the involvement of HBs and the unique role of S-S bonds in Cys. However, in this article the analysis of such nature for all seven structural domains will be exceptionally demanding and time consuming and deemed not necessary. In future, if specific case entails such need such as in the mutational process on D614G in SD2 that (to be discussed in Section 4.4), they will be separately studied and reported.
(4) SD2
The 4th domain in S1 in the S-protein is SD2 which also have a very elongated structure that can be roughly divided into an upper and the lower portion (see Fig. 3(b)). It has a total 132 AAs, slightly smaller than RBD and only one gap. The bar graph plot of the AABP for SD2 is shown in Fig. S5 (d)). There are 86AAs (65.2%) with off-diagonal contributions, and much smaller than 77.9% in NTD. Like the other domains, we identify those AAs with large off-diagonal contributions. They are Lys535, Lys557, Arg567, Asp571, Asp574, and Glu583. Again, it shows the propensity of involvements in the nonlocal contribution of the AABP from amino acids Lys, Asp and Glu. All these amino acids have large positive or large negative PC as indicted by blue or red arrows in Fig. S5 (d). One of the most important amino acid in SD2 is the aspartic acid residue Asp614, which has AABP value of 0.964 e− (shown in Table S12, 0.827 e− from NN and 0.137 e− from off-diagonal AABP) and has a large negative PC of −0.825 e− shown in Table S4. This residue mutates to glycine in D614G which occurs in many cases in HIV and SARS-CoV-2 virus [39] and will be discussed in Section 4.4.
(5) FP
We now proceed to the first domain FP in S2 of the S-protein. Compare with the four structural domains in S1 discussed above, they are much less studied but are more important than those in S1 except RBD. This is because FP initiates contact between protein and host membrane. FP also have an elongated structure at the lower end of the Chain A inter-twisted with SD2 in S1 and HR1-CH in S2. Fig. S5 (e) shows the bar graph plot of the AABP for FP which has 139 AAs and only one gap. Among the 139 AAs, 84 (60.4%) have off-diagonal contribution to the AABP, which is lower than NTD, RBD and SD2. The AAs with large contribution of off-diagonal AABP in FP are Lys733, Asp775, Lys776, Glu780, Asp808, Lys811, Ser816, and Gly819. All of them except Ser816 have large positive or large negative PC. It also shows the dominance of the same AAs Asp, Glu and Lys in nonlocal contribution to AABP. One interesting thing we noticed is the presence of four Cys residue (Cys738, Cys743, Cys749, and Cys760) in FP with significant off-diagonal part and the total AABP. This is a solid evidence that Cys residues play a unique role in making 3D non-local off-diagonal contribution to the total AABP, yet they do not have large partial charge. This can be related to the unique role of the S atom with electronic configuration of 3s2-3p4 present in Cys. In case of Met, the other S-containing residue, some has modest off-diagonal contribution (Met731), and some do not (Met697, Met740). It is not clear what other electronic factors attribute to this difference between Cys and Met.
(6) HR1-CH
The HR1-CH in S2 is a large domain having183 AAs with no gap in the sequence. It has a double-helix type of structure on the lower part of Chain A heavily intervened with FP and CD (see Fig. 3). Like NTD in Fig. S5 (a), we present the AABP bar graph plot in two rows which is shown in Fig. S5 (f). There are far more AAs in HR1-CH with contributions from the off-diagonal AAs (156 AAs out of 183 or 85.2%). HR1-CH is the structural domain with maximum percentage of AAs involved in the off-diagonal AABP contribution. The AAs that make large off-diagonal contributions to AABP are: Thr866, Asn914, Ser929, Lys933, Asn953, Asp979, Asp994, Thr998, Arg1000, Arg1014, Ser1021. Among these 11 AA, 5 AA have large positive or large negative PC. Here some new amino acids making substantial AABP contributions besides Asp and Lys appear. They are Thr, Asn, and Arg. We would like to point out Met residues (Met869, Met900, Met902, and Met1029,) in this structural domain has modest off-diagonal AABP and significant sum of AABP.
(7) CD
CD is the last structure domain for the seven structural domains in this paper. Most of its elongated structure is located at the bottom of the Chain A far from RBD but intimately mixed with FP and HR1-CH. It has 111 AAs and no gap. The bar graph distribution of AABP in CD is presented in Fig. S5 (g). Among the 111 AAs, 72 AAs (64.9%) make off-diagonal contribution to the AABP. The AAs that make large off-diagonal contributions to AABP are: Gln1036, Lys1038, Asp1084, Lys1086, Thr1116. All of these AAs except Thr1116 fall under the category of AAs with large positive or large negative PC. We would like to point out Met1048 is the other AA which contains S similar to Cys and has a significant AABP of 0.899 e−.
4. Discussion
4.1. Advocating for larger-scale modeling
We advocate the large-scale ab initio computational modeling in complex biomolecular systems as an important branch of materials science. The combination of multi-scale complexity and atomic-scale interaction that requires an enormous computational resources and efficient methods is a tall order. Our vision, based on the present work, is that multiscale modeling and ab initio computation in biosciences, biomaterials and bioengineering could benefit from ambitious attempts to tackle some of the most outstanding scientific problems and thus validate its strength compared to other possibly more empirical methodologies [40], [41].
Ab initio computation at the atomistic level based on DFT occupies the lowest ladder of the multi-scale complexity of biomolecular systems, but it offers many insights and provides fundamental understanding few other approaches can achieve. As demonstrated in this work, highly accurate structural optimization can significantly improve the structures obtained by the state-of-the-art experimental techniques and provide the missing details of specific interactions such as accurate partial charge and detailed bonding distributions. Such extraordinary claims demand extraordinary evidences. It is our hope that the research community will appreciate such computational efforts that expand the fundamental understanding of complex biological systems in different environments. This is easier said than done since multidisciplinary effort requires expertise from varied scientific disciplines including but not limited to biology, chemistry, physics, computer science, medicine, materials science, pharmacology, virology to name explicitly just a few. In what follows, we discuss several areas where the current methodology can be directly applied to some of the urgent issues related to SARS-CoV2 virus and biomedical science in general.
4.2. Interaction between seven structural domains in spike protein
The interaction and connection between the seven structural domains in the spike protein are the “conquer” part of the divide and conquer strategy on which this study is based. The complete Chain A in S-protein shown in Fig. 3 and Fig. 4 with these seven structural domains is obviously a spectacularly complex biomolecular system. Although the RBD and SD1 parts have been extensively discuss by us recently [33], [34], discussions of the other structural domains NTD, SD2, and FP, HR1-CH and CD in S2 were relatively limited despite the results presented in Section 3. Here we will relate and attempt to connect these structural domains. The subunit S1 is involved in the binding of virus particles to the receptors of the host cell and initiates the virus infection hence is the target for the drug design. In comparison, the membrane fusing subunit S2 is more conserved [42] but relatively less studied. It is therefore a timely target for focused study in relation to antiviral development [43], [44]. To understand its mechanism, it is necessary to understand their interaction.
NTD is the largest unit among the seven structural domains of S-protein, with 226 AAs and 3459 atoms. It is located at the lower left of RBD (Fig. 3). However, NTD does not share HBs with RBD. NTD does not have HB with SD2 but have one HB with SD1 (Table S8) between Lys300 of NTD and Ser305 of SD1. Lys300 has large positive PC and Ser305 has large negative PC implies potential strong interaction between them. Our NTD model ends with Lys304, which is the NN of Ser305 of SD1. Lys300 falls under AAs with large off-diagonal contribution in AABP discussed in Section 3.6.
The subdomains SD2 and SD1 in S1 are located between NTD from S1 and FP, HR1-CH in S2. The proximity of SD2 and SD1 results in the largest number of HBs. SD2 and RBD have 8 HBs (Table S8). We would like to point out that there are three HBs between Asn360-Thr523, Cys361-Thr523, and Ala363-Cys525 from Table S8 and the fact that Asn360, Cys361, and Ala363 are close to Ser359 in RBD. Ser359, located in RBD, and Pro561, located in SD2, are involved in the hinge-like conformational movement in S1, which is crucial for viral infection [15], [33], [34]. In our recent work [34], we have identified the HB network Ser359-Asn394-Glu516-Thr393-Ala520 provoking the hinge-like movement in Ser359. Here, we have identified a few more AAs potentially involved in the hinge-like movement. Among them Cys361 has a large contribution in off-diagonal AABP. Pro561 located in SD2, has large AABP of 0.968 e− including a modest off-diagonal AABP of 0.023 e−. This careful HB analysis between two domains is one level deeper in explaining the hinge movement between RBD and SD2 in S-protein during viral infections.
Fusion peptides are initiators of protein and host membrane contact [45]. Their lengths can vary but usually shows intermediate hydrophobicity and contains glycine (Gly) and alanine (Ala) [46]. In SARS-CoV, several regions were identified for stronger membrane interacting regions or FP as discussed by Sainz et al [47] using Wimley and White hydrophobicity scale [48]. Further regions 873–888 and 1185–1202 were identified as strong membrane interacting regions that work synergistically with 770–788 for fusion [49], [50]. Similarly, in SARS-CoV-2 there are different speculations on the location of FP. In SARS-CoV-2, Xia et al [51] identified FP to be 788–806 whereas Wrapp et al [10] listed FP to be 816–833. Both sequences from Xia et al and Wrapp et al., fall under our FP model (687–828), a relatively larger model with 139 AAs available in 6VSB.
In SARS-CoV, a region 798–835 was identified as FP based on single-point mutagenesis studies signifying its importance in fusion [52], [53]. This region consists a highly conserved region 798–808 with the AAs sequence Ser-Phe-Ile-Glu-Asp-Leu-Leu-Phe-Asn-Lys-Val. This highly conserved region in the case of SAR-CoV-2 (6VSB) falls under sequence number 816–824. According to our calculation in FP, Glu819 and Asp820 in this highly conserved region have large negative PC of −0.680 e− and −0.724 e− respectively (see Table S5). In addition, Lys825 in this highly conserved region have larger positive PC of 0.867 e− (see Table S5 and is marked in solvent accessible surface Fig. 7(e), Fig. S2 (e), and Fig. S2 (l)). These AAs could play a key role in the fusion process due to electrostatic interaction between AAs with higher PC. Glu819 and Ser816 in the highly conserved region of FP have large off-diagonal contribution to AABP (shown in Table S13 and Fig. S5 (e)).
Similar to SARS-CoV, SARS-CoV-2 (PDB 6VSB) could have more potential FP regions, which could be located in HR1-CH region and could aid the fusion synergistically. During the fusion process, the HR1-CH forms a long helix inserting FP into the cell membrane thus triggering HR1 and consequently bringing virial and cell membrane closer for their fusion [54]. It can be speculated that this process originates from a large number of HBs between FP and HR1-CH (shown in Table S8). Among those HBs shown in Table S8, we would like to point out some of HBs between AAs: Lys733-Pro862, Asp775-Leu864, Glu780-Arg1019, and Tyr756-Asp994. These HBs could play significant role in triggering and bringing virial and cell membrane closer since Lys733, Asp775, and Glu780 of FP and Asp994 of HR1-CH have large off-diagonal contribution in AABP. We would like to further point out these residues, Asp, Glu and Lys are more dominant in the off-diagonal contribution as discussed in Section 3.6. HR1-CH structural domain is a conserved site and is used as a target for protein inhibitors, neutralizing antibodies [54], [55]. CH is followed by CD at the bottom of Chain A far form RBD at the top with FP and HR1-CH sandwiched between. After FP inserts into the cell membrane, HR1-CH and CD act synergistically to stabilize the spike. One of the HB between AAs of HR1-CH and CD, Gly908-Lys1038, could play role in this process. Since Lys1038 has large contribution in off-diagonal AABP. The synergic nature of interaction can be traced to the HB interaction among FP, HR1-CH and CD.
4.3. Role of partial charge and solvent effect in electrostatic interaction
At present the inclusion of explicit solvent molecules, not to even mention the self-dissociation of water and the effect of local dielectric and solution environment, is beyond atomistic computational reach, and the PC values obtained in the ab initio computation in the divide and conquer strategy are the only game in town. Still, there are interesting details emerging by comparing the computed PCs with the expectations of the canonical values obtained from the Henderson-Hasselbalch equation for AAs in the bulk solution [56]. The comparison leads to a tentative conclusion that the positively charged AAs show less effect of the local solvent environment then the negatively charged ones, meaning that more of the positively charged AAs conform to the canonical prediction. This could have further repercussions and could signify that there is a hidden structural charge stability, genetically encoded, confined preferentially to positively charged AA residues. It remains to be seen if the positively charged Arg246 and Lys304 in NTD, Arg454 in RBD, Arg328 in SD1 and Lys811 in FP as well as the negatively charged Tyr449 in RBD could play in some aspects a prominent role also in the S-protein RBD-ACE2 complex, possibly through HB bridges or possibly even salt bridges in the presence of a bathing solution salt ions. It thus seems to be worthwhile to focus on these particular AAs in the future computational endeavors that would hopefully take into account the S-protein-ACE2 interactions also in the presence of strategically positioned water molecules.
The distribution of the structural PCs on the solvent accessible surface of the S-protein, shown in Section 3.3. Fig. 7 is of course only one component of electrostatic interactions in intermolecular binding [57]. The second component deals with the equilibrium distribution of the mobile charges in the bathing solution to the structural charge on the protein [58], which we did not address here and is intimately related to the intra-protein stability and inter-protein interactions through the consideration of the aqueous solvent and its [pH]. In fact, presently almost all published work on SARS-CoV-2 seldom addresses the effect of the aqueous bathing solution at the ab initio level, with MD approach based on model force fields prone to its own limitations [59].
Aqueous solvent is of course present at different scales of the viral infection, starting from drops or aerosols, and is crucial in mitigating the spread, infection and transmission of the virus [60]. The fundamental role of water follows first from the protonation-deprotonation equilibrium of dissociable of AAs, but also from the fact that the lipid membrane envelope of the pleomorphic SARS-CoV-2 is composed of phospholipids and embedded protein amphiphilic moieties, that both strongly interact with water [61]. Water mediated interactions are still far from being brought into the modeling fold, as the PDB deposited SARS-CoV-2 PDB data completely lack the assignments of H atoms, or the water molecules for that matter. Nevertheless, inclusion of water is not an insurmountable obstacle. If one is to preserve the scale of ab initio computations the solvation effects can only be studied by adding water molecules at strategic locations along the SAS of the protein structure and investigations of various aspects related to hydration can only be performed at the expense of a much larger scale of computation.
4.4. Extension to mutation and drug design
Mutation is an important part in evolution of biological systems over time, intimately related to the grand and controversial topics such as the origin of life [62]. It features prominently in virology, connected in particular with the infectivity of COVID-19, itself related to the structural components of the viral proteins. Generally speaking, a mutation refers to an error in the DNA or RNA code [63], which can be both positive or negative, depending on the effect it imposes on the proteome. SARS-CoV-2 is a positive ss-RNA virus and has in principle high mutation rates, allowing it to adapt to local environmental conditions. In this regard, SARS-CoV-2 is far more dangerous than other virus we faced in the past and definitely not the last. It is not clear at this point if there exist any mutationally conserved sites, and one would need to probe the differences in binding efficacy between the viral spike and the many recombinant ACE2s. Recently, there have been rapid advances in the study of mutations in SARS-CoV-2, such as the specific mutation D614G in the spike protein [64] and/or many other single or multiple mutations such as: A475V, L452R, V483A, and F490L [65], [66]. RBD of SARS-CoV-2 is mainly responsible to attach onto the host cell and is thus a target for neutralizing antibodies. Studying mutation and its effect is therefore considered important for its biological significance [66] and large-scale computational modeling could provide useful insights.
Another urgent topic in relation to COVID-19 pandemic is the vaccine and drug development as well as the screening and monitoring of the infection. Safety is the primary concern when addressing the antibody dependent enhancement (ADE) of infection as was observed in previous studies investigating SARS and MERS vaccine candidates [67], [68], [69], [70]. For example, clinical data from SARS-CoV-2 patient serum suggest disease severity is positively correlated with IgG titer [68], [71], [72]. In this regard, detailed structural information and understanding of how neutralizing antibodies interact with SARS-CoV-2 is highly desirable and critical. Computational modeling can certainly help to differentiate between targets that are neutralizing vs. those that induce undesired ADE or other adverse immune responses. To this end, the concerted computational informatics and immunological screening of antibodies derived from patient sera may facilitate the prediction of various B- and T-cell epitopes of the SARS-CoV-2 S-protein. The invasion of the human host cell by SARS-CoV-2 virus starts with direct binding of the S-protein to the ACE2 receptor, so targeting the S-protein RBD-ACE2 complex is a promising therapeutic strategy for combating COVID-19 infection.
5. Conclusions
We report the results of detailed ab initio computations performed on one of the most complex biomolecular system to date, viz., the spike protein of the SARS-CoV-2. Using a divide and conquer methodology, separate calculations on seven large structural domains yield information on the inter-domain interactions within the S-protein. Such ultra-large-scale ab initio calculations are unprecedented and could be a game changer in computational research on COVID-19. The new features brought to the computational research by our methodology, including the new focused areas that are being currently under investigation, can be succinctly summarized as follows:
(1) Accurate structural refinement of all seven key domains in the spike protein of SARS-CoV-2 and the undisputable demonstration of its critical importance in analyzing the atomic-scale interactions in complex biomolecular systems.
(2) Calculation and detailed analysis of intramolecular bonding based on their electronic structure in different structural domains including the key information on the partial charge distribution.
(3) Extending the ab initio calculation to include the three dimensional non-local interactions between amino acids using AABP, not just those in the primary sequence with implications on the accepted norm of sequence conservation.
(4) Interaction among structural domains are discussed with several new insights such as crucial hinge-like movement and fusion process.
(5) Devise the divided and conquer strategy to extend to the properties and interaction or the entire Chain A of the spike protein containing a total of 14,488 atoms demonstrating the possible 3- dimensional hydrogen bonding network.
(6) Suggestion of extending the current methodology to systematic incorporation and analysis of mutations in key amino acids and interfacial modeling targeting the drug design.
(7) The supplementary materials contain all the data on the structures and properties of this massive investigation that will be extremely useful to the research community.
(8) In all these tasks the availability of supercomputer facility at the NERSC of the Lawrence Berkeley National Laboratory or other national facilities for COVID-19 research is a sine qua non.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This research used the resources of the National Energy Research Scientific Computing Center supported by DOE under Contract No. DE-AC03-76SF00098 and the Research Computing Support Services (RCSS) of the University of Missouri System. This project is funded by the National Science Foundation of USA: RAPID DMR/CMMT-2028803. RP would like to acknowledge the support from the 1000-Talents Program of the Chinese Foreign Experts Bureau, the School of Physics, the University of the Chinese Academy of Sciences, Beijing, and the Institute of Physics of the Chinese Academy of Sciences, Beijing.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.02.004.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Impact of COVID-19 on people's livelihoods, their health and our food systems. Available from: https://www.who.int/news/item/13-10-2020-impact-of-covid-19-on-people's-livelihoods-their-health-and-our-food-systems.
- 2.Nicola M. The socio-economic implications of the coronavirus pandemic (COVID-19): a review. Int J Surgery (London, England) 2020;78:185. doi: 10.1016/j.ijsu.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKee M., Stuckler D. If the world fails to protect the economy, COVID-19 will damage health not just now but also in the future. Nat Med. 2020;26(5):640–642. doi: 10.1038/s41591-020-0863-y. [DOI] [PubMed] [Google Scholar]
- 4.Hagerty S.L., Williams L.M. The impact of COVID-19 on mental health: the interactive roles of brain biotypes and human connection. Brain, Behav Immun-Health. 2020 doi: 10.1016/j.bbih.2020.100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seitz B.M. The pandemic exposes human nature: 10 evolutionary insights. Proceed Natl Acad Sci. 2020:1–10. doi: 10.1073/pnas.2009787117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.WHO, Coronavirus Disease 2019 (COVID-19): Situation Report 98 (WHO, 2020). Available from: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports.
- 7.Huang Y. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. 2020:1–9. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lan J. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 9.Anatomy of a killer Understanding SARS-CoV-2 and the drugs that might lessen its power.
- 10.Wrapp D. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapp M, Shapiro L, Ho DD. Cryo-EM structure of the SARS-CoV-2 spike glycoprotein bound to Fab 2-4. 2020; Available from: https://www.rcsb.org/structure/6XEY.
- 12.Wang QH, Song H, Qi JX. Structure of novel coronavirus spike receptor-binding domain complexed with its receptor ACE2. 2020; Available from: https://www.rcsb.org/structure/6lzg.
- 13.Huo J, et al. Structure of the SARS-CoV-2 spike S1 protein in complex with CR3022 Fab. 2020; Available from: https://www.rcsb.org/structure/6YOR.
- 14.Pinto D. et al. Structure of the SARS-CoV-2 spike glycoprotein in complex with the S309 neutralizing antibody Fab fragment (open state). 2020; Available from: https://www.rcsb.org/structure/6WPT.
- 15.Roy S. Dynamical asymmetry exposes 2019-nCoV prefusion spike. bioRxiv. 2020:1–30. doi: 10.1101/2020.04.20.052290. [DOI] [PubMed] [Google Scholar]
- 16.Bai C., Warshel A. Critical differences between the binding features of the spike proteins of SARS-CoV-2 and SARS-CoV. J Phys Chem B. 2020;124(28):5907–5912. doi: 10.1021/acs.jpcb.0c04317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grant O.C. 3D Models of glycosylated SARS-CoV-2 spike protein suggest challenges and opportunities for vaccine development. bioRxiv. 2020 doi: 10.1101/2020.04.07.030445. [DOI] [Google Scholar]
- 18.Mercurio I. Protein structure analysis of the interactions between SARS-CoV-2 spike protein and the human ACE2 receptor: from conformational changes to novel neutralizing antibodies. bioRxiv. 2020 doi: 10.1101/2020.04.17.046185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D'Annessa I., Marchetti F., Colombo G. Binding epitopes of 2019-nCoV proteins for perspective diagnostic and therapeutic applications. Insights Comput Approaches. 2020 doi: 10.20944/preprints202003.0221.v1. [DOI] [Google Scholar]
- 20.Hohenberg P., Kohn W. Inhomogeneous electron gas physical review 136. Phys Rev. 1964;136:B864–B871. [Google Scholar]
- 21.Kohn W., Sham L.J. Self-consistent equations including exchange and correlation effects. Phys Rev. 1965;140(4A):A1133–A1138. [Google Scholar]
- 22.Frisch, M.J., et al., Gaussian 16 Rev. C.01. 2016: Wallingford, CT.
- 23.Pettersen E.F. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 24.VASP – Vienna Ab initio Simulation Package. Available from: https://www.vasp.at/.
- 25.Perdew J.P., Burke K., Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 26.Ching W-Y, Rulis P. Electronic Structure Methods for Complex Materials: The orthogonalized linear combination of atomic orbitals. 2012: Oxford University Press.
- 27.Poudel L. Implication of the solvent effect, metal ions and topology in the electronic structure and hydrogen bonding of human telomeric G-quadruplex DNA. PCCP. 2016;18(31):21573–21585. doi: 10.1039/c6cp04357g. [DOI] [PubMed] [Google Scholar]
- 28.Poudel L. Impact of hydrogen bonding in the binding site between capsid protein and MS2 bacteriophage ssRNA. J Phys Chem B. 2017;121(26):6321–6330. doi: 10.1021/acs.jpcb.7b02569. [DOI] [PubMed] [Google Scholar]
- 29.Adhikari P. Electronic structure, dielectric response, and surface charge distribution of RGD (1FUV) peptide. Sci Rep. 2014;4:5605. doi: 10.1038/srep05605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eifler J. Charge distribution and hydrogen bonding of a collagen α2-chain in vacuum, hydrated, neutral, and charged structural models. Int J Quantum Chem. 2016;116(9):681–691. doi: 10.1002/qua.25089. [DOI] [Google Scholar]
- 31.Mulliken R.S. Electronic population analysis on LCAO–MO molecular wave functions I. J Chem Phys. 1955;23(10):1833–1840. doi: 10.1063/1.1740588. [DOI] [Google Scholar]
- 32.Mulliken R. Electronic population analysis on LCAO–MO molecular wave functions. II. Overlap populations, bond orders, and covalent bond energies. J Chem Phys. 1955;23(10):1841–1846. doi: 10.1063/1.1740589. [DOI] [Google Scholar]
- 33.Adhikari P. Intra- and intermolecular atomic-scale interactions in the receptor binding domain of SARS-CoV-2 spike protein: implication for ACE2 receptor binding. PCCP. 2020;22(33):18272–18283. doi: 10.1039/D0CP03145C. [DOI] [PubMed] [Google Scholar]
- 34.Adhikari P., Ching W.-Y. Amino acid interacting network in the receptor-binding domain of SARS-CoV-2 spike protein. RSC Adv. 2020;10:39831–39841. doi: 10.1039/d0ra08222h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Systèmes D. Dassault Systèmes; San Diego: 2020. BIOVIA, discovery studio visualizer, release 2019. [Google Scholar]
- 36.Yip K.M. Atomic-resolution protein structure determination by cryo-EM. Nature. 2020;587:157–161. doi: 10.1038/s41586-020-2833-4. [DOI] [PubMed] [Google Scholar]
- 37.Nakane T. Single-particle cryo-EM at atomic resolution. Nature. 2020;587:152–155. doi: 10.1038/s41586-020-2829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson D.L., Cox M.M. Fourth ed. W. H. Freeman and Company; New York: 2004. Lehninger principles of biochemistry. [Google Scholar]
- 39.Callaway E. Making sense of coronavirus mutations. Nature. 2020;585:174–177. doi: 10.1038/d41586-020-02544-6. [DOI] [PubMed] [Google Scholar]
- 40.Han Y., Král P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano. 2020;14(4):5143–5147. doi: 10.1021/acsnano.0c02857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiao B., Olvera de la Cruz M. Enhanced binding of SARS-CoV-2 spike protein to receptor by distal polybasic cleavage sites. ACS Nano. 2020 doi: 10.1021/acsnano.0c04798. p. A-H. [DOI] [PubMed] [Google Scholar]
- 42.Lai A.L. The SARS-CoV fusion peptide forms an extended bipartite fusion platform that perturbs membrane order in a calcium-dependent manner. J Mol Biol. 2017;429(24):3875–3892. doi: 10.1016/j.jmb.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xia, S., et al., A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci Adv, 2019. 5(4): p. eaav4580, 10.1126/sciadv.aav4580. [DOI] [PMC free article] [PubMed]
- 44.Tang T. Coronavirus membrane fusion mechanism offers as a potential target for antiviral development. Antiviral Res. 2020 doi: 10.1016/j.antiviral.2020.104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ou X. Identification of the fusion peptide-containing region in betacoronavirus spike glycoproteins. J Virol. 2016;90(12):5586–5600. doi: 10.1128/JVI.00015-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Epand R.M. Fusion peptides and the mechanism of viral fusion. Biochim Biophys Acta (BBA)-Biomembranes. 2003;1614(1):116–121. doi: 10.1016/S0005-2736(03)00169-X. [DOI] [PubMed] [Google Scholar]
- 47.Sainz B. Identification and characterization of the putative fusion peptide of the severe acute respiratory syndrome-associated coronavirus spike protein. J Virol. 2005;79(11):7195–7206. doi: 10.1128/JVI.79.11.7195-7206.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wimley W.C., White S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3(10):842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 49.Guillén J., Kinnunen P.K., Villalaín J. Membrane insertion of the three main membranotropic sequences from SARS-CoV S2 glycoprotein. Biochim Biophys Acta (BBA)-Biomembranes. 2008;1778(12):2765–2774. doi: 10.1016/J.BBAMEM.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guillén J. A second SARS-CoV S2 glycoprotein internal membrane-active peptide. Biophysical characterization and membrane interaction. Biochemistry. 2008;47(31):8214–8224. doi: 10.1021/bi800814q. [DOI] [PubMed] [Google Scholar]
- 51.Xia S. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30(4):343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madu I.G., Belouzard S., Whittaker G.R. SARS-coronavirus spike S2 domain flanked by cysteine residues C822 and C833 is important for activation of membrane fusion. Virology. 2009;393(2):265–271. doi: 10.1016/j.virol.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Madu I.G. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein S2 domain with characteristics of a viral fusion peptide. J Virol. 2009;83(15):7411–7421. doi: 10.1128/jvi.00079-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xia S. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol. 2020:765–767. doi: 10.1038/s41423-020-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan Y. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun. 2017;8:1–9. doi: 10.1038/ncomms15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meister A. Academic Press Inc.; New York: 1965. Biochemistry of the amino acids. [Google Scholar]
- 57.French R.H. Long range interactions in nanoscale science. Rev Mod Phys. 2010;82(2):1887. doi: 10.1103/RevModPhys.82.1887. [DOI] [Google Scholar]
- 58.Nap R.J. The role of solution conditions in the bacteriophage PP7 capsid charge regulation. Biophys J. 2014;107(8):1970–1979. doi: 10.1016/j.bpj.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jawad B. Thermodynamic dissection of the intercalation binding process of doxorubicin to dsDNA with implications of ionic and solvent effects. J Phys Chem B. 2020;124(36):7803–7818. doi: 10.1021/acs.jpcb.0c05840. [DOI] [PubMed] [Google Scholar]
- 60.Jayaweera M. Transmission of COVID-19 virus by droplets and aerosols: a critical review on the unresolved dichotomy. Environ Res. 2020;188 doi: 10.1016/j.envres.2020.109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leikin S. Hydration forces. Annu Rev Phys Chem. 1993;44(1):369–395. doi: 10.1146/annurev.pc.44.100193.002101. [DOI] [PubMed] [Google Scholar]
- 62.Sutherland J.D. Opinion: studies on the origin of life—the end of the beginning. Nat Rev Chem. 2017;1(2):1–7. doi: 10.1038/s41570-016-0012. [DOI] [Google Scholar]
- 63.Löwdin P-O. Quantum genetics and the aperiodic solid: Some aspects on the biological problems of heredity, mutations, aging, and tumors in view of the quantum theory of the DNA molecule. 1962, UPPSALA UNIV (SWEDEN).
- 64.Zhang L. et al., The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv, 2020: p. 1-25, 10.1101/2020.06.12.148726.
- 65.Li, Q., et al., The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell, 2020. 182(5): p. 1284-1294. e9, 10.1016/j.cell.2020.07.012. [DOI] [PMC free article] [PubMed]
- 66.Starr TN. et al., Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. BioRxiv, 2020: p. 1-40, 10.1101/2020.06.17.157982. [DOI] [PMC free article] [PubMed]
- 67.Quinlan B.D. The SARS-CoV-2 receptor-binding domain elicits a potent neutralizing response without antibody-dependent enhancement. Immunity. 2020:1–24. doi: 10.2139/ssrn.3575134. [DOI] [Google Scholar]
- 68.Wang Q. Immunodominant SARS coronavirus epitopes in humans elicited both enhancing and neutralizing effects on infection in non-human primates. ACS Infect Dis. 2016;2(5):361–376. doi: 10.1021/acsinfecdis.6b00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen W.-H. Optimization of the production process and characterization of the yeast-expressed SARS-CoV recombinant receptor-binding domain (RBD219-N1), a SARS vaccine candidate. J Pharm Sci. 2017;106(8):1961–1970. doi: 10.1016/j.xphs.2017.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao J. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin Infect Dis. 2020 doi: 10.1093/cid/ciaa344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang B. Immune phenotyping based on neutrophil-to-lymphocyte ratio and IgG predicts disease severity and outcome for patients with COVID-19. medRxiv. 2020 doi: 10.1101/2020.03.12.20035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ma H. COVID-19 diagnosis and study of serum SARS-CoV-2 specific IgA, IgM and IgG by a quantitative and sensitive immunoassay. medRxiv. 2020 doi: 10.1101/2020.04.17.20064907. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.