

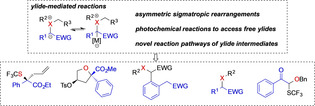
Abstract
Among the available methods to increase the molecular complexity, sigmatropic rearrangements occupy a distinct position in organic synthesis. Despite being known for over a century sigmatropic rearrangement reactions of ylides via carbene transfer reaction have only recently come of age. Most of the ylide mediated rearrangement processes involve rupture of a σ‐bond and formation of a new bond between π‐bond and negatively charged atom followed by simultaneous redistribution of π‐electrons. This minireview describes the advances in this research area made in recent years, which now opens up metal‐catalyzed enantioselective sigmatropic rearrangement reactions, metal‐free photochemical rearrangement reactions and novel reaction pathways that can be accessed via ylide intermediates.
Keywords: carbenes, diazoalkanes, rearrangements, sigmatropic rearrangements, ylides
The development of sigmatropic rearrangement reactions via ylide intermediates has witnessed significant advances in recent years. In this minireview, we summarize these advances that have contributed to the fundamental understanding of sigmatropic rearrangement reactions and that opened up new avenues in organic synthesis methodology beyond classic rearrangements.
Introduction
In 1912, Rainer Ludwig Claisen reported on the reaction of allyl vinyl ether 1 a under thermal reaction conditions to deliver γ,δ‐unsaturated carbonyl compound 2 a, which is now textbook knowledge in undergraduate course and commonly known as the Claisen reaction (Scheme 1 a). [1] The Claisen rearrangement is an example of a [3,3]‐sigmatropic rearrangement reaction. Sigmatropic rearrangement reactions are characterized by the migration of a σ‐bond, flanked by at least one π‐system, to a new position and the order of rearrangement reactions is determined by the original and terminal position of the migratory group. Sigmatropic rearrangements thus involve the reorganization of multiple σ‐bonds and consequently enable the access of complex molecular systems through well‐defined predictable transition states. [1] These features make sigmatropic rearrangement an important toolbox in synthesis methodology. This process can be classified into two key categories (a) ylide mediated or neutral rearrangements (3 to 4) and (b) anionic or nonbonding electron pair mediated rearrangements (5 to 6) (Scheme 1 b, c). [2]
Scheme 1.
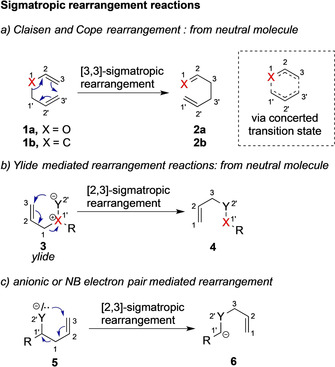
Rearrangement reactions.
Ylide mediated rearrangement reactions are accessed in general by two methods, either (a) by deprotonation of corresponding onium salts with a base or (b) by reaction of electrophilic metal‐carbene or free‐carbene intermediates with a nucleophile. [2] The latter is one of the most important strategies to conduct efficient rearrangement reactions and the required electrophilic carbene intermediate, is most generally obtained through metal‐catalyzed or photochemical decomposition of diazoalkanes 8. The reaction of the electron‐deficient carbene intermediate 8’ or 8’’ with a nonbonding lone pair of heteroatom nucleophiles 7 generates a metal‐bound ylide 10 or a free ylide 9 that participates in the rearrangement step, which proceeds, depending on the substituents pattern, via [2,3]‐ or [1,2]‐sigmatropic rearrangement reactions (Scheme 2). [2]
Scheme 2.
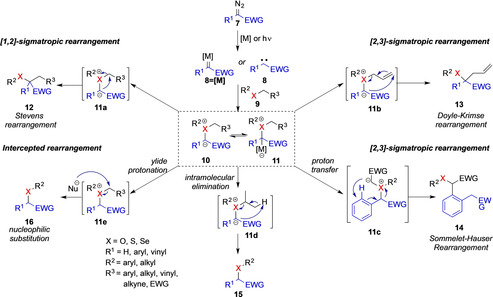
Ylide intermediates obtained via carbene transfer reaction and reactivity modes of rearrangement.
The stereochemical configuration of the newly formed σ‐bond can be controlled by the prevailing stereocenters within the starting materials, chiral ligands or chiral auxiliaries. These strategies have been widely employed with limited success,[ 3 , 4 ] and only recently the first reports on highly enantioselective sigmatropic rearrangement reactions via carbene transfer reactions and ylide intermediate have been reported.[ 5 , 6 ] The lack of existing methods and understanding can, in some part, be rationalized by missing knowledge on the rearrangement step itself. With the resurgence of photochemical carbene transfer reactions and detailed DFT calculations, the importance of free ylide intermediates in rearrangement was recently recognized. [7]
An intricacy of ylide‐mediated rearrangement reactions lies within the chemoselectivity of the rearrangement step itself and different types of rearrangement processes find common application, depending on the substitution pattern of the ylide intermediate (Scheme 2).[ 8 , 9 , 10 ] However, recent advances in this research area now demonstrate initial application that permit the control of the reaction pathway by choice of reaction conditions and therefore allow for new reactivity modes of ylide intermediates.[ 11 , 12 , 13 , 14 ]
This minireview aims at providing the reader with an overview on the advances made in the past five years that have significantly developed the field of sigmatropic rearrangement via carbene transfer reactions. These developments include the realization of highly enantioselective, metal‐catalyzed sigmatropic rearrangement reactions, metal‐free photochemical rearrangement reactions and novel reaction pathways that can be accessed via ylide intermediates and that are not covered in previous reviews. [2] In each section, we will discuss the advances that were made and give an overview on the respective reaction mechanism to provide a detailed understanding.
Catalytic, Enantioselective Sigmatropic Rearrangements
Without doubt, asymmetric catalysis is one of the key advances in the chemical sciences and was honored with the Nobel Prize in chemistry in 2001. [15] Many important milestones, ranging from asymmetric organocatalysis, [16] catalysis with chiral‐at‐metal complexes, [17] or biocatalysis [18] have been achieved and today a plethora of different approaches towards catalytic, asymmetric synthesis have been developed and revolutionized modern synthesis. [19] While asymmetric sigmatropic rearrangement reaction of allylic alcohols and derivatives thereof, have been extensively studied by the Davies group and high levels of enantioselectivity could be obtained, [20] the development of asymmetric sigmatropic rearrangement reactions proceeding via ylide intermediates of non‐alcohol substrates remained elusive in organic synthesis.
The [2,3]‐sigmatropic rearrangement reaction of allyl sulfides 17 with diazoalkanes 8 was initially discovered in 1968 by Kirmse and Kapps, who reported on the copper‐catalyzed reaction of an allylic thiol with diazomethane. [21a] However, this reaction only found its way into the organic synthesis repertoire after Doyle and co‐workers rediscovered this transformation in 1981. [21b] Since then, [2,3]‐sigmatropic rearrangement of allyl sulfides 17, or the Doyle–Kirmse reaction, has found widespread application in organic synthesis to access highly functionalized building blocks with applications in total synthesis. [22] Many groups reported their efforts using chiral catalysts [3] or auxiliary mediated approaches (19, Scheme 3 a), [4] yet with limited enantioselectivity or applicability. The missing enantioselectivity might be a result of lack of available chiral catalysts, understanding of the reaction mechanism, or simply reasoned by the choice of substrate, as side‐differentiation of allyl phenyl sulfide 17 a is intrinsically challenging due to the similar steric demand of both substituents. The latter then results in poor differentiation of the si‐ and re‐face of the allyl sulfide in the enantiodiscriminating step (formation of 22, Scheme 3 b).
Scheme 3.
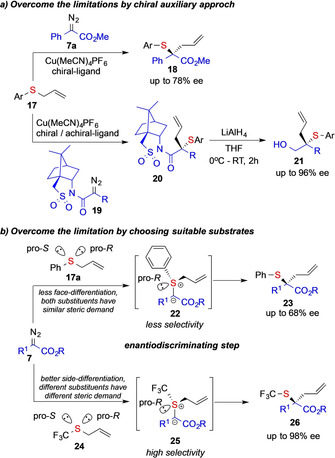
Strategies to enable asymmetric sigmatropic rearrangement reactions via carbene transfer reaction.
As a consequence, different strategies have been developed over recent years to overcome the challenges of stereoselective sigmatropic rearrangement reactions. One strategy employs the use of better suitable substrates 24, that facilitate face differentiation (formation of 25) and thus allows enantiospecific ylide formation (Scheme 3 b),[ 5 , 6 ] the other strategy harnesses latest developments in catalyst design to improve the face differentiation and thus allows for high asymmetric induction.[ 6 , 23 ]
In 2017, Wang and co‐workers reported on the first highly enantioselective [2,3]‐sigmatropic rearrangement reaction of allyl trifluoromethylsulfides 24. In the presence of the Rh2(DOSP)4 catalyst, donor/acceptor diazoalkanes 7 readily undergo formation of an electrophilic intermediate rhodium‐carbene complex 27, that can react with allyl trifluoromethylsulfides 24 with a high degree of stereocontrol to form the pivotal ylide intermediate 25. Subsequent [2,3]‐sigmatropic rearrangement then furnishes the corresponding trifluoromethyl thioether 26 reaction products. Importantly, the stereo‐determining step lies within the addition reaction of the sulfide 24 to the rhodium‐carbene intermediate 27. The high degree of stereocontrol is presumably due to both steric and electronic properties of the RhII catalyst, but also due to better face‐ differentiation of allyl trifluoromethylsulfides 24 as the substituents of sulfur differ in steric and electronic properties (Scheme 4). [5]
Scheme 4.
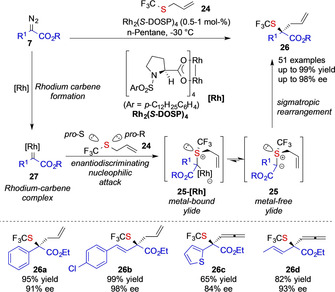
Enantioselective Doyle–Krimse reaction developed by the Wang group.
Almost at the same time, the Tambar group, reported the enantioselective [2,3]‐sigmatropic rearrangement reaction of allylic iodides 29 and acceptor‐only diazoalkanes 28 a using a Cu(box) complex (Scheme 5). This reaction proceeds via formation of a copper–carbene complex, followed by enantiospecific addition of the allylic iodide to give an intermediate iodonium ylide 30, which undergoes the sigmatropic rearrangement reaction to furnish the corresponding rearrangement products 31. Importantly, this reaction is limited to the reaction of cis‐configured allylic iodides 29; the corresponding trans‐configured allylic iodides gave significantly reduced enantioselectivity (up to 30 % ee). [23] It is important to note that the rearrangement reaction of halonium ylides was initially described by Kirmse in 1966, [24] yet this reaction only received very little attention in the past years. [25]
Scheme 5.
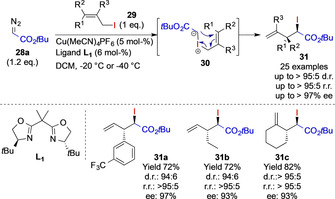
Enantioselective sigmatropic rearrangement of allylic iodides by the Tambar group.
Only shortly after these reports, Feng and co‐workers reported the use of chiral NiII catalysts (NiCl2 + ligand L2) in the reaction of pyrazoleamides 32. This strategy now involves the use of a carbene intermediate that can act as a chelating ligand of a NiII complex, which is important to lock conformation and as a consequence block one face of the carbene intermediate to deliver chiral NiII‐bound ylide 33. Following this concept, the authors could now demonstrate that even challenging substrates, such as allyl phenyl sulfide 17, can be subjected to highly asymmetric rearrangement reactions. In control experiments, the authors revealed the importance of the pyrazoleamide moiety as conventional diazoesters only gave very poor enantioinduction (Scheme 6). [6]
Scheme 6.
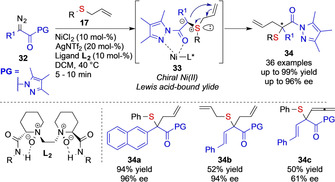
Chiral NiII complex catalyzed enantioselective Doyle–Krimse reaction developed by the Feng group.
The Fasan group reported a different strategy to achieve high enantioselectivities in asymmetric [2,3]‐sigmatropic rearrangement reactions. [26] They focused on the recently emerging concept of biocatalytic carbene transfer reactions and the mutagenesis of iron‐heme containing enzymes to engineer enzymes that can be tailored to specific non‐natural reactions. [27] In 2015 Fasan et al. reported the enzymatic Doyle–Kirmse reaction and could demonstrate proof‐of‐principle studies on enantioselective Doyle–Kirmse reactions using the Myoglobin variant Mb(L29A, H64V) with turnovers of 390. [26a] The same authors thereafter expanded this approach in a systematic study on [2,3]‐sigmatropic rearrangement using allylic (17) and propargylic sulfides. The most active variant they identified was the triple‐mutant Myoglobin (L29S, H64V, V68F) which yielded 36 in up to 8820 turnovers and yields of >99 % and up to 71 % ee. [26b] It is important to note that even with enzymatic catalysts that can often be engineered towards high selectivity in carbene transfer reactions of acceptor‐only diazoalkanes 28 b, a truly asymmetric variant with general applicability remains elusive (Scheme 7).
Scheme 7.
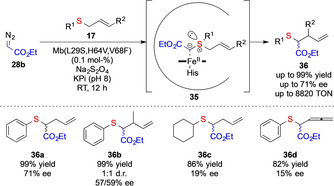
Biocatalytic sigmatropic rearrangement reaction of allyl sulfides with ethyl diazoacetate by the Fasan group.
These major strategies represent important milestones in the recent development of highly asymmetric [2,3]‐sigmatropic rearrangement reactions. It is expected that future research directions will further expand currently available methods[ 2d , 8 , 9 , 28 ] in the direction of catalytic, asymmetric [1,2]‐sigmatropic rearrangement reactions. The recent advances in the search of new chiral RhII catalysts, for example, by the Davies group,[ 6 , 20 , 25 ] clearly set the stage from the perspective of the catalyst to now address the next challenges in this research area.
Towards an Understanding of the Reaction Mechanism
The previously described advances have now opened up pathways to conduct asymmetric rearrangement reactions of sulfur or iodonium ylides. Yet, for further developments of asymmetric transformations, it is essential to understand differences in the reaction pathway of ylide intermediates generated via metal‐catalyzed carbene transfer reactions.
As outlined above, the widely accepted model involves the addition of nucleophilic substrates 17 to a metal–carbene intermediate 8=[M] to form ylide intermediate 37‐[M] or 37. The intricacies of the downstream reaction lie within the reactivity of this ylide intermediate. Currently, two different reaction mechanism for the subsequent rearrangement step are generally accepted: a) via a metal‐bound ylide 37‐[M] and b) via a free‐ylide 37 without further participation of the metal complex (Scheme 8).[ 3 , 4 , 20 ] It is important to note that two reaction steps need to be differentiated. The formation of the ylide proceeds via the enantiospecific nucleophilic attack of the heteroatom nucleophile 17 to the electrophilic metal–carbene intermediate 8=[M]. [20] The rearrangement step itself accounts for the diastereoselectivity in case multiple substituents are present at the allyl moiety 17 (Scheme 8).[ 5 , 20 ]
Scheme 8.
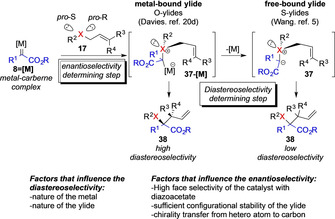
Enantio‐ versus diastereoselectivity determining steps and different reaction mechanisms for the rearrangement of onium ylides.
In this context, the Davies group studied the reaction of metal–carbene complexes with oxygen nucleophiles 40 in detail. In general, this reaction proceeds with a high degree of stereocontrol and DFT studies reveal the involvement of a metal‐bound ylide intermediate 41 in the course of the rearrangement step and subsequent dissociation of the metal complex. These calculations are supported by control experiment, which reveal a significant influence of RhII complexes on both enantio‐ and diastereoselectivity (Scheme 9). [20d]
Scheme 9.
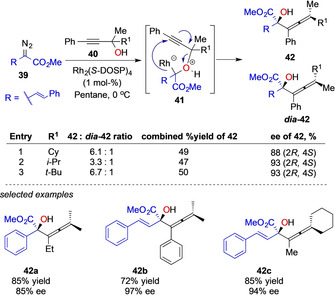
Enantioselective sigmatropic rearrangement reactions of propargylic alcohols by the Davies group.
The diastereoselectivity of the rearrangement reaction of allyl sulfides 17 with metal carbenes proceeds in striking difference. A remarkable example is the rearrangement reaction of cinnamyl sulfides 43. This particular reaction has been reported with a variety of different metal catalysts or carbene transfer conditions and diazoalkane reaction partners, yet not a single report states a high control on diastereoselectivity, which hints at a free ylide reaction mechanism (Scheme 10).[ 3c , 5a , 7a ]
Scheme 10.
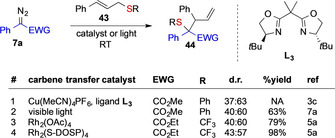
Influence of carbene transfer conditions on the diastereoselectivity of the rearrangement of cinnamyl sulfide 43.
In their 2017 report, Wang and co‐workers studied the rearrangement reaction of allyl sulfides with detailed control experiments to probe, whether [2,3]‐sigmatropic rearrangement reactions of sulfur ylides proceed via a metal‐bound or free ylide mechanism. In a first set of experiments, different chiral and achiral RhII complexes were studied in the reaction with a cinnamyl‐substituted thioether 43 or 24 b. Yet, all catalysts gave comparable d.r. values (Scheme 10, entries 3, 4 and Scheme 11 a), thus revealing only a small influence of the catalyst geometry on the selectivity of the rearrangement step. In a second set of control experiments, the reaction of symmetric allylic sulfide 45 was studied. The resulting free ylide 47 would thus be achiral and should result in a racemic reaction product 46, while a chiral metal‐bound ylide would give an enantioenriched reaction product 46. Indeed, a racemic reaction product 46 was observed, when using chiral RhII based catalysts (Scheme 11 b), which is supportive of a free ylide intermediate. In another control experiment, Wang and co‐workers studied the reaction of a phenyl propyl sulfide 48, which cannot undergo a rearrangement reaction. Yet, only the decomposition of the diazo compound 7 b was observed (Scheme 11 c). [5]
Scheme 11.
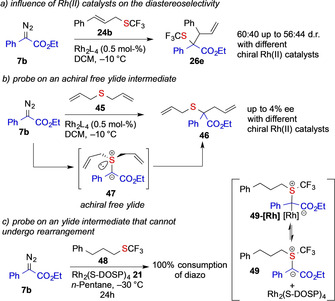
Control experiments conducted by Wang and co‐workers to probe the free ylide reaction mechanism.
In brief, the mechanistic insight from these experiments is now indicative of a free ylide mechanism in [2,3]‐sigmatropic rearrangement reactions of sulfur ylides and provides a very important basis for the understanding of differences in reactivity of oxygen and sulfur ylides. Yet, as reactions are carried out in the presence of metal catalysts, the influence of metals on the rearrangement step cannot be completely ruled out.[ 5 , 20 ]
Metal‐Free Sigmatropic Rearrangement: from Free Ylide Intermediates towards a Mechanistic Understanding
The photolysis of diazoalkanes represents a classic, metal‐free approach towards free carbene intermediates. Initial applications on the UV photolysis reaction of diazomethane in cyclopropanation reactions with olefins date back to 1959, [29] yet limited in selectivity of the carbene transfer reaction and in efficiency due to detrimental side‐reactions caused by the high‐energy UV light. [30] In 2018, several groups uncovered the potential of visible light promoted carbene transfer reactions to enable highly efficient cycloaddition, X−H insertion or olefination reactions. In all cases, the irradiation with low‐energy visible light enables an operationally simple and straightforward access to carbenes without the need to exclude air or moisture and applicability in a broad variety of different organic solvents.[ 7a , 31 ]
This metal‐free method towards carbene intermediates now offers an interesting approach to evaluate the relevance of metal‐free sulfur ylides in rearrangement reactions under photochemical conditions. As only substrate molecules are present in the reaction mixtures, this approach not only allows a metal‐free approach towards free ylide intermediates, but also no other additives, such as bases, are required to promote the rearrangement reaction. In this context, different groups studied [2,3]‐sigmatropic rearrangement reactions of sulfides and amines.[ 2 , 32 ] The Koenigs group could demonstrate the reaction of allyl sulfides and allyl amines in the photochemical reaction with aryldiazoacetates 7, to give homoallylic products 50 or 51, respectively.[ 7a , 33 ] The Xiao group reported on a difluoroallylation reaction that proceeds via a similar reaction mechanism to yield 52. [34] More recently, the Gryko group further expanded the scope of photochemical [2,3]‐sigmatropic rearrangement reactions towards propargyl sulfides to give allenes 53 (Scheme 12). [35] From the perspective of the reaction mechanism, the above sigmatropic rearrangement reactions can only proceed via a free ylide intermediate 11 and thus provide important experimental evidence that sigmatropic rearrangement reactions of sulfur ylides can indeed proceed without participation of a metal complex. This is further underlined by experimental evidence of the reaction of cinnamyl sulfides, which react in low diastereoselectivity to the rearrangement product. Importantly, the selectivity of this reaction is in a similar range as the corresponding metal‐catalyzed reactions (see Scheme 10), which now further supports a free ylide mechanism, even in the case of metal‐catalyzed [2,3]‐sigmatropic rearrangement reactions.[ 5 , 7 ]
Scheme 12.
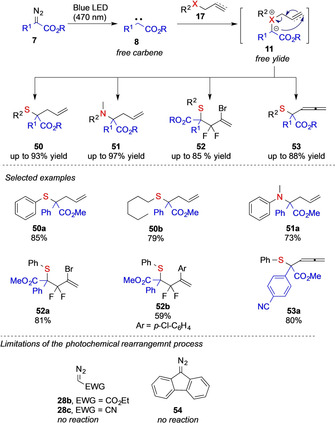
Overview on photochemical [2,3]‐sigmatropic rearrangement reactions.
In a further report on sigmatropic rearrangement reactions under photochemical carbene transfer conditions the Koenigs group recently showcased [1,2]‐sigmatropic rearrangement reactions of N‐sulfenyl phthalimides benzylic sulfides, and [2,3]‐sigmatropic rearrangement reactions of mercaptoacetates to further demonstrate the relevance of free sulfur ylide intermediates not only in the rearrangement of allyl sulfides (Scheme 13). [10]
Scheme 13.
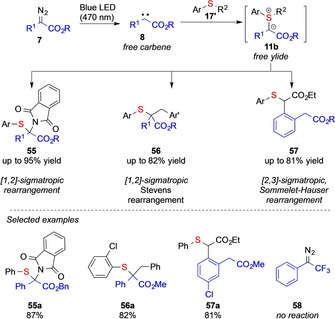
Overview on photochemical Stevens and Sommelet–Hauser rearrangement reactions by the Koenigs group.
This photochemical, approach now also opens up pathways towards ring expansion reactions of 4‐membered ring heterocycles 59 that proceed via [1,2]‐sigmatropic rearrangement. The Koenigs and Xu groups studied this ring expansion in detail using different oxetane or thietane heterocycles as substrates to give ring‐expanded products 60/61 in high yields (Scheme 14). [7b]
Scheme 14.
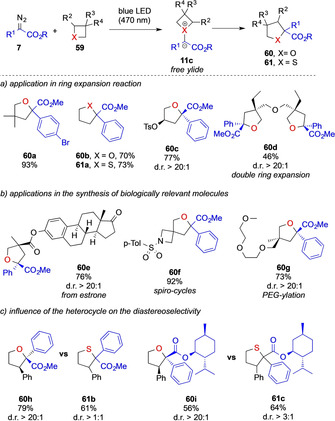
Photochemical ring expansion reaction of four membered heterocycles by the Koenigs and Xu group.
Experiments using 2‐phenyl oxetane or 2‐phenyl thietane as substrate revealed an interesting difference in the reactivity of oxygen and sulfur ylides. While a highly diastereoselective ring expansion was observed in the case of 2‐phenyl oxetane to give product 60 h; only little diastereoselectivity was observed the reaction of 2‐phenyl thietane to give the sulfur analogue 61 b. A similar difference was observed in the reaction of diazoacetates bearing a chiral auxiliary group (for 60 i and 61 c). This unexpected observation now shows that differences in the reactivity of oxygen and sulfur ylides lie not only in the nature of the ylide (metal‐bound vs. free ylide), but also in the rearrangement step itself (Scheme 14).
In subsequent DFT studies on the reaction mechanism of this [1,2]‐sigmatropic rearrangement reaction, the Koenigs and Xu groups could identify that this [1,2]‐sigmatropic rearrangement reaction proceeds via a diradical pathway INT2, similar to the [1,2]‐sigmatropic rearrangement of ammonium ylides. [36] In detailed studies, the authors mapped different reaction pathways, yet a concerted migration pathway or ionic pathway were ruled out due to high activation free energies of relevant transition states. Contrarily, a diradical pathway that involves the homolytic cleavage of a C−X bond of oxetane or thietane via TSrad2 followed by a rapid radical‐radical coupling mechanism (TS3) was identified as a suitable reaction mechanism with low activation free energies to give the desired ring expanded product for both oxygen and sulfur ylides (Scheme 15). This remarkable observation can now rationalize differences in reactivity of oxygen and sulfur ylides. Both ylides react along the same reaction pathway; however differences in the reaction outcome are not driven by different reaction mechanism, but are a consequence of different bond length of C−S versus C−O bonds that influence the stereochemical outcome in the case of 60 h/61 b or 60 i/61 c.
Scheme 15.
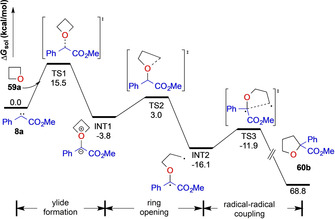
Reaction mechanism of the ring expansion of oxetane and thietane heterocycles at the (U)B3LYP/6–311+G(d,p)(chloroform)//(U)B3LYP/6‐31G(d)(chloroform) level of theory.
The recently emerging applications of photochemical carbene transfer reactions have thus provided important mechanistic into the reaction mechanism of rearrangement reactions of sulfur and oxygen ylides. The relevance of free ylide intermediates could be showcased and DFT studies now provide first evidence at the reaction mechanism. [7b]
En Route to New Reaction Pathways of Ylide Intermediates
In the previous sections, we focused on the discussion of recent advances in typical rearrangement reactions of the central ylide intermediate. In recent years, several groups however reported on novel reactivity of the ylide intermediate.[ 11 , 12 , 13 , 14 ] The ylide intermediate is, by the nature of this zwitterionic species, a reactant that can also undergo nucleophilic substitution reactions, via addition of the anionic carbon to appropriate electrophiles, the requirements for which are discussed in this section.
The Hu group made seminal contributions already in 2008, when uncovering the potential of trapping metal‐carbene complexes with heteroatom nucleophiles to form ylides that cannot undergo rearrangement. Instead, these ylides undergo addition reaction to various electrophiles. [37] This fundamental concept was studied by different groups in the past decade in detail and has been reviewed previously and will be not part of this article. [11]
In 2016, the Szabo group could show that α‐diazoketones 61 react in the presence of a RhII catalyst with THF solvent and NFSI 63 in a fluoro alkoxylation reaction. This reaction proceeds via formation of a rhodium carbene intermediate 62 that is trapped by THF under ylide formation INT1F. This ylide cannot under rearrangement reaction and thus reacts in a α‐fluorination reaction with NFSI 63, followed by ring opening of the pendant oxoniumion ion INT2F by the sulfonimide anion (Scheme 16). [12a] In the year after, the same group accounted for the application of different oxygen nucleophiles such as alcohols, acetals, or ethers and the NFSI analogue SCF3‐dibenzenesulfonimide 65, which now provides a basis for generalized α‐difunctionalization reactions of α‐diazoketones 61 via rhodium catalyzed carbene transfer reactions (Scheme 16). Phenyldiazoacetates 7 however, proved unreactive in this reaction. [12b] In 2020, the Koenigs group uncovered the reaction of aryldiazoacetates 7 under photochemical conditions, which react with 1,4‐dioxane and other 6‐ or 7‐membered oxygen heterocycles in a similar fluoro alkoxylation reaction (Scheme 16). [12d] Most notably, no reaction could be observed using THF solvent. DFT studies by both the Szabo and Koenigs group show that two similar reaction mechanisms are operating, which differ in the nature of the ylide intermediate. While in the case of RhII‐catalyzed fluoro alkoxylation a metal‐bound ylide is a key intermediate INT1F (Scheme 16), the photochemical reaction proceeds via a free ylide intermediate INT1P (Scheme 16). Both ylide intermediates undergo fluorination, followed by ring opening of the oxonium salt INT2F and INT2 p.[ 12c , 12d ]
Scheme 16.
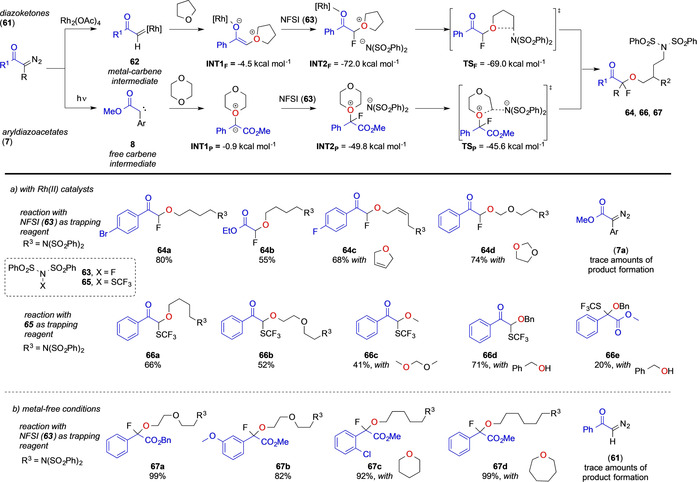
Fluoroalkoxylations and trifluoromethylthiolation reactions by the Szabo and Koenigs groups.
The reactivity of ylides that cannot undergo classic rearrangement reactions was recently further expanded by the Koenigs and Pan groups.[ 12d , 13 , 14 ] In their reports, these groups showcased that ylide intermediate can be trapped by either nucleophilic substitution with an external nucleophile or by intramolecular elimination reaction.
In this context, the Koenigs group studied the in situ formation of acceptor‐only diazoalkanes from 68 a–c via diazotization in a biphasic reaction mixture. In the presence of an iron catalyst, the carbene transfer reaction of diazoacetonitrile and other acceptor‐only diazoalkanes furnishes ylide intermediate 70, that first undergoes protonation reaction with water molecules 71 followed by substitution reaction with chloride ions to give the product 72 of a dealkylative, intercepted rearrangement reaction (Scheme 17). [13]
Scheme 17.
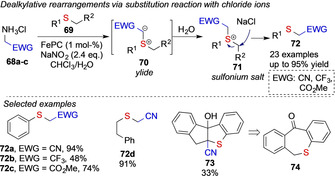
Iron‐catalyzed dealkylative rearrangements by the Koenigs group.
The Pan group studied the reaction of isopropyl sulfides 75, which form ylides 76 upon iron catalyzed carbene transfer. This ylide cannot undergo rearrangement reaction and consequently reacts in a protonation‐substitution reaction and formal dealkylation reaction (Scheme 18). [14a] The Koenigs group made a similar observation, when studying cyclohexyl‐substituted sulfides. [13]
Scheme 18.
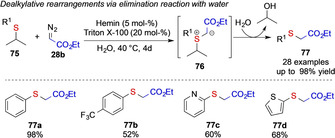
Iron‐catalyzed dealkylative rearrangement by the Pan group.
Pan and co‐workers recently observed a remarkable reactivity. While acceptor‐only diazoalkanes, such as ethyl diazoacetate and diazoacetonitrile undergo classic [2,3]‐sigmatropic rearrangement reactions with allyl sulfides 17, [38] dealkylative rearrangement reaction products 79 were observed when using trifluoro diazoethane under aqueous reaction conditions. [14b] The differences in reactivity of the fluorinated diazoalkane are yet not fully understood and it should be noted that [2,3]‐sigmatropic rearrangement products 78 were obtained in a DCM/water solvent mixture under very similar reaction conditions (Scheme 19). [39]
Scheme 19.
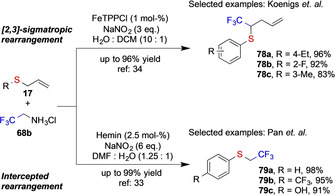
Comparison of reactivity of trifluoro diazoethane in different solvent mixture studied by the Koenigs and Pan group.
This unexpected influence of the reaction solvent was also observed by Koenigs and co‐workers in rhodium‐catalyzed rearrangements of methyl‐pyridin‐2‐yl substituted sulfides 80. Only by changing the solvent, the intermediate ylide underwent either selective [1,2]‐ sigmatropic (83) or [2,3]‐sigmatropic (82) rearrangement. This remarkable influence of solvent is not yet fully understood and detailed studies on the influence of solvent on the reactivity of the ylide are required to better account for the observed reactivity (Scheme 20). [8]
Scheme 20.
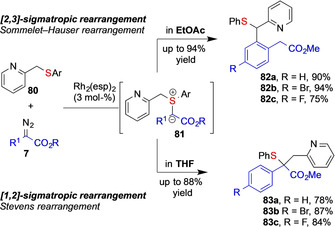
Influence of solvent on the rearrangement of ylide intermediates studied by the Koenigs group.
These pathways now open up new perspectives in the reactivity of carbene transfer reactions involving ylide intermediates and we expect that the reactivity of ylides that are unable to undergo classic rearrangement reactions or the effect of solvent, will be studied in the future in more detail.
Summary and Outlook
Sigmatropic rearrangements are of fundamental interest in organic synthesis. They are broadly applied as an efficient protocol for carbon−carbon/carbon−heteroatom bond forming reaction. However, compared to other enantioselective metal–carbene reactions, the development of asymmetric sigmatropic rearrangements via carbene transfer reactions are still challenging. However, the recent reports by different groups on asymmetric [2,3]‐sigmatropic rearrangement reaction now open up new perspectives. They furthermore provide the mechanistic insight into the reaction mechanism of [2,3]‐sigmatropic rearrangement for oxonium and sulfonium ylides, which can guide future developments. There is an expectation that future research in this area will aim, for example, at catalytic asymmetric [1,2]‐sigmatropic rearrangement reaction.
With the reemergence of photochemical carbene transfer reactions in 2018, the ylide intermediate can now be accessed in a metal‐free pathway that allows investigations on the reactivity of free ylide intermediates. Different groups reported on their efforts to open up this toolbox, which now gives insight into the rearrangement processes of free ylide intermediates, revealing that [1,2]‐sigmatropic rearrangement processes of sulfur and oxygen ylides proceeds via a radical mechanism. This observation can now account for the differences in diastereoselectivity of the rearrangement reaction of sulfur ylides.
In recent years, the reactivity pattern of ylides was significantly expanded towards intercepting this ylide intermediate by reaction with different electrophiles or by intramolecular elimination reaction. In this context oxyaminofluorination or trifluoromethylthiolation have been developed as well as intercepted rearrangement process offers a new reactivity mode of carbene transfer reaction.
It is expected that in the coming years research of ylide chemistry will experience further important advances. The authors of this minireview are of the opinion that these advances may include detailed mechanistic investigations involving experiments and calculations to get further insight into the reaction mechanism and to understand, in which cases ionic, concerted or diradical mechanism occur specifically. In terms of new reactivity, the newly developing area of intercepted or unusual rearrangement reactions are in our opinion an exciting area that will surprise chemists with unconventional reaction outcomes and that can further help expanding the scope of reactivity of sulfur ylides.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Sripati Jana obtained his B.Sc. (2016) from RKMVC College Rahara, Kolkata, and M.Sc. (2018) from Indian Institute of Technology Kharagpur, India. In 2018, Sripati moved to RWTH Aachen University, Germany, for his doctoral study in the group of Prof. Dr. Rene M. Koenigs. His research interest is focused on the metal catalyzed and photochemical carbene transfer reactions.

Biographical Information
Yujing Guo obtained her B.Sc. (2015) from Zhoukou Normal University, China, and M.Sc. (2018) from Zhengzhou University, China. She then moved to RWTH Aachen University, Germany, for her doctoral study. She is working in the group of Prof. Dr. Rene M. Koenigs since September 2018. He research interest is focused on metal‐catalyzed and photochemical cyclopropanation reactions.

Biographical Information
Rene M. Koenigs obtained his PhD in 2011 from RWTH Aachen University under the guidance of Prof. Magnus Rueping. He subsequently moved to Grünenthal GmbH working as a medicinal chemist on GPCR and ion channel targets in pain and inflammation research under the supervision of Dr. Paul Ratcliffe and Dr. Henning Steinhagen. In 2015 he was appointed as junior professor at RWTH Aachen University. His research interests focus on the discovery of new reactivity using metal‐catalyzed and photochemical carbene transfer reactions, continuous‐flow chemistry and fluorine chemistry.

Acknowledgements
R.M.K. thanks the German Science Foundation and Dean's Seed Fund of RWTH Aachen Foundation for financial support. Y.G. thanks the China Scholarship Council for financial support. Open access funding enabled and organized by Projekt DEAL.
S. Jana, Y. Guo, R. M. Koenigs, Chem. Eur. J. 2021, 27, 1270.
Contributor Information
Sripati Jana, www.koenigslab.rwth‐aachen.de.
Prof. Dr. Rene M. Koenigs, Email: rene.koenigs@rwth-aachen.de.
References
- 1.Selected references of [3,3]-sigmatropic rearrangement:
- 1a. Claisen L., Chem. Ber. 1912, 45, 3157–3166; [Google Scholar]
- 1b. Tarbell D. S., Chem. Rev. 1940, 27, 495–546; [Google Scholar]
- 1c. Ilardi E. A., Stivala C. E., Zakarian A., Chem. Soc. Rev. 2009, 38, 3133–3148; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Martín Castro A. M., Chem. Rev. 2004, 104, 2939–3002; [DOI] [PubMed] [Google Scholar]
- 1e. Gonda J., Angew. Chem. Int. Ed. 2004, 43, 3516–3524; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3600–3608; [Google Scholar]
- 1f. Majumdar K. C., Alam S., Chattopadhyay B., Tetrahedron 2008, 64, 597–643; [Google Scholar]
- 1g. Hoffmann R., Woodward R. B., Acc. Chem. Res. 1968, 1, 17–22. [Google Scholar]
- 2.For general reviews of sigmatropic rearrangement reaction:
- 2a. Zhang Y., Wang J., Coord. Chem. Rev. 2010, 254, 941–953; [Google Scholar]
- 2b. Sheng Z., Zhang Z., Chu C., Zhang Y., Wang J., Tetrahedron 2017, 73, 4011–4022; [Google Scholar]
- 2c. West T. H., Spoehrle S. S. M., Kasten K., Taylor J. E., Smith A. D., ACS Catal. 2015, 5, 7446–7479; [Google Scholar]
- 2d. Sweeney J. B., Chem. Soc. Rev. 2009, 38, 1027–1038; [DOI] [PubMed] [Google Scholar]
- 2e. Braverman S., Cherkinsky M., Top. Curr. Chem. 2006, 275, 67–101; [DOI] [PubMed] [Google Scholar]
- 2f. Reggelin M., Top. Curr. Chem. 2007, 275, 1–65; [DOI] [PubMed] [Google Scholar]
- 2g. Neuhaus J. D., Oost R., Merad J., Maulide N., Top. Curr. Chem. 2018, 376, 15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2h. Roiser L., Zielke K., Waser M., Asian J. Org. Chem. 2018, 7, 852–864; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2i. Li A.-H., Dai L.-X., Aggarwal V. K., Chem. Rev. 1997, 97, 2341–2372. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Fukuda T., Katsuki T., Tetrahedron Lett. 1997, 38, 3435–3438; [Google Scholar]
- 3b. Liao M., Wang J., Green Chem. 2007, 9, 184–188; [Google Scholar]
- 3c. Zhang X., Qu Z., Ma Z., Shi W., Jin X., Wang J., J. Org. Chem. 2002, 67, 5621–5625; [DOI] [PubMed] [Google Scholar]
- 3d. McMillen D. W., Barga N., Reed B. A., King C., J. Org. Chem. 2000, 65, 2532–2536; [DOI] [PubMed] [Google Scholar]
- 3e. Zhang X., Ma M., Wang J., Chin. J. Chem. 2010, 21, 878–882. [Google Scholar]
- 4. Ma M., Peng L., Li C., Zhang X., Wang J., J. Am. Chem. Soc. 2005, 127, 15016–15017. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Zhang Z., Sheng Z., Yu W., Wu G., Zhang R., Chu W.-D., Zhang Y., Wang J., Nat. Chem. 2017, 9, 970–976; [DOI] [PubMed] [Google Scholar]
- 5b. Hock K. J., Koenigs R. M., Angew. Chem. Int. Ed. 2017, 56, 13566–13568; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13752–13754. [Google Scholar]
- 6. Lin X., Tang Y., Yang W., Tan F., Lin L., Liu X., Feng X., J. Am. Chem. Soc. 2018, 140, 3299–3305. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Hommelsheim R., Guo Y., Yang Z., Empel C., Koenigs R. M., Angew. Chem. Int. Ed. 2019, 58, 1203–1207; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1216–1220; [Google Scholar]
- 7b. Jana S., Yang Z., Pei C., Xu X., Koenigs R. M., Chem. Sci. 2019, 10, 10129–10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Z., Guo Y., Koenigs R. M., Chem. Commun. 2019, 55, 8410–8413. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Jana S., Aseeva P., Koenigs R. M., Chem. Commun. 2019, 55, 12825–12828; [DOI] [PubMed] [Google Scholar]
- 9b. Jana S., Koenigs R. M., Org. Lett. 2019, 21, 3653–3657. [DOI] [PubMed] [Google Scholar]
- 10. Yang Z., Guo Y., Koenigs R. M., Chem. Eur. J. 2019, 25, 6703–6706. [DOI] [PubMed] [Google Scholar]
- 11.Selected reviews and book chapters:
- 11a. Padwa A., Hornbuckle S. F., Chem. Rev. 1991, 91, 263–309; [Google Scholar]
- 11b. Guo X., Hu W., Acc. Chem. Res. 2013, 46, 2427–2440; [DOI] [PubMed] [Google Scholar]
- 11c. Rueping M., Koenigs R. M., Atodiresei I., Chem. Eur. J. 2010, 16, 9350–9365; [DOI] [PubMed] [Google Scholar]
- 11d. Shao Z., Zhang H., Chem. Soc. Rev. 2009, 38, 2745–2755; [DOI] [PubMed] [Google Scholar]
- 11e. Allen A. E., MacMillan D. W. C., Chem. Sci. 2012, 3, 633–658; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Xing D., Hu W., Diazoacetate and Related Metal-Stabilized Carbene Species in MCR s, Wiley-VCH, Weinheim, 2014, pp. 183–206. [Google Scholar]
- 12.
- 12a. Yuan W., Szabó K. J., ACS Catal. 2016, 6, 6687–6691; [Google Scholar]
- 12b. Lübcke M., Yuan W., Szabó K. J., Org. Lett. 2017, 19, 4548–4551; [DOI] [PubMed] [Google Scholar]
- 12c. Mai B. K., Szabó K. J., Himo F., Org. Lett. 2018, 20, 6646–6649; [DOI] [PubMed] [Google Scholar]
- 12d. He F., Pei C., Koenigs R. M., Chem. Commun. 2020, 56, 599–602. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Empel C., Hock K. J., Koenigs R. M., Chem. Commun. 2019, 55, 338–341. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Yan X., Li C., Xu X., He Q., Zhao X., Pan Y., Tetrahedron 2019, 75, 3081–3087; [Google Scholar]
- 14b. Yan X., Li C., Xu X., Zhao X., Pan Y., Adv. Synth. Catal. 2020, 362, 2005–2011. [Google Scholar]
- 15. Ault A., J. Chem. Educ. 2002, 79, 572–577. [Google Scholar]
- 16.Selected references on asymmetric organocatalysis:
- 16a. Parmar D., Sugiono E., Raja S., Rueping M., Chem. Rev. 2014, 114, 9047–9153; [DOI] [PubMed] [Google Scholar]
- 16b. Volla C. M. R., Atodiresei I., Rueping M., Chem. Rev. 2014, 114, 2390–2431; [DOI] [PubMed] [Google Scholar]
- 16c. Doyle A. G., Jacobsen E. N., Chem. Rev. 2007, 107, 5713–5743. [DOI] [PubMed] [Google Scholar]
- 17.Selected references on chiral-at-metal complexes:
- 17a. Zhang L., Meggers E., Chem. Asian J. 2017, 12, 2335–2342; [DOI] [PubMed] [Google Scholar]
- 17b. Zhang L., Meggers E., Acc. Chem. Res. 2017, 50, 320–330. [DOI] [PubMed] [Google Scholar]
- 18. Brandenberg O. F., Fasan R., Arnold F. H., Curr. Opin. Biotechnol. 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 2024–2032; [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2126–2135; [Google Scholar]
- 19b. González-Sebastián L., Morales D. M., J. Organomet. Chem. 2019, 893, 39–91; [Google Scholar]
- 19c. Hooshmand S. E., Heidari B., Sedghi R., Varma R. S., Green Chem. 2019, 21, 381–405. [Google Scholar]
- 20.
- 20a. Li Z., Davies H. M. L., J. Am. Chem. Soc. 2010, 132, 396–401; [DOI] [PubMed] [Google Scholar]
- 20b. Parr B. T., Li Z., Davies H. M. L., Chem. Sci. 2011, 2, 2378–2382; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20c. Li Z., Parr B. T., Davies H. M. L., J. Am. Chem. Soc. 2012, 134, 10942–10946; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20d. Li Z., Boyarskikh V., Hansen J. H., Autschbach J., Musaev D. G., Davies H. M. L., J. Am. Chem. Soc. 2012, 134, 15497–15504; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20e. Parr B. T., Davies H. M. L., Nat. Commun. 2014, 5, 4455; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20f. Parr B. T., Davies H. M. L., Org. Lett. 2015, 17, 794–797; [DOI] [PubMed] [Google Scholar]
- 20g. Chepiga K. M., Feng Y., Brunelli N. A., Jones C. W., Davies H. M. L., Org. Lett. 2013, 15, 6136–6139. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Kirmse W., Kapps M., Chem. Ber. 1968, 101, 994–1003; [Google Scholar]
- 21b. Doyle M. P., Tamblyn W. H., Bagheri V. J., J. Org. Chem. 1981, 46, 5094–5102. [Google Scholar]
- 22.
- 22a. Jones A. C., May A. J., Sarpong R., Stoltz B. M., Angew. Chem. Int. Ed. 2014, 53, 2556–2591; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2590–2628; [Google Scholar]
- 22b. Nakai T., Mikami K., Chem. Rev. 1986, 86, 885–902; [Google Scholar]
- 22c. Murphy G. K., Stewart C., West F. G., Tetrahedron 2013, 69, 2667–2686. [Google Scholar]
- 23. Xu B., Tambar U. K., Angew. Chem. Int. Ed. 2017, 56, 9868–9871; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10000–10003. [Google Scholar]
- 24. Kirmse W., Kapps M., Hager R. B., Chem. Ber. 1966, 99, 2855–2868. [Google Scholar]
- 25.
- 25a. Doyle M. P., Forbes D. C., Vasbinder M. M., Peterson C. S., J. Am. Chem. Soc. 1998, 120, 7653–7654; [Google Scholar]
- 25b. Deng Q.-H., Chen J., Huang J.-S., Chui S. S.-Y., Zhu N., Li G.-Y., Che C.-M., Chem. Eur. J. 2009, 15, 10707–10712; [DOI] [PubMed] [Google Scholar]
- 25c. Xu B., Tambar U. K., J. Am. Chem. Soc. 2016, 138, 12073–12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Tyagi V., Bonn R. B., Fasan R., Chem. Sci. 2015, 6, 2488–2494; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Tyagi V., Sreenilayam G., Bajaj P., Tinoco A., Fasan R., Angew. Chem. Int. Ed. 2016, 55, 13562–13566; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13760–13764. [Google Scholar]
- 27.
- 27a. Coelho P. S., Brustad E. M., Kannan A., Arnold F. H., Science 2013, 339, 307–310; [DOI] [PubMed] [Google Scholar]
- 27b. Tyagi V., Fasan R., Angew. Chem. Int. Ed. 2016, 55, 2512–2516; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2558–2562; [Google Scholar]
- 27c. Sreenilayam G., Fasan R., Chem. Commun. 2015, 51, 1532–1534; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27d. Bordeaux M., Tyagi V., Fasan R., Angew. Chem. Int. Ed. 2015, 54, 1744–1748; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1764–1768. [Google Scholar]
- 28.
- 28a. Tayama E., Chem. Rec. 2015, 15, 789–800; [DOI] [PubMed] [Google Scholar]
- 28b. Song Z., Wu Y., Xin T., Jin C., Wen X., Sun H., Xu Q.-L., Chem. Commun. 2016, 52, 6079–6082; [DOI] [PubMed] [Google Scholar]
- 28c. Yuan H., Nuligonda T., Gao H., Tung C.-H., Xu Z., Org. Chem. Front. 2018, 5, 1371–1374; [Google Scholar]
- 28d. Li Y., Huang Z., Wu X., Xu P., Wang J., Zhang Y., Org. Lett. 2011, 13, 1210–1213; [DOI] [PubMed] [Google Scholar]
- 28e. Huang Y., Li X., Wang X., Yu Y., Zheng J., Wu W., Jiang H., Chem. Sci. 2017, 8, 7047–7051; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28f. Xu X., Li C., Xiong M., Tao Z., Pan Y., Chem. Commun. 2017, 53, 6219–6222; [DOI] [PubMed] [Google Scholar]
- 28g. Rao S., Prabhu K. R., Org. Lett. 2017, 19, 846–849. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. DeMore W. B., Pritchard H. O., Davidson N., J. Am. Chem. Soc. 1959, 81, 5874–5879; [Google Scholar]
- 29b. von E. Doering W., Mole T., Tetrahedron 1960, 10, 65–70. [Google Scholar]
- 30.Selected references:
- 30a. Ciszewski L. W., Jasińska K. R., Gryko D., Org. Biomol. Chem. 2019, 17, 432–448; [DOI] [PubMed] [Google Scholar]
- 30b. Empel C., Koenigs R. M., Synlett 2019, 30, 1929–1934; [Google Scholar]
- 30c. Candeias N. R., Afonso C. A. M., Curr. Org. Chem. 2009, 13, 763–787; [Google Scholar]
- 30d. Galkina O. S., Rodina L. L., Russ. Chem. Rev. 2016, 85, 537–555; [Google Scholar]
- 30e. Brunner J., Senn H., Richards F. M., J. Biol. Chem. 1980, 255, 3313–3318; [PubMed] [Google Scholar]
- 30f. Liang Y., Jiao L., Zhang S., Xu J., J. Org. Chem. 2005, 70, 334–337; [DOI] [PubMed] [Google Scholar]
- 30g. Wang J., Burdzinski G., Kubicki J., Platz M. S., J. Am. Chem. Soc. 2008, 130, 11195–11209; [DOI] [PubMed] [Google Scholar]
- 30h. Nakatani K., Maekawa S., Tanabe K., Saito I., J. Am. Chem. Soc. 1995, 117, 10635–10644. [Google Scholar]
- 31.
- 31a. Jurberg I., Davies H. M. L., Chem. Sci. 2018, 9, 5112–5118; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31b. Xiao T., Mei M., He Y., Zhou L., Chem. Commun. 2018, 54, 8865–8868; [DOI] [PubMed] [Google Scholar]
- 31c. He F., Koenigs R. M., Chem. Commun. 2019, 55, 4881–4884; [DOI] [PubMed] [Google Scholar]
- 31d. Empel C., Patureau F. W., Koenigs R. M., J. Org. Chem. 2019, 84, 11316–11322; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31e. Guo Y., Nguyen T. V., Koenigs R. M., Org. Lett. 2019, 21, 8814–8818; [DOI] [PubMed] [Google Scholar]
- 31f. He F., Li F., Koenigs R. M., J. Org. Chem. 2020, 85, 1240–1246. [DOI] [PubMed] [Google Scholar]
- 32.Selected references on [2,3]-sigmatropic rearrangement reactions of ammonium ylides:
- 32a. Clark J. S., Middleton M. D., Org. Lett. 2002, 4, 765–768; [DOI] [PubMed] [Google Scholar]
- 32b. Zhou C.-Y., Yu W.-Y., Chan P. W. H., Che C.-M., J. Org. Chem. 2004, 69, 7072–7082; [DOI] [PubMed] [Google Scholar]
- 32c. Lahm B., Pacheco J. C. O., Opatz T., Synthesis 2014, 46, 2413–2421; [Google Scholar]
- 32d. Workman J. A., Garrido N. P., Sancon J., Roberts E., Wessel H. P., Sweeney J. B., J. Am. Chem. Soc. 2005, 127, 1066–1067; [DOI] [PubMed] [Google Scholar]
- 32e. Aggarwal V. K., Fang G. Y., Charmant J. P. H., Meek G., Org. Lett. 2003, 5, 1757–1760; [DOI] [PubMed] [Google Scholar]
- 32f. Aviv I., Gross Z., Chem. Eur. J. 2008, 14, 3995–4005. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Li F., He F., Koenigs R. M., Synthesis 2019, 51, 1578; [Google Scholar]
- 33b. Empel C., Koenigs R. M., J. Flow Chem. 2019, 9, 157–160; [Google Scholar]
- 33c. Jana S., Koenigs R. M., Asian J. Org. Chem. 2019, 8, 683–686. [Google Scholar]
- 34. Yang J., Wang J., Huang H., Qin G., Jiang Y., Xiao T., Org. Lett. 2019, 21, 2654–2657. [DOI] [PubMed] [Google Scholar]
- 35. Orłowska K., Jasińska K. R., Krajewski P., Gryko D., Org. Lett. 2020, 22, 1018–1021. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Biswas B., Collins S. C., Singleton D. A., J. Am. Chem. Soc. 2014, 136, 3740–3743; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36b. Ghigo G., Cagnina S., Maranzana A., Tonachini G., J. Org. Chem. 2010, 75, 3608–3617; [DOI] [PubMed] [Google Scholar]
- 36c. Bott T. M., Vanecko J. A., West F. G., J. Org. Chem. 2009, 74, 2832–2836; [DOI] [PubMed] [Google Scholar]
- 36d. Hodgson D. M., Pierard F. Y. T. M., Stupple P. A., Chem. Soc. Rev. 2001, 30, 50–61. [Google Scholar]
- 37.Selected references:
- 37a. Hu W., Xu X., Zhou J., Liu W.-J., Huang H., Hu J., Yang L., Gong L.-Z., J. Am. Chem. Soc. 2008, 130, 7782–7783; [DOI] [PubMed] [Google Scholar]
- 37b. Li J., Zhang D., Chen J., Ma C., Hu W., ACS Catal. 2020, 10, 4559–4565; [Google Scholar]
- 37c. Che J., Niu L., Jia S., Xing D., Hu W., Nat. Commun. 2020, 11, 1511–1519; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37d. Zhang D., Zhou J., Xia F., Kang Z., Hu W., Nat. Commun. 2015, 6, 5801–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu X., Li C., Tao Z., Pan Y., Green Chem. 2017, 19, 1245–1249. [Google Scholar]
- 39. Hock K. J., Mertens L., Hommelsheim R., Spitzner R., Koenigs R. M., Chem. Commun. 2017, 53, 6577–6580. [DOI] [PubMed] [Google Scholar]