

Abstract
The parasitic worm, Schistosoma mansoni, expresses unusual fucosylated glycans in a stage‐dependent manner that can be recognized by the human innate immune receptor DC‐SIGN, thereby shaping host immune responses. We have developed a synthetic approach for mono‐ and bis‐fucosylated LacdiNAc (LDN‐F and LDN‐DF, respectively), which are epitopes expressed on glycolipids and glycoproteins of S. mansoni. It is based on the use of monosaccharide building blocks having carefully selected amino‐protecting groups, facilitating high yielding and stereoselective glycosylations. The molecular interaction between the synthetic glycans and DC‐SIGN was studied by NMR and molecular modeling, which demonstrated that the α1,3‐fucoside of LDN‐F can coordinate with the Ca2+‐ion of the canonical binding site of DC‐SIGN allowing for additional interactions with the underlying LDN backbone. The 1,2‐fucoside of LDN‐DF can be complexed in a similar manner, however, in this binding mode GlcNAc and GalNAc of the LDN backbone are placed away from the protein surface resulting in a substantially lower binding affinity. Glycan microarray binding studies showed that the avidity and selectivity of binding is greatly enhanced when the glycans are presented multivalently, and in this format Lex and LDN‐F gave strong responsiveness, whereas no binding was detected for LDN‐DF. The data indicates that S. mansoni has developed a strategy to avoid detection by DC‐SIGN in a stage‐dependent manner by the addition of a fucoside to a number of its ligands.
Keywords: chemical synthesis, glycans, immune modulation, molecular recognition, NMR spectroscopy
Chemically synthesized oligosaccharides derived from the parasite worm S. mansoni made it possible to investigate by a combination of analytical techniques molecular interactions with the innate immune receptor DC‐SIGN. It showed that by further fucosylation of antigens such as LDN‐NF, detection by DC‐SIGN is prevented, which in turn may be important for shaping immune responses during different phases of the life cycle of the parasite.
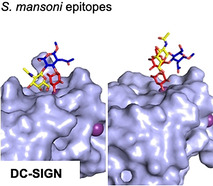
Introduction
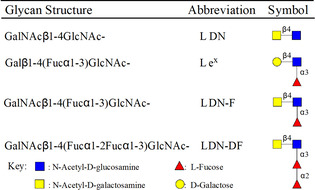
Schistosomes are parasitic helminths that infect over 250 million people worldwide and are responsible for 280 000 deaths annually. [1] They can manipulate the host's immune system to establish chronic infections. Although the molecular basis of these immune‐modulatory mechanisms remains poorly understood, it has been established that glycans of schistosomes can induce specific innate immune responses in the infected host, which in turn affects adaptive immunity. This occurs through an interplay between Toll like receptors (TLRs) and C‐Type lectin receptors (CLRs) of dendritic cells (DCs) thereby tuning immune responses toward an immune activation or tolerant state. [2] Schistosomes can biosynthesize a vast array of glycoconjugates, many of which are expressed at specific stages of their complex life cycle. The glycoproteins and glycolipids of schistosomes lack sialic acid and contain a variety of terminal glycan epitopes, including fucosylated antigens such as, Lewisx (Lex), pseudo‐Lewisy (pseudo_Ley), GalNAcβ1,4(Fucα1,3)GlcNAc (LDN‐F), Fucα1,3GalNAcβ1,4(Fucα1,3)GlcNAc (F‐LDN‐F), GalNAcβ1,4(Fucα1,2Fucα1,3)GlcNAc (LDN‐DF) and Fucα1,2Fucα1,3GalNAcβ1,4(Fucα1,2Fucα1,3)GlcNAc (DF‐LDN‐DF) (Table 1). [3] These glycan motifs are rarely found in mammalian glycoconjugates but are typical signatures of schistosomes. [4]
Table 1.
Major glycan structures of Schistosoma mansoni egg proteins.
|
|
Dendritic cell‐specific ICAM‐3 grabbing nonintegrin (DC‐SIGN), which is a C‐type lectin expressed by immature dendritic cells, has been implicated in schistosomiasis. [5] DC‐SIGN can recognize fucosylated glycan moieties presented by S. mansoni, such as Lex,[ 3b , 6 ] LDN‐F,[ 3a , 5a ] and pseudo_Ley. [7] Further fucosylation of these motifs to give LDN‐DF or DF‐LDN‐DF appears to abolish detection by DC‐SIGN. [3a]
The carbohydrate binding site of DC‐SIGN is composed of a wide solvent exposed surface flanked by a flexible loop, conferring broad glycan binding selectivity. [8] As a matter of fact, in addition to fucosylated glycans, high‐mannose structures can also be recognized by DC‐SIGN. Most structural studies dealing with DC‐SIGN‐glycan binding have focused on high mannose‐containing oligosaccharides. [9] Conversely, a structural understanding of binding of fucosyl containing glycans derived from S. mansoni is limited to Lex. [6b] A structural model, based on docking, indicates that the binding of pseudo_Ley occurs through rearrangement of the protein to accommodate the additional fucoside at galactoside, and supports substantial plasticity of DC‐SIGN. [7] Thus, it is surprising that bis‐fucosylated glycans such as LDN‐DF are not recognized by DC‐SIGN calling for further studies.
Here, we report the first chemical synthesis of LDN‐DF and LDN‐F by a convergent block synthetic approach. The compounds were obtained in ample quantities making it possible to examine their molecular interactions with DC‐SIGN using STD‐NMR, chemical shift perturbation, trNOESY and molecular modeling studies. It was found that DC‐SIGN can interact with the terminal fucoside of LDN‐DF, however, no further interactions are possible with the underlying glycan resulting in a low affinity binding. On the other hand, the binding of LDN‐F leads to interactions of the fucoside, the GalNAc moiety and the acetyl group of GlcNAc of the underlying LDN motif, leading to a substantial higher affinity. The binding of DC‐SIGN with LDN‐DF, LDN‐F, Lex and Lex‐Lex and SLex‐Lex was also examined in a glycan microarray format, which showed strong responsiveness of Lex and LDN‐F but no binding to LDN‐DF was observed, indicating that through multivalent interactions avidity and selectivity of binding is substantially enhanced. The data indicates that S. mansoni has developed a strategy to avoid detection by DC‐SIGN by the addition of a fucoside to its ligands.
Results and Discussion
Chemical synthesis
The preparation of Schistosoma‐derived glycans has received relatively little attention, [10] and the chemical synthesis of LDN‐DF has not been reported. The latter compound represents a challenging synthetic target because it requires careful selection of amino protecting groups to construct an LDN derivative that can be glycosylated with a fucosyl donor having a temporary protecting group at C‐2 to allow installation of the subsequent 1,2‐linked fucoside. The amino protecting groups need to be selected in such a way that they promote the introduction of β‐glycosides, but do not sterically block the introduction of the 1,3‐fucoside. Furthermore, the target compounds need to be modified by an aminopentyl linker, which is required for glycan microarray printing.
We have found that the monosaccharide building blocks 1–4 are appropriate for the assembly of aminopentyl modified LDN‐F (12) and LDN‐DF (20) (Scheme 1). In this respect, the NHTroc protecting group of 1 assures that a β‐glycoside is formed when glycosylated with acceptor 2, which has its C‐2 amino group masked as azide. The presence of the latter functional group is important because after oxidative removal of the Nap ether, an acceptor is formed (6) that sterically is sufficiently unencumbered for glycosylation with fucosyl donor 3. The latter compound also has a removable PMB ether allowing the installation of an 1,2‐fucoside using donor 4. After assembly of a tetrasaccharide, the azide can be converted into Troc and a glycosylation with properly protected aminopentanol will give linker modified compound LDN‐DF (19) as only the β‐anomer.
Scheme 1.
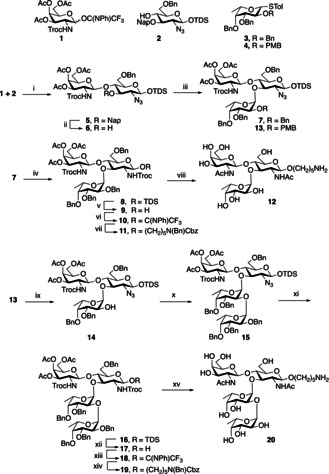
Synthesis of LDN‐F and LDN‐DF epitopes. Reagents and conditions: i) TMSTOf, DCM, −30 °C, 91 %; ii) DDQ, DCM, DCM/H2O, 84 %; iii) DPS/Tf2O, DCM, −60 °C to −40 °C, 68 % for 7 and 73 % for 13; iv) (a) PPh3, THF/H2O (b) trocCl, NEt3, DCM, 63 % (over two steps); v) HF/py in pyridine; vi) 2,2,2‐Trifluoro‐N‐phenylacetimidoyl chloride, DBU, DCM; vii) HO(CH2)5 N(Bn)Cbz, TMSTOf, DCM, −30 °C, 66 % (over 3 steps); viii) (a) Zn dust, AcOH, Ac2O, THF (b) NaOMe, MeOH (c) Pd/C, H2, H2O/MeOH, 45 % (over three steps); ix) DDQ, DCM/H2O, 74 %; x) TMSTOf, DCM/DMF, −30 °C to +5 °C, 83 %; xi) (a) PPh3, THF/H2O (b) TrocCl, NEt3, DCM; xii) HF/py in pyridine; xiii) 2,2,2‐Trifluoro‐N‐phenylacetimidoyl chloride, DBU, DCM; xiv) HO(CH2)5 N(Bn)Cbz, TMSTOf, DCM, −30 °C, 31 % (over five steps); xv) (a) Zn dust, AcOH, Ac2O, THF (b) NaOMe, MeOH (c) Pd/C, H2, H2O/MeOH, 37 % (over three steps).
A TMSOTf‐mediated glycosylation of N‐phenyl trifluo‐racetimidate donor 1 with acceptor 2 afforded disaccharide 5 in a yield of 91 %. The Nap ether of 5 was removed using DDQ in a mixture of DCM and H2O to give acceptor 6 in a yield of 84 %. Next, fucosyl donors 3 and 4, having Bn and PMB ether at C‐2, respectively, were preactivated with DPS/ Tf2O in the presence of TTBP at −60 °C, [11] which was followed by the addition of acceptor 6. The resulting reaction mixture was allowed to slowly warm to −40 °C resulting in formation of trisaccharides 7 and 13, which were isolated as mainly the α‐anomers in good yield (7, 68 %; J1,2=3.5 Hz; 1JC,H=174.9 Hz) and (13, 73 %; J1,2=3.8 Hz; 1JC,H=175.4 Hz). Alternative glycosylation conditions resulted in poor anomeric selectivity (Table S1). Trisaccharide 7 was further modified with an anomeric aminopentyl linker and to attain β‐anomeric selectivity, the azide was converted into NHTroc (→8) by a two‐step procedure entailing reduction using triphenyl phosphine followed by reaction of the resulting amine with TrocCl. Next, glycosyl donor 10 was prepared by removal of the anomeric TDS ether of 8 with HF/pyridine followed by reaction with N‐phenyl trifluoroacetimidate in the presence of DBU. As anticipated, a TMSOTf mediated glycosylation of 10 with N‐benzyloxycarbonyl‐N‐benzyl‐5 aminopentanol gave 11 as only the β‐anomer in 87 % yield. The latter trisaccharide was globally deprotected by a three‐step procedure in which the NHTroc was converted to NHAc using zinc dust, followed by global deacetylation and hydrogenation to provide spacer modified LDN‐F 12. A number of alternative strategies were explored to prepare LND‐F, and it was found that judicious selection of amino protecting groups was critical, and the use of bulky protecting groups such as N‐phthalimido resulted in a donor‐acceptor reactivity mismatch, resulting in almost immediate hydrolysis or degradation of donor (Table S1). In addition to donor 4 having a temporary PMB ether, several alternatives were examined but these gave disappointing results (Table S1). Next, attention was focused on the preparation of spacer modified LDN‐DF 20. Thus, treatment of trisaccharide 13 with DDQ in DCM/H2O afforded acceptor 14. Several reaction conditions were explored (Table S2) to install a fucoside, and tetrasaccharide 15 was obtained with high α‐anomeric selectivity in good yield (83 %) when the glycosylation was performed in DCM in the presence of DMF [12] and promoted by 1 equivalent of TMSTOf at −20 °C followed by slow warming to +5 °C (J1,2=3.9 Hz; 1JC,H=173.3 Hz). The anomeric TDS group of 15 was removed and the resulting lactol (17) converted into a N‐phenyl trifluoroacetimidate 18, which was coupled with N‐benzyloxycarbonyl‐N‐benzyl‐5‐aminopentanol to give 19. Global deprotection of the latter compounds gave target tetrasaccharide 20 in an overall good yield (37 % over three steps).
Molecular basis of binding of LDN‐F and LDN‐DF to DC‐SIGN
The molecular basis of the interactions of DC‐SIGN with LDN‐F (12) and LDN‐DF (20) in solution was examined by combining saturation transference difference (STD‐NMR), chemical shift perturbation analysis of the protein backbone, nuclear Overhauser effect (NOE) evaluation, and molecular modeling. The NMR‐based strategy combines molecular recognition analysis from the perspective of the protein and ligand thereby providing a detailed atomic view of the interactions. 1H‐STD‐NMR experiments were performed to establish which parts of LDN‐F and LDN‐DF make direct contacts with the lectin. 1H–15N HSQC experiments on the 15N‐labeled carbohydrate recognition domain (CRD) of DC‐SIGN were conducted to identify protein residues involved in ligand binding and to establish whether differences exist between the two epitopes. Furthermore, 1H–15N HSQC titration experiments provided binding affinity estimations. Finally, trNOESY experiments characterized the ligand conformation in the bound state and additionally provided short intermolecular distances between the ligand and the protein, thus defining the precise binding pose.
1H Saturation transfer difference (STD) NMR
1H‐STD NMR spectra resulting from the interaction of LDN‐F and LDN‐DF with DC‐SIGN extracellular domain (ECD) which tetramerizes in solution, along with the corresponding reference spectra are shown in Figures 1 a and b, respectively. It demonstrates that DC‐SIGN can bind both glycans but with substantial differences in binding mode. Both ligands bind through the terminal fucoside. The STD data for LDN‐F are similar to those previously reported for the structurally related Lex tri‐saccharide, [6b] indicating a similar mode of binding. As for Lex, LDN‐F is recognized by DC‐SIGN through the fucoside and additional contacts are made with the GalNAc pyranosyl ring and the N‐acetyl group of the GlcNAc moiety. The N‐acetyl moiety of GalNAc exhibited only a weak STD effect, indicating that it does not contribute substantially to binding (Figure 1 a). Detectable STD effects of the interaction of DC‐SIGN with LDN‐DF were restricted to H2, H4 and Me of the terminal 1,2‐fucoside with a remarkable absence of STD signals from the internal 1,3‐fucoside and the GalNAc and GlcNAc moieties (Figure 1 b). The much lower intensities of the STD signals of the LDN‐DF compared to those acquired for the LDN‐F suggests weaker binding of the former. The blank 1H‐STD NMR experiments of LDN‐F and LDN‐DF ligands alone are shown in the Supporting Information (Figures S1 and S2).
Figure 1.
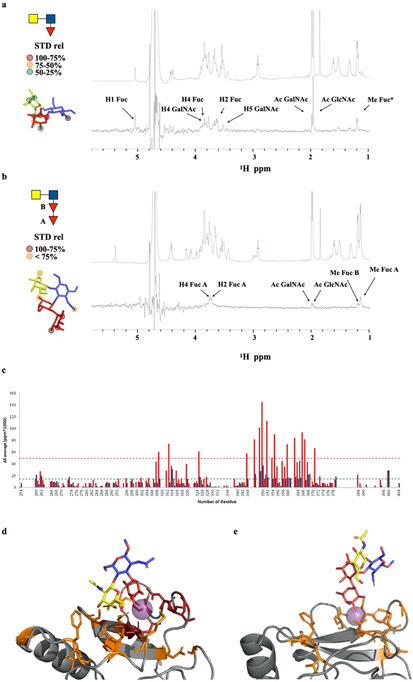
1H‐STD‐NMR spectra for the interaction of full‐length DC‐SIGN with (a) LDN‐F and (b) LDN‐DF. Relative STD and epitope mapping are shown on the left side for both ligands and refer to double difference between the STD spectrum in the presence of the protein and that in the absence of it. STD spectra were acquired in the same conditions, however, drastic difference in absolute STD intensities between the two ligands exist. Specifically, the maximum STD signal detected for LDN‐DF ligand corresponds to just the 25 % of the strongest STD signal detected for the LDN‐F. (c) Average chemical shift perturbation upon the addition of 100 equivalents of LDN‐F (red) and LDN‐DF (blue) respect to the protein. The cut‐offs values were determined as “mean±SD” and are indicated with dotted lines. Structural models for the complexes of CRD DC‐SIGN with the LDN‐F ligand (d) and the LDN‐DF ligand (e) obtained from MD simulations.
Chemical shift perturbation analysis
The primary carbohydrate binding site of DC‐SIGN is composed of an extended loop (from W343 to D355) and residues in β‐strand‐4 (from N363 to D367), which surround the principal Ca+2 ion. [9b] Additionally, residues in β‐strands‐3 and −2 (from E356 to G361 and F313, respectively) shape a secondary binding region. Chemical shift perturbation analysis using the 15N‐labeled protein was performed to examine which residues of the CRD of DC‐SIGN interact with the glycans. Thus, the 15N‐labeled CRD DC‐SIGN was titrated with LDN‐F and LDN‐DF and 1H‐15N‐HSQC spectra were acquired at every titration point (Figure S3). Both ligands provided similar chemical shift perturbation (CSP) profiles involving amino acids of the primary and secondary binding site. However, upon addition of the same number of equivalents, the CSP of the lectin backbone NH resonances were much larger for LDN‐F than for LDN‐DF (Figure 1 c), indicating weaker binding of the latter compound. In‐deed, fitting of the CSP to the corresponding binding isotherms for LDN‐F yielded a kD of 1.5±0.4 mm (Figure S3). Protein saturation was not possible with LDN‐DF providing an imprecise kD estimation, but substantially larger than 6 mm.
Transferred NOESY
Recently, [13] the molecular complexes of DC‐SIGN with A and B histo blood group antigens have been studied by trNOESY experiments. Key intermolecular NOEs between the Hγ protons of V351 with H1 and H2 of the fucoside moiety were observed. The importance of the van der Waals stabilizing contacts involving the Me groups of V351 for the recognition of branched fucosylated epitopes has also been shown by X‐ray crystallography, [6a] and is in agreement with mutagenesis studies, demonstrating that substitution of this residue has important implications for the binding of Lewis type epitopes. [14] Therefore, a trNOESY experiment was carried out for LDN‐F in the presence of 0.2 equivalents of the CRD of DC‐SIGN. After addition of the protein to the NMR tube, strong negative NOE were observed for the ligand protons, which contain information on the bound ligand conformation. Inter‐molecular NOE correlations between the methyl groups of V351 and H1‐Fuc, H2‐Fuc, and the methyl group of GlcNAc were observed (Figure 2). Consistent with the STD analysis, Fuc H1 and H2 are in close contact with the lectin. Moreover, the strong STD described above for the methyl moiety of the GlcNAc residue is supported by the tr‐NOE correlation between this group and V351. As control, the NOESY spectrum of the free ligand was measured which displayed very weak negative NOE effects. Unfortunately, a good tr‐NOESY spectrum could not be recorded for the LDN‐DF complex, probably due to its rather low binding affinity.
Figure 2.
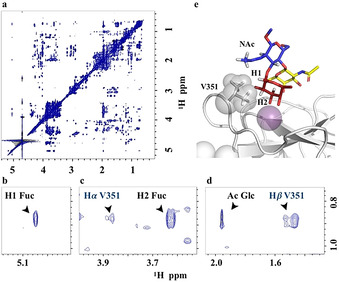
(a) NOESY spectrum of the complex of DC‐SIGN with LDN‐F. (b, c and d) The quest of protein/ligand intermolecular NOEs. (e) Three‐dimensional model from the MD simulation showing the key intermolecular NOEs between the ligand and the V351 residue of the lectin.
Molecular modeling
The NMR data were employed to derive three‐dimensional models for the complexes of DC‐SIGN with LDN‐F and LDN‐DF. For LDN‐F, the experimental STD and intermolecular NOE data showed close contacts of H1‐Fuc, H2‐Fuc and Ac‐GlcNAc with the methyl group of V351 of the protein. For the histo blood A and B antigens, [15] it has been demonstrated that DC‐SIGN binds the fucosyl ring exclusively through coordination of the C‐3 and C‐4 hydroxyls with the Ca2+ ion. The crystallographic structure of DC‐SIGN complexed with Lex (pdb code 1SL5) also fulfills this requirement. [6a] Therefore, the pyranosyl ring of LDN‐F was superimposed onto the corresponding monosaccharide in the deposited 1SL5 structure. The resulting binding pose was minimization by MD simulation that resulted in a structure that is in excellent agreement with the experimental data, including HSQC chemical shift perturbation and epitope mapping through STD. For LDN‐DF, a similar approach was used by superimposing the terminal 1,2‐linked fucoside in the primary Ca2+ binding site. This starting binding pose placed H2 of the terminal fucoside in close proximity to the protein backbone, which is in agreement with the STD data. The alternative binding pose through the inner fucoside was discarded due to steric clashes. Analysis of the MD trajectory showed that for the complex of LDN‐F with DC‐SIGN, the ligand conformation remained fairly well defined as revealed by the low dispersion of the ϕ and ψ angles (Figure S4). This arrangement favors hydrophobic contacts between V351 and the Fuc and GlcNAc moieties, which were maintained throughout the entire MD run (Figure 1 d). Additionally, the bound geometries were validated by simulating the STD spectrum with CORCEMA‐ST. [16] The match between the expected and the experimental STD intensities for LDN‐F was excellent, further supporting the proposed binding model (Figure S5).
The derived bound structure for LDN‐DF was very different (Figure 1 e). In this case, the LDN backbone was far from the protein with only the terminal fucoside making interactions. In this case, the contacts between the Ac of GlcNAc and Hγ protons of V351 were only transient and there was not a preferential spatial arrangement of both groups to make substantial van der Waals contacts. Although the LDN‐F moiety of LDN‐DF preserved conformational rigidity, the α1,2‐fucoside linkage was rather flexible (Figure S1) providing a loosely defined epitope presentation around the primary binding site. In this case, the fitting between the CORCEMA‐ST simulations with the experimental STD was less accurate, which is probably due to the weak STD signals and the inability of the MD simulation to reproduce the flexibility of the complex.
Glycan microarray binding studies
The CRDs of DC‐SIGN are clustered in tetramers, and such an arrangement can greatly amplify the avidity and specificity when interacting with glycans epitopes that are present in a multivalent arrangement. [8b–17] To examine the importance of multivalency, LDN‐F (12) and LDN‐DF (20) and a number of control glycans including Lex (21), Lex‐Lex (22), SLex (23) and SLex‐Lex (24), which all are equipped with an anomeric aminopentyl moiety, were immobilized on N‐hydroxysuccinimide (NHS)‐activated glass slides in replicates of 6 by piezoelectric printing. After incubation overnight in a saturated NaCl chamber, unreacted esters were quenched with ethanolamine. First, the glycan microarray was probed with biotinylated Aleuria aurantia lectin, which recognizes α1,2‐ α1,3‐ and α1,6‐fucosides, and Streptavidin‐AlexaFluor635. As anticipated all compounds showed strong responsiveness (Figure S3) confirming proper spot morphology and printing. Next, sub‐arrays were incubated with various concentrations of recombinant human DC‐SIGN‐Fc chimera premixed with anti‐IgG Fc‐biotin and Streptavidin‐AlexaFluor635 in TSM binding buffer containing Ca2+. After incubation for 1 h, the slide was washed, dried by centrifugation and scanned for fluorescence intensity. LDN‐F (12), Lex (21) and Lex‐Lex (22) exhibited strong responsiveness whereas no binding was detected for LDN‐DF (20) (Figure 3). This observation indicates that the avidity and selectivity of binding is greatly enhanced when the binding is probed on a multivalent surface. At a higher concentration of DC‐SIGN, SLex‐Lex (24) also exhibited responsiveness whereas this was not the case for SLex, indicating that the internal Lex moiety can be recognized by DC‐SIGN. When the microarray binding studies were performed in the absence of Ca2+, no binding was observed confirming specificity of binding.
Figure 3.
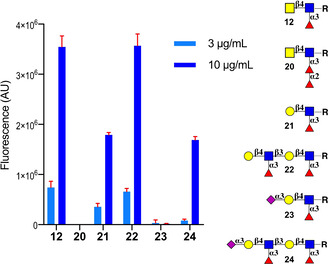
Microarray results of the glycan library printed at 100 μm for binding to DC‐SIGN (3 and 10 μg mL−1). Bars represent the mean±SD.
Conclusions
The recognition of fucosylated structures such as Lex, LDN‐F and pseudo_Ley, by DC‐SIGN has been implicated in schistosomiasism, [5a–18] resulting in modulation of innate and adaptive immune responses. [19] Further fucosylation of these epitopes to give structures such LDN‐DF or DF‐LDN‐DF abolishes binding. [3a] During the life cycle of S. mansoni, fucosylated glycans such as LDN, Lex, LDN‐F and LDN‐DF are expressed in a stage‐dependent manner, thereby shaping host immune responses. [3c–20] We have investigated, at a molecular level, in which way the terminal 1,2‐fucoside of LDN‐DF influences recognition by DC‐SIGN. Such a study required well‐defined glycans, which were obtained by a chemical approach in which amino protecting groups were carefully selected to facilitate high yielding and stereoselective chemical glycosylations. The molecular recognition of LDN‐F and LDN‐DF by DC‐SIGN was studied by NMR assisted by molecular modeling, which revealed that in solution it can recognize both glycans but with substantial differences in affinity and binding mode. The HSQC titration experiments provided a dissociation constant for LDN‐F of 1.5±0.4 mm at the monovalent level, whereas the kD for LDN‐DF could only be estimated but is substantially larger than 6 mm. In the case of the LDN‐F, the α1,3‐fucoside coordinates with the Ca2+‐ion of the CRD of DC‐SIGN, placing the GlcNAc residue in close proximity to the protein surface thereby allowing for additional interactions. The affinity and structural model for LDN‐F and Lex are very similar, [6b] indicating that the presence of a β4GalNAc vs. a β4Gal moiety does not substantially alter binding. The terminal α1,2‐linked fucoside of LDN‐DF can also bind into the canonical binding site of DC‐SIGN but in this case, the GlcNAc and GalNAc residues are placed away from the protein surface preventing additional contacts, and as a result the binding affinity is approximately an order of magnitude lower.
Previous studies have shown that antibodies directed against Lex and LDN‐F can block the binding of DC‐SIGN to soluble egg antigen of S. mansoni whereas an antibody against LDN‐DF had no effect on binding.[ 3a , 21 ] These observations lead to the conclusion that DC‐SIGN can recognize Lex and LDN‐F but not LDN‐DF. Our studies have shown that LDN‐DF can interact with DC‐SIGN albeit with a substantial lower affinity than for Lex and LDN‐F. The glycan microarray studies demonstrated that the avidity and selectivity of binding is greatly enhanced when the glycans are presented in a multivalent manner, and in this format Lex and LDN‐F gave strong responsiveness whereas no binding was detected for LDN‐DF. The extracellular domain of DC‐SIGN occurs as a tetramer. Furthermore, the glycans of S. mansoni are presented on its cell surface as glycoproteins and glycolipids, and thus it is anticipated that such assemblies can make multivalent interactions with DC‐SIGN, resulting in enhancement in avidity [8b] and magnify selectivities. [22]
Recently, it was shown that schistosomula extracellular vesicles (EVs) carry surface glycoproteins and glycolipids with a specific subset of fucosylated structures, such as Lex, pseudo_Ley and LDN‐F motifs that mediate internalization by moDCs in a DC‐SIGN dependent manner. [23] It is also known that DC‐SIGN signaling via fucosides decreases pro‐inflammatory responses, [24] indicating that S. mansoni may exploit these structures to dampen host immune response. Fucosylated glycans such as LDN, Lex, LDN‐F and LDN‐DF are expressed in a stage‐dependent manner during the life cycle of S. mansoni. [25] Interestingly, an increase in di‐fucosylated N‐glycans and glycolipids occurs during the transition from immature to mature eggs and in the miracidia stage.[ 20 , 26 ] During these stages, ligands, for DC‐SIGN such as Lex, are expressed at low levels. Thus, it is like that these changes result in a lack of detection by DC‐SIGN, thereby modulating the host's immune system.
Further studies are required to determine the importance of the density of specific glycans during the different life stages of Schistosomes and their influence on DC‐SIGN detection and subsequent skewing of host immune responses. Structures such as LDN‐DF and LDN‐F are part of complex oligosaccharides in which multiple of these epitopes can be presented. Potentially, such structures can make multivalent interactions leading to high avidity of binding. These features of molecular recognition require further investigation. In addition to DC‐SIGN, other C‐type lectins have been implicated in sensing helminth glycans by human DCs. Among those, the macrophage galactose‐type lectin MGL exhibits high specificity for S. mansoni glycans terminating in Gal‐NAc. [27] It is conceivable that these lectins act in concert to detect patterns of glycans thereby shaping immune responses. Finally, the synthetic approaches of LDN‐F and LDN‐DF will promote further biological studies to address the role of uniquely fucosylated glycans in S. mansoni infectivity and may lead to the development of immune‐modulatory compounds.
Experimental Section
General procedure for glycosylations for the synthesis of 7 and 13: Thioglycoside donor (4 equiv), diphenyl sulfoxide (4 equiv) and 2,4,6‐tri‐tert‐butylpyrimidine (4 equiv) were dissolved in DCM and stirred in the presence of pre‐activated molecular sieves (4 Å) for 30 min. Next, the temperature was lowered (−60 °C), followed by the addition of trifluromethanesulfonic anhydride (4 equiv). A solution of acceptor (1 equiv) in anhydrous DCM was added dropwise along the wall of the flask and the reaction was left stirring while the temperature was slowly raised to −40 °C. The reaction was quenched with triethyl amine, the molecular sieves were filtered off, and DCM was removed in vacuo. The residue was purified by silica gel column chromatography.
General procedure for conversion of azide (N3) into NHTroc: Compounds 7 and 13 (1 equiv) were dissolved in THF and water was added. Next trimethylphosphine (5 equiv) was added. The reaction mixture was stirred under an atmosphere of Ar for 2 h, after which the solvent was evaporated in vacuo and the residue was co‐evaporated with toluene twice. The residue was dissolved in DCM, followed by the addition of 2,2,2‐trichloroethyl chloroformate (2 equiv) and triethylamine (2 equiv). The reaction mixture was stirred for 1 h after which it was diluted by DCM and washed with water. The organic layer was dried (MgSO4), filtered, and the filtrate concentrated in vacuo. The residue was purified by silica gel column chromatography.
General procedure for the glycosylation of 5‐(N‐benzyloxycarbonyl,N‐benzyl)aminopentyl linker: The donors 10 and 18 (1 equiv) and 5‐(N‐benzyloxycarbonyl,N‐benzyl)aminopentyl linker (5 equiv) were dissolved in DCM, and stirred in the presence of pre‐activated molecular sieves (4 Å) under an atmosphere of Ar for 30 min. The reaction mixture was cooled (−50 °C), followed by the addition of trifluoromethanesulfonic acid (0.2 equiv). The temperature was slowly warmed up to −30 °C, after which TLC showed complete consumption of donor and the formation of a new product. The reaction mixture was quenched with triethyl amine, the molecular sieves were filtered off, and the solvent was evaporated. The residue was purified by silica gel column chromatography.
Protein expression: The extracellular domain of DC‐SIGN was obtained as previously de‐scribed. [15] The carbohydrate recognition domain of DC‐SIGN in its 15N labelled form was obtained as previously described. [13]
1 H Saturation transfer difference (STD) NMR: The samples for saturation‐transfer difference (STD) NMR experiments were prepared using the extracellular domain of DC‐SIGN at 10 μm concentration in 25 mm Tris‐d11, 150 mm NaCl, 4 mm CaCl2 in D2O (pD 8) using lectin/ligand ratios of 1:60. The temperature was set to 298 K. STD experiments were performed at 600 MHz Bruker spectrometer, using standard Bruker pulse sequences without water suppression nor protein spin‐lock filter. Protein saturation was achieved with a Gaussian‐shaped pulse of 49 ms. The on‐resonance frequency was set at aliphatic regions (0.76 ppm) and the off‐resonance frequency at 100 ppm. Blank STD experiments of the ligands alone were acquired in the same conditions. The results of blank 1H‐STD NMR experiments for ligands 12 and 20 are shown in Figures S1 and S2, respectively.
Chemical shift perturbation analysis: 1H‐15N‐HSQC‐based experiments were performed using 15N‐labeled CRD DC‐SIGN at 50 μm, with 2 mm DTT‐d10, at 800 MHz Bruker spectrometer equipped with a cryoprobe, at 310 K. Eight and ten titration points were acquired for ligands 12 and 20 respectively, with ligand concentrations varying from 0 to 0.5 mm for the former and from 0 to 1.5 mm for latter. Averaged chemical shift perturbation (CSP) and dissociation constants (kD) were calculated using the CcpNmr Analysis 2.4.2.3 The chemical shift perturbation analysis was performed based on the protein backbone assignment deposited in the BMRB database with the code 27854. The results from this analysis are shown in Figure S3.Transferred NOESY spectrum for glycan 12, was acquired at 800 MHz Bruker spectrometer equipped with a cryoprobe in the presence of 0.2 equivalents of DC‐SIGN (180 μm of protein), with a mixing time of 400 ms, at 298 K.
Molecular modelling: Initial geometries of ligands 12 and 20 were built in the Glycam web (http://glycam.org). Proton–proton distances derived from NOESY spectra (using the isolated spin‐pair ap‐proximation) were used to check the goodness of the minimized structures. The initial pdb coordinates for CRD of DC‐SIGN were derived from the crystal structure Protein Database (PDB) 1SL5. The magnesium ion was replaced by calcium, and the fucose pyranose ring of glycans 12 and 20 was superimposed onto the corresponding sugar in the deposited 1SL5 structure. The resulting binding poses were used as starting points for molecular dynamics (MD) simulations. The MD simulations were performed using the Amber16 program4 with the ff99SB force field parameters for protein and GLYCAM 06 h for the saccharides. Thereafter, the starting 3D geometries were placed into a 12 Å octahedral box of explicit TIP3P waters, and counterions were added to maintain electroneutrality. Two consecutive minimization stages were performed involving (1) only the water molecules and ions and (2) the whole system with a higher number of cycles, using the steepest de‐scent algorithm. The system was subjected to two rapid molecular dynamic simulations (heating and equilibration) before starting the real dynamic simulation. The equilibrated structures were the starting points for the final MD simulations at constant temperature (300 K) and pressure (1 atm). 500 ns Molecular dynamics simulations without constraints were recorded, using an NPT ensemble with periodic boundary conditions, a cut‐off of 10 Å, and the particle mesh Ewald method. A total of 250 000 000 molecular dynamics steps were run with a time step of 1 fs per step. Coordinates and energy values were recorded every 10 000 steps (10 ps) for a total of 25 000 MD models. A detailed analysis of the glycosydic linkages for glycans 12 and 20 was performed along the MD trajectory using the cpptraj module included in Amber‐Tools 16 package and are represented in Figure S4.
Microarray: The synthetic compounds were printed at 100 μm on activated glass slides by piezoelectric non‐contact printing (sciFLEXARRAYER S3, Scienion Inc). Printing was validated by assaying the binding to biotinylated Aleuria aurantia lectin (AAL) and detection by Streptavidin‐AlexaFluor635 (see Figure S6). Recombinant human DC‐SIGN‐Fc Chimera was assayed premixed with anti‐IgG Fc‐biotin and Streptavidin‐AlexaFluor635. The fluorescence was measured using a GenePix 4000 B microarray scanner (Molecular Devices) and data were processed with GenePix Pro 7 software and further analyzed using our home written Microsoft Excel macro. Data were fitted using Prism software (GraphPad Software, Inc). Further details are given in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the Netherlands Organization for Scientific Research (NWO; TOP‐PUNT grant 718.015.003 to G.‐J.B.), the Human Frontier Science Program Organization (HFSP; grant LT000747/2018‐C to L.U.), the European Research Council (ERC‐2017‐AdG, project number 788143‐RECGLYC‐ANMR to J.J.‐B.), the Agencia Estatal Investigación of Spain (AEI; grant RTI2018‐094751‐B‐C21 to J.J.‐B.) and the Severo Ochoa Excellence Accreditation (SEV‐2016‐0644 to J.J.‐B.).
A. D. Srivastava, L. Unione, M. A. Wolfert, P. Valverde, A. Ardá, J. Jiménez-Barbero, G.-J. Boons, Chem. Eur. J. 2020, 26, 15605.
References
- 1.
- 1a. Kalantari P., Bunnell S. C., Stadecker M. J., Front. Immunol. 2019, 10, 26; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. LoVerde P. T., Adv. Exp. Med. Biol. 2019, 1154, 45–70. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Geijtenbeek T. B. H., van Vliet S. J., Engering A., ′t Hart B. A., van Kooyk Y., Annu. Rev. Immunol. 2004, 22, 33–54. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. van Die I., van Vliet S. J., Nyame A. K., Cummings R. D., Bank C. M. C., Appelmelk B., Geijtenbeek T. B. H., van Kooyk Y., Glycobiology 2003, 13, 471–478; [DOI] [PubMed] [Google Scholar]
- 3b. Hokke C. H., Deelder A. M., Hoffmann K. F., Wuhrer M., Exp. Parasitol. 2007, 117, 275–283; [DOI] [PubMed] [Google Scholar]
- 3c. Hokke C. H., van Diepen A., Mol. Biochem. Parasitol. 2017, 215, 47–57. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Mickum M. L., Prasanphanich N. S., Heimburg-Molinaro J., Leon K. E., Cummings R. D., Front. Genet. 2014, 5, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Geijtenbeek T. B. H., den Dunnen J., Gringhuis S. I., Future Microbiol. 2009, 4, 879–890; [DOI] [PubMed] [Google Scholar]
- 5b. van Kooyk Y., Geijtenbeek T. B. H., Nat. Rev. Immunol. 2003, 3, 697–709. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Guo Y., Feinberg H., Conroy E., Mitchell D. A., Alvarez R., Blixt O., Taylor M. E., Weis W. I., Drickamer K., Nat. Struct. Mol. Biol. 2004, 11, 591–598; [DOI] [PubMed] [Google Scholar]
- 6b. Pederson K., Mitchell D. A., Prestegard J. H., Biochemistry 2014, 53, 5700–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Meyer S., van Liempt E., Imberty A., van Kooyk Y., Geyer H., Geyer R., van Die I., J. Biol. Chem. 2005, 280, 37349–37359. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. van Liempt E., Bank C. M. C., Mehta P., García-Vallejo J. J., Kawar Z. S., Geyer R., Alvarez R. A., Cummings R. D., van Kooyk Y., van Die I., FEBS Lett. 2006, 580, 6123–6131; [DOI] [PubMed] [Google Scholar]
- 8b. Mitchell D. A., Fadden A. J., Drickamer K., J. Biol. Chem. 2001, 276, 28939–28945. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Feinberg H., Castelli R., Drickamer K., Seeberger P. H., Weis W. I., J. Biol. Chem. 2006, 282, 4202–4209; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Feinberg H., Mitchell D. A., Drickamer K., Weis W. I., Science 2001, 294, 2163–2166; [DOI] [PubMed] [Google Scholar]
- 9c. Angulo J., Díaz I., Reina J. J., Tabarani G., Fieschi F., Rojo J., Nieto P. M., ChemBioChem 2008, 9, 2225–2227. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Brzezicka K., Echeverria B., Serna S., van Diepen A., Hokke C. H., Reichardt N. C., ACS Chem. Biol. 2015, 10, 1290–1302; [DOI] [PubMed] [Google Scholar]
- 10b. Ágoston K., Kerékgyártó J., Hajkó J., Batta G., Lefeber D. J., Kamerling J. P., Vliegenthart J. F. G., Chemistry 2002, 8, 151–161. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Gagarinov I. A., Fang T., Liu L., Srivastava A. D., Boons G.-J., Org. Lett. 2015, 17, 928–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Lu S. R., Lai Y. H., Chen J. H., Liu C. Y., Mong K. K., Angew. Chem. Int. Ed. 2011, 50, 7315–7320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7453–7458. [Google Scholar]
- 13.
- 13a. Valverde P., Delgado S., Martínez J. D., Vendeville J. B., Malassis J., Linclau B., Reichardt N. C., Cañada F. J., Jiménez-Barbero J., Arda A., ACS Chem. Biol. 2019, 14, 1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Geijtenbeek T. B. H., van Duijnhoven G. C. F., van Vliet S. J., Krieger E., Vriend G., Figdor C. G., van Kooyk Y., J. Biol. Chem. 2002, 277, 11314–11320. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Martínez J. D., Valverde P., Delgado S., Romanò C., Linclau B., Reichardt N. C., Oscarson S., Ardá A., Jiménez-Barbero J., Cañada F. J., Molecules 2019, 24, 2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Krishna N. R., Jayalakshmi V., Top. Curr. Chem. 2007, 273, 15–54. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Kiessling L. L., Splain R. A., Annu. Rev. Biochem. 2010, 79, 619–653. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Rodríguez E., Schetters S. T. T., van Kooyk Y., Nat. Rev. Immunol. 2018, 18, 204–211. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Anthony R. M., Rutitzky L. I., J. F. Urban, Jr. , Stadecker M. J., Gause W. C., Nat. Rev. Immunol. 2007, 7, 975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Smit C. H., van Diepen A., Nguyen D. L., Wuhrer M., Hoffmann K. F., Deelder A. M., Hokke C. H., Mol. Cell. Proteomics 2015, 14, 1750–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. den Dunnen J., Gringhuis S. I., Geijtenbeek T. B. H., Cancer Immunol. Immunother. 2009, 58, 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Mann D. A., Kanai M., Maly D. J., Kiessling L. L., J. Am. Chem. Soc. 1998, 120, 10575–10582; [Google Scholar]
- 22b. Lee R. T., Lee Y. C., Glycoconjugate J. 2000, 17, 543–551; [DOI] [PubMed] [Google Scholar]
- 22c. Dam T. K., Gerken T. A., Brewer C. F., Biochemistry 2009, 48, 3822–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Kuipers M. E., Nolte-′t Hoen E. N. M., van der Ham A. J., Ozir-Fazalalikhan A., Nguyen D. L., de Korne C. M., Koning R. I., Tomes J. J., Hoffmann K. F., Smits H. H., Hokke C. H., J. Extracell. Vesicles 2020, 9, 1753420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Gringhuis S. I., Kaptein T. M., Wevers B. A., Mesman A. W., Geijtenbeek T. B., Nat. Commun. 2014, 5, 3898. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Cummings R. D., Nyame A. K., Biochim. Biophys. Acta Mol. Basis Dis. 1999, 1455, 363–374; [DOI] [PubMed] [Google Scholar]
- 25b. Khoo K. H., Dell A., Adv. Exp. Med. Biol. 2001, 491, 185–205; [DOI] [PubMed] [Google Scholar]
- 25c. Hokke C. H., Deelder A. M., Glycoconjugate J. 2001, 18, 573–587. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Jang-Lee J., Curwen R. S., Ashton P. D., Tissot B., Mathieson W., Panico M., Dell A., Wilson R. A., Haslam S. M., Mol. Cell. Proteomics 2007, 6, 1485–1499. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. van Vliet S. J., van Liempt E., Saeland E., Aarnoudse C. A., Appelmelk B., Irimura T., Geijtenbeek T. B., Blixt O., Alvarez R., van Die I., van Kooyk Y., Int. Immunol. 2005, 17, 661–669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary