

Abstract
Throughout the nervous system, the convergence of two or more presynaptic inputs on a target cell is commonly observed. The question we ask here is to what extent converging inputs influence each other's structural and functional synaptic plasticity. In complex circuits, isolating individual inputs is difficult because postsynaptic cells can receive thousands of inputs. An ideal model to address this question is the Drosophila larval neuromuscular junction (NMJ) where each postsynaptic muscle cell receives inputs from two glutamatergic types of motor neurons (MNs), known as 1b and 1s MNs. Notably, each muscle is unique and receives input from a different combination of 1b and 1s MNs; we surveyed multiple muscles for this reason. Here, we identified a cell-specific promoter that allows ablation of 1s MNs postinnervation and measured structural and functional responses of convergent 1b NMJs using microscopy and electrophysiology. For all muscles examined in both sexes, ablation of 1s MNs resulted in NMJ expansion and increased spontaneous neurotransmitter release at corresponding 1b NMJs. This demonstrates that 1b NMJs can compensate for the loss of convergent 1s MNs. However, only a subset of 1b NMJs showed compensatory evoked neurotransmission, suggesting target-specific plasticity. Silencing 1s MNs led to similar plasticity at 1b NMJs, suggesting that evoked neurotransmission from 1s MNs contributes to 1b synaptic plasticity. Finally, we genetically blocked 1s innervation in male larvae and robust 1b synaptic plasticity was eliminated, raising the possibility that 1s NMJ formation is required to set up a reference for subsequent synaptic perturbations.
SIGNIFICANCE STATEMENT In complex neural circuits, multiple convergent inputs contribute to the activity of the target cell, but whether synaptic plasticity exists among these inputs has not been thoroughly explored. In this study, we examined synaptic plasticity in the structurally and functionally tractable Drosophila larval neuromuscular system. In this convergent circuit, each muscle is innervated by a unique pair of motor neurons. Removal of one neuron after innervation causes the adjacent neuron to increase neuromuscular junction outgrowth and functional output. However, this is not a general feature as each motor neuron differentially compensates. Further, robust compensation requires initial coinnervation by both neurons. Understanding how neurons respond to perturbations in adjacent neurons will provide insight into nervous system plasticity in both healthy and disease states.
Keywords: co-innervation, compensation, neuromuscular junction, neurotransmission, plasticity, synaptic growth
Introduction
The nervous system is characterized by complex wiring patterns that include different neurons converging onto the same postsynaptic cell. This wiring paradigm is found in pyramidal neurons that receive input from excitatory and inhibitory contacts (Megías et al., 2001), and in esophageal striated muscles that receive enteric and vagal nerve inputs (Kallmünzer et al., 2008; Neuhuber and Wörl, 2016). While dynamic regulation of individual synapses has been examined (Berry and Nedivi, 2017; Kruijssen and Wierenga, 2019), interplay between convergent neurons has been predominantly studied by monitoring postsynaptic spine changes (Oh et al., 2015; Hedrick et al., 2016; Jungenitz et al., 2018; Chistiakova et al., 2019). Understanding how convergent neurons respond to perturbations will shed light on the etiologies of neurodegenerative disorders, such as amyotrophic lateral sclerosis, which display progressive neuronal cell death and devastating functional consequences (Cluskey and Ramsden, 2001).
Analysis of individual inputs in the CNS is complicated by the high density of converging inputs on the same target cell. The Drosophila larval neuromuscular circuit, however, circumvents this with a simple, hard-wired connectivity map (Fig. 1A). The body plan is segmentally repeated, and each hemisegment is bilaterally symmetrical and composed of 35 motor neurons (MNs) and 30 postsynaptic muscles. Like most vertebrate central synapses, the individual glutamatergic neuromuscular contacts consist of axon terminal swellings, called boutons, and elaborate postsynaptic complexes (Collins and DiAntonio, 2007; Van Vactor and Sigrist, 2017). Each bouton houses several active zones (AZs) that enable neurotransmission. Larval muscles are innervated by two main MNs, type 1b (big) and 1s (small), which resemble tonic and phasic neurons, respectively (Schaefer et al., 2010), unlike vertebrate skeletal muscles that are monoinnervated. Most 1b MNs innervate a single muscle, whereas 1s MNs innervate functional muscle groups (Landgraf et al., 1997; Lnenicka and Keshishian, 2000; Hoang and Chiba, 2001), highlighting important structural and functional distinctions between these classes of neurons.
Figure 1.

1b and 1s MNs differentially contribute to the total EPSPs. A, Schematic of the innervation pattern of a subset of 1s MNs (vCE: dark green; dCE: light green) and 1b MNs (m6-1b: rust; m12-1b: orange; m4-1b: peach). Muscles analyzed in this study are marked by the red boxes. B, Representative frames showing the baseline fluorescence (top), 1b + 1s firing event (middle), and 1b alone firing event (bottom) of m6 in MHC-CD8::GCaMP6f-Sh larvae (1b: red; 1s: blue; also see Movie 1). C–E, Representative EPSP traces of 1b + 1s and 1b alone on m6 (C), m12 (D), and m4 (E). Traces and graphs are color coded as indicated in the color key. F–H, Paired EPSP amplitudes of m6 (F, t(14)=18.60, p < 0.0001, paired t test), m12 (G, t(14)=15.73, p < 0.0001, paired t test), and m4 (H, t(15)=7.43, p < 0.0001, paired t test). I, Calculated EPSP ratios of 1b/1b + 1s of m6, m12, and m4 (F(2,43)=26.03, p < 0.0001, one-way ANOVA, Tukey post hoc test). Error bars indicate ±SEM ****p < 0.0001. n values (NMJs/larva) are 15/12, 15/12, and 16/15, respectively.
In the larval neuromuscular circuit, 1b and 1s MNs are required for normal locomotion. Each MN type has unique electrophysiological properties: 1b MNs display a higher rate of spontaneous neurotransmitter release but the quantal size is smaller compared with 1s MNs (Nguyen and Stewart, 2016; Newman et al., 2017). In traditional neuromuscular junction (NMJ) electrophysiology experiments, EPSPs in muscles represent simultaneous stimulation of both neurons. However, several studies have isolated 1b-derived and 1b + 1s-derived EPSPs from different muscles (Table 1) and demonstrated that convergent 1b and 1s MNs do not equally contribute to the EPSP amplitude. Several forms of synaptic plasticity have been observed in the larval NMJ, including facilitation and depression (Hallermann et al., 2010). As each muscle is coinnervated and individual synaptic activities can be distinguished, this system provides an ideal platform to investigate structural and physiological plasticity changes that enable one MN to compensate for perturbations of a convergent MN. A recent study found that ablating the 1s MN on muscle 1 (m1) elevated evoked neurotransmission of the corresponding 1b NMJ (Aponte-Santiago et al., 2020). However, each muscle is innervated by a unique 1b–1s MN pair, so it is unclear whether this plasticity is consistent across all muscles.
Table 1.
Comparative 1b and 1s physiology from prior NMJ studies
| Muscle | 1b EPSP (mV) | 1s EPSP (mV) | 1b + 1s EPSP (mV) | [Ca2+] | Reference |
|---|---|---|---|---|---|
| 1 | 11.8 | 24.2 | 0.3 mm | Aponte-Santiago et al., 2020 | |
| 6 | 14.9 | 24.3 | 1.0 mm | Li et al., 2002 | |
| 6 | ∼10 | ∼35 | 1.8 mm | Kurdyak et al., 1994 | |
| 6 | 15.6 | 25.7 | 0.5 mm | Genc and Davis, 2019a | |
| 6 | 15.6 | 23.1 | 1.5 mm | Lnenicka and Keshishian, 2000 | |
| 7 | 12.1 | 15.5 | 1.5 mm | Lnenicka and Keshishian, 2000 |
aThis study used optogenetics, instead of voltage injection, to differentially activate 1b or 1s.
In this study, we examined three separate muscles to test structural and functional plasticity. First, we confirmed muscle-specific 1b activities. Next, we ablated 1s MNs after innervation and examined corresponding 1b NMJ compensatory responses. We found that all three 1b NMJs increased their bouton numbers and rates of spontaneous neurotransmitter release without a corresponding upregulation of AZs and some 1b NMJs also partially increased their evoked neurotransmission. An important factor for 1b synaptic plasticity is the 1s EPSP as silencing 1s evoked activity caused similar 1b NMJ responses. Additionally, robust 1b synaptic plasticity does not occur if the 1s NMJ is never formed, indicating that initial 1s innervation is required. These results demonstrate a novel, target-specific synaptic plasticity that may be a common feature of convergent neural circuits.
Materials and Methods
Fly and antibody reagents
Fly lines and antibodies used in this study are listed in Tables 2 and 3.
Table 2.
Fly strains used in this study
| Fly strain | Source |
|---|---|
| w1118 | Carrillo et al. (2015) |
| A8-GAL4 | Venkatasubramanian et al. (2019) |
| 10XUAS-mCD8::GFP | Bloomington Drosophila Stock Center, stock #32184 |
| 10XUAS-mCD8::GFP,A8-GAL4 | This article |
| DIP-αnull | (Xu et al., 2018) |
| UAS-hid,rpr | Zhou et al. (1997) |
| UAS-TNT | Bloomington Drosophila Stock Center, stock #28838 |
| MHC-CD8::GCaMP6f-Sh | Newman et al. (2017) |
Table 3.
Antibodies used in this study
| Antibody | Source | Concentration |
|---|---|---|
| Rabbit anti-GFP | Thermo Fisher Scientific, catalog #A11122 | 1:500 |
| Chicken anti-GFP | Thermo Fisher Scientific, catalog #A10262 | 1:500 |
| Rabbit anti-Fas2 | Budnik Laboratory (Koh et al., 1999) | 1:3500 |
| Rabbit anti-DLG | Budnik Laboratory (Koh et al., 1999) | 1:40,000 |
| Mouse anti-DLG | 4F3, DSHB | 1:100 |
| Mouse anti-Eve | 3C10, DSHB | 1:50 |
| Mouse anti-Bruchpilot (BRP) | nc82, DSHB | 1:50 |
| Mouse anti-GluRIIA | 8B4D2 (MH2B), DSHB | 1:50 |
| Goat anti-HRP 405 | Jackson ImmunoResearch, catalog #123–475-021 | 1:50 |
| Goat anti-HRP-Alexa Fluor 647 | Jackson ImmunoResearch, catalog #123–605-021 | 1:50 |
| Goat anti-mouse-Alexa Fluor 568 | Thermo Fisher Scientific, catalog #A11031 | 1:500 |
| Goat anti-chicken-Alexa Fluor 488 | Thermo Fisher Scientific, catalog #A11039 | 1:500 |
| Goat anti-rabbit-Alexa Fluor 405 | Thermo Fisher Scientific, catalog #A31556 | 1:500 |
| Goat anti-rabbit-Alexa Fluor 568 | Thermo Fisher Scientific, catalog #A11036 | 1:500 |
| Goat anti-rabbit-Alexa Fluor 647 | Thermo Fisher Scientific, catalog #A32733 | 1:500 |
DSHB, Developmental Studies Hybridoma Bank.
Dissection and immunocytochemistry
Embryonic dissections were performed as previously described (Lee et al., 2009). Egg-laying chambers were set up with adult flies (15–20 females and 10–15 males) and capped with grape juice plates (3% agar, 1.3% sucrose, 25% grape juice in water). After 6 h laying periods, grape plates covered in embryos were collected. Embryos were staged on double-sided tape using the autofluorescence and shape of the gut (Bownes, 1975; Hartenstein et al., 1987) under a Zeiss V20 stereoscope, and then dechorionated with a sharpened metal probe and placed on grape juice agar. Embryos were transferred to double-sided tape on a Superfrost Plus slides (catalog #22–037-246, Thermo Fisher Scientific) with the dissecting area outlined by a PAP pen (catalog #195506, Research Products International), and then covered with 0.22 µm filtered PBS (0.01 m sodium phosphate, 150 mm sodium chloride). Embryos were opened with an electrolytically sharpened tungsten wire, removed from the vitellin membrane, and then adhered to the charged slide. Dissected embryos were washed once with PBS, and then fixed for 30 min at room temperature using 4% paraformaldehyde (Electron Microscopy Sciences). Samples were then washed three times in 0.05% PBST (PBS with 0.05% Triton X-100), and then blocked for 1 h in 5% normal goat serum (5% goat serum diluted in 0.05% PBST). Samples were incubated in primary antibody solutions overnight at 4°C and washed three times in PBST. Samples were then incubated with secondary antibody solutions at room temperature for 2 h and washed three times with PBST. Finally, samples were mounted in Vectashield (Vector Laboratories), and the coverslip was sealed with nail polish.
First and third instar larval dissections were performed as previously described (Ashley et al., 2019). Wandering third instar larvae were dissected in PBS on Sylgard-184 (Dow) dishes and pinned down using sharpened 0.1 mm insect pins (catalog #26 002–10, Fine Science Tools). For first instar larvae, electrolytically sharpened tungsten pins were used to accommodate the size of smaller larvae. Samples were then fixed for 30 min using 4% paraformaldehyde [or 5 min in Bouin's solution for glutamate receptor IIA (GluRIIA)] and then transferred to 0.5 ml Eppendorf tubes. Samples were blocked and treated with primary and secondary antibodies as the embryo samples described above. All larval washes and antibody incubations were performed with mild agitation on a nutator.
Image acquisition
All imaging was acquired on a Zeiss LSM800 confocal microscope with either a 20× plan-apo 0.8 numerical aperture (NA) objective, a 40× plan-neofluar 1.3 NA objective, or a 63× plan-apo 1.4 NA objective. Laser intensity, pinhole, and gain were adjusted to increase the signal but avoid overexposure. All samples from the same experiment were imaged under identical conditions. Representative images are the maximum projection of the corresponding confocal z-stack (ImageJ).
Image analysis
Dorsal common exciter and ventral common exciter identification
The existence of ventral common exciter (vCE) and dorsal common exciter (dCE) MNs (two 1s type MNs) was confirmed in embryos and first instars. dCEs were identified by the expression of GFP, the transcription factor Even-skipped (Eve), and their positions in the ventral nerve cord (VNC). vCEs were identified by the expression of GFP and their positions in the VNC. Final confirmation was done by identifying the muscle innervation patterns in larval abdominal hemisegments.
1b bouton counting
1b bouton counting was performed in third instar samples. Boutons were examined using HRP and scored as 1s or 1b based on the Discs large (DLG) signal, as 1s boutons have a smaller and dimmer DLG signal than 1b boutons (Guan et al., 1996). Satellite boutons were identified as small bud-like structures emerging from “parent” boutons (Lee and Wu, 2010).
Bruchpilot and GluRIIA quantification
Bruchpilot (BRP) and GluRIIA signals at 1b NMJs were quantified in a manner similar to that described in the study by Han et al. (2020). Briefly, BRP and GluRIIA in 1s NMJs were excluded in controls by using the GFP signal (A8>GFP) to create a surface boundary (Imaris, Oxford Instruments) and setting the BRP and GluRIIA signal intensities inside 1s NMJs to zero. Next, a 1b NMJ surface was created based on the masked BRP, or GluRIIA, channel, and the total intensities of horseradish peroxidase (HRP), DLG, and BRP or GluRIIA were collected. For normalization, A8>hid,rpr and control samples were normalized to corresponding mean control values. For BRP/HRP and GluRIIA/DLG ratios, each sample was normalized to the sum intensity of either HRP or DLG, respectively.
Electrophysiology and analysis
Current-clamp recordings were performed as previously described (Meng et al., 2020). Third instar larvae were dissected in modified HL3 saline (70 mm NaCl, 5 mm KCl, 10 mm MgCl2, 10 mm NaHCO3, 5 mm trehelose, 115 mm sucrose, 5 mm HEPES) with 0.3 mm calcium. Segmental nerves were cut near the ventral nerve cord to remove VNC input and then the larval fillet was perfused with modified HL3 saline containing 0.5 mm calcium. Body-wall muscle 6, 12, or 4 in abdominal segment 3 were impaled with a 15–30 MΩ sharp electrode filled with 3 m KCl and recorded for miniature EPSPs (mEPSPs) for 2 min. All mEPSP amplitudes from the same genotype were pooled together and binned with at 0.01 mV increments to calculate the cumulative probability.
Nerves were drawn into a suction electrode and stimulated to elicit EPSPs. Specifically, for muscle 6 and 12 EPSP recordings, the whole segmental nerve bundle was stimulated, whereas for muscle 4 EPSP recording, the intersegmental nerve (ISN) above muscle 5 was stimulated. For each muscle, 24 EPSPs were elicited at 0.2 Hz and the largest 12 EPSPs were averaged to indicate the mean EPSP (while measuring EPSPs in A8>hid,rpr, we sometimes observed smaller EPSPs, similar to those reported by Lnenicka and Keshishian (2000)). All resting potentials are reported in Table 4.
Table 4.
Recorded resting potentials
| Resting potential (−mV) | |
|---|---|
| Figure 1 | |
| m6, 1b + 1s | 65.31 ± 3.119 |
| m6, 1b | |
| m12 1b + 1s | 62.60 ± 3.942 |
| m12, 1b | |
| m4, 1b + 1s | 61.50 ± 3.778 |
| m4, 1b | |
| Figure 6 | |
| m6, A8>GFP | 63.46 ± 2.824 |
| m6, A8>GFP,hid,rpr | 67.03 ± 3.247 |
| m12, A8>GFP | 64.13 ± 2.586 |
| m12, A8>GFP,hid,rpr | 63.71 ± 4.947 |
| m4, A8>GFP | 63.32 ± 2.933 |
| m4, A8>GFP,hid,rpr | 64.76 ± 2.985 |
| Figure 10 | |
| A8>GFP | 63.67 ± 1.732 |
| A8>GFP,TNT | 64.56 ± 3.046 |
| Figure 11 | |
| A8>GFP | 62.07 ± 2.154 |
| DIP-alphanull,A8>GFP | 62.62 ± 3.160 |
| Figure 12 | |
| m4 with 1s | 62.70 ± 2.263 |
| m4 with no 1s | 62.30 ± 3.310 |
Values are mean ± SD.
Because of nonlinear summation of quantal units of large EPSPs, we calculated corrected EPSPs using the equation defined by Martin (1955) and elaborated by Feeney et al. (1998). Quantal content (QC) was then calculated by dividing the corrected mean EPSP amplitude by the mean mEPSP amplitude for each muscle. Specifically, in genotypes where 1b and 1s NMJs were both present, the mean of the smaller two-thirds of mEPSP amplitudes was used to represent the 1b-derived mEPSP. This assumption was based on the published spontaneous frequencies of 1b and 1s MNs (Newman et al., 2017). Then the 1b-derived EPSP was divided by the 1b-derived mEPSP amplitude to estimate the 1b-derived QC. Similarly, the 1b + 1s EPSP was divided by the overall mEPSP amplitude to represent 1b + 1s QC. Finally, the 1b-derived QC was normalized to the 1b + 1s QC to indicate the estimated 1b baseline QC (1b QC/1b + 1s QC).
Paired-pulse recordings were performed under two-electrode voltage-clamp configuration (Li et al., 2018) in modified HL3 saline with 0.5 mm calcium. A second sharp electrode with 10–15 MΩ resistance was used to inject current. Muscle 6 was clamped at −70 mV, and the nerve was stimulated by two pulses 20 ms apart at 0.1 Hz. Only muscles with a leak current <10 nA were subjected to analysis. Paired-pulse ratios (EPSC2/EPSC1) were calculated by dividing the second EPSC by the first EPSC.
Signals were amplified using a MultiClamp 700B amplifier (Molecular Devices) for EPSP and mEPSP recordings or a Geneclamp 500B amplifier (Molecular Devices) for paired-pulse recordings and digitized with a Digidata 1550B (Molecular Devices). Stimulus was triggered via a Master-9 stimulator (A.M.P.I.). Data were acquired in pCLAMP 10 software (Molecular Devices) and analyzed using Mini Analysis software (Synaptosoft).
GCaMP imaging coupled with electrophysiology
Third instar MHC-CD8::GCaMP6f-Sh larvae were dissected in modified HL3 saline with 0.5 mm calcium and followed the same procedure of electrophysiology. Larval fillets were visualized using a Nikon FS microscope with a 40× long-working distance objective and GCaMP-positive 1b and 1s NMJs of muscles 6, 12, and 4 were illuminated by an Aura II solid-state illuminator. Together with electrophysiology stimulation, each stimulus triggered a GCaMP firing event with the corresponding EPSP. GCaMP signals were scanned and recorded by a PCO Edge 4.2 camera and NIS-Elements Imaging Software. Electrophysiology data were collected as described above. Given that 1b MNs have a lower evoked threshold than 1s MNs, stimulating voltages were tuned to isolate 1b alone firing events and 1b + 1s firing events (Movie 1). If a stimulation only triggered a GCaMP firing event at 1b NMJs but not 1s NMJs, the corresponding EPSP was counted as a 1b alone EPSP. If a stimulation triggered GCaMP fluorescence changes at both 1b and 1s NMJs, the corresponding EPSP was counted as a 1b + 1s EPSP. For each sample, 1b alone EPSPs and 1b + 1s EPSPs were averaged respectively. 1b alone/1b + 1s was calculated by dividing the mean 1b alone EPSP by the mean 1b + 1s EPSP of each sample.
SynapGCaMP allows 1b and 1b+1s-derived EPSPs from larval muscles to be differentiated. Nerves were stimulated at 0.5 Hz using different voltages for the two stimulus conditions. Frame rate is 30.
Experimental design and statistical analysis
In all experiments, we included A8>GFP to ensure that 1s NMJs were present in control animals (Ashley et al., 2019; Aponte-Santiago et al., 2020). For experiments with DIP-αnull;A8>GFP larvae, males were selected as DIP-α on the X chromosome. For A8>GFP/+ controls, males were also selected. All statistical analyses were performed using Prism 8 software (GraphPad). Average and SEM are reported. Outliers are determined by Q-test and excluded from the sample pools. For each data point, at least eight animals per genotype were dissected and at least two biological replicates were examined. All data were assumed to follow a Gaussian distribution. As we were making comparisons within specific target cells, and not between targets, most comparisons were performed by Student's t test (Welch's correction was used in cases of unequal variance) or Kolmogorov–Smirnov (K-S) test. Comparisons between target cells was performed with one-way ANOVA followed by Tukey's test.
Results
1b and 1s inputs contribute to postsynaptic activity in a target-specific manner
Converging inputs contribute to the overall postsynaptic response. In this study, we examined to what extent one input can influence the structure and function of a convergent input. To address this, we first determined the activity contribution of each MN on the postsynaptic muscle target in a wild-type condition. We chose m6, m12, and m4 because (1) prior studies showed that each 1b contributes a unique percentage of the total postsynaptic activity, (2) these muscles have been frequently analyzed in NMJ studies (Nose, 2012; Menon et al., 2013), and (3) the 1b and 1s innervation patterns on these muscles enabled the identification of common and muscle-specific principles (all muscles have unique 1b MNs, but m6 and m12 are innervated by the same 1s MN).
We measured the 1b MN contribution to the total EPSP on a muscle by muscle basis. 1b and 1s axons fasciculate into nerve bundles as they exit the VNC, which impeded us from physically stimulating each neuron independently without patching each cell body (Choi et al., 2004). To circumvent this, we combined NMJ electrophysiology with a postsynaptically targeted genetically encoded calcium indicator, SynapGCaMP6f (MHC-CD8::GCaMP6f-Sh; Newman et al., 2017). Some of the earliest evidence for the existence of two distinct MN populations was uncovered >40 years ago (Jan and Jan, 1976) through observations that different voltage injections elicited two populations of muscle EPSPs. It was later found that lower voltage injection generated action potentials in 1b MNs (Lnenicka and Keshishian, 2000); therefore, we varied the stimulus protocol to independently elicit and record EPSPs from 1b alone and 1b + 1s together (see Materials and Methods). SynapGCaMP6f fluorescence changes at 1b and 1s NMJs confirmed whether the recorded EPSPs were because of 1b alone or 1b + 1s activity (Fig. 1B, Movie 1). Using this procedure, the average total EPSP amplitude (1b + 1s) in m6 was 31.55 mV and the average 1b-derived EPSP was 17.17 mV (Fig. 1C,F). Thus, the m6-1b MN contributes 54% of the total m6 EPSP (Fig. 1I). Interestingly, at m12 the 1b MN contributes 31% of the total EPSP, and at m4 the 1b MN represents 62% (Fig. 1D,E,G–I). Thus, we determined the contribution of each MN (1b and 1s) to the postsynaptic muscle activity in wild-type larvae and found that the relative strength of each MN differed between muscles, with the m4-1b MN contributing the most and the m12-1b MN the least. Notably, m6-1b MN and m12-1b MN contributions are different although they are innervated by the same 1s MN. These data established a model in which to introduce perturbations and examine synaptic plasticity.
Cell-specific genetic ablation of 1s MNs by ectopic hid,rpr expression
To begin to examine whether one input can respond to perturbations in an adjacent input, we needed to disrupt one MN and monitor the impact on the convergent MN. Additionally, being able to disrupt one input before or after innervation can shed light on whether initial coinnervation is required for synaptic plasticity. We needed to identify drivers that are expressed specifically within subsets of the convergent neurons. In a previous study, we showed that DIP-α is expressed in subsets of neurons in the larval VNC, including interneurons and two 1s MNs, and that the removal of DIP-α impeded 1s innervation of m4 (Ashley et al., 2019). To gain genetic access specifically to the 1s MNs, we examined a GAL4 driver derived from the DIP-α promoter. In the adult neuromuscular circuit, this driver [hereafter referred to as A8-GAL4 or (A8)] was found to be expressed in a small subset of MNs (Venkatasubramanian et al., 2019). We used A8-GAL4 to drive UAS-GFP and found expression in only two pairs of segmentally repeated neurons in the third instar VNC (Fig. 2A,B, arrows). The labeled neurons located ventrally in the VNC have axons that project medially and dorsally toward the neuropil (Fig. 2C, arrow). The other A8-expressing neurons are located in the dorsal region of the VNC and showed an ipsilateral projection with a large dendritic arbor (Fig. 2D, arrowhead). Examination of nerves exiting the VNC showed two axons in each bundle (Fig. 2B,D asterisks), suggesting that these A8-positive neurons are MNs. The dorsal neurons were colabeled with the transcription factor Eve, which labels three medial neurons, aCC, pCC, and MNISN-1s (Doe et al., 1988; Broadus et al., 1995), and thus these neurons were confirmed as MNISN-1s (also called the dCE) based on their location (Fig. 2B). Drosophila larval MNs make connections with their muscle targets with very high fidelity, allowing us to unequivocally determine the identity of the MNs based on their innervation pattern. In A8>GFP third instar larvae, one axon innervates the ventral muscles including muscles 6, 7, 12, and 13, similar to the connectivity pattern of MNISNb/d-1s (also called the vCE). The other axon innervates the dorsal muscles, corresponding to dCE (Figs. 1A, 2E,F). To distinguish 1b and 1s NMJs, we also stained for DLG as 1b NMJs are surrounded by significantly more DLG (Guan et al., 1996; Fig. 2E,F). A8 is not expressed in the third 1s MN that innervates lateral transverse muscles. Importantly, A8 does not label any other MNs but only a few additional cells in one segment of the VNC (Fig. 2A, carets). In summary, A8 labels two 1s MNs (the vCE and dCE) within each hemisegment.
Figure 2.

A8-GAL4 drives expression in 1s MNs and can be used to ablate 1s MNs. A, B, Representative third instar larval VNC z-sections showing ventral (A) and dorsal (B) cell bodies labeled with GFP (green) and Eve (magenta; labels nuclei of dCE and other neurons) in A8>GFP. Arrows indicate GFP-positive vCE and dCE cell bodies. Carets indicate other cells that express A8. Asterisks indicate two axons exiting the VNC. C, D, 3D representations of A8-expressing neurons in the VNC viewed from lateral (C) and dorsal (D) sides (same VNC as A and B). Arrow in C shows a ventral cell (left, green) that projects an axon to the dorsal midline. Arrowhead in D shows an ipsilateral projection from a dorsal cell. Heat map colors are as follows: red denotes the dorsal-most region; and blue denotes the ventral most region. Asterisks indicate axons exiting the VNC similar to B. E, F, Representative third instar larval abdominal hemisegment labeled with GFP (green) and the postsynaptic marker DLG (magenta) in A8>GFP. Ventral muscles (m6, m7, m13, and m12) innervated by vCE (E) and dorsal muscles (m4, m3, m2 and m1) innervated by dCE (F). Arrows indicate 1s NMJs and arrowheads indicate 1b NMJs. G, H, Representative third instar larval VNCs lacking vCE (G) and dCE (H) labeled with GFP (green) and Eve (magenta) in A8>GFP,hid,rpr. Dashed circles mark the absence of vCE and dCE. Note that both vCE and dCE cell bodies are ablated by the third instar stage. Asterisk marks a GFP-positive cell that remained. I, J, Representative third instar larval abdominal hemisegment showing ventral muscles (I) and dorsal muscles (J), labeled with GFP (green) and DLG (magenta) in A8>GFP,hid,rpr. Note that all NMJs from vCE and dCE are absent (no GFP). Arrowheads indicate 1b NMJs.
We used A8-GAL4 to drive head involution defective (hid) and reaper (rpr) in vCE and dCE. hid and rpr have important functions in programmed cell death, and ectopic expression of both genes more reliably induces neuronal death than either gene alone (Zhou et al., 1997; Pauls et al., 2015). As shown in Figure 2A–F, A8>GFP robustly labels 1s MNs but in third instar larvae that ectopically express hid,rpr (A8>GFP,hid,rpr), all vCE or dCE cell bodies and NMJs are absent (Fig. 2G–J). Thus, the expression of hid,rpr is sufficient to genetically ablate 1s MNs.
To determine whether cell death occurred before or after innervation, we examined earlier developmental stages and visualized GFP in 1s MN cell bodies and NMJs in A8>GFP and A8>GFP,hid,rpr. Neuromuscular innervation is established at embryonic stage 16 (Halpern et al., 1991; Broadie and Bate, 1993; Yoshihara et al., 1997), so we focused on stage 15 and later stages including stage 17 and first instar larvae. In stage 15 embryo controls (A8>GFP), only a subset of 1s MNs are detected since not all dCE cell bodies (Eve positive) are costained with GFP and no vCE cell bodies are observed (Fig. 3A,B). Age-matched A8>GFP,hid,rpr embryos showed GFP expression patterns similar to those of controls (Fig. 3C,D); thus, no cell death occurred before neuromuscular innervation. By embryonic stage 17, more vCE and dCE cell bodies expressed A8 but the lack of GFP in some cells suggests that A8 expression has not reached maximal levels (Fig. 3E,F). In stage 17 A8>GFP,hid,rpr embryos, cells were undergoing apoptosis, as revealed by significant decreases in GFP and by the loss of Eve staining (suggesting nuclear degradation; Fig. 3G,H). Finally, in A8>GFP early first instar larvae, all vCE and dCE cell bodies were labeled (Fig. 3I,J) and NMJs were present (Fig. 3M). Age-matched A8>GFP,hid,rpr larvae completely lacked vCE and dCE cell bodies (Fig. 3K,L), although some 1s NMJs were still observed (Fig. 3M,N), suggesting that 1s NMJs were established before ablation in this genetic background. By late first instar stage, A8>GFP,hid,rpr larvae show no traces of 1s NMJs on m6, m12, and m4 (Fig. 3O). Together, A8-GAL4 is specifically expressed in vCE and dCE, and driving ectopic expression of cell death genes with A8 triggers apoptosis in 1s MNs after synaptic contacts are established.
Figure 3.
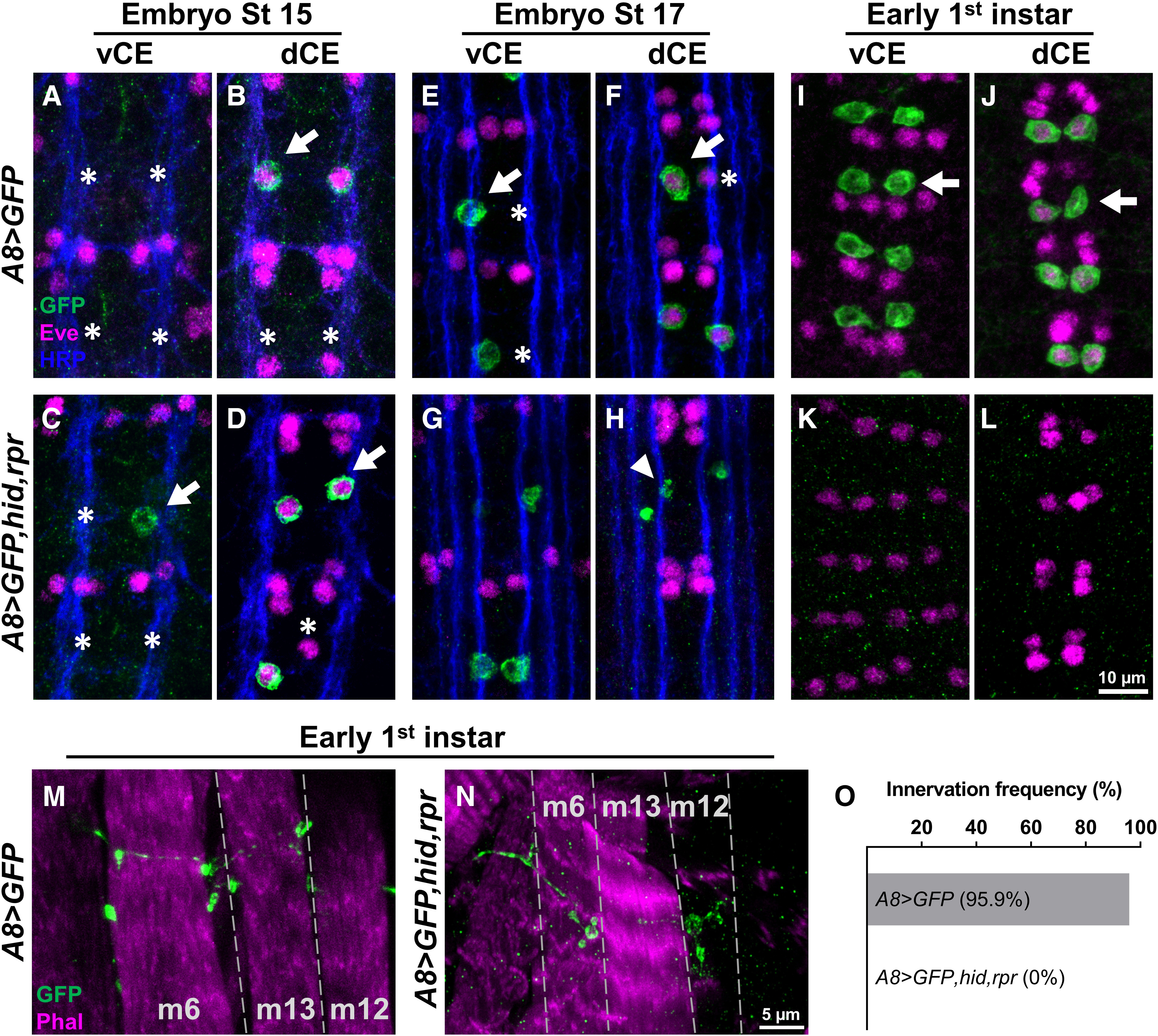
A8>hid,rpr-induced cell death occurs after 1s innervation. A–L, Representative images depicting the presence or absence of vCE and dCE cell bodies from embryonic stage 15 (A–D), stage 17 (E–H), and early first instar (I–L) larval VNCs labeled with GFP (green), Eve (magenta), and Fasciclin 2 (blue) in control (A8>GFP) and 1s-ablated (A8>GFP,hid,rpr) animals. Arrows and asterisks indicate the cells expressing or not expressing A8, respectively. A, B, A8 expression begins at embryonic stage 15. In A8>GFP,hid,rpr, vCEs, and dCEs undergo apoptosis starting at embryonic stage 17 (G, H), noted by the loss of Eve staining in dCE and GFP-positive debris (indicated by arrowhead), and are completely absent in early first instar larvae (K, L). M, N, Representative 1s NMJs labeled with GFP (green) and a muscle marker, phalloidin (magenta), in control (M) and 1s-ablated early first instar larvae (N). Note that 1s NMJs are labeled by GFP in control animals, and some 1s NMJs are still present in A8>GFP,hid,rpr animals, suggesting that A8>GFP,hid,rpr-induced cell death happens after 1s NMJ formation. O, Innervation frequency of 1s MNs in control and 1s-ablated late first instar larvae. Three muscles (m6, m12, and m4) were pooled and analyzed together. All 1s NMJs were eliminated in 1s-ablated animals by this stage. n values (NMJs/larva) are 76/5 and 86/5 for O.
1b NMJs expand upon loss of adjacent 1s MNs
NMJ structural plasticity can be induced by the perturbation of synaptic function (Budnik et al., 1990; Jarecki and Keshishian, 1995; Sigrist et al., 2003; Goel et al., 2019a; Perry et al., 2020). Here, we examined whether the loss of 1s MNs can induce structural changes at adjacent 1b NMJs. The size of each NMJ is well characterized (represented by the number of boutons), and this allows us to observe structural changes because of perturbations. We genetically ablated 1s MNs (vCE and dCE) and counted the number of boutons on m6, m12, and m4 in wandering third instar larvae (NMJ expansion is complete; Li et al., 2002). We observed an increase in the number of 1b boutons on all three muscles (Fig. 4A–G) when comparing them to A8>GFP controls, suggesting that 1b NMJs expanded when adjacent 1s MNs were ablated. Because m4-1s innervation frequency is ∼80% in wild type (Ashley et al., 2019; Aponte-Santiago et al., 2020), we used A8>GFP to confirm the presence of m4-1s in controls and excluded muscles lacking 1s innervation. Interestingly, we also found an increase in small budding boutons, called satellite boutons, emanating from 1b boutons (Fig. 4D–F, insets, H). These structures represent immature but functional boutons as they contain all synaptic machinery and postsynaptic receptors (Torroja et al., 1999; Dickman et al., 2006; O'Connor-Giles et al., 2008; O'Connor-Giles and Ganetzky, 2008; Lee and Wu, 2010; Carrillo et al., 2015). Thus, all 1b NMJs initiate structural plasticity mechanisms to respond to the loss of adjacent 1s MNs.
Figure 4.

1b NMJs expand upon ablation of 1s MNs. A–F, Representative NMJ arbors (1b arbors and 1s arbors, arrowheads and arrows, respectively) of m6 (A, D), m12 (B, E), and m4 (C, F) labeled with DLG (green) and HRP (magenta) in control (A8>GFP) and 1s-ablated (A8>GFP,hid,rpr) third instar larvae. Insets are 5× zoomed images of corresponding dashed regions in D–F. Satellite boutons are indicated by asterisks. Note that 1s NMJs are absent in 1s-ablated animals. Images and graphs are color coded as indicated in the color key. G, Quantification of 1b bouton number of m6 (t(30)=2.458, p = 0.02, unpaired t test), m12 (t(26)=4.449, p = 0.0001, unpaired t test), and m4 (t(28)=4.431, p = 0.0001, unpaired t test) in control and 1s-ablated animals (satellite 1b boutons were not included). H, Quantification of satellite boutons of m6 (t(30)=2.359, p = 0.025, unpaired t test), m12 (t(19.05)=2.397, p = 0.0269, unpaired t test with Welch's correction), and m4 (t(16.41)=3.682, p = 0.0019, unpaired t test with Welch's correction) in control and 1s-ablated animals. Error bars indicate ±SEM. *p < 0.05, **p < 0.01, ***p < 0.001. n values (NMJs/larva) are 16/8, 16/8, 16/8, 15/8, 15/8, and 15/8, respectively.
1b NMJs elevate their rate of spontaneous release upon loss of 1s MNs
To determine to what extent a synaptic input can influence the functional synaptic plasticity of a converging input, we first examined 1b spontaneous neurotransmitter release at muscles where 1s MNs were ablated after innervation. 1b and 1s MNs have unique spontaneous release properties. For example, 1b-derived spontaneous events (stimulus-independent release of neurotransmitter vesicles; also referred to as mEPSPs) have smaller amplitudes compared with 1s-derived mEPSPs (Nguyen and Stewart, 2016; Newman et al., 2017). Therefore, the ablation of 1s inputs should shift the mean mEPSP amplitude toward the smaller 1b-like amplitude if there is no compensation. We performed current-clamp recordings from m6, m12, and m4. Indeed, A8>GFP,hid,rpr revealed decreased mEPSP amplitudes compared with A8>GFP controls, and a significant shift in cumulative amplitude probability distribution (Fig. 5A–G). Because of the inability of standard NMJ electrophysiology experiments to distinguish between 1b and 1s mEPSPs in controls, we are unable to unambiguously determine whether the 1b mEPSP amplitudes are affected by the loss of 1s inputs. Nonetheless, we can conclude that 1b NMJs cannot fully restore the average mEPSP amplitudes to wild-type levels.
Figure 5.

Loss of 1s MNs decreases overall mEPSP amplitudes and increases 1b mEPSP frequencies. A–C, Representative mEPSP recordings of m6 (A), m12 (B), and m4 (C) in control (A8>GFP) and 1s-ablated (A8>GFP,hid,rpr) animals. Traces and graphs are color coded as indicated in the color key. D–F, Pooled cumulative probability distributions of m6 (D; p < 0.0001, K-S test), m12 (E; p < 0.0001, K-S test), and m4 (F; p < 0.0001, K-S test). G, Quantification of mEPSP amplitude of m6 (t(22) = 2.630, p = 0.0153, unpaired t test), m12 (t(22) = 2.294, p = 0.0317, unpaired t test), and m4 (t(29) = 3.700, p = 0.0009, unpaired t test) in control and 1s-ablated animals. Each data point represents the average mEPSP amplitude from one sample. H, Quantification of mEPSP frequencies of m6 (t(22) = 1.224, p = 0.2339, unpaired t test), m12 (t(22) = 2.331, p = 0.0293, unpaired t test), and m4 (t(29) = 0.8369, p = 0.4095, unpaired t test) in control and 1s-ablated animals. Each data point represents the average mEPSP frequency from one sample. Error bars indicate ±SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001, ns - not significant. n values (NMJs/larva) are 12/9, 12/11, 12/9, 12/12, 14/11, and 17/14, respectively.
Another measure of stimulus-independent activity is the rate of spontaneous neurotransmitter release. In prior studies, mEPSP frequencies were found to be higher at 1b NMJs than 1s NMJs (e.g., 2.3 Hz at m4-1b NMJ and 1 Hz at m4-1s NMJ; Newman et al., 2017); thus, if the elimination of 1s MNs does not affect the rate of 1b spontaneous release, overall mEPSP frequencies should decrease by about one-third. However, we did not observe any reduction of mEPSP frequencies on m6, m12, and m4 when comparing A8>GFP and A8>GFP,hid,rpr animals, and m12 even showed an increased rate of spontaneous release (Fig. 5A–C,H). Importantly, these spontaneous events correspond to 1b-derived mEPSPs since (1) the mean amplitudes decrease (Fig. 5G), and (2) there are no remaining 1s NMJs (Fig. 2I,J). These results strongly suggest that 1b MN synaptic plasticity mechanisms detect the loss of convergent 1s MNs and elevate their rate of spontaneous neurotransmitter release. Spontaneous neurotransmitter release has been implicated in synaptic maturation (Joseph et al., 2012; Choi et al., 2014; Andreae and Burrone, 2015, 2018; Cho et al., 2015; Kavalali, 2015), and, thus, the increased mEPSP frequencies we observe could underlie the increased 1b bouton numbers.
1b MNs elevate evoked neurotransmission upon loss of 1s MNs in a target-specific manner
Having demonstrated 1b synaptic plasticity of spontaneous activity, we next examined whether 1b-evoked neurotransmission could also be modified by the loss of 1s MNs. Unlike spontaneous neurotransmitter release, EPSPs require stimulation to depolarize the presynaptic neuron above threshold. This suprathreshold stimulation triggers an action potential to induce neurotransmitter release and elicit a postsynaptic response. To examine whether 1b MNs can compensate for the loss of 1s synaptic drive, we recorded EPSPs in both A8>GFP and A8>GFP,hid,rpr (Fig. 6A–F). To determine whether 1b-derived EPSP amplitudes are affected by the loss of 1s inputs, we normalized the EPSPs (A8>GFP,hid,rpr EPSP/A8>GFP EPSP) and compared these values to the calculated 1b/1b + 1s ratio in Figure 1I. This analysis allows for the comparison of 1b-derived EPSPs with and without convergent 1s MNs.
Figure 6.

1b NMJs elevate evoked neurotransmission in a target-specific manner in the absence of 1s inputs. A–C, Representative EPSP traces of m6 (A), m12 (B), and m4 (C) in control (A8>GFP) and 1s-ablated (A8>GFP,hid,rpr) animals. Traces and graphs are color coded as indicated in the color key. D–F, Quantification of EPSP amplitudes in m6 (D; t(22) = 8.306, p < 0.0001, unpaired t test), m12 (E; t(22) = 13.82, p < 0.0001, unpaired t test), and m4 (F; t(29) = 3.057, p = 0.0048, unpaired t test) in control and 1s-ablated animals. G–I, Normalized EPSPs of m6 (G; t(25) = 2.301, p = 0.0300, unpaired t test), m12 (H; t(25) = 1.552, p = 0.1332, unpaired t test), and m4 (I; t(31) = 4.605, p < 0.0001, unpaired t test) in control and 1s-ablated third instar larvae. Normalized EPSPs in A8>GFP,hid,rpr are compared with the EPSP ratio of 1b/1b + 1s calculated from corresponding muscles in Figure 1I, indicated by gray dashed line. Note m6-1b slightly compensates, m12-1b does not compensate and m4 largely compensates the loss of 1s MNs. J–L, Normalized QC of m6 (J; t(25) = 1.239, p = 0.2268, unpaired t test), m12 (K; t(25) = 2.119, p = 0.0442, unpaired t test), and m4 (L; t(31) = 4.639, p < 0.0001, unpaired t test) in control and 1s-ablated larvae. Normalized QCs were compared with 1b baseline QC (see Materials and Methods). Note m4-1b shows an increased QC, while m12-1b shows a decrease. Error bars indicate ±SEM. *p < 0.05, **p < 0.01, ****p < 0.0001, ns - not significant. n values (NMJs/larva) are 12/9, 12/11, 12/9, 12/12, 14/11, and 17/14, respectively.
We observed target-specific changes in 1b-derived EPSPs (Fig. 6G–I). At m4, we observed a significant increase in 1b-derived EPSPs compared with the control 1b/1b + 1s ratio (Fig. 6I). By repeating the analysis at other muscles, we found a mild increase in 1b-derived EPSPs at m6 (Fig. 6G) and, surprisingly, no change at m12 (Fig. 6H). When comparing the degree of EPSP compensations (Fig. 6G–I) to the wild-type 1b contribution to total EPSP at each muscle (Fig. 1I), a pattern arises whereby the m4-1b has both the most compensation and the largest 1b contribution. Meanwhile, m12-1b did not compensate and displayed the smallest 1b contribution.
Next, we examined QC, a calculation of the approximate amount of neurotransmitter released per stimulation. We found that the normalized QC of m4-1b in A8>GFP,hid,rpr was significantly increased compared with the m4-1b baseline QC (see Materials and Methods), whereas there was no change at m6-1b, and even a decrease at m12-1b (Fig. 6J–L). To test whether the loss of 1s MNs perturbed short-term 1b NMJ plasticity, we examined paired-pulse facilitation (PPF) as it is a measure of evoked release probability (Pr). We measured m6 PPF in A8>GFP,hid,rpr and found an enhancement compared with A8>GFP controls (Fig. 7). Although these data suggest an increase in 1b paired-pulse ratio (PPR), this can also be explained by the loss of 1s-specific synaptic depression (Newman et al., 2017) as both MNs are stimulated in controls. Furthermore, we were unable to definitively isolate 1b-specific PPF in controls as the second EPSC could be either 1b derived or 1b + 1s derived.
Figure 7.

Overall paired-pulse ratio is increased upon ablation of 1s MNs. A, Representative paired-pulse recordings of m6 in control (A8>GFP) and 1s-ablated (A8>GFP,hid,rpr) animals. B, Quantification of paired-pulse ratio (EPSC2/EPSC1) in control (red) and 1s-ablated animals (orange; t(26) = 3.120, p = 0.0044, unpaired t test). n values (NMJs/larva) are 15/10, 13/10, respectively. Error bars indicate ±SEM. **p < 0.01.
Overall, when ablating 1s MNs after NMJ formation, 1b MNs that innervate the same muscle upregulated their rate of spontaneous release, and, importantly, the m4-1b MN significantly compensated the total EPSP by increasing neurotransmitter release.
Active zone and glutamate receptor levels are unaltered by ablation of 1s MNs
One way to increase both spontaneous and evoked release is by the addition of more AZs. We stained for BRP, an AZ scaffolding protein (Kittel et al., 2006), to visualize 1b AZs in the presence or absence of convergent 1s MNs. Despite elevated 1b synaptic drive, we observed no change in overall 1b AZ intensity (Fig. 8A–G), although the larger NMJ sizes (Fig. 4G) led to reduced AZ densities (BRP/HRP; Fig. 8H). These data suggest that the number of AZs remains constant although NMJ size increases, similar to previous studies (Goel et al., 2019a, b; Aponte-Santiago et al., 2020; Goel et al., 2020).
Figure 8.
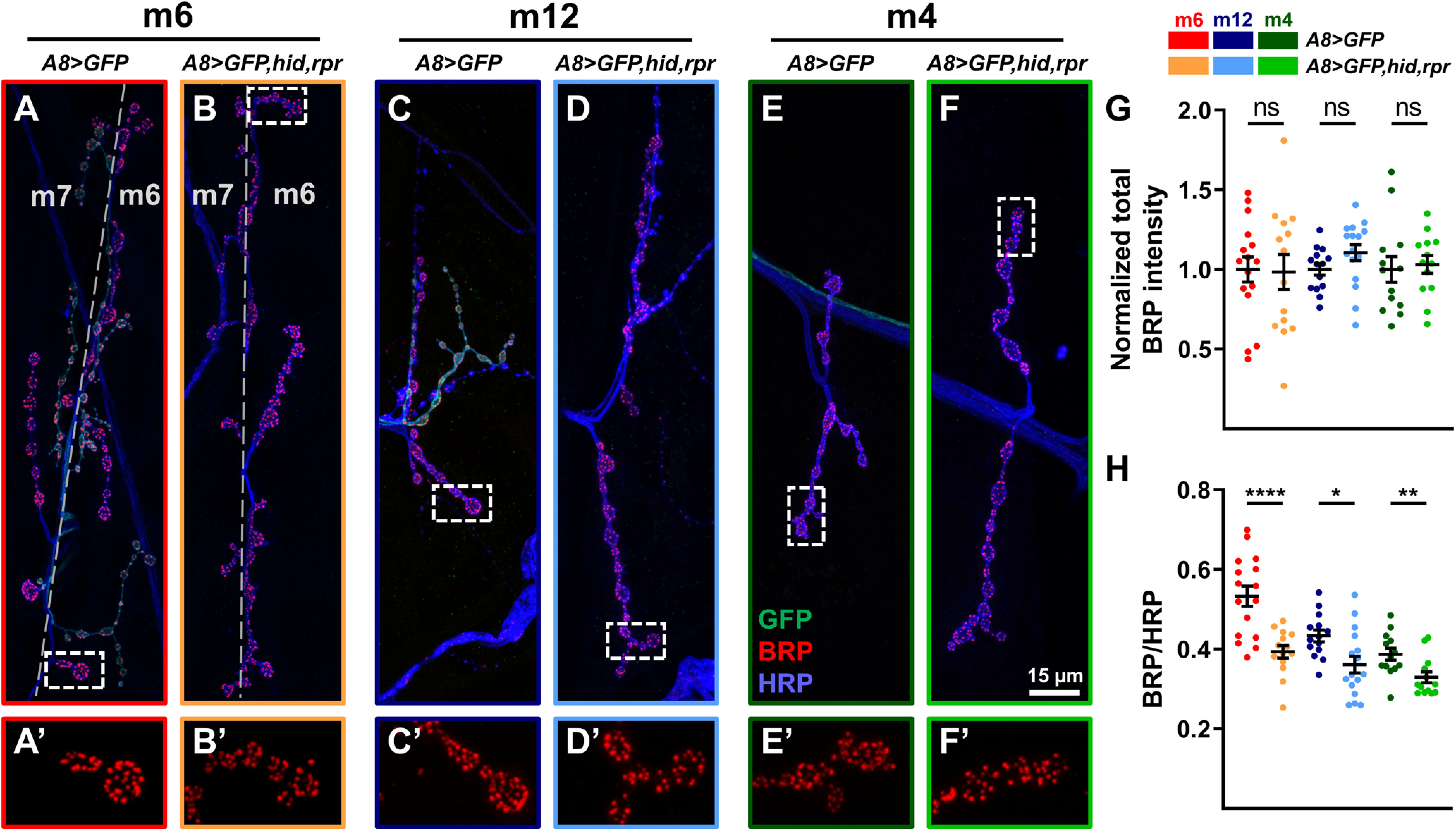
Quantification of BRP levels at 1b NMJs. A–F, Representative BRP immunostaining from m6 (A, B), m12 (C, D), and m4 (E, F) in control (A8>GFP) and 1s-ablated (A8>GFP,hid,rpr) animals labeled with GFP (green), BRP (red), and HRP (blue). A′–F′, 2.5× zoomed 1b boutons corresponding to dashed regions in A–F showing BRP staining. G, Quantification of normalized total BRP intensity of 1b NMJs on m6 (t(28) = 0.1199, p = 0.9054, unpaired t test), m12 (t(28) = 1.627, p = 0.1148, unpaired t test), and m4 (t(24) = 0.3189, p = 0.7525, unpaired t test). Images and graphs are color coded as indicated in the color key. H, Quantification of BRP/HRP sum intensity ratio (density) from m6 (t(24.31) = 4.705, p < 0.0001, unpaired t test with Welch's correction), m12 (t(28) = 2.736, p = 0.0107, unpaired t test), and m4 (t(24) = 2.847, p = 0.0089, unpaired t test). Error bars indicate ±SEM. *p < 0.05, **p < 0.01, ****p < 0.0001, ns - not significant. n values (NMJs/larva) are 16/8, 14/8, 14/8, 16/8, 13/7, and 13/7, respectively.
Changes in the postsynaptic glutamate receptor density or localization can contribute to functional responses. To test whether the functional synaptic plasticity we observed was because of changes in glutamate receptor levels, we visualized the GluRIIA (Petersen et al., 1997). m6-1b NMJs showed a significant increase in GluRIIA intensity, whereas m12-1b and m4-1b did not show a significant increase (Fig. 9A–G). Interestingly, 1b NMJ GluRIIA density (Fig. 9H) was unaffected, suggesting that receptor abundance may not contribute to 1b synaptic plasticity.
Figure 9.

Quantification of GluRIIA levels at 1b NMJs. A–F, Representative GluRIIA immunostaining from m6 (A, B), m12 (C, D), and m4 (E, F) in control (A8>GFP) and 1s-ablated animals (A8>GFP,hid,rpr) labeled with GFP (green), GluRIIA (red), and DLG (blue). A′–F′, 2.5× zoomed 1b boutons corresponding to dashed regions in A–F showing GluRIIA staining. G, Quantification of normalized total GluRIIA intensity of m6 (t(18.38) = 3.812, p = 0.0012, unpaired t test with Welch's correction), m12 (t(28) = 1.337, p = 0.1921, unpaired t test), and m4 (t(26) = 2.049, p = 0.0507, unpaired t test). Images and graphs are color coded as indicated in the color key. H, Quantification of GluRIIA/DLG sum intensity ratio (density) of m6 (t(29) = 1.108, p = 0.2771, unpaired t test), m12 (t(28) = 0.8544, p = 0.4002, unpaired t test), and m4 (t(26) = 0.2679, p = 0.7909, unpaired t test). Error bars indicate ±SEM. **p < 0.01, ns - not significant. n values (NMJs/larva) are 16/8, 15/8, 14/7, 16/8, 15/8, and 13/7, respectively.
Overall, these data indicate that presynaptic BRP or postsynaptic GluRIIA levels are not an integral component of 1b synaptic plasticity induced by the ablation of 1s MNs.
Loss of the 1s-derived EPSP triggers 1b synaptic plasticity
Ablation is a crude perturbation that completely removes all 1s MN synaptic activity and signaling pathways. Here, we focus on 1s evoked neurotransmission to examine whether its removal mimics the ablation of 1s MNs. Tetanus toxin light chain (TNT) is a clostridial toxin that cleaves the vesicle SNARE synaptobrevin, which is required for calcium-dependent vesicle release (Sweeney et al., 1995). Ectopic expression of TNT blocks evoked neurotransmission without affecting spontaneous release and synaptic morphology (Sweeney et al., 1995; Aponte-Santiago et al., 2020). Therefore, we expressed TNT with A8-GAL4 to specifically block 1s-derived EPSPs and measured 1b responses. We focused on m4-1b NMJs as these showed robust EPSP compensation, and we observed similar 1b synaptic plasticity when compared with ablation of 1s MNs. Structurally, the m4-1b NMJ was larger (Fig. 10A–C) and displayed more satellite boutons (Fig. 10D). Functionally, we observed an increase in overall mEPSP frequency (Fig. 10E,F) and 1b-derived EPSP amplitude (when comparing the normalized EPSP to the 1b/1b + 1s ratio; Fig. 10G,H). The compensatory 1b-derived EPSP, however, was less than that observed with 1s ablation (Fig. 6I). QC in A8>GFP,TNT was calculated as before (see Materials and Methods; 1b baseline QC), then normalized and compared with 1b baseline QC. Importantly, we observed a similar increase in QC (Fig. 10I). These results suggest that the loss of 1s-derived EPSPs was sufficient to trigger 1b synaptic plasticity, although other 1s-derived signals likely contribute since the ablation of 1s induces a more robust 1b response.
Figure 10.
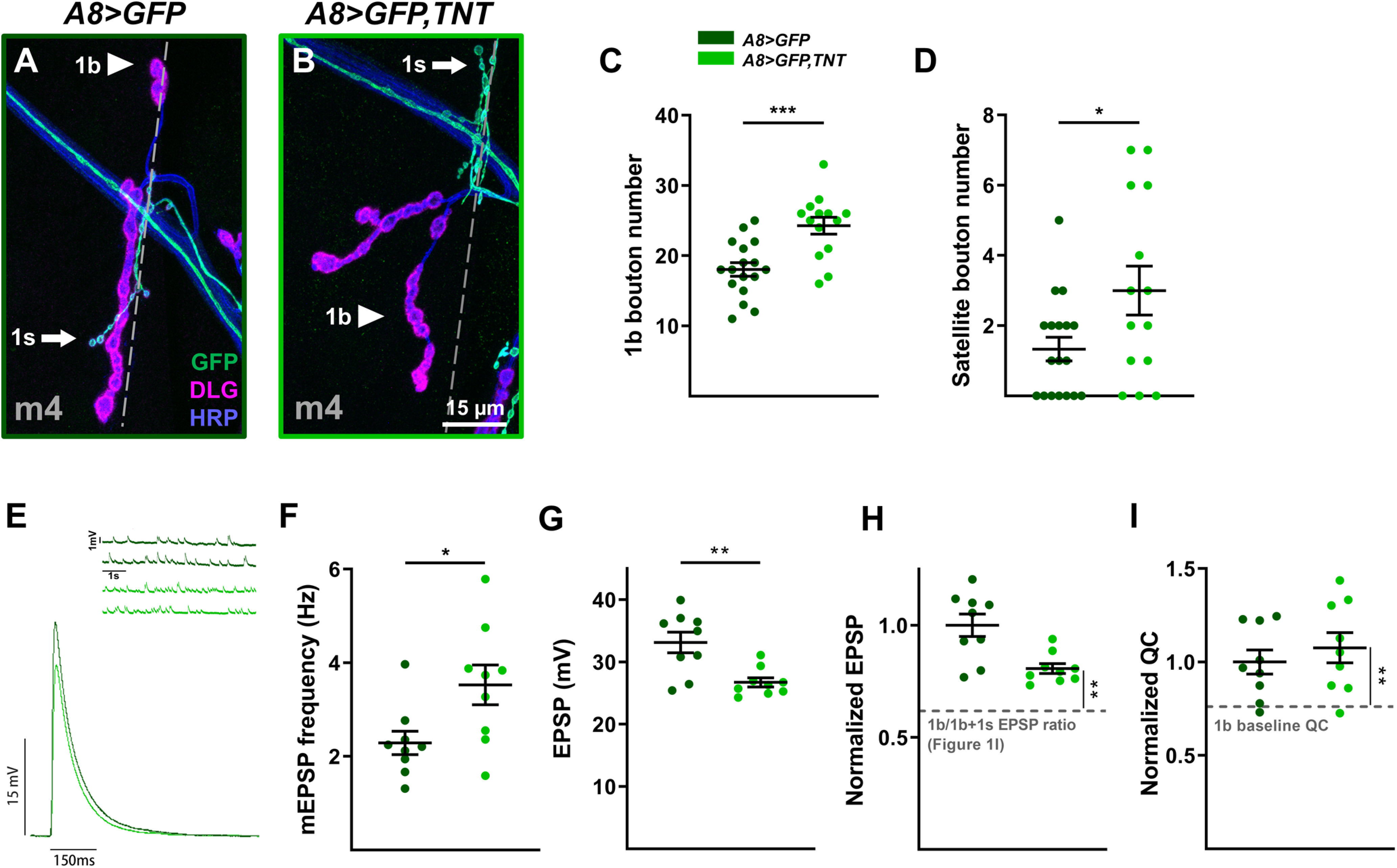
Loss of 1s evoked neurotransmission contributes to 1b synaptic plasticity. A, B, Representative m4 NMJs labeled with GFP (green), DLG (magenta), and HRP (blue) in control (A; A8>GFP: dark green) and 1s-silenced (B; A8>GFP,TNT: light green) animals. C, Quantification of 1b boutons on m4 in control and 1s-silenced animals (t(29) = 4.099, p = 0.0003, unpaired t test). Images, representative traces, and graphs are color coded as indicated in the color key. D, Quantification of satellite boutons from m4 in control and 1s-silenced animals (t(18.90) = 2.161, p = 0.0437, unpaired t test with Welch's correction). E, Representative mEPSP and EPSP recordings in control and 1s-silenced animals. F, Quantification of mEPSP frequencies from m4 in control and 1s-silenced animals (t(16) = 2.515, p = 0.0230, unpaired t test). G, Quantification of EPSP amplitudes in control and 1s-silenced animals (t(11.01) = 3.529, p = 0.0047, unpaired t test with Welch's correction). H, Normalized EPSP from m4 in control and 1s-silenced animals (t(20.75) = 3.736, p = 0.0012, unpaired t test with Welch's correction). Normalized EPSP of 1s-silenced animals is compared with the EPSP ratio of 1b/1b + 1s calculated from m4 in Figure 1I, indicated by gray dashed line. I, Normalized QC from m4 in control and 1s-silenced animals (t(23) = 3.159, p = 0.0044, unpaired t test). QC from 1s-silenced m4 was estimated using corrected EPSP and estimated 1b-derived mEPSP amplitude. Normalized QC was compared with 1b baseline QC (see Materials and Methods). Error bars indicate ±SEM. *p < 0.05, **p < 0.01, ***p < 0.001. n values (NMJs/larva) are 14/10, 17/9 for C and D, and 9/7, 9/6 for E–I.
Robust 1b synaptic plasticity requires initial 1s innervation
Above, we examined the structural and functional synaptic plasticity of one neuron when a converging neuron is ablated after innervation (A8>GFP,hid,rpr). To probe whether initial coinnervation is required for plasticity, we explored another context when the 1s NMJ never forms on the postsynaptic target. In a prior study, we found that DIP-α is required for the recognition of m4 by the dCE (Fig. 11A,B), and we confirmed that no 1s boutons are observed on m4 in DIP-αnull early first instar larvae (Fig. 11C). Quantification of m4-1b boutons and satellite boutons in DIP-αnull mutants showed no difference from controls (Fig. 11D,E). Also, in DIP-αnull mutants, the m4 mEPSP frequency was significantly reduced (Fig. 11F,G), in contrast to the 1s ablation (Fig. 5H). These results suggest that the m4-1b NMJ failed to compensate when the corresponding 1s NMJ never formed. Next, we examined EPSPs (Fig. 11F,H). Surprisingly, the lack of 1s innervation in DIP-αnull mutant larvae revealed only a slight increase in the 1b-derived EPSP amplitude on m4 (Fig. 11I) and no change in QC (Fig. 11J). Thus, m4-1b synaptic plasticity is not as robust in DIP-αnull larvae, compared with A8>GFP,hid,rpr.
Figure 11.
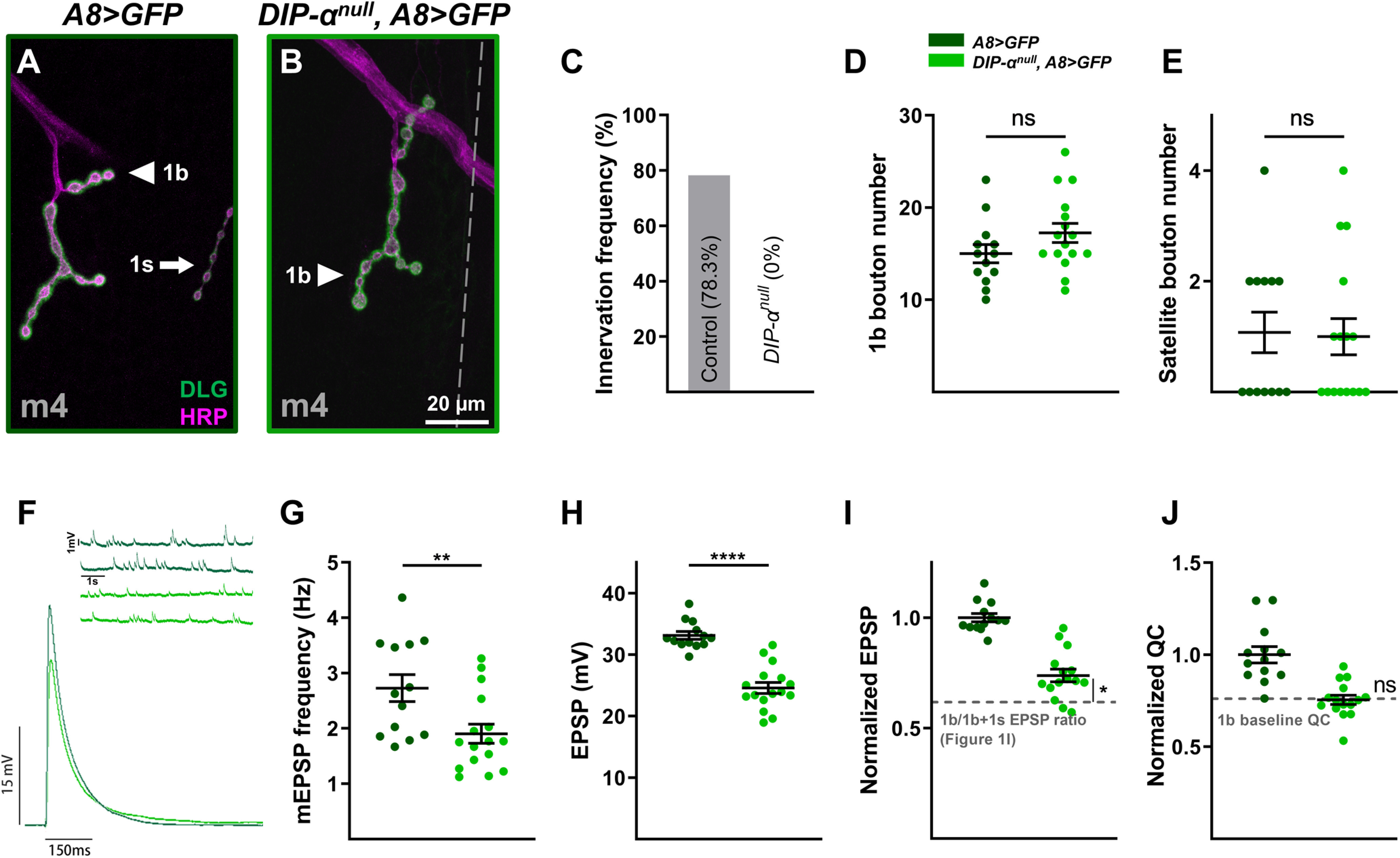
Robust 1b MN synaptic plasticity requires initial 1s innervation. A, B, Representative m4 NMJs labeled with DLG (green) and HRP (magenta) in control (A8>GFP: dark green; A) and mutant (DIP-αnull, A8>GFP: light green; B) animals. C, Quantification of 1s innervation frequency in control and mutant first instar larvae. No m4-1s NMJs observed in mutants. D, Quantification of 1b boutons from m4 in control and mutant animals (t(27) = 1.552, p = 0.1323, unpaired t test). Images, representative traces, and graphs are color coded as indicated in the color key. E, Quantification of satellite boutons from m4 in control and mutant animals (t(27) = 0.1563, p = 0.8770, unpaired t test). F, Representative mEPSP and EPSP recordings from control and mutant animals. G, Quantification of mEPSP frequencies from m4 in control and mutant animals (t(27) = 2.824, p = 0.0088, unpaired t test). H, Quantification of EPSP amplitudes in control and mutant animals (t(27) = 7.505, p < 0.0001, unpaired t test). I, Normalized EPSPs from m4 in control and mutant animals (t(29) = 2.215, p = 0.0348, unpaired t test). Normalized mutant EPSP is compared with the EPSP ratio of 1b/1b + 1s calculated from m4 in Figure 1I, indicated by gray dashed line. Note that m4-1b-derived EPSP increases 23% in DIP-α mutants but increases 45% in A8>GFP,hid,rpr (Fig. 6I). J, Normalized QC from m4 in control and mutant animals (t(20.14) = 0.086, p = 0.9326, unpaired t test with Welch's correction). Normalized QC was compared with 1b baseline QC (see Materials and Methods). Error bars indicate ±SEM. *p < 0.05, **p < 0.01, ****p < 0.0001, ns - not significant. n values (NMJs/larva) are 46/8 and 20/4 for B and C; 13/8 and 16/8 for D and E; and 13/10 and 16/10 for F–J.
Next, we examined 1b structural and functional plasticity in another context where m4 is naturally missing 1s innervation. In wild-type larvae, ∼20% of m4s lack 1s NMJs (Fig. 11C, control), which could underlie why m4-1b MNs are able to compensate for 1s MN perturbation. However, comparing wild-type m4s that naturally lack 1s innervation with those that have 1s innervation, we observed no change in 1b bouton numbers (Fig. 12A,B). Similarly, 1b-derived mEPSP, EPSP and QC were unaffected (Fig. 12C–F). This confirms that lack of synaptic plasticity in DIP-αnull larvae is not because of the genetic manipulation. Further, these data show that m4-1b MNs that naturally lack adjacent 1s MNs are unable to compensate, suggesting that robust m4-1b synaptic plasticity is not because of the variable 1s innervation observed on m4, but instead, induced when adjacent neurons are perturbed (for our model, see Discussion).
Figure 12.

m4s that naturally lack 1s innervation do not show 1b plasticity. A, Quantification of 1b boutons from m4 with and without 1s innervation (t(44) = 1.885, p = 0.0661, unpaired t test). B, Quantification of satellite boutons from m4 with and without 1s innervation (t(44) = 0.3895, p = 0.6988, unpaired t test). C, Quantification of mEPSP frequencies from m4 with and without 1s innervation (t(18) = 2.414, p = 0.0267, unpaired t test). D, Quantification of EPSP amplitudes from m4 with and without 1s innervation (t(18) = 4.576, p = 0.0002, unpaired t test). E, Normalized EPSPs from m4 without 1s innervation are compared with the EPSP ratio of 1b/1b + 1s calculated from m4 in Figure 1I, indicated by the gray dashed line (t(24) = 1.263, p = 0.2186, unpaired t test). F, Normalized QC from m4 without 1s innervation is compared with the 1b baseline QC (see Materials and Methods; t(24) = 0.1449, p = 0.8860, unpaired t test). Error bars indicate ±SEM. *p < 0.05, ***p < 0.001, ns - not significant. n values (NMJs/larva) are 23/17 and 23/17 for A and B, and 10/7 and 10/9 for C–F.
Discussion
The major gap that this article addresses is to what extent one synaptic input can influence the structural and functional plasticity of a converging input. Here, we examine the Drosophila neuromuscular circuit and demonstrate 1b synaptic plasticity induced by the loss of convergent 1s MNs. The muscles examined in this study, m6, m12, and m4, are coinnervated by unique 1b–1s MN pairs. First, we established an activity baseline in wild type and uncovered that 1b MNs contribute a unique percentage of the total EPSP in a muscle-specific manner. Genetic ablation of 1s MNs (vCE and dCE) after innervation led to the expansion of 1b NMJs and the elevation of their spontaneous release rates. Furthermore, some 1b MNs elevated evoked neurotransmission while others remained unchanged. A recent study examining m1 found similar 1b functional plasticity when m1-1s was ablated (Aponte-Santiago et al., 2020), but no structural compensation. This target specificity indicates heterogeneity in synaptic plasticity mechanisms. Silencing 1s MNs yielded partial m4-1b compensation, suggesting that 1s-evoked neurotransmission is an important factor in eliciting 1b NMJ plasticity. In mutant larvae where the m4-1s NMJs never form, we observed no changes in m4-1b bouton number or spontaneous release rate, and decreased EPSP compensation. These results suggest that initial 1s innervation may set up a reference point for robust 1b synaptic plasticity.
Potential mechanisms to regulate 1b synaptic plasticity
A well established form of plasticity at the larval NMJ is synaptic homeostasis, which can manifest in presynaptic and/or postsynaptic changes, including neurotransmitter release (Petersen et al., 1997; Davis and Goodman, 1998; Davis, 2013) and neurotransmitter receptor dynamics and abundance (Turrigiano et al., 1998; Wierenga et al., 2005; Frank et al., 2020), respectively. Presynaptic homeostatic plasticity (PHP) maintains EPSPs at baseline when postsynaptic glutamate receptor function is perturbed (Davis and Müller, 2015; Frank et al., 2020) and can be induced in a synapse-specific manner (Newman et al., 2017; Li et al., 2018). In our study, the loss of 1s MNs could be interpreted as a decrease in glutamate receptor function by the muscle and trigger similar PHP mechanisms to induce compensatory changes at adjacent 1b NMJs. Thus, further analysis of PHP pathways could reveal mechanisms underlying this convergent plasticity.
1b NMJs must adapt to accommodate the increased spontaneous and evoked neurotransmission. Our results do not reveal robust expansion of AZs or GluRs to support 1b synaptic plasticity, as observed in other studies where NMJ size is altered but total AZs or QCs remain the same (Goel et al., 2019a,b, 2020; Aponte-Santiago et al., 2020). Therefore, we propose that existing AZs may alter their properties. AZs exist in two different states, active or silent (Atwood and Wojtowicz, 1999; Ruiz et al., 2011), and a subset of AZs specializes in spontaneous release or evoked neurotransmission (Melom et al., 2013; Peled et al., 2014; Newman et al., 2017; Akbergenova et al., 2019). Thus, even without an increase in AZs, 1b plasticity mechanisms may modify AZ properties to respond to the loss of 1s inputs. Overall, our data suggest that silent AZs may become activated to increase the pools of spontaneous and evoked AZs as the ablation of 1s MNs led to enhanced 1b spontaneous release rates, target-specific compensation of EPSPs, and increased QC. Additionally, spontaneous and evoked activities may be independently regulated. Furthermore, the readily releasable pool size is under dynamic control during synaptic plasticity (Weyhersmüller et al., 2011; Müller et al., 2012) and could modulate m4-1b-evoked neurotransmitter release. Detailed examination of AZs will significantly bolster our understanding of the mechanism underlying 1b NMJ plasticity.
Prior studies reported that spontaneous neurotransmitter release regulates synaptic development in both mammals and Drosophila (Joseph et al., 2012; Choi et al., 2014; Andreae and Burrone, 2015, 2018; Cho et al., 2015; Kavalali, 2015). Thus, the expanded size of all 1b NMJs following 1s ablation may be caused by the elevated spontaneous activity. These data also suggest that all 1b MNs can detect and respond to the loss of adjacent 1s inputs. However, the ability to differentially compensate the spontaneous and evoked activity is likely because of independent mechanisms since only some 1b MNs elevate their EPSPs.
Correlation between synaptic weight and target-specific plasticity
In complex neural circuits, dissecting contributions of individual inputs to the total postsynaptic activity, also referred to as synaptic weight (Magee and Cook, 2000; Bhalla, 2008), remains difficult because of thousands of converging inputs on a single cell. The larval NMJ facilitates the partitioning of synaptic inputs as each muscle is innervated by few MNs. In this study, we combined electrophysiology with calcium imaging and found that 1b synaptic weights differ on m6, m12, and m4. Together with the degree of EPSP compensation after the ablation of 1s MNs, there was a direct correlation with the level of target-specific synaptic weight. Thus, robust 1b MNs that carry more synaptic drive may be endowed with certain synaptic plasticity mechanisms that respond to the loss of adjacent inputs. However, we cannot rule out regulatory roles for type II and type III MNs (Gorczyca et al., 1993; Chang and Keshishian, 1996; Koon et al., 2011) that are present on some muscles.
Interestingly, a similar correlation exists in Hebbian plasticity, where stronger synapses are more likely strengthened than weaker ones (Song et al., 2000; Babadi and Abbott, 2010). This correlation is also reflected in PHP. Two studies examined input-specific PHP on different muscles. On m4, PHP can be induced only at 1b NMJs (Newman et al., 2017); however, on m6, PHP can be induced on both 1b and 1s NMJs (Genc and Davis, 2019). This correlates with our observation that the m4-1b has more synaptic weight than m4-1s, whereas m6-1b and m6-1s have similar synaptic weights. Together, homeostatic plasticity varies in target-specific and input-specific manners, suggesting heterogeneous mechanisms.
Establishing an EPSP set point for 1b MN synaptic plasticity
Models of synaptic homeostasis rely on an activity set point to stabilize neurons when confronted with perturbations (LeMasson et al., 1993; Liu et al., 1998; Turrigiano, 2007; Davis, 2013; O'Leary et al., 2014). Each target neuron must account for all presynaptic inputs to produce a defined output (i.e., the set point). The structural and functional properties of each input thus determine not only its contribution to the postsynaptic activity but also its ability to respond to perturbations in synaptic function. For example, transcription factors not only regulate the temporal expression of ion channels that shape neuronal excitability, but also homeostatic mechanisms (Turrigiano, 2007; Davis, 2013; Parrish et al., 2014; Engelmann and Haenold, 2016; Diering et al., 2017; Schaukowitch et al., 2017). Like many activity-dependent processes (Ataman et al., 2008; Carrillo et al., 2010; Berke et al., 2013; Vonhoff and Keshishian, 2017a), the optimal set point may be established during a narrow time window of development. This hypothesis was tested in a Drosophila seizure mutant by inhibiting activity during embryonic development and observing the suppression of seizures in postembryonic stages (Giachello and Baines, 2015). Thus, manipulating activity during an embryonic critical period may alter the activity set point.
In this study, one intriguing hypothesis is that 1b + 1s coinnervation determines the EPSP set point during embryogenesis and is referenced by some 1b NMJs to compensate for the loss of 1s MNs. We propose a model to describe how 1b NMJs increase their sizes and spontaneous and evoked neurotransmission because of the loss of convergent 1s MNs. The neuromuscular innervation map is formed during late embryonic development (Prokop et al., 1996; Yoshihara et al., 1997; Vonhoff and Keshishian, 2017b). Since the maximum EPSP amplitude is established at the first instar stage and maintained throughout larval development (Davis and Goodman, 1998; Li et al., 2002), the set point is likely determined by the initial 1b + 1s coinnervation. Blocking the initial formation of 1s NMJs would create a set point that is devoid of 1s influence; and thus, the corresponding 1b NMJs would not compensate toward the wild-type EPSP but instead to this alternate EPSP set point. If the 1s MN is ablated after synaptogenesis, the correct set point is established and the 1b responds accordingly. Removing 1s innervation at various time points during larval development will provide a deeper understanding of the temporal dynamics of 1b plasticity and whether the EPSP set point is maintained by 1b + 1s activities.
Synaptic plasticity in other convergent neural circuits
The Drosophila neuromuscular circuit is a reductionist system with polyinnervated muscles. The loss of an input triggering a compensatory increase in synaptic growth or function of a convergent input could be viewed as synaptic competition. Synaptic competition is well characterized at the vertebrate neuromuscular junction, where neonatal muscles are innervated by several motor axons, but only one survives to adulthood (Sanes and Lichtman, 1999; Tapia et al., 2012). Neuromuscular activity is critical for this pruning process but the signaling pathway is not completely understood (Schuldiner and Yaron, 2015; Lee, 2020). Studying convergent synaptic plasticity at the Drosophila NMJ may shed light on this process.
Mechanisms uncovered at the NMJ can act in more complex circuits in vertebrates and invertebrates. For example, in the fly VNC, each MN receives sensory information from many interneurons to regulate motor behaviors (Heckscher et al., 2015; Schneider-Mizell et al., 2016; Kohsaka et al., 2019). The convergence of inputs on one MN may establish a homeostatic set point in the MN, similar to the muscle. The complex interneuron–MN connectivity more closely resembles that of vertebrate CNS neurons with polysynaptic dendrites (Kim et al., 2009; Heckscher et al., 2015; Kohsaka et al., 2019; Zarin et al., 2019). Additionally, age-related (Bergado and Almaguer, 2002; Mattson and Magnus, 2006; Mostany et al., 2013; Griffith et al., 2014; Petralia et al., 2014) and disease-related (Gorman, 2008; Milnerwood and Raymond, 2010; Lepeta et al., 2016; Salvadores et al., 2017; Smith-Dijak et al., 2019) changes in synaptic function and cell survival have been observed in CNS and neuromuscular circuits. In patients and animal models of Alzheimer's disease, while some neurons are depressed in amyloid-β plaque-enriched regions, other neurons show a compensatory hyperactivation (Busche et al., 2012; Merlo et al., 2019). Thus, future studies at the NMJ and other circuits will elucidate the mechanisms governing how and when the activity set point is defined in a target-specific manner and how neurons respond to dysfunctional neighbors.
Footnotes
Stocks obtained from the Bloomington Drosophila Stock Center [National Institutes of Health (NIH) Grant P40-OD-018537] were used in this study. The monoclonal antibodies 3C10, 4F3, and 8B4D2 were deposited to the Developmental Studies Hybridoma Bank (DSHB) by C. Goodman, while the nc82 antibody was deposited by E. Buchner and were obtained from the DSHB, created by the Eunice Kennedy Shriver National Institute of Child Health and Development of the NIH and maintained at The University of Iowa, Department of Biology (Iowa City, IA). This work was supported by NIH Grants K01-NS-102342 (to R.A.C.) and T32-GM-007183 (to M.L.-R.), funding from the University of Chicago Biological Sciences Division (BSD) International Student Fellowship (to Y.W.), and funding from the BSD Office of Diversity and Inclusion (to R.A.C.) and the Grossman Institute for Neuroscience, Quantitative Biology and Human Behavior (to R.A.C.). We thank Edwin “Chip” Ferguson, Ellie Heckscher, Kai Zinn, Kai Wang, Hong Liu, Richard Fehon, David Pincus, and members of the Carrillo laboratory for helpful discussions and comments. We also thank Vytas Bindokas for helpful guidance for Imaris analysis.
The authors declare no competing financial interests.
References
- Akbergenova Y, Cunningham KL, Zhang YV, Weiss S, Littleton JT (2018) Characterization of developmental and molecular factors underlying release heterogeneity at Drosophila synapses. eLife 7:e38268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreae LC, Burrone J (2015) Spontaneous neurotransmitter release shapes dendritic arbors via long-range activation of NMDA receptors. Cell Rep 10:873–882. 10.1016/j.celrep.2015.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreae LC, Burrone J (2018) The role of spontaneous neurotransmission in synapse and circuit development. J Neurosci Res 96:354–359. 10.1002/jnr.24154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte-Santiago NA, Ormerod KG, Akbergenova Y, Littleton JT (2020) Synaptic plasticity induced by differential manipulation of tonic and phasic motoneurons in Drosophila. J Neurosci 40:6270–6288. 10.1523/JNEUROSCI.0925-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley J, Sorrentino V, Lobb-Rabe M, Nagarkar-Jaiswal S, Tan L, Xu S, Xiao Q, Zinn K, Carrillo RA (2019) Transsynaptic interactions between IgSF proteins DIP-α and Dpr10 are required for motor neuron targeting specificity. Elife 8:e42690 10.7554/eLife.42690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataman B, Ashley J, Gorczyca M, Ramachandran P, Fouquet W, Sigrist SJ, Budnik V (2008) Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron 57:705–718. 10.1016/j.neuron.2008.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood HL, Wojtowicz JM (1999) Silent synapses in neural plasticity: current evidence. Learn Mem 6:542–571. 10.1101/lm.6.6.542 [DOI] [PubMed] [Google Scholar]
- Babadi B, Abbott LF (2010) Intrinsic stability of temporally shifted spike-timing dependent plasticity. PLOS Comput Biol 6:e1000961. 10.1371/journal.pcbi.1000961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergado JA, Almaguer W (2002) Aging and synaptic plasticity: a review. Neural Plast 9:217–232. 10.1155/NP.2002.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke B, Wittnam J, McNeill E, Van Vactor DL, Keshishian H (2013) Retrograde BMP signaling at the synapse: a permissive signal for synapse maturation and activity-dependent plasticity. J Neurosci 33:17937–17950. 10.1523/JNEUROSCI.6075-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry KP, Nedivi E (2017) Spine dynamics: are they all the same? Neuron 96:43–55. 10.1016/j.neuron.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla US (2008) How to record a million synaptic weights in a hippocampal slice. PLoS Comput Biol 4:e1000098. 10.1371/journal.pcbi.1000098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bownes M (1975) A photographic study of development in the living embryo of Drosophila melanogaster. J Embryol Exp Morphol 33:789–801. [PubMed] [Google Scholar]
- Broadie KS, Bate M (1993) Development of the embryonic neuromuscular synapse of Drosophila melanogaster. J Neurosci 13:144–166. 10.1523/JNEUROSCI.13-01-00144.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadus J, Skeath JB, Spana EP, Bossing T, Technau G, Doe CQ (1995) New neuroblast markers and the origin of the aCC/pCC neurons in the Drosophila central nervous system. Mech Dev 53:393–402. 10.1016/0925-4773(95)00454-8 [DOI] [PubMed] [Google Scholar]
- Budnik V, Zhong Y, Wu CF (1990) Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci 10:3754–3768. 10.1523/JNEUROSCI.10-11-03754.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A (2012) Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 109:8740–8745. 10.1073/pnas.1206171109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo RA, Olsen DP, Yoon KS, Keshishian H (2010) Presynaptic activity and CaMKII modulate retrograde semaphorin signaling and synaptic refinement. Neuron 68:32–44. 10.1016/j.neuron.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo RA, Özkan E, Menon KP, Nagarkar-Jaiswal S, Lee PT, Jeon M, Birnbaum ME, Bellen HJ, Garcia KC, Zinn K (2015) Control of synaptic connectivity by a network of Drosophila IgSF cell surface proteins. Cell 163:1770–1782. 10.1016/j.cell.2015.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TN, Keshishian H (1996) Laser ablation of Drosophila embryonic motoneurons causes ectopic innervation of target muscle fibers. J Neurosci 16:5715–5726. 10.1523/JNEUROSCI.16-18-05715.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakova M, Ilin V, Roshchin M, Bannon N, Malyshev A, Kisvárday Z, Volgushev M (2019) Distinct heterosynaptic plasticity in fast spiking and non-fast-spiking inhibitory neurons in rat visual cortex. J Neurosci 39:6865–6878. 10.1523/JNEUROSCI.3039-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RW, Buhl LK, Volfson D, Tran A, Li F, Akbergenova Y, Littleton JT (2015) Phosphorylation of complexin by PKA regulates activity-dependent spontaneous neurotransmitter release and structural synaptic plasticity. Neuron 88:749–761. 10.1016/j.neuron.2015.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BJ, Imlach WL, Jiao W, Wolfram V, Wu Y, Grbic M, Cela C, Baines RA, Nitabach MN, McCabe BD (2014) Miniature neurotransmission regulates Drosophila synaptic structural maturation. Neuron 82:618–634. 10.1016/j.neuron.2014.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JC, Park D, Griffith LC (2004) Electrophysiological and morphological characterization of identified motor neurons in the Drosophila third instar larva central nervous system. J Neurophysiol 91:2353–2365. 10.1152/jn.01115.2003 [DOI] [PubMed] [Google Scholar]
- Cluskey S, Ramsden DB (2001) Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. Mol Pathol 54:386–392. [PMC free article] [PubMed] [Google Scholar]
- Collins CA, DiAntonio A (2007) Synaptic development: insights from Drosophila. Curr Opin Neurobiol 17:35–42. 10.1016/j.conb.2007.01.001 [DOI] [PubMed] [Google Scholar]
- Davis GW (2013) Homeostatic signaling and the stabilization of neural function. Neuron 80:718–728. 10.1016/j.neuron.2013.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Goodman CS (1998) Genetic analysis of synaptic development and plasticity: homeostatic regulation of synaptic efficacy. Curr Opin Neurobiol 8:149–156. 10.1016/S0959-4388(98)80018-4 [DOI] [PubMed] [Google Scholar]
- Davis GW, Müller M (2015) Homeostatic control of presynaptic neurotransmitter release. Annu Rev Physiol 77:251–270. 10.1146/annurev-physiol-021014-071740 [DOI] [PubMed] [Google Scholar]
- Dickman DK, Lu Z, Meinertzhagen IA, Schwarz TL (2006) Altered synaptic development and active zone spacing in endocytosis mutants. Curr Biol 16:591–598. 10.1016/j.cub.2006.02.058 [DOI] [PubMed] [Google Scholar]
- Diering GH, Nirujogi RS, Roth RH, Worley PF, Pandey A, Huganir RL (2017) Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 355:511–515. 10.1126/science.aai8355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe CQ, Smouse D, Goodman CS (1988) Control of neuronal fate by the Drosophila segmentation gene even-skipped. Nature 333:376–378. 10.1038/333376a0 [DOI] [PubMed] [Google Scholar]
- Engelmann C, Haenold R (2016) Transcriptional control of synaptic plasticity by transcription factor NF-kappaB. Neural Plast 2016:7027949. 10.1155/2016/7027949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney CJ, Karunanithi S, Pearce J, Govind CK, Atwood HL (1998) Motor nerve terminals on abdominal muscles in larval flesh flies, Sarcophaga bullata: comparisons with Drosophila. J Comp Neurol 402:197–209. [DOI] [PubMed] [Google Scholar]
- Frank CA, James TD, Müller M (2020) Homeostatic control of Drosophila neuromuscular junction function. Synapse 74:e22133. 10.1002/syn.22133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genc O, Davis GW (2019) Target-wide induction and synapse type-specific robustness of presynaptic homeostasis. Curr Biol 29:3863–3873.e2. 10.1016/j.cub.2019.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachello CN, Baines RA (2015) Inappropriate neural activity during a sensitive period in embryogenesis results in persistent seizure-like behavior. Curr Biol 25:2964–2968. 10.1016/j.cub.2015.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel P, Dufour Bergeron D, Böhme MA, Nunnelly L, Lehmann M, Buser C, Walter AM, Sigrist SJ, Dickman D (2019a) Homeostatic scaling of active zone scaffolds maintains global synaptic strength. J Cell Biol 218:1706–1724. 10.1083/jcb.201807165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel P, Khan M, Howard S, Kim G, Kiragasi B, Kikuma K, Dickman D (2019b) A screen for synaptic growth mutants reveals mechanisms that stabilize synaptic strength. J Neurosci 39:4051–4065. 10.1523/JNEUROSCI.2601-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel P, Nishimura S, Chetlapalli K, Li X, Chen C, Dickman D (2020) Distinct target-specific mechanisms homeostatically stabilize transmission at pre- and post-synaptic compartments. Front Cell Neurosci 14:196. 10.3389/fncel.2020.00196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorczyca M, Augart C, Budnik V (1993) Insulin-like receptor and insulin-like peptide are localized at neuromuscular junctions in Drosophila. J Neurosci 13:3692–3704. 10.1523/JNEUROSCI.13-09-03692.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman AM (2008) Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J Cell Mol Med 12:2263–2280. 10.1111/j.1582-4934.2008.00402.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith WH, Dubois DW, Fincher A, Peebles KA, Bizon JL, Murchison D (2014) Characterization of age-related changes in synaptic transmission onto F344 rat basal forebrain cholinergic neurons using a reduced synaptic preparation. J Neurophysiol 111:273–286. 10.1152/jn.00129.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan B, Hartmann B, Kho YH, Gorczyca M, Budnik V (1996) The Drosophila tumor suppressor gene, dlg, is involved in structural plasticity at a glutamatergic synapse. Curr Biol 6:695–706. 10.1016/S0960-9822(09)00451-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallermann S, Heckmann M, Kittel RJ (2010) Mechanisms of short-term plasticity at neuromuscular active zones of Drosophila. HFSP J 4:72–84. 10.2976/1.3338710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern ME, Chiba A, Johansen J, Keshishian H (1991) Growth cone behavior underlying the development of stereotypic synaptic connections in Drosophila embryos. J Neurosci 11:3227–3238. 10.1523/JNEUROSCI.11-10-03227.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TH, Vicidomini R, Ramos CI, Wang Q, Nguyen P, Jarnik M, Lee C-H, Stawarski M, Hernandez RX, Macleod GT, Serpe M (2020) Neto-α controls synapse organization and homeostasis at the drosophila neuromuscular junction. Cell Rep 32:107866. 10.1016/j.celrep.2020.107866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenstein V, Rudloff E, Campos-Ortega JA (1987) The pattern of proliferation of the neuroblasts in the wild-type embryo of Drosophila melanogaster. Rouxs Arch Dev Biol 196:473–485. 10.1007/BF00399871 [DOI] [PubMed] [Google Scholar]
- Heckscher ES, Zarin AA, Faumont S, Clark MQ, Manning L, Fushiki A, Schneider-Mizell CM, Fetter RD, Truman JW, Zwart MF, Landgraf M, Cardona A, Lockery SR, Doe CQ (2015) Even-Skipped(+) interneurons are core components of a sensorimotor circuit that maintains left-right symmetric muscle contraction amplitude. Neuron 88:314–329. 10.1016/j.neuron.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick NG, Harward SC, Hall CE, Murakoshi H, McNamara JO, Yasuda R (2016) Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 538:104–108. 10.1038/nature19784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang B, Chiba A (2001) Single-cell analysis of Drosophila larval neuromuscular synapses. Dev Biol 229:55–70. 10.1006/dbio.2000.9983 [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN (1976) Properties of the larval neuromuscular junction in Drosophila melanogaster. J Physiol 262:189–214. 10.1113/jphysiol.1976.sp011592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki J, Keshishian H (1995) Role of neural activity during synaptogenesis in Drosophila. J Neurosci 15:8177–8190. 10.1523/JNEUROSCI.15-12-08177.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J, Dunn FA, Stopfer M (2012) Spontaneous olfactory receptor neuron activity determines follower cell response properties. J Neurosci 32:2900–2910. 10.1523/JNEUROSCI.4207-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungenitz T, Beining M, Radic T, Deller T, Cuntz H, Jedlicka P, Schwarzacher SW (2018) Structural homo- and heterosynaptic plasticity in mature and adult newborn rat hippocampal granule cells. Proc Natl Acad Sci U S A 115:E4670–E4679. 10.1073/pnas.1801889115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallmünzer B, Sörensen B, Neuhuber WL, Wörl J (2008) Enteric co-innervation of striated muscle fibres in human oesophagus. Neurogastroenterol Motil 20:597–610. 10.1111/j.1365-2982.2007.01075.x [DOI] [PubMed] [Google Scholar]
- Kavalali ET (2015) The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16:5–16. 10.1038/nrn3875 [DOI] [PubMed] [Google Scholar]
- Kim MD, Wen Y, Jan YN (2009) Patterning and organization of motor neuron dendrites in the Drosophila larva. Dev Biol 336:213–221. 10.1016/j.ydbio.2009.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, Hell SW, Buchner E, Heckmann M, Sigrist SJ (2006) Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 312:1051–1054. 10.1126/science.1126308 [DOI] [PubMed] [Google Scholar]
- Kohsaka H, Zwart MF, Fushiki A, Fetter RD, Truman JW, Cardona A, Nose A (2019) Regulation of forward and backward locomotion through intersegmental feedback circuits in Drosophila larvae. Nat Commun 10:2654. 10.1038/s41467-019-10695-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YH, Popova E, Thomas U, Griffith LC, Budnik V (1999) Regulation of DLG localization at synapses by CaMKII-dependent phosphorylation. Cell 98:353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koon AC, Ashley J, Barria R, DasGupta S, Brain R, Waddell S, Alkema MJ, Budnik V (2011) Autoregulatory and paracrine control of synaptic and behavioral plasticity by octopaminergic signaling. Nat Neurosci 14:190–199. 10.1038/nn.2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruijssen DLH, Wierenga CJ (2019) Single synapse LTP: a matter of context? Front Cell Neurosci 13:496. 10.3389/fncel.2019.00496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdyak P, Atwood HL, Stewart BA, Wu CF (1994) Differential physiology and morphology of motor axons to ventral longitudinal muscles in larval Drosophila. J Comp Neurol 350:463–472. [DOI] [PubMed] [Google Scholar]
- Landgraf M, Bossing T, Technau GM, Bate M (1997) The origin, location, and projections of the embryonic abdominal motorneurons of Drosophila. J Neurosci 17:9642–9655. 10.1523/JNEUROSCI.17-24-09642.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Wright AP, Zinn K (2009) Live dissection of Drosophila embryos: streamlined methods for screening mutant collections by antibody staining. J Vis Exp (34):1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Wu CF (2010) Orchestration of stepwise synaptic growth by K+ and Ca2+ channels in Drosophila. J Neurosci 30:15821–15833. 10.1523/JNEUROSCI.3448-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YI (2020) Developmental neuromuscular synapse elimination: activity-dependence and potential downstream effector mechanisms. Neurosci Lett 718:134724. 10.1016/j.neulet.2019.134724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMasson G, Marder E, Abbott LF (1993) Activity-dependent regulation of conductances in model neurons. Science 259:1915–1917. 10.1126/science.8456317 [DOI] [PubMed] [Google Scholar]
- Lepeta K, Lourenco MV, Schweitzer BC, Martino Adami PV, Banerjee P, Catuara-Solarz S, de La Fuente Revenga M, Guillem AM, Haidar M, Ijomone OM, Nadorp B, Qi L, Perera ND, Refsgaard LK, Reid KM, Sabbar M, Sahoo A, Schaefer N, Sheean RK, Suska A, et al. (2016) Synaptopathies: synaptic dysfunction in neurological disorders—a review from students to students. J Neurochem 138:785–805. 10.1111/jnc.13713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Peng X, Cooper RL (2002) Development of Drosophila larval neuromuscular junctions: maintaining synaptic strength. Neuroscience 115:505–513. 10.1016/S0306-4522(02)00380-9 [DOI] [PubMed] [Google Scholar]
- Li X, Goel P, Chen C, Angajala V, Chen X, Dickman DK (2018) Synapse-specific and compartmentalized expression of presynaptic homeostatic potentiation. Elife 7:e34338 10.7554/eLife.34338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Golowasch J, Marder E, Abbott LF (1998) A model neuron with activity-dependent conductances regulated by multiple calcium sensors. J Neurosci 18:2309–2320. 10.1523/JNEUROSCI.18-07-02309.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lnenicka GA, Keshishian H (2000) Identified motor terminals in Drosophila larvae show distinct differences in morphology and physiology. J Neurobiol 43:186–197. [DOI] [PubMed] [Google Scholar]
- Magee JC, Cook EP (2000) Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nat Neurosci 3:895–903. 10.1038/78800 [DOI] [PubMed] [Google Scholar]
- Martin AR (1955) A further study of the statistical composition on the end-plate potential. J Physiol 130:114–122. 10.1113/jphysiol.1955.sp005397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Magnus T (2006) Ageing and neuronal vulnerability. Nat Rev Neurosci 7:278–294. 10.1038/nrn1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megías M, Emri Z, Freund TF, Gulyás AI (2001) Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 102:527–540. 10.1016/S0306-4522(00)00496-6 [DOI] [PubMed] [Google Scholar]
- Melom JE, Akbergenova Y, Gavornik JP, Littleton JT (2013) Spontaneous and evoked release are independently regulated at individual active zones. J Neurosci 33:17253–17263. 10.1523/JNEUROSCI.3334-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng JL, Wang Y, Carrillo RA, Heckscher ES (2020) Temporal transcription factors determine circuit membership by permanently altering motor neuron-to-muscle synaptic partnerships. Elife 9:e56898 10.7554/eLife.56898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon KP, Carrillo RA, Zinn K (2013) Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip Rev Dev Biol 2:647–670. 10.1002/wdev.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo S, Spampinato SF, Sortino MA (2019) Early compensatory responses against neuronal injury: a new therapeutic window of opportunity for Alzheimer's Disease? CNS Neurosci Ther 25:5–13. 10.1111/cns.13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA (2010) Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci 33:513–523. 10.1016/j.tins.2010.08.002 [DOI] [PubMed] [Google Scholar]
- Mostany R, Anstey JE, Crump KL, Maco B, Knott G, Portera-Cailliau C (2013) Altered synaptic dynamics during normal brain aging. J Neurosci 33:4094–4104. 10.1523/JNEUROSCI.4825-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Liu KS, Sigrist SJ, Davis GW (2012) RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J Neurosci 32:16574–16585. 10.1523/JNEUROSCI.0981-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhuber WL, Wörl J (2016) Enteric co-innervation of striated muscle in the esophagus: still enigmatic? Histochem Cell Biol 146:721–735. 10.1007/s00418-016-1500-1 [DOI] [PubMed] [Google Scholar]
- Newman ZL, Hoagland A, Aghi K, Worden K, Levy SL, Son JH, Lee LP, Isacoff EY (2017) Input-specific plasticity and homeostasis at the Drosophila larval neuromuscular junction. Neuron 93:1388–1404.e10. 10.1016/j.neuron.2017.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen CT, Stewart BA (2016) The influence of postsynaptic structure on missing quanta at the Drosophila neuromuscular junction. BMC Neurosci 17:53. 10.1186/s12868-016-0290-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose A (2012) Generation of neuromuscular specificity in Drosophila: novel mechanisms revealed by new technologies. Front Mol Neurosci 5:62. 10.3389/fnmol.2012.00062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor-Giles KM, Ganetzky B (2008) Satellite signaling at synapses. Fly (Austin) 2:259–261. 10.4161/fly.7133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor-Giles KM, Ho LL, Ganetzky B (2008) Nervous wreck interacts with thickveins and the endocytic machinery to attenuate retrograde BMP signaling during synaptic growth. Neuron 58:507–518. 10.1016/j.neuron.2008.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary T, Williams AH, Franci A, Marder E (2014) Cell types, network homeostasis, and pathological compensation from a biologically plausible ion channel expression model. Neuron 82:809–821. 10.1016/j.neuron.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh WC, Parajuli LK, Zito K (2015) Heterosynaptic structural plasticity on local dendritic segments of hippocampal CA1 neurons. Cell Rep 10:162–169. 10.1016/j.celrep.2014.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish JZ, Kim CC, Tang L, Bergquist S, Wang T, Derisi JL, Jan LY, Jan YN, Davis GW (2014) Krüppel mediates the selective rebalancing of ion channel expression. Neuron 82:537–544. 10.1016/j.neuron.2014.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls D, von Essen A, Lyutova R, van Giesen L, Rosner R, Wegener C, Sprecher SG (2015) Potency of transgenic effectors for neurogenetic manipulation in Drosophila larvae. Genetics 199:25–37. 10.1534/genetics.114.172023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled ES, Newman ZL, Isacoff EY (2014) Evoked and spontaneous transmission favored by distinct sets of synapses. Curr Biol 24:484–493. 10.1016/j.cub.2014.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry S, Goel P, Tran NL, Pinales C, Buser C, Miller DL, Ganetzky B, Dickman D (2020) Developmental arrest of Drosophila larvae elicits presynaptic depression and enables prolonged studies of neurodegeneration. Development 147:dev186312 10.1242/dev.186312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A (1997) Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19:1237–1248. 10.1016/S0896-6273(00)80415-8 [DOI] [PubMed] [Google Scholar]
- Petralia RS, Mattson MP, Yao PJ (2014) Communication breakdown: the impact of ageing on synapse structure. Ageing Res Rev 14:31–42. 10.1016/j.arr.2014.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokop A, Landgraf M, Rushton E, Broadie K, Bate M (1996) Presynaptic development at the Drosophila neuromuscular junction: assembly and localization of presynaptic active zones. Neuron 17:617–626. 10.1016/S0896-6273(00)80195-6 [DOI] [PubMed] [Google Scholar]
- Ruiz R, Cano R, Casañas JJ, Gaffield MA, Betz WJ, Tabares L (2011) Active zones and the readily releasable pool of synaptic vesicles at the neuromuscular junction of the mouse. J Neurosci 31:2000–2008. 10.1523/JNEUROSCI.4663-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadores N, Sanhueza M, Manque P, Court FA (2017) Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front Neurosci 11:451. 10.3389/fnins.2017.00451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22:389–442. 10.1146/annurev.neuro.22.1.389 [DOI] [PubMed] [Google Scholar]
- Schaefer JE, Worrell JW, Levine RB (2010) Role of intrinsic properties in Drosophila motoneuron recruitment during fictive crawling. J Neurophysiol 104:1257–1266. 10.1152/jn.00298.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaukowitch K, Reese AL, Kim SK, Kilaru G, Joo JY, Kavalali ET, Kim TK (2017) An intrinsic transcriptional program underlying synaptic scaling during activity suppression. Cell Rep 18:1512–1526. 10.1016/j.celrep.2017.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Mizell CM, Gerhard S, Longair M, Kazimiers T, Li F, Zwart MF, Champion A, Midgley FM, Fetter RD, Saalfeld S, Cardona A (2016) Quantitative neuroanatomy for connectomics in Drosophila. Elife 5:e12059 10.7554/eLife.12059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner O, Yaron A (2015) Mechanisms of developmental neurite pruning. Cell Mol Life Sci 72:101–119. 10.1007/s00018-014-1729-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist SJ, Reiff DF, Thiel PR, Steinert JR, Schuster CM (2003) Experience-dependent strengthening of Drosophila neuromuscular junctions. J Neurosci 23:6546–6556. 10.1523/JNEUROSCI.23-16-06546.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Dijak AI, Sepers MD, Raymond LA (2019) Alterations in synaptic function and plasticity in Huntington disease. J Neurochem 150:346–365. 10.1111/jnc.14723 [DOI] [PubMed] [Google Scholar]
- Song S, Miller KD, Abbott LF (2000) Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat Neurosci 3:919–926. 10.1038/78829 [DOI] [PubMed] [Google Scholar]
- Sweeney ST, Broadie K, Keane J, Niemann H, O'Kane CJ (1995) Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron 14:341–351. 10.1016/0896-6273(95)90290-2 [DOI] [PubMed] [Google Scholar]
- Tapia JC, Wylie JD, Kasthuri N, Hayworth KJ, Schalek R, Berger DR, Guatimosim C, Seung HS, Lichtman JW (2012) Pervasive synaptic branch removal in the mammalian neuromuscular system at birth. Neuron 74:816–829. 10.1016/j.neuron.2012.04.017 [DOI] [PubMed] [Google Scholar]
- Torroja L, Packard M, Gorczyca M, White K, Budnik V (1999) The Drosophila β-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci 19:7793–7803. 10.1523/JNEUROSCI.19-18-07793.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G (2007) Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol 17:318–324. 10.1016/j.conb.2007.04.004 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391:892–896. 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- Van Vactor D, Sigrist SJ (2017) Presynaptic morphogenesis, active zone organization and structural plasticity in Drosophila. Curr Opin Neurobiol 43:119–129. 10.1016/j.conb.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubramanian L, Guo Z, Xu S, Tan L, Xiao Q, Nagarkar-Jaiswal S, Mann RS (2019) Stereotyped terminal axon branching of leg motor neurons mediated by IgSF proteins DIP-α and Dpr10. Elife 8:e42692 10.7554/eLife.42692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonhoff F, Keshishian H (2017a) Activity-dependent synaptic refinement: new insights from Drosophila. Front Syst Neurosci 11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonhoff F, Keshishian H (2017b) In vivo calcium signaling during synaptic refinement at the Drosophila neuromuscular junction. J Neurosci 37:5511–5526. 10.1523/JNEUROSCI.2922-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyhersmüller A, Hallermann S, Wagner N, Eilers J (2011) Rapid active zone remodeling during synaptic plasticity. J Neurosci 31:6041–6052. 10.1523/JNEUROSCI.6698-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG (2005) Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci 25:2895–2905. 10.1523/JNEUROSCI.5217-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Xiao Q, Cosmanescu F, Sergeeva AP, Yoo J, Lin Y, Katsamba PS, Ahlsen G, Kaufman J, Linaval NT, Lee PT, Bellen HJ, Shapiro L, Honig B, Tan L, Zipursky SL (2018) Interactions between the Ig-Superfamily Proteins DIP-alpha and Dpr6/10 Regulate Assembly of Neural Circuits. Neuron 100:1369–1384 e1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Rheuben MB, Kidokoro Y (1997) Transition from growth cone to functional motor nerve terminal in Drosophila embryos. J Neurosci 17:8408–8426. 10.1523/JNEUROSCI.17-21-08408.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarin AA, Mark B, Cardona A, Litwin-Kumar A, Doe CQ (2019) A multilayer circuit architecture for the generation of distinct locomotor behaviors in Drosophila. Elife 8:e51781 10.7554/eLife.51781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Schnitzler A, Agapite J, Schwartz LM, Steller H, Nambu JR (1997) Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proc Natl Acad Sci U S A 94:5131–5136. 10.1073/pnas.94.10.5131 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SynapGCaMP allows 1b and 1b+1s-derived EPSPs from larval muscles to be differentiated. Nerves were stimulated at 0.5 Hz using different voltages for the two stimulus conditions. Frame rate is 30.