

Abstract
Transient signaling orchestrates complex spatiotemporal behaviour in living organisms via (bio)chemical reaction networks (CRNs). Compartmentalization of signal processing is an important aspect for controlling such networks. However, artificial CRNs mostly focus on homogeneous solutions to program autonomous self‐assembling systems, which limits their accessible behaviour and tuneability. Here, we introduce layered compartments housing antagonistic pH‐modulating enzymes and demonstrate that transient pH signals in a supernatant solution can be programmed based on spatial delays. This overcomes limitations of activity mismatches of antagonistic enzymes in solution and allows to flexibly program acidic and alkaline pH lifecycles beyond the possibilities of homogeneous solutions. Lag time, lifetime, and the pH minima and maxima can be precisely programmed by adjusting spatial and kinetic conditions. We integrate these spatially controlled pH flips with switchable peptides, furnishing time‐programmed self‐assemblies and hydrogel material system.
Keywords: chemical reaction networks, life-like materials, pH-feedback systems, self-regulation, systems chemistry
Chemical reaction networks based on antagonistic enzymes can show new behaviour when the enzymes are immobilized into layered compartments to impose spatial diffusive constraints. This allows to overcome mismatches of the enzymatic activities and reduces crosstalk in their pH‐dependent activity when targeting pH‐feedback mechanisms to program self‐assembly lifecycles.
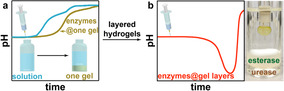
Introduction
Transient signal generation and processing in (bio)chemical reaction networks (CRNs) guide the behavior of living organisms from molecular level to macroscopic quorum sensing and collective movement. [1] Understanding transient signals in biology has been inspiring to develop analogously autonomous, adaptive, and interactive life‐like material systems. [2] Energy‐rich signal molecules or auxiliary fuel molecules are needed to process signals, and compartmentalization of network nodes [3] in, for example, biological condensates [4] is an important aspect to gain better control of such CRNs in living systems.
Inspired by biological CRNs and using energy‐rich signal and fuel molecules, artificial transient signal systems and transient structures sustained by light, [5] enzymatic reactions, [6] oscillatory reactions, [7] and small fuel molecules, [8] have emerged. For chemically fueled systems, based on how fuel molecules are used, such transient systems can be distinguished into active/dissipative structures or structures coupled to an active/dissipative environment. [9] In our previous work, we developed transient pH lifecycles by combining fast signal producers and slower signal deactivators using also feedback‐regulated processes in enzymatic reactions,[ 6g , 6h , 9d , 9f ] and demonstrated how to couple them to diverse self‐assembling systems and materials applications.
Despite tremendous achievements in artificial transient systems and their preliminary applications, most of them essentially work with homogeneous systems, only a few reports paid attention to confining CRNs into colloidal compartments for additional control mechanisms.[ 3b , 10 ] Herein, we move a distinct step beyond homogenous solutions and introduce how the spatial compartmentalization of two antagonistic enzymes allows to shape transient pH signals in autonomous systems in a very straight forward and highly tunable manner. To this end, we spatially separate two antagonistic pH‐modulating enzymes of a biocatalytic pH feedback system into hydrogel layers at the bottom of reaction vessels. Previously, using the same enzymes in homogeneous solutions, [6f] we were able to program transient alkaline pH profiles, but failed to produce acidic ones due to mismatch of the pH‐dependent enzymatic activities. Here, we overcome this mismatch by layered compartmentalization using diffusion delays to control the effect of enzymatic conversions in the spatially separated, supernatant solution. This enables fundamentally new control features for both transient acidic and alkaline pH lifecycles. Thereafter we couple the spatially controlled transient pH flips to pH‐sensitive peptides, producing transient fibrillization and gels with defined lifetimes—notably without the presence of the fuel conversion systems in the supernatant solution.
Results and Discussion
Scheme 1 a illustrates the concept toward autonomous transient acidic pH flips by spatial organization of two counteracting enzymes. The whole system has three layers. From bottom to top: A urease and an esterase gel layer based on photo‐crosslinked poly(poly(ethylene glycol) diacrylate) (PPEGDA), and a supernatant layer to which the fuels (urea and ethyl acetate (EA)) and buffer, as well as later peptides are added. The molecular weight of PEGDA was optimized to prevent leakage of the enzymes into the supernatant (Scheme S1 in Supporting Information). After injection of the chemical fuels, EA and urea diffuse from the supernatant into the hydrogel layers, where they are converted into acidic (acetic acid, HAc) and basic (NH3) products by their respective enzymes. Those diffuse into the supernatant and control the pH profiles. A transient acidic pH flip is achieved by taking advantage of having a top layer of esterase, while the urease layer is underneath. For both fuel conversions, the respective effector (pHup vs. pHdown) is a function of the enzyme concentrations and the diffusivities, v x, of fuels and products (v EA, v , v urea, and v in Scheme 1 b). Critically, the pHup effector experiences a spatially imposed delay, governed majorly by the thickness of the esterase top layer (h esterase). At moderate enzyme concentration and high substrate concentration, the height of the individual compartments also plays a role for the individual reaction as not all substrate is directly converted at the very top of the enzymatic layer. Experimentally, these conditions can be screened in a very facile manner, and we surmised that controlling the spatial (layer thickness) and activity (enzyme and fuel concentration) aspects would allow to program the depth of the pH‐cycle (pHmin), lag time (t lag, defined as the time that the pH profile needs to decrease to half of initial pH (pHi) and pHmin), and lifetime (t lf) of the pH‐cycle (Scheme 1 c,d).
Scheme 1.
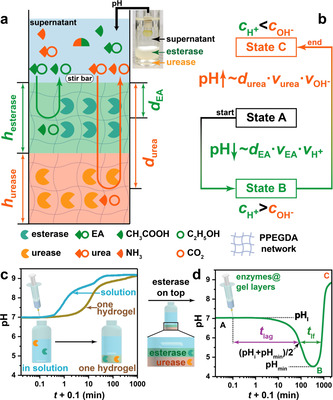
Transient pH flips shaped by layered compartmentalization of antagonistic enzymatic reactions. a) Schematic illustration of the transient acidic pH flip: esterase encapsulated in the top gel layer catalyzes the hydrolysis of EA to HAc, while urease embedded in the bottom layer converts urea to NH3 and CO2. b) The transient excess of H+ over OH− in the supernatant drives initial state A (neutral pH) to a temporal state B (acidic pH) because of the shorter diffusion distance of EA (d EA) and H+ than that of urea (d urea) and NH4 +/OH−. The overall generated OH− (c ) can be adjusted higher than H+ (c ) to reach an alkaline state C at the end. c,d) pH profiles of a homogeneous solution or a supernatant with enzymes embedded homogeneously in a single gel layer climb to alkaline regime, whereas layered compartmentalization leads to a transient acidic state. Conditions: All three vials with a constant total height of ≈24.5 mm contain 0.7 mg mL−1 esterase, 0.3 mg mL−1 urease, 5 mM PBS (pH 7.0), 0.5 mg mL−1 bovine serum albumin (BSA). The homogeneous PPEGDA gel layer (c) has a height of ≈7.5 mm, while the heights of esterase layer and urease layer (d) are 6 and ≈1.5 mm, respectively. To initiate the systems, 100 mM urea and 100 mM EA were added at 20 °C, detailed experimental conditions are listed in Table S1. The definitions of t lf and t lag of the transient pH flip are shown in (d). Diffusion speeds are abbreviated as v x and diffusion distances as d x.
Scheme 1 c shows the pH development for (i) a homogenous solution and (ii) a single hydrogel layer containing both enzymes. When reacting a homogeneous solution of esterase (0.7 mg mL−1) and urease (0.3 mg mL−1) buffered at pH 7.0 (5 mM phosphate buffer solution (PBS)) with their fuels (100 mM urea and 100 mM EA), a direct increase to high pH occurs due to the higher activity of urease. When immobilizing both enzymes into a single gel layer with a thickness of ca. 7.5 mm, a delay occurs due to diffusion of the substrates needed to reach the enzymes in the gel layer. Strikingly, when placing the esterase layer (6 mm) on top of a urease layer (≈1.5 mm), the pH profile stays around 7 for a period of 10 min, then dives down to the acidic regime with pH 4.5, and finally climbs back to alkaline regime (Scheme 1 d). This key demonstration highlights how simple spatial compartmentalization of two antagonistic enzymatic reactions can entirely change the system behaviour.
We then discuss details on programming such pH lifecycles. We first change the enzyme ratio at constant layer thickness (h esterase≈6 mm; h urease≈1.5 mm; h supernatant≈17 mm) at a starting pH of 7.0 (5 mM PBS, the inherent hydrolysis of EA is negligible without esterase in this buffer (Figure S1); Figure 1 a/a′). After injecting 100 mM urea and 100 mM EA, several trends are observed: Higher esterase concentrations cause that (i) the pHmin can be decreased from 5.7 to 4.4, (ii) the t lag is shortened from ≈140 to ≈90 min, and (iii) the t lf is extended from 450 to 860 min. Increasing the buffer capacity from 1 to 5 mM increases the t lag from ≈30 to ≈90 min (Figure 1 b/b′). This is simply due to the fact that the enzymatic reaction products (particularly HAc from the upper gel layer) first need to scavenge the buffer capacity. Nevertheless, the pHmin and the sum of t lag and t lf are almost constant. By increasing the amount of EA fuel (c EA) and decreasing the amount of its counter fuel (c urea), extended lifetimes are found from 5.6 h for c EA=20 mM to several days >48 h for c EA=180 mM (Figure 1 c/c′). Meanwhile the pHmin decreases from 5.0 to 4.1, but the t lag stays around 84±10 min. In summary, at constant gel layer thicknesses, the full lifecycle regarding t lag, t lf and pHmin can be widely tuned by regulating the activity of the antagonistic enzymatic reactions.
Figure 1.
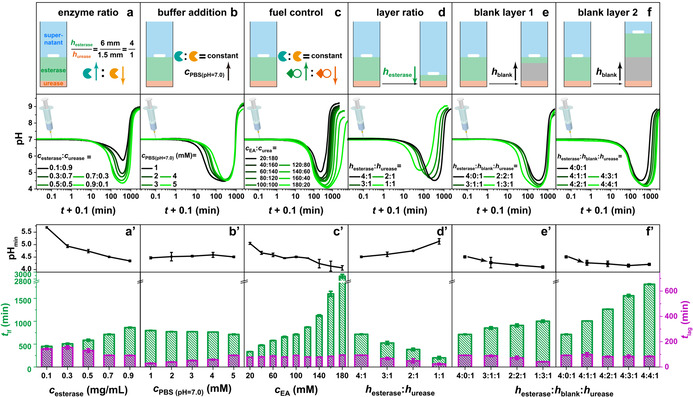
Programming the t lf, t lag and pHmin of transient acidic pH flips: a) Increasing the c esterase leads to a pronounced pHmin, longer t lf and shorter t lag. Conditions: h esterase:h urease=4:1, c EA=c urea=100 mM, c esterase + c urease=1.0 mg mL−1, and 5 mM PBS (pH 7.0; 0.5 mg mL−1 BSA; 20 °C). b) The t lag is controlled by the buffer concentration. Conditions: h esterase:h urease=4:1, 0.7 mg mL−1 esterase, 0.3 mg mL−1 urease, c EA=c urea=100 mM, 0.5 mg mL−1 BSA, and 20 °C. c) Adjusting the ratio of EA and urea results in a wide tuneability of t lf and pHmin. Conditions: h esterase:h urease=4:1, 0.7 mg mL−1 esterase, 0.3 mg mL−1 urease, c EA + c urea=200 mM, and 5 mM PBS (pH 7.0; 0.5 mg mL−1 BSA; 20 °C). d) The height ratio of esterase and urease layers regulates the pHmin, t lf and t lag. Conditions: 0.7 mg mL−1 esterase, 0.3 mg mL−1 urease, c EA=c urea=100 mM, and 5 mM PBS (pH 7.0; 0.5 mg mL−1 BSA; 20 °C). e) Effects of installing a blank hydrogel layer between esterase and urease layers on the expense of the esterase layer thickness. Conditions: 0.7 mg mL−1 esterase, 0.3 mg mL−1 urease, c EA=c urea=100 mM, and 5 mM PBS (pH 7.0; 0.5 mg mL−1 BSA; 20 °C). f) Effects of installing a blank layer while maintaining esterase and urease layer thickness. Conditions: 0.7 mg mL−1 esterase, 0.3 mg mL−1 urease, c EA=c urea=100 mM, and 5 mM PBS (pH 7.0; 0.5 mg mL−1 BSA; 20 °C). (a′–f′) The corresponding pHmin, t lag and t lf of the transient acidic pH flips in (a–f). All data are an average of three measurements, detailed experimental conditions are listed in Table S2.
We then focus on how the spatial domain influences the pH flips by regulating d EA and d urea. We first decrease the h esterase but keep other conditions constant (c esterase=0.7 mg mL−1, c urease=0.3 mg mL−1, c EA=c urea=100 mM, 5 mM PBS, Figure 1 d/d′). Upon decreasing the h esterase layer from previously used ≈6 mm to ≈1.5 mm, the t lf is shortened from ca. 710 to 200 min, and the pHmin is lifted from 4.5 to 5.1. This is qualitatively similar to decreasing the c esterase in Figure 1 a, but the origin is different. Here the key is that the d urea is shortened by 4.5 mm, resulting in a quicker return to alkaline regime for lower h esterase.
More interestingly, we added a non‐reactive PPEGDA layer between the two active layers to more strongly decouple the active layers to (i) reduce neutralization of H+ and OH− at the interface of esterase and urease layers, and (ii) limit the influence of pH changes of the top active layer on the activity of the bottom layer due to the intrinsic pH‐dependent activity profiles of the enzymes. [6f] Indeed, once a blank layer is inserted on expense of the esterase layer (Figure 1 e/e′), the pHmin falls slightly from 4.3 to 4.1, the t lag decreases from ≈90 to ≈40 min, and the t lf increases from 850 to 1000 min, due to a spatially delayed effect of the underlying urease layer. When embedding similar variable blank layers (from ≈1.5 to ≈6 mm) while keeping an esterase layer of ≈6 mm, the t lf can be further pushed from ca. 1000 to 1800 min due to an additional diffusion distance of d urea (Figure 1 f/f′). However, the pHmin and t lag remain constant around 4.2 and ≈90 min, respectively. This is clearly advantageous as compared to scenarios in Figure 1 a/a′ and c/c′, where the pHmin cannot be decoupled from t lf and t lag.
In short, the effective approaches to shortening the t lag are kinetically accelerating the hydrolysis of EA by adding more esterase (Figure 1 a), decreasing the concentration of buffer (Figure 1 b), and spatially shortening the d EA (Figure 1 d and e). The t lf and pHmin can also be programmed by regulating the kinetic domain via adding more esterase (Figure 1 a) and EA (Figure 1 c), and the spatial domain through shortening d EA (Figure 1 d and e) and prolonging d urea (Figure 1 f). Blank layers allow a more efficient decoupling and better orthogonal programmability.
After establishing the autonomous, transient acidic pH flip, we transferred the concept to achieve opposite alkaline pH flips by inverting the enzymatic layers. Although we have demonstrated the concept of a transient alkaline pH flip by regulating kinetic parameters in homogeneous solutions, [6f] we here chose a recipe (c esterase=0.7 mg mL−1, c urease=0.3 mg mL−1, c EA=195 mM, c urea=5 mM, 5 mM PBS (pH 7.0)) where the pH profile of a supernatant above a homogeneous enzyme gel layer (thickness ≈7.5 mm) directly descends to acidic regime (Figure 2 a). However, when placing the urease layer (≈1.5 mm) on top of an esterase layer (≈6 mm), the pH profile climbs first to alkaline regime with a pHmax of 8.2, and then dives down to acidic regime, generating a transient alkaline pH flip (Figure 2 b).
Figure 2.
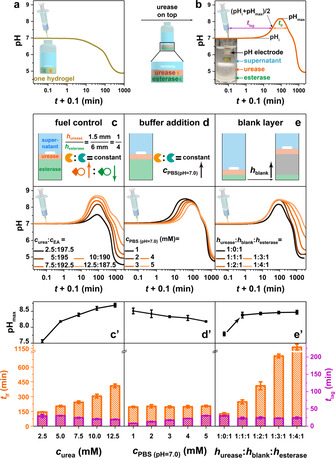
Transient alkaline pH flips and t lag, t lf, pHmax programming. a,b) The pH profile of a supernatant with enzymes homogeneously embedded in a single gel layer descends to acidic regime, whereas layered compartmentalization leads to a transient alkaline state. Conditions: Both vials with a constant total height of ≈24.5 mm contain 0.3 mg mL−1 urease, 0.7 mg mL−1 esterase, 5 mM PBS (pH 7.0), 0.5 mg mL−1 BSA, the homogeneous PPEGDA gel layer (a) has a height of ≈7.5 mm, while the heights of urease layer and esterase layer (b) are ≈1.5 and 6 mm, respectively. To initiate the systems, 5 mM urea and 195 mM EA were added at 20 °C. c) Programming the t lf and t lag by regulating the ratio of urea and EA. Conditions: h urease:h esterase=1:4, 0.3 mg mL−1 urease, 0.7 mg mL−1 esterase, and 5 mM PBS (pH 7.0; 0.5 mg mL−1 BSA; 20 °C). d) Higher buffer capacity generates a longer t lag. Conditions: h urease:h esterase=1:4, 0.3 mg mL−1 urease, 0.7 mg mL−1 esterase, 5 mM urea, 195 mM EA, and 0.5 mg mL−1 BSA at 20 °C. e) Inserting a blank layer while leaving the heights of the urease and esterase layer unaltered leads to a higher pHmax and longer t lf. Conditions: h urease:h esterase=1:1, 0.3 mg mL−1 urease, 0.7 mg mL−1 esterase, 5 mM urea, 195 mM EA, and 5 mM PBS (pH 7.0; 0.5 mg mL−1 BSA; 20 °C). (c′–e′) The corresponding pHmax, t lag and t lf of the transient alkaline pH flips in (c–e). All data are an average of three measurements, detailed experimental conditions are listed in Table S3. The definitions of t lf and t lag are shown in (b).
Following related considerations from above, we realized similar programmability of t lag, t lf and pHmax. We adjusted the ratio of chemical fuels (c urea≤12.5 mM) to assure that the pH eventually returns to acidic regime. Figures 2 c/c′ display the transient alkaline pH flips fueled by a series of c urea at otherwise constant parameters. As the c urea increases from 2.5 to 12.5 mM, the pHmax jumps from 7.6 up to 8.7, the t lag is shortened from ≈30 to ≈20 min, while the t lf is prolonged from 145 to 410 min. The variations of pHmax, t lag and t lf are attributed to more rapidly produced NH3. We need to emphasize that the end pH value at 3000 min increases from 4.5 to 6.6 for higher c urea. Therefore, it is necessary to balance the ratio of chemical fuels for obtaining a high pHmax but a relatively low end pH value, which is important for coupling such pH lifecycles to achieve transient self‐assemblies. Similar to the transient acidic pH flip, the buffer capacity programs t lag, which is extended from 8 to 30 min upon increasing PBS from 1 to 5 mM. This goes along with slightly lower pHmax (8.5 to 8.2) as more NH3 is needed to deplete the buffer capacity, while t lf remains almost constant (Figure 2 d/d′). Subsequently, we applied the concept of blank layers (Figure 2 e/e′). Qualitatively similar to above, it becomes clear that the pHmax can be reached rather independently of the enzyme amounts present in both layers, and that the t lf can be increased substantially by imposing additional diffusion pathways for EA and its products.
To showcase the application of such transient pH flips, we coupled the transient acidic pH flip to the self‐assembly of a dipeptide Fmoc‐Leu‐Gly‐OH (Figure 3 a), which is known to form well‐defined, twisted ribbon‐like nanofibrils (transmission electron microscopy (TEM) image in Figure 3 a) in acidic regime (pH<5.8) and gels at high concentration. [6h] As the pH dives down below 5.8, the supernatant starts to become opaque (Figure 3 b), indicating fibrillar self‐assembly. The opaque suspensions return to transparent when the pH profile returns to alkaline regime, confirming successful autonomous disassembly. The lifetime of the transient self‐assembly can be programmed from less than 24 h to more than 1 week by increasing the c EA from 60 to 120 mM (Figure 3 b, c). However, gelation only occurs with c EA higher than 140 mM (vial inversion test in Figure S2) and at high acidity.
Figure 3.
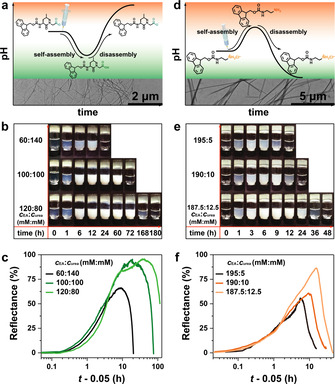
Temporal programming of the lifetime of transient fibrillization of peptides. a) Coupling the self‐assembly of Fmoc‐Leu‐Gly‐OH with transient acidic pH flips (Inserted TEM image shows self‐assembled peptide nanofibrils). b) Snapshots series of transient Fmoc‐Leu‐Gly‐OH solutions with different fuel ratios of EA and urea. Conditions: 0.7 mg mL−1 esterase, 0.3 mg mL−1 urease, h esterase:h urease=4:1 (total height of 7.5 mm), 1 mM PBS (pH 7.0; 0.5 mg mL−1 BSA), and 0.6 wt % dipeptide at 20 °C. c) Optical determination of the lifetime of transient Fmoc‐Leu‐Gly‐OH assembly by monitoring the reflectance (500 nm) of the supernatant in side view. d) Coupling the self‐assembly of Fmoc‐ethylenediamine with transient alkaline pH flips (Inserted TEM image shows peptide assemblies in alkaline condition). e) Snapshots of transient lifecycles of Fmoc‐ethylenediamine fueled by different fuel ratios of EA and urea. Conditions: 0.3 mg mL−1 urease, 0.7 mg mL−1 esterase, h urease:h esterase=1:4 (total height of 7.5 mm), 1 mM PBS (pH 7.0, 0.5 mg mL−1 BSA), and 0.3 wt % Fmoc‐ethylenediamine hydrochloride at 20 °C. f) Optical analysis of the lifecycles. Detailed experimental conditions are listed in Table S4,5.
We also coupled the opposite transient alkaline pH flips to the self‐assembly of Fmoc‐ethylenediamine hydrochloride. [11] The peptide is initially deprotonated, then assembles into fibrils at the transient pH jump (TEM in Figure 3 d), and the fibrils gradually disintegrate as the pH goes to the acidic state. The lifetime of the self‐assembled state can be programmed from less than 12 h to more than 36 h by adding more urea (Figure 3 e, f).
Conclusion
In conclusion, we introduced a new concept to achieve spatial control over CRNs powered by fuel conversion of antagonistic enzymes by layered compartmentalization of the two enzymes. In its application to pH feedback systems, this now allows to pre‐program transient acidic pH flips, which were otherwise impossible to reach in homogeneous solutions due to a kinetic mismatch of the enzymatic activities and their pH‐dependent activity. The lag time, lifetime, depth of the transient acidic pH dive, and the height of the transient alkaline pH jump can be precisely controlled by programming the spatial conditions including height of urease, esterase and blank hydrogel layers, as well as activity parameters such as enzyme ratio, buffer concentration, and ratio of chemical fuels. The addition of blank layers allows to decouple interference between both compartments. We showcased the application for the transient time‐programmed self‐assembly of suitable peptide gelators.
Our concept of integrating spatial domains into programming and shaping transient pH signal and autonomous transient material systems drives current artificial transient signaling a step closer to biological systems where spatial and temporal domains are orchestrated—albeit it is a first attempt and still in its infancy. We believe that spatial patterns using 3D printing will allow to increase structural control and system features. The concept should be widely applicable to other catalyst‐driven CRNs using the concept of temporally‐programmed environmental profiles (e.g. AMP/ADP/ATP[ 6a , 6b , 6c ] or redox[ 9c , 12 ]), and it may on an experimental level be very useful as the enzymes are removed from the solutions to allow for cleaner in situ analytics and material systems applications.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge funding through the DFG WA‐3084/4‐2, the ERC Starting Grant “TimeProSAMat” (ID: 677960). X.F. acknowledges financial support from China Scholarship Council (CSC) and Deutscher Akademischer Austauschdienst (DAAD). We thank Sebastian Löscher for TEM measurements. Open access funding enabled and organized by Projekt DEAL.
X. Fan, A. Walther, Angew. Chem. Int. Ed. 2021, 60, 3619.
Contributor Information
Dr. Xinlong Fan, https://www.walther‐group.com/
Prof. Dr. Andreas Walther, Email: andreas.walther@makro.uni-freiburg.de.
References
- 1.
- 1a. Merindol R., Walther A., Chem. Soc. Rev. 2017, 46, 5588–5619; [DOI] [PubMed] [Google Scholar]
- 1b. Karsenti E., Nat. Rev. Mol. Cell Biol. 2008, 9, 255–262. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Walther A., Adv. Mater. 2020, 32, 1905111; [Google Scholar]
- 2b. Jee E., T. Bánsági, Jr. , Taylor A. F., Pojman J. A., Angew. Chem. Int. Ed. 2016, 55, 2127–2131; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2167–2171; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Markovic V. M., T. Bánsági, Jr. , McKenzie D., Mai A., Pojman J. A., Taylor A. F., Chaos 2019, 29, 033130. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Grzybowski B. A., Huck W. T., Nat. Nanotechnol. 2016, 11, 585–592; [DOI] [PubMed] [Google Scholar]
- 3b. Nijemeisland M., Abdelmohsen L. K., Huck W. T., Wilson D. A., van Hest J. C., ACS Cent. Sci. 2016, 2, 843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Banani S. F., Lee H. O., Hyman A. A., Rosen M. K., Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Yucknovsky A., Mondal S., Burnstine-Townley A., Foqara M., Amdursky N., Nano Lett. 2019, 19, 3804–3810; [DOI] [PubMed] [Google Scholar]
- 5b. Singh D. P., Choudhury U., Fischer P., Mark A. G., Adv. Mater. 2017, 29, 1701328; [DOI] [PubMed] [Google Scholar]
- 5c. Kundu P. K., Samanta D., Leizrowice R., Margulis B., Zhao H., Börner M., Udayabhaskararao T., Manna D., Klajn R., Nat. Chem. 2015, 7, 646–652. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Deng J., Bezold D., Jessen H. J., Walther A., Angew. Chem. Int. Ed. 2020, 59, 12084–12092; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12182–12190; [Google Scholar]
- 6b. Deng J., Walther A., J. Am. Chem. Soc. 2020, 142, 685–689; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Heinen L., Walther A., Sci. Adv. 2019, 5, eaaw0590; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Dhiman S., Jain A., George S. J., Angew. Chem. Int. Ed. 2017, 56, 1329–1333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1349–1353; [Google Scholar]
- 6e. Postma S. G., Vialshin I. N., Gerritsen C. Y., Bao M., Huck W. T., Angew. Chem. Int. Ed. 2017, 56, 1794–1798; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1820–1824; [Google Scholar]
- 6f. Heinen L., Heuser T., Steinschulte A., Walther A., Nano Lett. 2017, 17, 4989–4995; [DOI] [PubMed] [Google Scholar]
- 6g. Heuser T., Merindol R., Loescher S., Klaus A., Walther A., Adv. Mater. 2017, 29, 1606842; [DOI] [PubMed] [Google Scholar]
- 6h. Heuser T., Weyandt E., Walther A., Angew. Chem. Int. Ed. 2015, 54, 13258–13262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13456–13460; [Google Scholar]
- 6i. Pappas C. G., Sasselli I. R., Ulijn R. V., Angew. Chem. Int. Ed. 2015, 54, 8119–8123; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8237–8241. [Google Scholar]
- 7.
- 7a. Leira-Iglesias J., Tassoni A., Adachi T., Stich M., Hermans T. M., Nat. Nanotechnol. 2018, 13, 1021–1027; [DOI] [PubMed] [Google Scholar]
- 7b. Semenov S. N., Wong A. S., Van Der Made R. M., Postma S. G., Groen J., Van Roekel H. W., De Greef T. F., Huck W. T., Nat. Chem. 2015, 7, 160–165. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Wang H., Wang Y., Shen B., Liu X., Lee M., J. Am. Chem. Soc. 2019, 141, 4182–4185; [DOI] [PubMed] [Google Scholar]
- 8b. della Sala F., Maiti S., Bonanni A., Scrimin P., Prins L. J., Angew. Chem. Int. Ed. 2018, 57, 1611–1615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1627–1631; [Google Scholar]
- 8c. Rieß B., Wanzke C., Tena-Solsona M., Grötsch R. K., Maity C., Boekhoven J., Soft Matter 2018, 14, 4852–4859; [DOI] [PubMed] [Google Scholar]
- 8d. Dhiman S., Jain A., Kumar M., George S. J., J. Am. Chem. Soc. 2017, 139, 16568–16575; [DOI] [PubMed] [Google Scholar]
- 8e. Van Ravensteijn B. G., Hendriksen W. E., Eelkema R., Van Esch J. H., Kegel W. K., J. Am. Chem. Soc. 2017, 139, 9763–9766; [DOI] [PubMed] [Google Scholar]
- 8f. Kariyawasam L. S., Hartley C. S., J. Am. Chem. Soc. 2017, 139, 11949–11955; [DOI] [PubMed] [Google Scholar]
- 8g. Boekhoven J., Hendriksen W. E., Koper G. J., Eelkema R., van Esch J. H., Science 2015, 349, 1075–1079; [DOI] [PubMed] [Google Scholar]
- 8h. Pezzato C., Prins L. J., Nat. Commun. 2015, 6, 7790; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8i. Boekhoven J., Brizard A. M., Kowlgi K. N., Koper G. J., Eelkema R., van Esch J. H., Angew. Chem. Int. Ed. 2010, 49, 4825–4828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4935–4938. [Google Scholar]
- 9.
- 9a. Guan Z. N., Ogden W. A., ChemSystemsChem 2019, 1, e1900030; [Google Scholar]
- 9b. Conte M. P., Sahoo J. K., Abul-Haija Y. M., Lau K. A., Ulijn R. V., ACS Appl. Mater. Interfaces 2018, 10, 3069–3075; [DOI] [PubMed] [Google Scholar]
- 9c. Wojciechowski J. P., Martin A. D., Thordarson P., J. Am. Chem. Soc. 2018, 140, 2869–2874; [DOI] [PubMed] [Google Scholar]
- 9d. Heinen L., Walther A., Chem. Sci. 2017, 8, 4100–4107; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Maiti S., Fortunati I., Ferrante C., Scrimin P., Prins L. J., Nat. Chem. 2016, 8, 725–731; [DOI] [PubMed] [Google Scholar]
- 9f. Heuser T., Steppert A.-K., Molano Lopez C., Zhu B., Walther A., Nano Lett. 2015, 15, 2213–2219; [DOI] [PubMed] [Google Scholar]
- 9g. Debnath S., Roy S., Ulijn R. V., J. Am. Chem. Soc. 2013, 135, 16789–16792. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Te Brinke E., Groen J., Herrmann A., Heus H. A., Rivas G., Spruijt E., Huck W. T., Nat. Nanotechnol. 2018, 13, 849–855; [DOI] [PubMed] [Google Scholar]
- 10b. Che H., Cao S., van Hest J. C., J. Am. Chem. Soc. 2018, 140, 5356–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Panja S., Adams D. J., Chem. Commun. 2019, 55, 47–50. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Singh N., Formon G. J., De Piccoli S., Hermans T. M., Adv. Mater. 2020, 32, 1906834; [DOI] [PubMed] [Google Scholar]
- 12b. Singh N., Lainer B., Formon G. J., De Piccoli S., Hermans T. M., J. Am. Chem. Soc. 2020, 142, 4083–4087; [DOI] [PubMed] [Google Scholar]
- 12c. Ogden W. A., Guan Z., ChemSystemsChem 2020, 2, e1900030. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary