

Abstract
Ciliopathy syndromes are a diverse spectrum of disease characterized by a combination of cystic kidney disease, hepatobiliary disease, retinopathy, skeletal dysplasia, developmental delay, and brain malformations. Though generally divided into distinct disease categories based on the pattern of system involvement, ciliopathy syndromes are known to display certain phenotypic overlap. We performed next‐generation sequencing panel testing, clinical exome sequencing, and research‐based exome sequencing reanalysis on patients with suspected ciliopathy syndromes with additional features. We identified biallelic pathogenic variants in BBS1 in a child with features of cranioectodermal dysplasia, and biallelic variants in BBS12 in a child with the clinical stigmata of Bardet‐Biedl syndrome, but also with anal atresia. We additionally identified biallelic pathogenic variants in WDR35 and DYNC2H1 in children with predominant liver disease and ductal plate malformation without skeletal dysplasia. Our study highlights the phenotypic and genetic diversity of ciliopathy syndromes, the importance of considering ciliopathy syndromes as a disease‐spectrum and screening for all associated complications in all patients, and describes exclusive extra‐skeletal manifestations in two classical skeletal dysplasia syndromes.
Keywords: Bardet‐Biedl syndrome, ciliopathy, cranioectodermal dysplasia, DYNC2H1, WDR35
1. INTRODUCTION
Ciliopathy syndromes are genetically and phenotypically diverse diseases caused by dysfunction of the primary cilium. Cilia are hair‐like projections that exist atop most cell types, and can be either motile or nonmotile. Motile cilia line the respiratory and reproductive tracts to facilitate mucociliary clearance, movement of the ovum through the fallopian tube, and sperm propulsion. Nodal cilia, a type of motile cilia, are expressed transiently during embryogenesis and beat in a rotary fashion to facilitate morphogen distribution and establishment of the left–right body axis. Nonmotile or sensory cilia are solitary projections that exist atop most cell types that house receptors and effector proteins for multiple growth factor signaling pathways and play a critical role in mechanochemotransduction, cell growth and organ patterning (Mitchison & Valente, 2017). Ciliopathy syndromes are caused by dysfunction of the primary cilium, and result in severe developmental abnormalities in multiple organ systems because of impaired cellular signaling.
Ciliopathy syndromes are classified into disease categories based on the pattern of system involvement. Specifically, the ciliopathy Bardet‐Biedl syndrome (BBS) is characterized by cystic kidney disease, retinopathy, obesity, polydactyly, and development delays. In contrast, Jeune Asphyxiating Thoracic Dystrophy (JATD) is characterized by thoracic insufficiency, polydactyly, cystic kidney disease, hepatobiliary disease, retinal disease, and congenital heart disease (Forsythe & Beales, 2013; Tüysüz et al., 2009). Another classic ciliopathy, cranioectodermal dysplasia (CED), is characterized by sagittal craniosynostosis with dolichocephaly, ectodermal dysplasia, skeletal dysplasia, kidney disease, hepatobiliary disease, and developmental delays (Walczak‐Sztulpa et al., 2010).
The wide clinical use of next generation sequencing and whole exome sequencing technology has facilitated the molecular diagnosis of multiple genetic conditions, including ciliopathies. Importantly, this has also allowed for expansion of the clinical phenotype of each ciliopathy syndrome and gene, as it was discovered that variants in the same gene are causal for multiple ciliopathies.
Here we report four children found by next‐generation sequencing to have pathogenic variants in well‐characterized ciliopathy genes who presented with atypical features. We additionally report a novel association between biallelic, pathogenic variants in DYNC2H1, previously associated exclusively with thoracic insufficiency syndromes, in a child with isolated hepatobiliary disease. We suggest that the clinical care of children with ciliopathies should not universally be restricted to the systems known to be involved in distinct ciliopathy syndromes, but instead attention should be given to all features associated with ciliopathy syndromes including retinal, kidney, hepatic, cardiac, skeletal, pulmonary, endocrine, and neurological disease to ensure that subclinical disease is detected and appropriately medically‐managed, as the boundaries that separate individual ciliopathy syndromes can be fluid.
2. METHODS
2.1. Editorial policies and ethical considerations
All individuals and families agreed to participate in this study and signed appropriate consent forms. Permission for clinical photographs was given separately. This study was approved by the institutional IRB.
2.2. Genetic testing methodology
Clinical exome sequencing was performed on DNA from Patients 1 and 2 and their available relatives at Children's Hospital of Philadelphia (CHOP) Division of Genomic Diagnostics (DGD). Testing was diagnostic for Patient 1. A clinically available next generation sequencing panel from Molecular Vision Laboratories was used for diagnosis of Patient 3. Patients 2 and 4 were recruited into the Center for Applied Genomics (CAG) at CHOP for exome reanalysis and exome sequencing, respectively.
2.3. Exome sequencing and analysis
Under an Institutional Review Board approved protocol, informed consent was from the patients' families. Exome sequencing was performed using the Twist Human Core Exome Capture Kit (TWIST Bioscience) and Illumina NovaSeq 6000 at the CAG at CHOP. Approximately 33 million 100 bp paired‐end reads were generated with median insert size of ~250 bp. Data were quality controlled and analyzed using a custom‐built pipeline that incorporates BWA‐mem v0.7.12 for alignment, Picard v1.97 for PCR duplication removal, and GATK v2.6.5 for variant calling. This resulted in ~97.4% of the patient's exome sequenced at a depth of at least 20X. ANNOVAR and SnpEff were used to functionally annotate the variants and collect minor allele frequency (MAF) data from 1,000 Genomes Projects, ESP6500SI, ExAC, gnomAD, and Kaviar. Relatively common variants for suspected autosomal dominant mode of inheritance were excluded based on MAF threshold of 0.1% in either population dataset, and functional annotation, such as, synonymous, non‐exonic, and non‐splicing‐altering were included. Subsequent gene prioritization was performed on the basis of deleterious prediction and biological relevance by referring to the Online Mendelian Inheritance in Man (OMIM) database and Human Gene Mutation Database (HGMD).
2.4. Exome reanalysis
Variant annotation, filtration, and prioritization was performed with Cyclo and GDCross. These tools were designed and built at CAG, and are both validated for clinical use. Variants with ≥5x coverage were initially filtered at 0.5% gnomAD MAF and annotated with a combination of multiple tools and databases, including Variant Effect Predictor, HGMD, ClinVar, dbSNP, OMIM, HPO, PolyPhen‐2 and SIFT, and a custom‐built splice‐site annotator. This combined dataset is updated quarterly and versioned for use by CYCLO. Subpopulation frequency data is obtained from gnomAD2.1.1 for whole exome sequencing (125,748 exomes). Multiple transcripts for each gene/variant are evaluated, and the one with the overall most deleterious annotation for the variant is chosen for further analysis. CYCLO includes an internal frequency table to reduce sequencing artifacts. The CYCLO‐generated list of patient variants is filtered against the pedigree and HPO terms describing the patient's phenotype, and GDCross assigns each variant a priority score of likelihood as the causal variant for the patient's disease. Variants are ranked using a weighted combination of (a) overlap with HPO terms, (b) patient and family genotypes, (c) predicted functional impact, (d) inheritance modeling, (e) presence in mutation databases such as HGMD and ClinVar; and other factors.
3. RESULTS
3.1. Patient cohort
3.1.1. Patient 1
Patient 1 was the product of a naturally conceived pregnancy to a 32‐year old G4P2➔3 mother. Pregnancy was complicated by maternal cholecystitis requiring cholecystectomy, and prenatal ultrasound findings concerning for enlarged kidneys. Amniocentesis and karyotype were performed, which were nondiagnostic. Patient was born via vaginal delivery at 39 weeks +5 days gestational age. Birth weight was 3.86 kg (70%), length was 54.6 cm (90%). He was noted to have bilateral lower extremity polydactyly and dolichocephaly. There were no complications, and he was discharged home on day of life two. Patient was evaluated by Nephrology, Plastic Surgery and Neurosurgery within the first month of life, and was noted to have bilateral cortical kidney cysts with poor corticomedullary differentiation as well as sagittal craniosynostosis. Chromosomal microarray was nondiagnostic. At 6 years of age, he was presented to Genetics clinic. At that time, history was additionally notable for Stage 1–2 chronic kidney disease, gross motor delay with independent walking achieved at ~2 years of age with persistent poor balance and coordination, and speech delay with first words emerging at 2 years of age. Physical examination was notable for a height of 127 cm (97%), weight of 31.7 kg (97%), mild dolichocephaly, deep set and hooded eyes, hypermobility (Beighton score 4), and bilateral lower extremity polydactyly (Figure 1). Laboratories were notable for mild dyslipidemia (total cholesterol 174 mg/dl, triglycerides 59 mg/dl, HDL 44 mg/dl, and LDL 118 mg/dl). Imaging studies were notable for bilateral increased kidney echogenicity with multiple cysts in the right kidney. Clinical exome sequencing through CHOP DGD was performed for concern for underlying ciliopathy syndrome.
FIGURE 1.
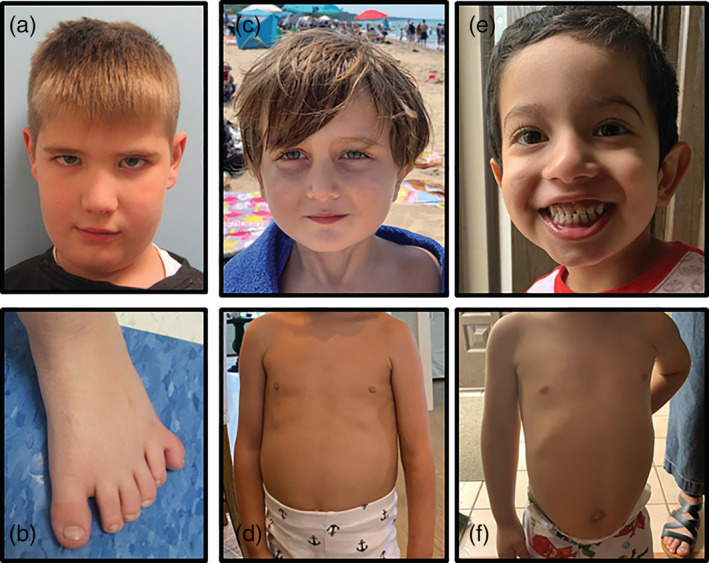
Photographs of Patient 1, 2, and 4 (photographs of Patient 3 not available) (a) Facial features in Patient 1 notable for mild dolichocephaly and deep‐set and hooded eyes (b) Lower extremity of Patient 1 notable for postaxial polydactyly (c) Photograph of Patient 2 highlighting non‐dysmorphic facial features (d) Chest view of Patient 2 highlighting normal chest circumference and prominent hepatosplenomegaly (e) Facial features of Patient 4, notable for normal teeth and non‐dysmorphic features (f) Abdominal view of Patient 4 highlighting prominent hepatosplenomegaly and normal chest circumference [Color figure can be viewed at wileyonlinelibrary.com]
3.1.2. Patient 2
Patient 2 was the product of a naturally conceived pregnancy to a 27‐year old G1P0➔1 mother. Pregnancy was uncomplicated. Patient was born via vaginal delivery at 37 weeks +5 days gestational age. Birth weight was 2.863 kg (14%), length was 49.5 cm (46%). At 5 months of age, he was noted to have splenomegaly and pancytopenia during an admission for RSV bronchiolitis and pneumonia. Evaluation was notable for a normal bone marrow biopsy and nondiagnostic metabolic screening laboratories including plasma amino acids, urine organic acids, acylcarnitine profile, lactate/pyruvate, carbohydrate deficient transferrin, very‐long chain fatty acids, and lysosomal enzyme testing. Abdominal ultrasound showed normal kidneys and normal biliary tree, but hepatosplenomegaly with a coarse‐appearing liver with abnormal echogenicity, concerning for fibrosis. Liver biopsy was concerning for ductal plate malformation and congenital hepatic fibrosis. At 30 months of age, he presented to Genetics clinic for evaluation. At that time, he displayed normal development, sitting independently at 5 months, walking independently at 11 months, first words at 11 months, and speaking in full sentences at 30 months. Physical examination was notable for normal growth parameters (height 91 cm, 50% and weight 13.6 kg, 53%) and splenomegaly (Figure 1). Laboratories were notable for persistently elevated aminotransferases and gamma‐glutamyltransferase (100–300 s U/L, reference values for aminotransferase 10–60 U/L and for gamma‐glutamyltransferase 5–16 U/L). Imaging studies were notable for hepatosplenomegaly with evidence of fibrosis and portal hypertension. Kidney ultrasound was normal. Whole exome sequencing performed through CHOP DGD was nondiagnostic. He was enrolled in the CAG for clinical exome reanalysis.
3.1.3. Patient 3
Patient 3 was the product of a naturally conceived pregnancy to a 19‐year old G1P0➔1 mother. Pregnancy was uncomplicated. Patient was born via spontaneous vaginal delivery at 40 weeks gestational age. Birth weight was 3.11 kg (25–50%) and birth length was 50 cm (50%). She was noted after birth to have bilateral lower extremity polydactyly and imperforate anus. Shortly thereafter she was diagnosed with bilateral cystic kidney dysplasia and vesicoureteral reflux. She was evaluated by Genetics at 4 years and 9 months of age. At that time, her history was notable for astigmatism, blocked tear ducts, and mild global developmental delay, with independent sitting achieved at 12 months, independent walking at 2 years, and first words at 2 years. Physical examination was notable for obesity (weight 27 kg, > 99%) with normal height (104 cm, 34%), and lower extremity polydactyly. Laboratories were notable for a chromosomal microarray showing a 364 kB deletion at 5q21.1 and a 518 kB duplication at Xp22.33, both of unclear clinical significance (arr(hg19) 5q21.1 (101,345,712–101,709,433) x1, Xp22.33 (3,426,104–3,944,402) x3). Clinical BBS testing was performed.
3.1.4. Patient 4
Patient 4 was the product of a naturally conceived pregnancy to a 36‐year old G2P1➔2 mother. Pregnancy was complicated by kidney stones requiring hospitalization for IV hydration. Patient was born full term via repeat caesarian section. Birth weight was 3.81 kg (70%) and birth length was 52 cm (78%). At 2 months of age, he was noted to have left anterior plagiocephaly with concern for left coronal craniosynostosis, and he was referred to Plastic Surgery for evaluation. No imaging was done, and after 2 months of observation his head shape normalized. At 6 months of age he was noted to have splenomegaly. Laboratories were notable for elevated aminotransferases (ALT 120 U/L, reference value 10–30 U/L and AST 152 U/L, reference value 20–60 U/L), gamma‐glutamyltransferase (367 U/L, reference value 5–16 U/L), and direct bilirubin (0.8 mg/dl, reference value 0–0.3 mg/dl). Abdominal ultrasound was notable for splenomegaly, cavernous transformation of the portal vein, abnormal liver echotexture, portal hypertension, and increased bilateral kidney echogenicity. He underwent liver biopsy, which showed periportal fibrosis and ductal plate malformation. He was referred to Genetics for evaluation at 30 months of age. History at that time was notable for hypertension and delayed development with independent walking achieved at 23 months and no speech. Physical examination was notable for normal growth parameters (height 89 cm, 28% and weight 13.7 kg, 54%), protuberant abdomen, splenomegaly and poor eye contact (Figure 1). Imaging was notable for an abdominal ultrasound showing hepatosplenomegaly and cirrhosis with paraesophageal, perigastric, and perisplenic varices. He was enrolled in the CAG for research exome sequencing.
3.2. Genetic testing
3.2.1. Patient 1
The combination of polydactyly, cystic kidney disease, developmental delay, and sagittal craniosynostosis was concerning for CED versus BBS. Clinical exome sequencing revealed homozygous, BBS1 pathogenic variants (c.1169 T > G; p.[Met390Arg]), consistent with a genetic diagnosis of BBS (Tables 1 and S1). No reportable variants were found in FGFR1, FGFR2, FGFR3, TWIST1, IFT122, IFT43, WDR19, or WDR35. With these genetic findings, the patient was referred to Endocrinology, Gastroenterology, and Ophthalmology and had a normal evaluation. He is currently 8‐years old, and completed first grade in a home‐schooling environment with an individualized education plan receiving physical, speech, and occupational therapies.
TABLE 1.
Clinical features and molecular diagnosis for Patients 1–4
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
|---|---|---|---|---|
| Gene | BBS1 | DYNC2H1 | BBS12 | WDR35 |
| RefSeq number | NM_024649.4 | NM_001080463.2 | NM_001178007.1 | NM_001006657.1 |
| cDNA | c.1169 T > G | c.7277G > T and c.7967G > A | c.695_731del37 | c.1633C > T and c.308G > T |
| Protein | p.(Met390Arg) | p.(Arg2426Leu) and p.(Arg2656His) | p.(Ile232Lysfsx3) | p.(Arg545*) and p.(Gly103Val) |
| Variant type | Homozygous missense | Compound heterozygous missense | Homozygous Frameshift | Compound heterozygous missense |
| Inheritance | Autosomal recessive | Autosomal recessive | Autosomal recessive | Autosomal recessive |
| Testing modality | Clinical exome sequencing | Research exome reanalysis | BBS gene panel | Research exome sequencing |
| Sex | Male | Male | Female | Male |
| Age at evaluation | 6 years | 30 months | 4 years 9 months | |
| Growth | Height and weight: 97% | Height and weight: 50% | Height 30%, weight > 99% | |
| Craniofacial | Sagittal craniosynostosis, dolichocephaly | Abnormal head shape as a neonate, self‐resolved | ||
| Eyes | Deep set and hooded eyes, no evidence of retinitis pigmentosa | Normal ophthalmology examination | No evidence of retinitis pigmentosa, astigmatism | Normal ophthalmology examination |
| Cardiac | Normal echocardiogram | Normal echocardiogram | ||
| Pulmonary | Negative | Negative | Negative | Recurrent otitis media, no pulmonary findings |
| GI | Normal liver ultrasound and aminotransferases | Ductal plate malformation, cirrhosis, portal hypertension | Imperforate anus | Ductal plate malformation, cirrhosis, portal hypertension |
| GU | Increased renal echogenicity and right sided renal cysts | Normal renal ultrasounds | Bilateral cystic renal dysplasia, vesicoureteral reflux | Abnormal renal echogenicity, hypertension |
| Endocrine | Hyperphagia, dyslipidemia | Obesity, normal hemoglobin A1c | ||
| Skeletal | Bilateral lower extremity polydactyly | Normal chest xray, chest circumference 50% | Bilateral lower extremity polydactyly | Normal skeletal survey |
| Skin and joints | No ectodermal findings, hypermobility | No ectodermal findings | No ectodermal findings | No ectodermal findings |
| Development | Mild global developmental delay | Normal | Mild global developmental delay | Mild global developmental delay |
3.2.2. Patient 2
Patient was enrolled in the CAG for research exome reanalysis given negative clinical testing. Reanalysis revealed biallelic variants of uncertain significance in DYNC2H1 (c.7277G > T; p.(Arg2426Leu) and c.7967G > A; p.(Arg2656His)) (Tables 1 and S1). Follow up kidney ultrasound, blood pressure, and kidney function panel were normal. Ophthalmology examination did not show evidence of retinitis pigmentosa. Chest xray did not show thoracic insufficiency or short ribs. Skeletal survey has not yet been performed. Patient is currently 5‐years old and has completed pre‐Kindergarten. There are no developmental concerns.
3.2.3. Patient 3
The combination of obesity, polydactyly, developmental delay, and kidney dysplasia was concerning for BBS. BBS gene panel testing was sent, and revealed homozygous, biallelic BBS12 pathogenic variants (c.695_731del37; p.[Ile232Lysfsx3]), consistent with a genetic diagnosis of BBS (Tables 1 and S1). She was referred to Endocrinology and Ophthalmology; hemoglobin A1C and thyroid studies were normal and retinal exam showed no evidence of retinitis pigmentosa. Patient is currently 8‐years old. She attends regular school, but has an individualized education plan and exhibits mild global developmental delay.
3.2.4. Patient 4
The combination of ductal plate malformation, cirrhosis, developmental delay, hypertension, and abnormal kidney echogenicity was concerning for an underlying ciliopathy syndrome. Patient was enrolled in the CAG for research exome sequencing, which was notable for biallelic WDR35 likely pathogenic variants, c.1633C > T; p.(Arg545*) (paternally inherited) and c.308G > T; p.(Gly103Val) (maternally inherited), consistent with a diagnosis of CED versus JATD (Tables 1 and S1). Follow up ophthalmology examination showed no evidence of retinitis pigmentosa. Skeletal survey was normal and showed no evidence of narrow thorax. Patient is now 3 years old, and has normal motor development; however, his speech remains mildly delayed.
4. DISCUSSION
Ciliopathy syndromes are characterized by dysfunction of the primary cilium, a mechanosensory organelle present atop most cell types that facilitates organized cellular growth and organogenesis. Clinical features include kidney disease, hepatic fibrosis, structural brain malformations, developmental delay, primary ciliary dyskinesia, obesity, and skeletal abnormalities (Ware, Aygun, & Hildebrandt, 2011). Ciliopathy syndromes are divided into specific disease categories based on the combination of clinical features present; however, it has become increasingly recognized that the boundaries that separate distinct syndromes are fluid.
Here were present four cases of children with clinical features suggestive of an underlying ciliopathy, but with patterns inconsistent with the named syndromes. Specifically, the combination of cystic kidney disease, polydactyly, and developmental delays in Patient 1 is consistent with BBS; however, sagittal craniosynostosis is a well‐described feature of the related ciliopathy syndrome, CED, which is also associated with cystic kidney disease and polydactyly, although with ectodermal and skeletal findings, which our patient did not have. Interestingly, genome‐wide association studies identified a susceptibility locus for sagittal craniosynostosis near BBS9; however, to date, there have been no reports of craniosynostosis in children with BBS (Justice et al., 2012). It is interesting to speculate why our patient had craniosynostosis whereas other children with the same BBS1 variant did not. One possibility is a hypomorphic or regulatory variant in a known or yet undiscovered ciliopathy or craniosynostosis gene or a copy number variation missed on exome sequencing due to technology limitations. Another possibility relates to fetal crowding, a known risk factor for craniosynostosis (Sanchez‐Lara PA et al., 2010). Specifically, it is possible that there was sub‐clinically decreased amniotic fluid from underlying kidney disease compounded by fluid removal for amniocentesis and exacerbated by mechanical forces exerted on the uterus during mother's cholecystectomy performed at 30 weeks gestational age that put pressure on the sagittal suture, and on the risk background of biallelic BBS1 variants caused craniosynostosis. No matter what the true underlying cause of craniosynostosis in our patient, this case highlights the phenotypic diversity of ciliopathy syndromes, and also supports broader testing methods for suspected ciliopathy patients.
Regarding Patient 2, ductal plate malformations are pathognomonic for underlying ciliopathy syndrome, most commonly autosomal recessive polycystic kidney disease (ARPKD); however, it also occurs in combination with other features including skeletal dysplasia, cystic kidney disease, and structural brain abnormalities. Patient 2 was found to have biallelic variants in DYNC2H1, which is associated with the skeletal ciliopathies JATD and short‐rib polydactyly syndrome. These skeletal dysplasias can have associated liver disease and ductal plate malformation; however, isolated liver disease without skeletal findings has not been reported for DYNC2H1. The variants detected in our patient have been reported in two individuals with short rib polydactyly syndrome, one of which had known liver involvement and the other who was lost to follow up at 3 months (Zhang et al., 2018). It is unknown at this time why our patient has no evidence of thoracic insufficiency or skeletal involvement and presented with isolated liver disease. Of note, biallelic DYNC2H1 variants have recently been reported in isolated retinal disease, consistent with isolated extra‐skeletal manifestations with DYNC2H1 variants (Vig et al., 2020). Our patient's unique presentation may be related to yet unidentified modifying genetic and/or environmental factors, or a yet undiscovered tissue‐specific splicing mechanism in which our patient's variants play a critical role in a splice isoform involved in ductal plate remodeling but not skeletogenesis. These possibilities remain speculative at this time.
Patient 3 presented with mild global developmental delay, polydactyly, obesity, kidney disease, and imperforate anus, which is highly consistent with her underlying diagnosis of BBS except for imperforate anus, which is a rare association (Bahceci et al., 2012; David et al., 1999; CRIBBS database, unpublished). Given the atypical finding of anal atresia, it is important to consider a potential contribution from her copy number variations detected on chromosomal microarray, namely her 364 kB deletion at 5q21.1 and her 518 kB duplication at Xp22.33. The deletion on 5q21.1 contains two genes, SLCO4C1 and SLCO6A1 (partial deletion), neither of which are associated with human disease or anal atresia. The duplication on Xp22.3 includes one gene (PRKX) and one pseudogene (LOC389906). PRKX is not associated with Mendelian disease, but is implicated in kidney development and is suspected to play a modulatory role in autosomal dominant polycystic kidney disease (Li et al., 2008). There is no known role for PRKX in anal atresia. Parental testing could not be performed to further clarify the clinical implications of these copy number variations.
Imperforate anus is seen in Pallister Hall syndrome and Townes‐Brock syndrome (TBS). Pallister‐Hall syndrome is caused by pathogenic variants in GLI3, a transcription factor known to be an effector in ciliary signaling (Johnston et al., 2010). TBS is caused by biallelic pathogenic variants in the transcriptional repressor SALL1, and is additionally associated with ear malformations and abnormal thumbs, most commonly triphalangeal or duplicated thumbs as well as kidney disease. SALL1 variants have recently been associated with impaired ciliogenesis and ciliary function. The phenotypic overlap between GLI3 and SALL1‐associated disease and ciliopathy syndromes suggests a possible role for disrupted ciliary signaling in the pathogenesis of imperforate anus (Bozal‐Basterra et al., 2018). Indeed, imperforate anus has been described in a lethal case of biallelic IFT27 pathogenic variants (Quélin et al., 2018), but is a rare association in BBS. Of course, the patient's variants are novel, and may affect BBS12 function more severely than other variants reported to date, causing the added feature of imperforate anus. Additionally, this patient was diagnosed by panel testing and not by exome sequencing, allowing for the possibility of pathogenic variants in other ciliopathy genes driving the anal atresia phenotype. While a second genetic diagnosis responsible for the anal atresia cannot be entirely excluded, this case still highlights a potential role of ciliary signaling in the pathogenesis of anal atresia.
Patient 4 has a history of ductal plate malformation, developmental delay, hypertension, and abnormal kidney echogenicity. There was initially concern for ARPKD given the combination of ductal plate malformation and evidence of kidney disease; however, delayed development is not a typical feature of ARPKD. Indeed, research testing confirmed biallelic variants in WDR35, which is causal for CED and JATD. Our patient showed no evidence of ectodermal or skeletal findings, even after careful phenotyping, which is atypical for CED and JATD, though has been reported in a large cohort of CED patients (Lin et al., 2013). It is possible that the novel variant found in our patient (c.308 G > T; p.(Gly103Val)) does not significantly compromise the role of WDR35 in skeletal development, but severely impairs the ability of WDR35 to appropriately pattern the ductal plate, thereby allowing appropriate skeletal patterning but resulting in significant liver disease. Interestingly, this patient did have a history of anterior plagiocephaly with concern for left coronal craniosynostosis during infancy, which spontaneously resolved, but may reflect craniosynostosis‐spectrum, consistent with a CED‐like presentation.
The overlapping clinical spectrum of ciliopathy syndromes has been reported previously (Braun & Hildebrandt, 2017; Grochowsky & Gunay‐Aygun, 2019; Shaheen et al., 2016; Ware et al., 2011 and Yamada et al., 2019). Indeed, there are several reports of individuals with features of both Joubert syndrome and oral‐facial‐digital syndrome and Joubert syndrome and Jeune syndrome (Bhardwaj, Sharma, & Ahluwalia, 2018; Franco & Thauvin‐Robinet, 2016; Johnston et al., 2017; Lehman et al., 2010; Parisi, 2019 and Wentzensen et al., 2015). There are also reports of disconcordant ciliopathy phenotypes within families (Shaheen et al., 2019; Zaki et al., 2011; and Parisi, 2019). Our cases additionally highlight the inability to neatly package ciliopathies within distinct disease categories, and suggest that there should be a low threshold for evaluating and monitoring for all complications associated with ciliary dysfunction, including kidney and liver disease, skeletal dysplasia, endocrinopathies, retinitis pigmentosa, primary ciliary dyskinesia, and varying degrees of developmental delays. We also highlight the importance of performing genetic testing more broadly when considering ciliopathy syndromes, as these cases can be missed by focusing on gene panels guided by clinical classification information.
CONFLICT OF INTEREST
Jessica Wen receives research support from Gilead, Abbvie and Alexion.
AUTHOR CONTRIBUTIONS
Alanna Strong helped conceptualize and design the study, evaluated the patients clinically, helped in variant interpretation and data analysis, and drafted the article. Dong Li facilitated data acquisition and analysis, and contributed to article preparation and review. Frank Mentch contributed to data acquisition and analysis and critically reviewed the article. Emma Bedoukian, Erum A. Hartung, Kevin Meyers, Cara Skraban, Jessica Wen, Livija Medne, and Ian Krantz each evaluated a subset of the patients, guided appropriate genetic evaluation, imaging studies and clinical care, and recognized each patients' atypical presentations. They also critically reviewed the article. Joseph Glessner and Deborah Watson facilitated genetic data collection and analysis. Hakon Hakonarson conceptualized and designed the study, helped in data analysis, critically reviewed and edited the article, and provided funding for the work. All authors approved the final article as submitted and agree to be accountable for all aspects of the work.
Supporting information
Supplementary Table 1 Details for genetic variants detected in our patient cohort.
ACKNOWLEDGMENT
The authors would like to thank the patients and their families for allowing us to participate in their care and permitting publication of their cases.
Strong A, Li D, Mentch F, et al. Ciliopathies: Coloring outside of the lines. Am J Med Genet Part A. 2021;185A:687–694. 10.1002/ajmg.a.62013
REFERENCES
- Bahceci, M. , Dolek, D. , Tutuncuoglu, P. , Gorgel, A. , Oruk, G. , & Yenen, I. (2012). A case series of Bardet‐Biedl syndrome in a large Turkish family and review of the literature. Eating and Weight Disorders, 17(1), e66–e69. [DOI] [PubMed] [Google Scholar]
- Bhardwaj, P. , Sharma, M. , & Ahluwalia, K. (2018). Joubert syndrome with Orofacial digital features. Journal of Neurosciences in Rural Practice, 9(1), 152–154. 10.4103/jnrp.jnrp_338_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozal‐Basterra, L. , Martín‐Ruíz, I. , Pirone, L. , Liang, Y. , Sigurðsson, J. O. , Gonzalez‐Santamarta, M. , … Barrio, R. (2018). Truncated SALL1 impedes primary cilia function in Townes‐brocks syndrome. American Journal of Human Genetics, 102(2), 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, D. A. , & Hildebrandt, F. (2017). Ciliopathies. Cold Spring Harbor Perspectives in Biology, 9(3), a028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, A. , Bitoun, P. , Lacombe, D. , Lambert, J. C. , Nivelon, A. , Vigneron, J. , & Verloes, A. (1999). Hydrometrocolpos and polydactyly: A common neonatal presentation of Bardet‐Biedl and McKusick‐Kaufman syndromes. Journal of Medical Genetics, 36(8), 599–603. [PMC free article] [PubMed] [Google Scholar]
- Forsythe, E. , & Beales, P. L. (2013). Bardet‐Biedl syndrome. European Journal of Human Genetics: EJHG, 21(1), 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco, B. , & Thauvin‐Robinet, C. (2016). Update on oral‐facial‐digital syndromes (OFDS). Cilia, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochowsky, A. , & Gunay‐Aygun, M. (2019). Clinical characteristics of individual organ system disease in non‐motile ciliopathies. Translational Science of Rare Diseases, 4(1–2), 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. J. , Lee, C. , Wentzensen, I. M. , Parisi, M. A. , Crenshaw, M. M. , Sapp, J. C. , … Biesecker, L. G. (2017). Compound heterozygous alterations in intraflagellar transport protein CLUAP1 in a child with a novel Joubert and oral‐facial‐digital overlap syndrome. Cold Spring Harbor Molecular Case Studies, 3(4), a001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. J. , Sapp, J. C. , Turner, J. T. , Amor, D. , Aftimos, S. , Aleck, K. A. , … Biesecker, L. G. (2010). Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Human Mutation, 31(10), 1142–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice, C. M. , Yagnik, G. , Kim, Y. , Peter, I. , Jabs, E. W. , Erazo, M. , … Boyadjiev, S. A. (2012). A genome‐wide association study identifies susceptibility loci for nonsyndromic sagittal craniosynostosis near BMP2 and within BBS9. Nature Genetics, 44(12), 1360–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman, A. M. , Eydoux, P. , Doherty, D. , Glass, I. A. , Chitayat, D. , Chung, B. Y. , … Trnka, P. (2010). Co‐occurrence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy. American Journal of Medical Genetics. Part A, 152A(6), 1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Burrow, C. R. , Polgar, K. , Hyink, D. P. , Gusella, G. L. , & Wilson, P. D. (2008). Protein kinase X (PRKX) can rescue the effects of polycystic kidney disease‐1 gene (PKD1) deficiency. Biochimica et Biophysica Acta, 1782(1), 1–9. [DOI] [PubMed] [Google Scholar]
- Lin, A. E. , Traum, A. Z. , Sahai, I. , Keppler‐Noreuil, K. , Kukolich, M. K. , Adam, M. P. , … Arts, H. H. (2013). Sensenbrenner syndrome (Cranioectodermal dysplasia): Clinical and molecular analyses of 39 patients including two new patients. American Journal of Medical Genetics. Part A, 161A(11), 2762–2776. [DOI] [PubMed] [Google Scholar]
- Mitchison, H. M. , & Valente, E. M. (2017). Motile and non‐motile cilia in human pathology: From function to phenotypes. The Journal of Pathology, 241(2), 294–309. [DOI] [PubMed] [Google Scholar]
- Parisi, M. A. (2019). The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Translational Science of Rare Diseases, 4(1–2), 25–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quélin, C. , Loget, P. , Boutaud, L. , Elkhartoufi, N. , Milon, J. , Odent, S. , … Attié‐Bitach, T. (2018). Loss of function IFT27 variants associated with an unclassified lethal fetal ciliopathy with renal agenesis. American Journal of Medical Genetics. Part A, 176(7), 1610–1613. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Lara, P. A. , Carmichael, S. L. , Graham, J. M., Jr. , Lammer, E. J. , Shaw, G. M. , Ma, C. , … National Birth Defects Prevention Study . (2010). Fetal constraint as a potential risk factor for craniosynostosis. American Journal of Medical Genetics. Part A, 152A(2), 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen, R. , Jiang, N. , Alzahrani, F. , Ewida, N. , Al‐Sheddi, T. , Alobeid, E. , … Alkuraya, F. S. (2019). Bi‐allelic mutations in FAM149B1 cause abnormal primary cilium and a range of Ciliopathy phenotypes in humans. American Journal of Human Genetics, 104(4), 731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen, R. , Szymanska, K. , Basu, B. , Patel, N. , Ewida, N. , Faqeih, E. , … Alkuraya, F. S. (2016). Characterizing the morbid genome of ciliopathies. Genome Biology, 17(1), 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tüysüz, B. , Bariş, S. , Aksoy, F. , Madazli, R. , Ungür, S. , & Sever, L. (2009). Clinical variability of asphyxiating thoracic dystrophy (Jeune) syndrome: Evaluation and classification of 13 patients. American Journal of Medical Genetics. Part A, 149A(8), 1727–1733. [DOI] [PubMed] [Google Scholar]
- Vig, A. , Poulter, J. A. , Ottaviani, D. , Tavares, E. , Toropova, K. , Tracewska, A. M. , … Heon, E. (2020). DYNC2H1 hypomorphic or retina‐predominant variants cause nonsyndromic retinal degeneration. Genetics in Medicine: Official Journal of the American College of Medical Genetics , 22(12), 2041–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak‐Sztulpa, J. , Eggenschwiler, J. , Osborn, D. , Brown, D. A. , Emma, F. , Klingenberg, C. , … Kuss, A. W. (2010). Cranioectodermal dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. American Journal of Human Genetics, 86(6), 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware, S. M. , Aygun, M. G. , & Hildebrandt, F. (2011). Spectrum of clinical diseases caused by disorders of primary cilia. Proceedings of the American Thoracic Society, 8(5), 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzensen, I. M. , Johnston, J. J. , Keppler‐Noreuil, K. , Acrich, K. , David, K. , Johnson, K. D. , … Biesecker, L. G. (2015). Exome sequencing identifies novel mutations in C5orf42 in patients with Joubert syndrome with oral‐facial‐digital anomalies. Human Genome Variation, 2, 15045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, M. , Uehara, T. , Suzuki, H. , Takenouchi, T. , Fukushima, H. , Morisada, N. , … Kosaki, K. (2019). IFT172 as the 19th gene causative of oral‐facial‐digital syndrome. American Journal of Medical Genetics. Part A, 179(12), 2510–2513. [DOI] [PubMed] [Google Scholar]
- Zaki, M. S., Sattar, S., Massoudi, R. A., & Gleeson, J. G. (2011). Co‐occurrence of distinct ciliopathy diseases in single families suggests genetic modifiers. American Journal of Medical Genetics Part A, 155(12), 3042–3049. 10.1002/ajmg.a.34173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Taylor, S. P. , Ennis, H. A. , Forlenza, K. N. , Duran, I. , Li, B. , … Cohn, D. H. (2018). Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Human Mutation, 39(1), 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Details for genetic variants detected in our patient cohort.