

Abstract
Pompe disease is an inherited disorder caused by disease‐associated variants in the acid α‐glucosidase gene (GAA). The Pompe disease GAA variant database (http://www.pompevariantdatabase.nl) is a curated, open‐source, disease‐specific database, and lists disease‐associated GAA variants, in silico predictions, and clinical phenotypes reported until 2016. Here, we provide an update to include 226 disease‐associated variants that were published until 2020. We also listed 148 common GAA sequence variants that do not cause Pompe disease. GAA variants with unknown severity that were identified only in newborn screening programs were listed as a new feature to indicate the reason why phenotypes were still unknown. Expression studies were performed for common missense variants to predict their severity. The updated Pompe disease GAA variant database now includes 648 disease‐associated variants, 26 variants from newborn screening, and 237 variants with unknown severity. Regular updates of the Pompe disease GAA variant database will be required to improve genetic counseling and the study of genotype–phenotype relationships.
Keywords: database, disease‐associated variants, GAA, NBS, Pompe disease, SNP
This article describes an update of the Pompe disease GAA variant database (http://www.pompevariantdatabase.nl) providing new variants and findings related to the genetics of Pompe disease. The update includes genotypes and phenotypes obtained from clinical and/or research studies published between 2016 and 2020, as well as data from large registries and newborn screening programs. Variants that were already present in the previous version of the database have been retrospectively analyzed and updated. This information is freely accessible for clinicians, councilors and researchers in an open‐access database in order to improve the diagnostic process and to aid in decision making on the treatment of patients with Pompe disease worldwide.
1. INTRODUCTION
Pompe disease (glycogen storage disease type II; MIM #232300) is an autosomal recessive disorder caused by disease‐associated variants in the acid α‐glucosidase (GAA) gene, resulting in a deficiency of the GAA enzyme, accumulation of lysosomal glycogen, and progressive muscle weakness. The clinical spectrum of Pompe disease is broad (Güngör & Reuser, 2013). The most severe classic infantile phenotype presents shortly after birth with hypertrophic cardiomyopathy and generalized muscle weakness. These patients die in the first year of life due to cardiorespiratory insufficiency if left untreated. The slower progressing phenotype is characterized by muscle weakness that can appear at any age from <1 year into adulthood. These patients are generally spared from cardiac symptoms (Kohler et al., 2018; van der Ploeg & Reuser, 2008). Enzyme replacement therapy (ERT) with intravenously applied recombinant human GAA is available since 2006. ERT normalizes hypertrophic cardiomyopathy, improves motor function, and extends survival.
The differences between phenotypes in Pompe disease can, in part, be attributed to the severity of the disease‐associated variants present in the GAA gene. Classic infantile patients carry two disease‐associated variants that completely disrupt the function of GAA (i.e., null alleles). This group of patients can be subdivided based on their cross‐reactive immunological material (CRIM) status, which is defined by the disease‐associated variants involved. When two GAA variants are present that do not result in GAA protein expression, the patient is classified as CRIM‐negative. When at least one GAA variant gives rise to GAA protein expression (in which the GAA protein can be enzymatically inactive), the patient is classified as CRIM‐positive. The clinical importance of CRIM status is highlighted by the fact that CRIM‐negative classic infantile patients have a poorer prognosis compared with CRIM‐positive classic infantile patients, possibly due to the formation of high sustained anti‐GAA antibody titers upon treatment with ERT (Bali et al., 2012; van Gelder et al., 2015). Patients who do not have the classic infantile phenotype carry at least one disease‐associated variant that allows some residual enzymatic activity. These patients are, by definition, CRIM‐positive (Kroos et al., 2012b; Kulessa et al., 2020).
The “Pompe disease GAA variant database” (http://www.pompevariantdatabase.nl) is an open‐access database that lists and classifies all reported variants in the GAA gene. We recently revised this database to include clinical data from patients collected from the literature, adapted the classification system for variant severity, and added (predicted) CRIM status for disease‐associated variants. The database included literature up to May 2016, resulting in a total of 561 variants (Niño et al., 2019). In recent years, many new patients and GAA variants have been reported. These include findings from large patient populations, such as the French nationwide study (246 patients with late‐onset Pompe disease) and the Pompe registry (1079 patients from 26 countries; Reuser et al., 2019; Semplicini et al., 2018).
In addition, various countries, including Taiwan, the United States, Italy, Brazil, and Japan, have implemented newborn screening (NBS) programs for Pompe disease, resulting in an increase of variants of unknown significance (VUS; Bravo et al., 2017; Burlina et al., 2018; Chien et al., 2019; Elliott et al., 2016; Momosaki et al., 2019; Yang et al., 2014). For variants associated with late onset, the associated phenotypes from NBS cases are still unknown as symptom onset could, in principle, be delayed until (late) adulthood. It will be important to monitor the onset and progress of symptoms in patients identified via NBS programs closely to determine the severity of the newly identified genetic variants.
Public databases, such as dbSNP (https://www.ncbi.nlm.nih.gov/snp) and gnomAD (https://gnomad.broadinstitute.org), provide a source of variants that have been detected in various genome‐wide studies (Karczewski et al., 2020; Sherry et al., 2001). A large percentage of these variants represent common sequence variants that have a minor allele frequency (MAF) ≥ 1%. Several of these variants have already been reported for the GAA gene and have been ruled out to cause Pompe disease (Kroos et al., 2007; Labrousse et al., 2010; Turaça et al., 2015). However, most of the common sequence variants in these databases are listed as VUSs and may lead to misinterpretation during molecular diagnostics.
In this study, we provide an update of the Pompe disease GAA variant database with variants and patients described in the literature up to January 2020. We included information on novel GAA variants that were identified via NBS and for which no phenotype was yet known. Known common sequence variants in the GAA gene that do not cause Pompe disease have now also been added to prevent misdiagnosis. In addition, selected common missense variants were tested in expression studies and also this information was added to the updated database. The database provides a curated up‐to‐date reference source for the molecular diagnosis of Pompe disease.
2. METHODS
The Pompe disease GAA variant database is publicly available at http://www.pompevariantdatabase.nl. The previous version of the database included literature until 2016; the update described here contains variants from publications up to January 2020. Additionally, NBS studies that screened for Pompe disease were now included if the authors provided the genotypes of the described cases. Novel variants were analyzed as described in Niño et al. (2019). Variants were annotated based on the reference sequences NM_000152.3 for GAA messenger RNA (mRNA), LRG_673 genomic sequence for describing variants in intronic sequences, and NP_000143.2 for GAA protein. Exon annotations were based on the human genomic build (GRCH37/hg19) for exons 2–20; however, changes were made to the annotation of exon 1 to reflect the findings of (GRCH38/hg38). Within this region, a new 195‐bp intron was identified at positions c.−112 and c.−113. Therefore, the region that was previously annotated as exon 1 has been split between exons 1A and 1B, which are separated by intron 1A. Intron 1 has been renamed to intron 1B. This numbering was made to maintain the same numbering of subsequent exons compared with existing literature.
Common sequence variants in the GAA gene (hg38 Chr17:80,101,556‐80,119,881) were extracted from gnomAD and were categorized as “not disease‐associated.” Combined Annotation‐Dependent Depletion (CADD) in silico predictions were performed using the CADD (https://cadd.gs.washington.edu) platform, which compiles different tools for analysis of intronic insertion and deletion variants (Rentzsch et al., 2019). The MAF and CADD scores were obtained in April 2020. Predictions of effect on pre‐mRNA splicing were performed using Alamut Visual v.2.15 (Interactive Biosoftware).
Functional studies were performed using site‐directed mutagenesis (SDM) to generate complementary DNA (cDNA) expression constructs containing the missense variant of interest as described (in 't Groen et al., 2020). The activity of the GAA protein produced by the constructs was measured using 4‐methylumbelliferyl‐α‐d‐glucopyranoside (4‐MU) as a substrate in transfected COS‐7 cells, as described in Kroos et al. (2008). Statistical analysis was performed using one‐way analysis of variance with Tukey honestly significant difference post hoc multiple testing corrections. p < .05 was considered significant.
3. RESULTS AND DISCUSSION
Table 1 provides an overview of the novel variants. We performed a literature search covering the past 4 years and identified 80 publications (listed in the updated database and Table S1) that described 350 novel variants, of which 226 were considered to be disease‐associated (Table 1 and Figure 1a). Seventy‐six novel variants (33%) were present in combination with a null allele, which allowed prediction of the clinical severity of these variants (Table 1 and Figure 1b). In addition, the inclusion of new patient information allowed us to classify the severity of 55 variants that were already present in the database. This resulted in a new total of 911 GAA variants, of which 648 were disease‐associated (71%). In total, 336 out of 648 disease‐associated variants (52%) could be associated with a clinical phenotype. The geographical or ethnical distribution of reported patients remained similar to what was described previously. The majority of patients had a Caucasian background or were of Caucasian descent (data not shown). This introduces a bias in the current version of the database and indicates the necessity of extending the database to patients of other descent. Mapping of missense variants to GAA protein domains revealed an even stronger enrichment in the catalytic core compared with the mapping we performed previously (Niño et al., 2019; Figure 1c).
Table 1.
Novel disease‐associated variants added to the Pompe variant database
| DNA nomenclature | Phenotype combined with a null allele | DNA nomenclature | Phenotype combined with a null allele |
|---|---|---|---|
| Ch37/hg19 chr17:78,059,821_ 78,076,592del | Unknown (disease‐associated) | c.1057C>T | Unknown (disease‐associated) |
| c.−113+2T>A | Unknown (disease‐associated) | c.1057del | Unknown (disease‐associated) |
| c.−32‐17_−32‐10delins(30) | Classic infantile | c.1099T>G | Unknown (disease‐associated) |
| c.−32‐1G>C | Unknown (disease‐associated) | c.1106T>A | Unknown (disease‐associated) |
| c.40_47del | Classic infantile | c.1109G>A | Unknown (disease‐associated) |
| c.104T>C | Classic infantile | c.1114C>G | Unknown (disease‐associated) |
| c.169C>T | Classic infantile | c.1114C>T | Unknown (disease‐associated) |
| c.205C>T | Unknown (disease‐associated) | c.1121G>A | Unknown (disease‐associated) |
| c.258C>A | Unknown (disease‐associated) | c.1127_1130del | Unknown (disease‐associated) |
| c.265C>T | Unknown (disease‐associated) | c.1129G>A | Unknown (disease‐associated) |
| c.295_314del | Unknown (disease‐associated) | c.1153del | Unknown (disease‐associated) |
| c.323G>C | Unknown (disease‐associated) | c.1192del | Unknown (disease‐associated) |
| c.365del | Unknown (disease‐associated) | c.1193del | Unknown (disease‐associated) |
| c.380G>A | Unknown (disease‐associated) | c.1201C>A | Unknown (disease‐associated) |
| c.397T>G | Unknown (disease‐associated) | c.1209C>A | Unknown (disease‐associated) |
| c.437del | Classic infantile | c.1211A>C | Unknown (disease‐associated) |
| c.445A>C | Unknown (disease‐associated) | c.1211A>T | Classic infantile |
| c.484A>C | Classic infantile | c.1212C>G | Unknown (disease‐associated) |
| c.502C>T | Unknown (disease‐associated) | c.1216G>A | Childhood |
| c.505C>A | Unknown (disease‐associated) | c.1219T>C | Unknown (disease‐associated) |
| c.517_519del | Childhood | c.1221C>A | Classic infantile |
| c.541_545del | Classic infantile | c.1221del | Unknown (disease‐associated) |
| c.547‐1G>C | Unknown (disease‐associated) | c.1226_1227insG | Classic infantile |
| c.568C>T | Unknown (disease‐associated) | c.1231del | Unknown (disease‐associated) |
| c.665T>G | Classic infantile | c.1240T>C | Unknown (disease‐associated) |
| c.686G>C | Unknown (disease‐associated) | c.1241del | Classic infantile |
| c.691C>T | Unknown (disease‐associated) | c.1242C>A | Unknown (disease‐associated) |
| c.692T>C | Unknown (disease‐associated) | c.1249A>C | Unknown (disease‐associated) |
| c.692+1G>T | Unknown (disease‐associated) | c.1281G>T | Classic infantile |
| c.693‐2A>C | Classic infantile | c.1292_1295dup | Classic infantile |
| c.693‐1G>C | Unknown (disease‐associated) | c.1293_1326+57del | Unknown (disease‐associated) |
| c.715_716del | Unknown (disease‐associated) | c.1298A>C | Classic infantile |
| c.730C>T | Classic infantile | c.1311_1312ins(26) | Classic infantile |
| c.736del | Unknown (disease‐associated) | c.1320_1322del | Classic infantile |
| c.756_757insT | Unknown (disease‐associated) | c.1327‐54_1437+178del | Classic infantile |
| c.759del | Unknown (disease‐associated) | c.1358_1361del | Classic infantile |
| c.766_784del | Unknown (disease‐associated) | c.1378G>T | Unknown (disease‐associated) |
| c.781G>A | Classic infantile | c.1388_1406del | Unknown (disease‐associated) |
| c.784G>C | Unknown (disease‐associated) | c.1396dup | Unknown (disease‐associated) |
| c.796C>A | Childhood | c.1402A>T | Unknown (disease‐associated) |
| c.799_803delinsA | Unknown (disease‐associated) | c.1409A>G | Unknown (disease‐associated) |
| c.837G>C | Unknown (disease‐associated) | c.1431del | Classic infantile |
| c.841C>T | Unknown (disease‐associated) | c.1441del | Unknown (disease‐associated) |
| c.876C>G | Classic infantile | c.1447G>T | Unknown (disease‐associated) |
| c.878G>T | Unknown (disease‐associated) | c.1456G>T | Unknown (disease‐associated) |
| c.883C>A | Unknown (disease‐associated) | c.1464dup | Classic infantile |
| c.930_932del | Classic infantile | c.1470C>A | Childhood |
| c.942C>A | Unknown (disease‐associated) | c.1477C>T | Unknown (disease‐associated) |
| c.947A>G | Classic infantile | c.1493G>A | Classic infantile |
| c.950C>T | Unknown (disease‐associated) | c.1501_1515del | Unknown (disease‐associated) |
| c.955+1G>A | Classic infantile | c.1507del | Classic infantile |
| c.971dup | Classic infantile | c.1526A>T | Unknown (disease‐associated) |
| c.982_988del | Classic infantile | c.1531C>A | Unknown (disease‐associated) |
| c.983T>C | Classic infantile | c.1537G>A | Unknown (disease‐associated) |
| c.994_995insTT | Unknown (disease‐associated) | c.1538A>G | Classic infantile |
| c.1000G>T | Classic infantile | c.1551+3A>T | Unknown (disease‐associated) |
| c.1004_1005dup | Unknown (disease‐associated) | c.1551+5G>A | Unknown (disease‐associated) |
| c.1047del | Unknown (disease‐associated) | c.1559A>G | Unknown (disease‐associated) |
| c.1560C>G | Unknown (disease‐associated) | c.2096T>C | Unknown (disease‐associated) |
| c.1579_1580del | Classic infantile | c.2109del | Unknown (disease‐associated) |
| c.1583G>C | Unknown (disease‐associated) | c.2131A>C | Classic infantile |
| c.1594G>A | Adult | c.2146G>C | Unknown (disease‐associated) |
| c.1597T>G | Classic infantile | c.2153_2156delinsACGCCG | Classic infantile |
| c.1602_1605delinsAGG | Classic infantile | c.2182_2183del | Unknown (disease‐associated) |
| c.1610del | Unknown (disease‐associated) | c.2190‐345A>G | Unknown (disease‐associated) |
| c.1627T>G | Unknown (disease‐associated) | c.2205dup | Classic infantile |
| c.1629C>G | Unknown (disease‐associated) | c.2213G>A | Classic infantile |
| c.1636G>C | Unknown (disease‐associated) | c.2221G>A | Classic infantile |
| c.1636+5G>A | Classic infantile | c.2222A>T | Unknown (disease‐associated) |
| c.1650del | Unknown (disease‐associated) | c.2234T>C | Classic infantile |
| c.1657C>T | Classic infantile | c.2235dup | Classic infantile |
| c.1681_1699dup | Unknown (disease‐associated) | c.2237G>T | Unknown (disease‐associated) |
| c.1688A>T | Unknown (disease‐associated) | c.2240G>A | Unknown (disease‐associated) |
| c.1716C>A | Classic infantile | c.2261dup | Unknown (disease‐associated) |
| c.1721T>C | Unknown (disease‐associated) | c.2294G>A | Classic infantile |
| c.1753_2799del | Classic infantile | c.2296T>A | Classic infantile |
| c.1754+1dup | Unknown (disease‐associated) | c.2297A>C | Classic infantile |
| c.1754+2T>C | Unknown (disease‐associated) | c.2304del | Unknown (disease‐associated) |
| c.1780C>T | Unknown (disease‐associated) | c.2320G>A | Unknown (disease‐associated) |
| c.1784C>T | Unknown (disease‐associated) | c.2331+5G>C | Classic infantile |
| c.1799G>C | Unknown (disease‐associated) | c.2331+102del | Unknown (disease‐associated) |
| c.1822del | Unknown (disease‐associated) | c.2334_2335dup | Unknown (disease‐associated) |
| c.1825T>G | Unknown (disease‐associated) | c.2377_2378insAC | Classic infantile |
| c.1835A>C | Unknown (disease‐associated) | c.2380dup | Unknown (disease‐associated) |
| c.1835A>G | Unknown (disease‐associated) | c.2395C>T | Unknown (disease‐associated) |
| c.1837T>G | Unknown (disease‐associated) | c.2407C>T | Unknown (disease‐associated) |
| c.1839G>C | Unknown (disease‐associated) | c.2411G>A | Classic infantile |
| c.1844_1846del | Unknown (disease‐associated) | c.2459_2461del | Unknown (disease‐associated) |
| c.1844G>T | Classic infantile | c.2460dup | Unknown (disease‐associated) |
| c.1844G>A | Classic infantile | c.2474C>G | Unknown (disease‐associated) |
| c.1847dup | Unknown (disease‐associated) | c.2480A>G | Unknown (disease‐associated) |
| c.1859C>A | Unknown (disease‐associated) | c.2515C>T | Unknown (disease‐associated) |
| c.1879_1881del | Classic infantile | c.2537C>A | Unknown (disease‐associated) |
| c.1888+2_1888+15del | Classic infantile | c.2544del | Unknown (disease‐associated) |
| c.1895T>C | Unknown (disease‐associated) | c.2563G>C | Classic infantile |
| c.1895T>G | Classic infantile | c.2578G>A | Unknown (disease‐associated) |
| c.1903A>G | Unknown (disease‐associated) | c.2584G>A | Childhood |
| c.1913G>A | Classic infantile | c.2585del | Classic infantile |
| c.1944_1950del | Unknown (disease‐associated) | c.2596del | Unknown (disease‐associated) |
| c.1952dup | Unknown (disease‐associated) | c.2619C>G | Unknown (disease‐associated) |
| c.1961C>G | Unknown (disease‐associated) | c.2636T>C | Classic infantile |
| c.2004C>A | Unknown (disease‐associated) | c.2655_2656del | Unknown (disease‐associated) |
| c.2015G>T | Unknown (disease‐associated) | c.2716G>A | Unknown (disease‐associated) |
| c.2020C>G | Unknown (disease‐associated) | c.2720T>C | Unknown (disease‐associated) |
| c.2020C>T | Unknown (disease‐associated) | c.2725G>A | Unknown (disease‐associated) |
| c.2024A>G | Classic infantile | c.2740dup | Unknown (disease‐associated) |
| c.2040+2dup | Unknown (disease‐associated) | c.2742dup | Classic infantile |
| c.2040+29_2190‐270del | Classic infantile | c.2757del | Unknown (disease‐associated) |
| c.2041‐2A>G | Classic infantile | c.2799+5G>A | Unknown (disease‐associated) |
| c.2051C>A | Unknown (disease‐associated) | c.2800‐1G>C | Classic infantile |
| c.2051C>G | Unknown (disease‐associated) | c.2843dup | Classic infantile |
| c.2051C>T | Classic infantile | c.2845_2847del | Unknown (disease‐associated) |
| c.2056_2057delinsCC | Unknown (disease‐associated) | ||
| c.2084dup | Unknown (disease‐associated) |
Figure 1.
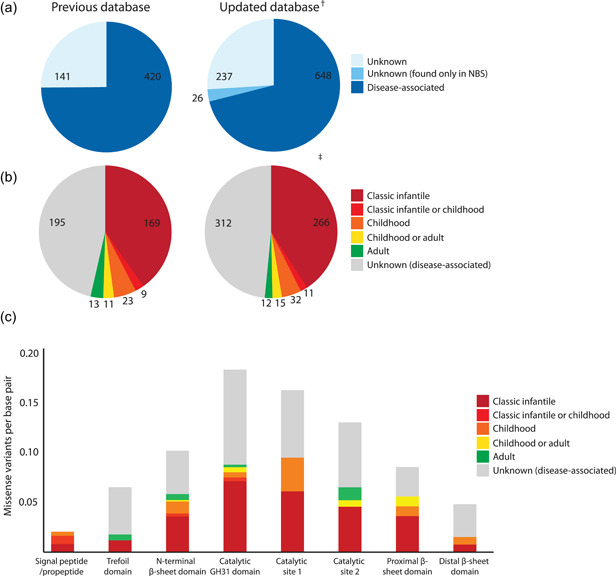
Overview of variants, comparing the previous (Niño et al., 2019) and updated version of the Pompe disease GAA variant database (http://www.pompevariantdatabase.nl). (a) Number of disease‐associated and unknown variants in the previous database (left) and the updated version of the database (right). (b) Number of disease‐associated variants classified based on the predicted clinical phenotype when combined with a null allele in the previous database (left) and in the updated version of the database (right). (c) Distribution of disease‐associated missense variants listed in the updated database, based on the protein domains of GAA and the predicted clinical phenotype when combined with a null allele. Numbers are corrected for the length of each domain. †Two entries in the previous version of the database were removed as the variants were described twice using different nomenclatures. ‡For 36 variants listed in the previous version of the database, a reclassification of the phenotypic severity was performed due to the addition of novel patients included in this update
We included in the current version of the database common sequence variants that have a MAF ≥ 1% and do not cause Pompe disease. This resulted in a relative increase in the number of nondisease‐associated variants (Table 2). We decided to include common sequence variants in response to the misreporting of these variants as the principal cause of disease in several patients. Examples of this are the c.547‐67C>G (rs8069491) and 547‐39T>G (rs12452721) variants, which were reported as the cause of disease while having an allele frequency of 67% in the global population (Bekircan‐Kurt et al., 2017; Guevara‐Campos et al., 2019). In total, the database now includes 148 variants with a MAF ≥ 1%. All variants had a low CADD score (<10; Table 2) and were classified as “unknown.” We note that while these common sequence variants do not result in clinical manifestation of Pompe disease, it remains possible that they might modify disease progression when present in cis with a disease‐associated variant. In Pompe disease, this is the case for the Asian pseudodeficiency allele (c.[1726G>A (p.Gly576Ser);2065G>A (p.Glu689Lys)]) and GAA2 (c.271G>A, (p.Asp91Asn)), which have a MAF of 14% for c.1726G>A, 23.5% for c.2065G>A (both East Asian), and 3.2% for GAA2 (European), and can be present in cis with known disease‐associated variants (Kroos et al., 2006; Labrousse et al., 2010). Also, a variant with a low MAF in the general population, c.510C>T (p.=) (rs564758226), is known to be linked to the late‐onset variant c.−32‐13T>G (p.[=,0]) (IVS1). c.510C>T has a global MAF of 0.005%, but a MAF of 27.3% in compound heterozygous IVS1 patients with symptom onset at childhood. It worsens aberrant splicing caused by IVS1 and causes lower levels of leaky wild‐type splicing and lower GAA enzyme activity, resulting in accelerated disease onset (Bergsma et al., 2019).
Table 2.
List of common sequence variants located within the boundaries of the GAA gene
| Location | Variant | Variant ID | Global allele frequency (GnomAD) | Predictions of pre‐mRNA splicing | CADD score PHRED |
|---|---|---|---|---|---|
| Exon 1A, 5ʹ UTR | c.−338C>G | rs144639114 | 2% | No effect on splicing | 6.524 |
| Exon 1A, 5ʹ UTR | c.−260G>C | rs2304849 | 16% | No effect on splicing | 8.996 |
| Exon 1A, 5ʹ UTR | c.−178G>A | rs77514632 | 2% | No effect on splicing | 9.948 |
| Exon 1B, 5ʹ UTR | c.−75C>G | rs80020206 | 0.9% (3% in African population) | No effect on splicing | 9.989 |
| Intron 1B | c.−33+219G>C | rs4889961 | 75% | No effect on splicing | 0.866 |
| Intron 1B | c.−33+316C>A | rs8077055 | 20% | No effect on splicing | 9.079 |
| Intron 1B | c.−33+317C>T | rs8077056 | 20% | No effect on splicing | 8.579 |
| Intron 1B | c.−33+671A>C | rs55751636 | 31% | No effect on splicing | 1.456 |
| Intron 1B | c.−33+757G>A | rs28413147 | 5% | No effect on splicing | 4.974 |
| Intron 1B | c.−33+903A>C | rs12450199 | 34% | No effect on splicing | 8.196 |
| Intron 1B | c.−33+1104A>G | rs11150841 | 75% | No effect on splicing | 6.976 |
| Intron 1B | c.−33+1172G>A | rs1442315 | 5% | No effect on splicing | 0.064 |
| Intron 1B | c.−33+1190G>T | rs12602593 | 10% | No effect on splicing | 1.784 |
| Intron 1B | c.−33+1309T>C | rs1442314 | 76% | No effect on splicing | 1.752 |
| Intron 1B | c.−32‐1298G>C | rs12602610 | 33% | No effect on splicing | 2.604 |
| Intron 1B | c.−32‐1124C>T | rs58959690 | 20% | No effect on splicing | 5.825 |
| Intron 1B | c.−32‐884T>C | rs145362066 | 0.9% (3% in African population) | No effect on splicing | 3.993 |
| Intron 1B | c.−32‐793C>G | rs55666739 | 2% | No effect on splicing | 4.041 |
| Intron 1B | c.−32‐721G>C | rs75754966 | 2% | Generates a new cryptic splice accepter site | 1.008 |
| Intron 1B | c.−32‐686A>G | rs147264695 | 0.3% (1% in Finnish population) | No effect on splicing | 4.349 |
| Intron 1B | c.−32‐640C>T | rs12600845 | 51% | No effect on splicing | 0.136 |
| Intron 1B | c.−32‐521G>T | rs115060925 | 1% | Generates a new cryptic splice donor site | 0.639 |
| Intron 1B | c.−32‐494C>G | rs140325572 | 2% | No effect on splicing | 0.036 |
| Intron 1B | c.−32‐462G>A | rs74003606 | 5% | No effect on splicing | 0.226 |
| Exon 2 | c.271G>A | rs1800299 | 2% | No effect on splicing | 0.256 |
| Exon 2 | c.324T>C | rs1800300 | 72% | No effect on splicing | 8.391 |
| Exon 2 | c.447G>A | rs2289536 | 0.5% (3% in East Asian population) | No effect on splicing | 1.252 |
| Intron 2 | c.546+293G>A | rs34746710 | 20% | No effect on splicing | 1.899 |
| Intron 2 | c.547‐243C>G | rs8065426 | 67% | No effect on splicing | 2.529 |
| Intron 2 | c.547‐238T>C | rs12452263 | 20% | No effect on splicing | 5.667 |
| Intron 2 | c.547‐67C>G | rs8069491 | 67% | No effect on splicing | 1.337 |
| Intron 2 | c.547‐39T>G | rs12452721 | 67% | Loss of cryptic splice donor site | 2.78 |
| Intron 2 | c.547‐4C>G | rs3816256 | 67% | No effect on splicing | 4.721 |
| Exon 3 | c.596A>G | rs1042393 | 67% | No effect on splicing | 0.548 |
| Exon 3 | c.642C>T | rs1800301 | 18% | No effect on splicing | 1.805 |
| Exon 3 | c.668G>A | rs1042395 | 67% | No effect on splicing | 1.46 |
| Intron 3 | c.692+38C>T | rs2304848 | 3% | Generates a new cryptic splice donor site | 5.574 |
| Intron 3 | c.692+144A>G | rs2304847 | 67% | No effect on splicing | 3.653 |
| Intron 3 | c.692+509T>C | rs8082405 | 66% | No effect on splicing | 3.271 |
| Intron 3 | c.692+674G>C | rs8078350 | 67% | No effect on splicing | 4.501 |
| Intron 3 | c.692+751T>C | rs8068051 | 67% | No effect on splicing | 2.363 |
| Intron 3 | c.693‐586G>A | rs112308142 | 3% | No effect on splicing | 2.71 |
| Intron 3 | c.693‐585T>C | rs8068555 | 67% | No effect on splicing | 4.133 |
| Intron 3 | c.693‐559C>T | rs12602422 | 67% | No effect on splicing | 1.879 |
| Intron 3 | c.693‐491G>A | rs12948631 | 67% | No effect on splicing | 3.629 |
| Intron 3 | c.693‐441C>G | rs12602440 | 67% | Loss of a cryptic splice acceptor site | 7.559 |
| Intron 3 | c.693‐434C>A | rs12941269 | 66% | No effect on splicing | 4.416 |
| Intron 3 | c.693‐414C>G | rs12941289 | 66% | Loss of a cryptic splice acceptor site | 0.077 |
| Intron 3 | c.693‐413A>G | rs12937590 | 67% | Loss of a cryptic splice acceptor site | 1.544 |
| Intron 3 | c.693‐216T>A | rs11150844 | 67% | No effect on splicing | 4.13 |
| Intron 3 | c.693‐94C>T | rs79849256 | 0.2% (3% in East Asian population) | No effect on splicing | 9.666 |
| Intron 3 | c.693‐78C>T | rs74003611 | 6% | No effect on splicing | 0.06 |
| Intron 3 | c.693‐49C>T | rs78855075 | 7% | No effect on splicing | 2.374 |
| Exon 4 | c.852G>A | rs142626724 | 0.6% (1% in European population) | No effect on splicing | 1.095 |
| Intron 4 | c.858+30T>C | rs2304845 | 66% | No effect on splicing | 0.067 |
| Exon 5 | c.921A>T | rs1800303 | 8% | No effect on splicing | 9.101 |
| Intron 5 | c.955+12G>A | rs2252455 | 69% | No effect on splicing | 0.981 |
| Intron 5 | c.955+155C>A | rs9901190 | 5% | No effect on splicing | 7.196 |
| Intron 5 | c.955+167C>T | rs77717164 | 0.7% (6% in East Asian population) | No effect on splicing | 6.348 |
| Intron 5 | c.956‐107G>A | rs2241888 | 73% | No effect on splicing | 5.835 |
| Intron 5 | c.956‐84C>T | rs2241887 | 67% | No effect on splicing | 0.061 |
| Intron 6 | c.1075+13C>T | rs41292402 | 1% | No effect on splicing | 7.496 |
| Exon 8 | c.1203G>A | rs1800304 | 67% | No effect on splicing | 5.972 |
| Exon 8 | c.1286A>G | rs200294882 | 0.07% (1% in East Asian population) | Loss of cryptic splice acceptor site and generates a new cryptic splice donor site | 0.068 |
| Intron 8 | c.1326+132G>A | rs894306 | 67% | No effect on splicing | 1.999 |
| Intron 8 | c.1326+459C>T | rs74679377 | 0.7% (6% in East Asian population) | No effect on splicing | 0.435 |
| Intron 8 | c.1326+460G>A | rs12150323 | 2% | No effect on splicing | 0.322 |
| Intron 8 | c.1327‐514G>A | rs72850826 | 5% | No effect on splicing | 1.914 |
| Intron 8 | c.1327‐356G>T | rs6565640 | 73% | No effect on splicing | 0.258 |
| Intron 8 | c.1327‐321del | rs140385114 | 7% | No effect on splicing | 0.888 |
| Intron 8 | c.1327‐269A>G | rs6565641 | 67% | No effect on splicing | 4.207 |
| Intron 8 | c.1327‐209C>T | rs76604157 | 0.3% (6% in East Asian population) | No effect on splicing | 0.471 |
| Intron 8 | c.1327‐179G>A | rs2278620 | 20% | No effect on splicing | 0.643 |
| Intron 8 | c.1327‐118A>G | rs74003628 | 7% | No effect on splicing | 0.184 |
| Intron 8 | c.1327‐18A>G | rs2278619 | 72% | No effect on splicing | 0.124 |
| Exon 9 | c.1374C>T | rs1800305 | 7% | No effect on splicing | 0.206 |
| Intron 9 | c.1438‐220A>G | rs2278618 | 67% | No effect on splicing | 6.607 |
| Intron 9 | c.1438‐108G>A | rs12944802 | 67% | No effect on splicing | 0.013 |
| Intron 9 | c.1438‐19G>C | rs2304844 | 67% | No effect on splicing | 3.529 |
| Intron 10 | c.1551+42G>A | rs115427918 | 0.9% (3% in African population) | No effect on splicing | 5.792 |
| Intron 10 | c.1551+49C>A | rs2304843 | 67% | No effect on splicing | 7.131 |
| Exon 11 | c.1581G>A | rs1042396 | 23% | No effect on splicing | 6.758 |
| Intron 11 | c.1636+43G>T | rs2304842 | 5% | Generates a new cryptic splice accepter site | 6.859 |
| Intron 11 | c.1636+117del | rs199788201 | 59% | No effect on splicing | 0.045 |
| Intron 11 | c.1636+117C>T | rs12945868 | 11% | No effect on splicing | 0.181 |
| Intron 11 | c.1636+118G>T | rs4889817 | 59% | No effect on splicing | 3.161 |
| Intron 11 | c.1636+205C>T | rs79673008 | 3% | No effect on splicing | 0.013 |
| Intron 11 | c.1636+210G>A | rs79487884 | 5% | No effect on splicing | 1.463 |
| Intron 11 | c.1636+269C>T | rs111625854 | 2% | No effect on splicing | 3.828 |
| Intron 11 | c.1636+284G>C | rs111551014 | 2% | No effect on splicing | 1.81 |
| Intron 11 | c.1636+389C>G | rs7221675 | 63% | No effect on splicing | 0.573 |
| Intron 11 | c.1636+390A>G | rs7209921 | 63% | No effect on splicing | 1.829 |
| Intron 11 | c.1636+404A>G | rs4889818 | 74% | No effect on splicing | 1.902 |
| Intron 11 | c.1637‐185A>G | rs12951255 | 55% | No effect on splicing | 0.576 |
| Exon 12 | c.1726G>A | rs1800307 | 2% | Generates a new cryptic splice acceptor | 0.268 |
| Intron 12 | c.1754+12G>A | rs2304840 | 6% | No effect on splicing | 4.325 |
| Intron 12 | c.1754+100C>T | rs113688685 | 0.9% (3% in African population) | No effect on splicing | 8.142 |
| Intron 12 | c.1754+104C>G | rs2304839 | 5% | No effect on splicing | 0.763 |
| Intron 12 | c.1754+144C>T | rs2304838 | 61% | No effect on splicing | 1.787 |
| Intron 12 | c.1755‐186A>G | rs62075593 | 2% | No effect on splicing | 2.032 |
| Intron 13 | c.1888+21G>A | rs2304837 | 6% | No effect on splicing | 3.378 |
| Intron 14 | c.2040+20A>G | rs2304836 | 72% | No effect on splicing | 2.163 |
| Intron 14 | c.2040+66C>T | rs2304835 | 7% | No effect on splicing | 3.54 |
| Intron 14 | c.2040+69A>G | rs2304834 | 6% | No effect on splicing | 0.027 |
| Intron 14 | c.2041‐64G>A | rs2304833 | 27% | No effect on splicing | 0.371 |
| Exon 15 | c.2065G>A | rs1800309 | 6% | No effect on splicing | 1.783 |
| Exon 15 | c.2133A>G | rs1800310 | 27% | No effect on splicing | 1.134 |
| Intron 15 | c.2189+95C>T | rs72850840 | 5% | No effect on splicing | 3,771 |
| Intron 15 | c.2189+263G>A | rs7221604 | 66% | Generates a new cryptic splice donor site | 0.563 |
| Intron 15 | c.2189+510T>G | rs4889963 | 5% | No effect on splicing | 1.444 |
| Intron 15 | c.2189+607G>A | rs112710614 | 7% | No effect on splicing | 0.189 |
| Intron 15 | c.2189+616T>C | rs139307163 | 5% | No effect on splicing | 1.94 |
| Intron 15 | c.2189+723G>A | rs4889819 | 20% | No effect on splicing | 0.367 |
| Intron 15 | c.2189+729A>G | rs74737410 | 5% | No effect on splicing | 0.498 |
| Intron 15 | c.2189+859A>G | rs4889964 | 5% | No effect on splicing | 1.503 |
| Intron 15 | c.2189+884G>A | rs4889965 | 5% | No effect on splicing | 0.355 |
| Intron 15 | c.2189+1153A>G | rs72850844 | 5% | No effect on splicing | 3.687 |
| Intron 15 | c.2189+1201C>A | rs72850846 | 5% | No effect on splicing | 2.352 |
| Intron 15 | c.2189+1208A>G | rs72850847 | 5% | No effect on splicing | 0.367 |
| Intron 15 | c.2189+1263A>G | rs74700450 | 5% | No effect on splicing | 2.97 |
| Intron 15 | c.2189+1290A>G | rs74003630 | 5% | No effect on splicing | 6.015 |
| Intron 15 | c.2189+1600C>T | rs60668271 | 5% | No effect on splicing | 0.481 |
| Intron 15 | c.2190‐1531G>A | rs74702528 | 0.9% (3% in African population) | No effect on splicing | 0.489 |
| Intron 15 | c.2190‐1463G>A | rs116416508 | 0.9% (3% in African population) | No effect on splicing | 0.328 |
| Intron 15 | c.2190‐1139A>G | rs184803352 | 0.7% (2% in African population | No effect on splicing | 0.095 |
| Intron 15 | c.2190‐1005A>G | rs4889820 | 5% | No effect on splicing | 2.452 |
| Intron 15 | c.2190‐686G>A | rs12452616 | 19% | No effect on splicing | 2.725 |
| Intron 15 | c.2190‐647G>A | rs59362713 | 10% | No effect on splicing | 0.227 |
| Intron 15 | c.2190‐536G>A | rs60429724 | 10% | No effect on splicing | 0.454 |
| Intron 15 | c.2190‐490G>A | rs111477580 | 1% | No effect on splicing | 3.101 |
| Intron 15 | c.2190‐444A>G | rs4889967 | 73% | No effect on splicing | 1.059 |
| Intron 15 | c.2190‐336C>T | rs76178719 | 3% | No effect on splicing | 1.566 |
| Intron 16 | c.2331+20G>A | rs2304832 | 75% | No effect on splicing | 5.346 |
| Intron 16 | c.2331+24T>C | rs2304831 | 15% | No effect on splicing | 0.204 |
| Intron 16 | c.2331+151C>T | rs111537160 | 2% | No effect on splicing | 0.608 |
| Intron 16 | c.2332‐198A>T | rs2304830 | 73% | No effect on splicing | 3.363 |
| Exon 17 | c.2338G>A | rs1126690 | 72% | No effect on splicing | 2.675 |
| Exon 17 | c.2446G>A | rs1800314 | 5% | No effect on splicing | 5.793 |
| Intron 17 | c.2482‐132C>T | rs113824706 | 0.9% (3% in African population) | No effect on splicing | 0.066 |
| Exon 18 | c.2553G>A | rs1042397 | 57% | Weakens a cryptic splice donor site | 1.241 |
| Intron 18 | c.2647‐71G>C | rs4889821 | 5% | No effect on splicing | 3.473 |
| Exon 19 | c.2780C>T | rs1800315 | 2% | No effect on splicing | 0.222 |
| Intron 19 | c.2800‐227C>G | rs9890469 | 66% | No effect on splicing | 0.661 |
| Intron 19 | c.2800‐60G>A | rs55662462 | 0.7% (11% in Latino population) | No effect on splicing | 2.209 |
| Exon 20, 3ʹ UTR | c.*3G>A | rs1800317 | 5% | No effect on splicing | 0.03 |
| Exon 20, 3ʹ UTR | c.*91G>A | rs2229221 | 12% | No effect on splicing | 6.887 |
| Exon 20, 3ʹ UTR | c.*223C>T | rs8132 | 22% | No effect on splicing | 3.025 |
| Exon 20, 3ʹ UTR | c.*419G>T | rs7567 | 19% | No effect on splicing | 4.17 |
Abbreviations: CADD, Combined Annotation‐Dependent Depletion; mRNA, messenger RNA; UTR, untranslated region.
Figure 2a,b shows the results on the GAA variants we subjected to a more in‐depth investigation. We selected the common missense variants c.307T>G (p.Cys103Gly), c.655G>A (p.Gly219Arg), c.670C>T (p.Arg224Trp), c.1655T>C (p.Leu552Pro), and c.1798C>T (p.Arg600Cys) and performed in vitro analysis of their severity using SDM of GAA cDNA expression constructs. In addition, c.1597T>C (p.Cys533Arg) and c.309C>G (p.Cys103Trp) were tested due to a request for diagnostic purposes. All of these variants fully abrogated GAA enzymatic activity following transfection in COS‐7 cells (Figure 2, compare mutant GAA with mock transfections). The c.309C>G variant was included because the patient that harbored this variant in combination with c.525del p.(Glu176Argfs*45) showed an atypical Pompe disease phenotype (Mori et al., 2017). This case report described an adult patient with cardiomyopathy. Molecular analysis of primary skin fibroblasts identified a reduction in GAA activity, although not at pathogenic levels, and GAA activity was in the normal range for skeletal muscle tissue (Mori et al., 2017). We note that the c.309C>G variant was not detected in DNA from either parent and was described as a de novo variant (Mori et al., 2017). This variant might have been introduced during embryonic development, resulting in mosaicism similar to, as described previously in Labrijn‐Marks et al. (2019) and in 't Groen et al. (2020). This might explain the “uneven pattern” of glycogen accumulation in histological sections derived from cardiac tissue (Mori et al., 2017). The in vitro analysis indicated that the c.309C>G variant is fully deleterious and has a predicted classic infantile phenotype in combination with a null allele. A comprehensive genetic analysis would be necessary to confirm this hypothesis.
Figure 2.
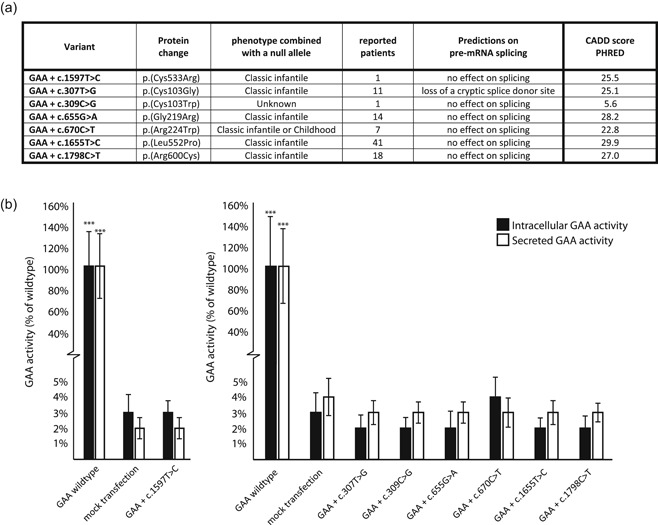
Expression study of seven disease‐associated missense variants in the GAA gene. (a) Overview of basic information regarding the pathogenicity of selected variants. (b) Measured GAA activity in both cells and medium of COS‐7 cultures after transfection with the generated constructs. Findings for the c.1597T>C variants are plotted separately as this was performed in a separate experiment. Data represent means, error bars represent SD (n = 3 biological replicates), ***p < .001. CADD, Combined Annotation‐Dependent Depletion; mRNA, messenger RNA
Novel variants that have been reported only through NBS studies, but for which no clinical phenotype has been provided, were classified as “Unknown (found only in NBS)”. In the current version of the database, 26 variants have been classified as such (Table 3). Seven out of 26 variants were also present in cis with the Asian pseudodeficiency allele, indicating that additional testing is required because the Asian pseudodeficiency is known to result in false‐positive outcomes in dried blood spot‐based assays (Liao et al., 2014; Momosaki et al., 2019). It is currently unknown at what age symptoms will develop in neonates diagnosed with disease‐associated variants that are potentially associated with a late‐onset phenotype. Symptoms might be delayed until late adulthood or, for some genetic variants, might not even lead to disease. In these cases, further research on the effect of the genetic variants is essential to better inform patients, families, and doctors. As reported, in these cases, the uncertainty of the diagnosis, the possibility of an emerging disease, and the doubt on when to start treatment with ERT could lead to emotional stress (Bodamer et al., 2017). This underscores the importance of phenotype prediction for disease‐associated variants, especially in the case of asymptomatic patients identified through NBS programs.
Table 3.
Variants of unknown significance that were found only through newborn screening programs
| Variant | Protein change | Location | Type of variant (protein) | MAF | Predictions on splicing–Align GVGD–SIFT–Mutation taster–[CADD score] | Experimental data | Country and reference |
|---|---|---|---|---|---|---|---|
| c.317G>A* | p.(Arg106His) | Exon 2 | Missense | MAF <1% | No effect on splicing–Class C0–Deleterious–Disease causing–[25.9] | Japan; Momosaki et al. (2019) | |
| c.365T>A | p.(Met122Lys) | Exon 2 | Missense | MAF not reported | No effect on splicing–Class C0–Tolerated–Polymorphism–[14.17] | USA; Scott et al. (2013) | |
| c.424_440del | p.(Ser142Leufs*29) | Exon 2 | Frameshift | MAF not reported | No effect on splicing–Results in an out of frame product–[32] | Taiwan; Chien et al. (2011) | |
| c.533G>A* | p.(Arg178His) | Exon 2 | Missense | MAF <1% | No effect on splicing–Class C0–Tolerated–Disease causing–[31] | No effect on splicing of exon 2 in minigene construct (Goina, et al., 2019) | Taiwan; Chien et al. (2011) |
| c.546+5G>T* | p.? | Intron 2 | No category (splicing) | MAF <1% | Weakens exon 2 splice donor and generates a cryptic splice donor–[23.7] | Affects splicing of exon 2 in minigene construct (Goina, et al., 2019) | Taiwan; Labrousse et al. (2010) |
| c.705G>A | p.(=) | Exon 4 | Silent | MAF <1% | No effect on splicing–[0.534] | Japan; Momosaki et al. (2019) | |
| c.811A>G* | p.(Thr271Ala) | Exon 4 | Missense | MAF not reported | No effect on splicing–Class C0–Tolerated–Polymorphism–[16.93] | 71% residual activity of GAA in expression study (Kroos, et al., 2012a) | Taiwan; Labrousse et al. (2010) |
| c.1054C>T | p.(Gln352*) | Exon 6 | Nonsense | MAF not reported | No effect on splicing–Introduces an early stop codon–[43] | Taiwan; Liao et al. (2014) | |
| c.1080C>G | p.(Tyr360*) | Exon 7 | Nonsense | MAF not reported | No effect on splicing–Introduces an early stop codon–[39] | Taiwan; Chien et al. (2011) | |
| c.1082C>A | p.(Pro361Arg) | Exon 7 | Missense | MAF <1% | No effect on splicing–Class C65–Deleterious–Disease causing–[25.5] | Japan; Momosaki et al. (2019) | |
| c.1220A>G | p.(Tyr407Cys) | Exon 8 | Missense | MAF <1% | No effect on splicing–Class C65–Deleterious–Disease causing–[25.9] | Mexico; Navarrete‐Martínez et al. (2017) | |
| c.1244C>T | p.(Thr415Met) | Exon 8 | Missense | MAF <1% | No effect on splicing–Class C15–Deleterious–Disease causing–[24.6] | Japan; Momosaki et al. (2019) | |
| c.1324G>A* | p.(Val442Met) | Exon 8 | Missense | MAF <1% | No effect on splicing–Class C0–Deleterious–Disease causing–[23.8] | Taiwan; Chien et al. (2011) | |
| c.1409A>C | p.(Asn470Thr) | Exon 9 | Missense | MAF <1% | No effect on splicing–Class C25–Deleterious–Disease causing–[23.2] | Hungary; Witmann et al. (2012) | |
| c.1574T>A | p.(Phe525Tyr) | Exon 11 | Missense | MAF not reported | No effect on splicing–Class C15–Deleterious–Disease causing–[28.8] | 10% residual activity of GAA in expression study (Kroos, et al., 2012a) | Taiwan; Chien et al. (2011) |
| c.1805C>T | p.(Thr602Ile) | Exon 13 | Missense | MAF not reported | No effect on splicing–Class C0–Tolerated–Disease causing–[24.1] | USA; Elliott et al. (2016) | |
| c.1840A>G | p.(Thr614Ala) | Exon 13 | Missense | MAF not reported | No effect on splicing–Class C55–Deleterious–Disease causing–[24.3] | Taiwan; Liao et al. (2014) | |
| c.1925T>A | p.(Val642Asp) | Exon 14 | Missense | MAF not reported | No effect on splicing–Class C45–Deleterious–Disease causing–[29.2] | USA; Scott et al. (2013) | |
| c.1958C>A | p.(Thr653Asn) | Exon 14 | Missense | MAF <1% | No effect on splicing–Class C15–Tolerated–Disease causing–[25.4] | Taiwan; Chien et al. (2011) | |
| c.2003A>G* | p.(Tyr668Cys) | Exon 14 | Missense | MAF not reported | No effect on splicing–Class C65–Deleterious–Disease causing–[31] | Japan; Momosaki et al. (2019) | |
| c.2055C>G | p.(Tyr685*) | Exon 15 | Nonsense | MAF not reported | No effect on splicing–Introduces an early stop codon–[36] | Japan; Momosaki et al. (2019) | |
| c.2174G>A | p.(Arg725Gln) | Exon 15 | Missense | MAF <1% | No effect on splicing–Class C0–Tolerated–Disease causing–[32] | Hungary; Witmann et al. (2012) | |
| c.2482‐5T>C* | p.? | Intron 17 | No category (splicing) | MAF not reported | No effect on splicing–[8.409] | Taiwan; Liao et al. (2014) | |
| c.2482‐2A>G | p.? | Intron 17 | No category (splicing) | MAF <1% | Loss of exon 18 splice acceptor site–[35] | Hungary; Witmann et al. (2012) | |
| c.2647‐23del | p.? | Intron 18 | No category (intron variant) | MAF <1% | No effect on splicing–[0.451] | Taiwan; Liao et al. (2014) | |
| c.2843dup | p.(Val949Argfs*69) | Exon 20 | Frameshift | MAF not reported | No effect on splicing–Results in an out of frame product–[23.1] | Taiwan; Liao et al. (2014) |
Abbreviations: CADD, Combined Annotation‐Dependent Depletion; MAF, minor allele frequency.
*Variants found in cis with the Asian pseudodeficiency allele c.[1726G>A; 2065G>A].
The sharp increase in reports on patients with Pompe disease and GAA disease‐associated variants highlights the need for regular updates of the Pompe disease GAA variant database. Increased awareness and improved diagnostic technology with exome and genome sequencing and NBS programs are expected to further increase the number of entries in the database in the coming years. It will be important to link variants to clinical information and to test their deleterious effect in vitro using expression and splicing assays. Curated disease‐specific databases such as the Pompe disease GAA variant database will be important to provide guidance to clinicians and clinical geneticists to establish an accurate molecular diagnosis.
CONFLICT OF INTERESTS
Ans T. van der Ploeg has provided consulting services for various industries in the field of Pompe disease under an agreement between these industries and Erasmus MC, Rotterdam, the Netherlands. The remaining authors declare that there are no conflict of interests.
WEB RESOURCES
Pompe disease GAA variant database: http://www.pompevariantdatabase.nl/
LOVD: http://gaa.lovd.nl/
GnomAD: https://gnomad.broadinstitute.org/
dbSNP: https://www.ncbi.nlm.nih.gov/snp/
CADD score: https://cadd.gs.washington.edu/
Supporting information
Supporting information.
ACKNOWLEDGMENTS
We thank the members of the Molecular Stem Cell Biology group for the critical discussions. This study was funded through Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasil; grant number: 234407/2014‐0), Sophia Foundation for Medical Research (SSWO; project number: s17‐32); Metakids (project number: 2016‐063), and Departamento Administrativo de Ciencia, Tecnología e Innovación (Colciencias). The collaboration project is cofunded by the PPP Allowance made available by Health‐Holland, Top Sector Life Sciences & Health, to the Prinses Beatrix Spier fonds to stimulate public–private partnerships (project number: LSHM19015).
de Faria DOS, in ‘t Groen SLM, Hoogeveen‐Westerveld M, et al. Update of the Pompe variant database for the prediction of clinical phenotypes: Novel disease‐associated variants, common sequence variants, and results from newborn screening. Human Mutation. 2021;42:119–134. 10.1002/humu.24148
Douglas O. S. de Faria and Stijn L. M. in't Groen contributed equally to this study.
Contributor Information
Atze J. Bergsma, Email: a.bergsma@erasmusmc.nl.
W. W. M. Pim Pijnappel, Email: w.pijnappel@erasmusmc.nl.
DATA AVAILABILITY STATEMENT
The data described in this study is available upon request from the corresponding authors, and new variants have been added to the Pompe disease GAA variant database (http://www.pompevariantdatabase.nl/) and LOVD (http://gaa.lovd.nl/).
REFERENCES
- Bali, D. S. , Goldstein, J. L. , Banugaria, S. , Dai, J. , Mackey, J. , Rehder, C. , & Kishnani, P. S. (2012). Predicting cross‐reactive immunological material (CRIM) status in Pompe disease using GAA mutations: Lessons learned from 10 years of clinical laboratory testing experience. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 160C(1), 40–49. 10.1002/ajmg.c.31319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekircan‐Kurt, C. E. , Güneş, H. N. , Yildiz, F. G. , Saka, E. , Tan, E. , & Erdem‐Özdamar, S. (2017). New mutations and genotype–phenotype correlation in late‐onset Pompe patients. Acta Neurologica Belgica, 117, 269–275. 10.1007/s13760-016-0738-7 [DOI] [PubMed] [Google Scholar]
- Bergsma, A. J. , in 't Groen, S. L. M. , van den Dorpel, J. J. A. , van den Hout, H. J. M. P. , van der Beek, N. A. M. E. , Schoser, B. , Toscano, A. , Musumeci, O. , Bembi, B. , Dardis, A. , Morrone, A. , Tummolo, A. , Pasquini, E. , van der Ploeg, A. T. , & Pijnappel, W. W. M. P. (2019). A genetic modifier of symptom onset in Pompe disease. EBioMedicine, 43, 553–561. 10.1016/j.ebiom.2019.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodamer, O. A. , Ronald Scott, C. , & Giugliani, R. (2017). Newborn screening for Pompe disease. Pediatrics, 140, S4–S13. 10.1542/peds.2016-0280C [DOI] [PubMed] [Google Scholar]
- Bravo, H. , Neto, E. C. , Schulte, J. , Pereira, J. , Filho, C. S. , Bittencourt, F. , Sebastião, F. , Bender, F. , de Magalhães, A. P. S. , Guidobono, R. , Trapp, F. B. , Michelin‐Tirelli, K. , Souza, C. F. M. , Rojas Málaga, D. , Pasqualim, G. , Brusius‐Facchin, A. C. , & Giugliani, R. (2017). Investigation of newborns with abnormal results in a newborn screening program for four lysosomal storage diseases in Brazil. Molecular Genetics and Metabolism Reports, 12, 92–97. 10.1016/j.ymgmr.2017.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlina, A. B. , Polo, G. , Salviati, L. , Duro, G. , Zizzo, C. , Dardis, A. , Bembi, B. , Cazzorla, C. , Rubert, L. , Zordan, R. , Desnick, R. J. , & Burlina, A. P. (2018). Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. Journal of Inherited Metabolic Disease, 41, 209–219. 10.1007/s10545-017-0098-3 [DOI] [PubMed] [Google Scholar]
- Chien, Y.‐H. , Hwu, W.‐L. , & Lee, N.‐C. (2019). Newborn screening: Taiwanese experience. Annals of Translational Medicine, 7, 281 10.21037/atm.2019.05.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien, Y.‐H. , Lee, N.‐C. , Huang, H.‐J. , Thurberg, B.‐L. , Tsai, F.‐J. , & Hwu, W.‐L. (2011). Later‐onset Pompe disease: Early detection and early treatment initiation enabled by newborn screening. Journal of Pediatrics, 158(6), 1023–1027. 10.1016/j.jpeds.2010.11.053 [DOI] [PubMed] [Google Scholar]
- Elliott, S. , Buroker, N. , Cournoyer, J. J. , Potier, A. M. , Trometer, J. D. , Elbin, C. , Schermer, M. J. , Kantola, J. , Boyce, A. , Turecek, F. , Gelb, M. H. , & Scott, C. R. (2016). Pilot study of newborn screening for six lysosomal storage diseases using tandem mass spectrometry. Molecular Genetics and Metabolism, 118, 304–309. 10.1016/j.ymgme.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goina, E. , Musco, L. , Dardis, A. , & Buratti, E. (2019). Assessment of the functional impact on the pre‐mRNA splicing process of 28 nucleotide variants associated with Pompe disease in GAA exon 2 and their recovery using antisense technology. Human Mutation, 40(11), 2121–2130. 10.1002/humu.23867 [DOI] [PubMed] [Google Scholar]
- Guevara‐Campos, J. , González‐Guevara, L. , & Cauli, O. (2019). Skeletal alterations, developmental delay and new mutations in juvenile‐onset Pompe disease. Neuromuscular Disorders, 29, 192–197. 10.1016/j.nmd.2018.11.013 [DOI] [PubMed] [Google Scholar]
- Güngör, D. , & Reuser, A. J. (2013). How to describe the clinical spectrum in Pompe disease? American Journal of Medical Genetics, Part A, 161, 399–400. 10.1002/ajmg.a.35662 [DOI] [PubMed] [Google Scholar]
- in't Groen S. L. M., de Faria, D. O. S. , Iuliano, A. , van den Hout, J. M. P. , Douben, H. , Dijkhuizen, T. , Cassiman, D. , Witters, P. , Barba Romero, M.‐Á. , de Klein, A. , Somers‐Bolman, G. M. , Saris, J. J. , Hoefsloot, L. H. , van der Ploeg, A. T. , Bergsma, A. J. , & Pijnappel, W. W. M. P. (2020). Novel GAA variants and mosaicism in Pompe disease identified by extended analyses of patients with an incomplete DNA diagnosis. Molecular Therapy—Methods & Clinical Development, 17, 337–348. 10.1016/j.omtm.2019.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , Gauthier, L. D. , Brand, H. , Solomonson, M. , Watts, N. A. , Rhodes, D. , Singer‐Berk, M. , England, E. M. , Seaby, E. G. , Kosmicki, J. A. , … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler, L. , Puertollano, R. , & Raben, N. (2018). Pompe disease: From basic science to therapy. Neurotherapeutics, 15, 928–942. 10.1007/s13311-018-0655-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroos, M. , Hoogeveen‐Westerveld, M. , Michelakakis, H. , Pomponio, R. , van der Ploeg, A. , Halley, D. , & Reuser, A. (2012a). GAA Database Consortium. Update of the Pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Human Mutation, 33(8), 1161–1165. 10.1002/humu.22108 [DOI] [PubMed] [Google Scholar]
- Kroos, M. , Hoogeveen‐Westerveld, M. , van der Ploeg, A. , & Reuser, A. J. (2012b). The genotype‐phenotype correlation in Pompe disease. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 160C, 59–68. 10.1002/ajmg.c.31318 [DOI] [PubMed] [Google Scholar]
- Kroos, M. , Manta, P. , Mavridou, I. , Muntoni, F. , Halley, D. , Van der Helm, R. , Zaifeiriou, D. , Van der Ploeg, A. , Reuser, A. J. , & Michelakakis, H. (2006). Seven cases of Pompe disease from Greece. Journal of Inherited Metabolic Disease, 29, 556–563. 10.1007/s10545-006-0280-5 [DOI] [PubMed] [Google Scholar]
- Kroos, M. , Pomponio, R. J. , van Vliet, L. , Palmer, R. E. , Phipps, M. , Van der Helm, R. , Halley, D. , Reuser, A. J. , & GAA Database Consortium . (2008). Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Human Mutation, 29, E13–E26. 10.1002/humu.20745 [DOI] [PubMed] [Google Scholar]
- Kroos, M. A. , Pomponio, R. J. , Hagemans, M. L. , Keulemans, J. L. M. , Phipps, M. , DeRiso, M. , Palmer, R. E. , Ausems, M. G. E. M. , Van der Beek, N. A. M. E. , Van Diggelen, O. P. , Halley, D. J. J. , Van der Ploeg, A. T. , & Reuser, A. J. J. (2007). Broad spectrum of Pompe disease in patients with the same c.−32‐13T→G haplotype. Neurology, 68, 110–115. 10.1212/01.wnl.0000252798.25690.76 [DOI] [PubMed] [Google Scholar]
- Kulessa, M. , Weyer‐Menkhoff, I. , Viergutz, L. , Kornblum, C. , Claeys, K. G. , Schneider, I. , Plöckinger, U. , Young, P. , Boentert, M. , Vielhaber, S. , Mawrin, C. , Bergmann, M. , Weis, J. , Ziagaki, A. , Stenzel, W. , Deschauer, M. , Nolte, D. , Hahn, A. , Schoser, B. , & Schänzer, A. (2020). An integrative correlation of myopathology, phenotype and genotype in late onset Pompe disease. Neuropathology and Applied Neurobiology, 46, 359–374. 10.1111/nan.12580 [DOI] [PubMed] [Google Scholar]
- Labrijn‐Marks, I. , Somers‐Bolman, G. M. , In't Groen, S. L. M. , Hoogeveen‐Westerveld, M. , Kroos, M. A. , Ala‐Mello, S. , Amaral, O. , Miranda, C. , Mavridou, I. , Michelakakis, H. , Naess, K. , Verheijen, F. W. , Hoefsloot, L. H. , Dijkhuizen, T. , Benjamins, M. , van den Hout, H. J. M. , van der Ploeg, A. T. , Pijnappel, W. W. M. P. , Saris, J. J. , & Halley, D. J. (2019). Segmental and total uniparental isodisomy (UPiD) as a disease mechanism in autosomal recessive lysosomal disorders: Evidence from SNP arrays. European Journal of Human Genetics, 27, 919–927. 10.1038/s41431-019-0348-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrousse, P. , Chien, Y. H. , Pomponio, R. J. , Keutzer, J. , Lee, N. C. , Akmaev, V. R. , Scholl, T. , & Hwu, W. L. (2010). Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. Molecular Genetics and Metabolism, 99, 379–383. 10.1016/j.ymgme.2009.12.014 [DOI] [PubMed] [Google Scholar]
- Liao, H. C. , Chiang, C. C. , Niu, D. M. , Wang, C. H. , Kao, S. M. , Tsai, F. J. , Huang, Y. H. , Liu, H. C. , Huang, C. K. , Gao, H. J. , Yang, C. F. , Chan, M. J. , Lin, W. D. , & Chen, Y. J. (2014). Detecting multiple lysosomal storage diseases by tandem mass spectrometry—A national newborn screening program in Taiwan. Clinica Chimica Acta, 431, 80–86. 10.1016/j.cca.2014.01.030 [DOI] [PubMed] [Google Scholar]
- Momosaki, K. , Kido, J. , Yoshida, S. , Sugawara, K. , Miyamoto, T. , Inoue, T. , Okumiya, T. , Matsumoto, S. , Endo, F. , Hirose, S. , & Nakamura, K. (2019). Newborn screening for Pompe disease in Japan: Report and literature review of mutations in the GAA gene in Japanese and Asian patients. Journal of Human Genetics, 64, 741–755. 10.1038/s10038-019-0603-7 [DOI] [PubMed] [Google Scholar]
- Mori, M. , Bailey, L. A. , Estrada, J. , Rehder, C. W. , Li, J. S. , Rogers, J. G. , Bali, D. S. , Buckley, A. F. , & Kishnani, P. S. (2017). Severe cardiomyopathy as the isolated presenting feature in an adult with late‐onset Pompe disease: A case report. JIMD Reports, 31, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete‐Martínez, J. I. , Limón‐Rojas, A. E. , Gaytán‐García, M. J. , Reyna‐Figueroa, J. , Wakida‐Kusunoki, G. , Delgado‐Calvillo, M. D. R. , Cantú‐Reyna, C. , Cruz‐Camino, H. , & Cervantes‐Barragán, D. E. (2017). Newborn screening for six lysosomal storage disorders in a cohort of Mexican patients: Three‐year findings from a screening program in a closed Mexican health system. Molecular Genetics and Metabolism, 121(1), 16–21. 10.1016/j.ymgme.2017.03.001 [DOI] [PubMed] [Google Scholar]
- Niño, M. Y. , Groen, S. L. M. , in't Bergsma, A. J. , van der Beek, N. A. M. E. , Kroos, M. , Hoogeveen‐Westerveld, M. , van der Ploeg, A. T. , Pijnappel, W. W. M. P. , & (2019). Extension of the Pompe mutation database by linking disease‐associated variants to clinical severity. Human Mutation, 40, 1954–1967. 10.1002/humu.23854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch, P. , Witten, D. , Cooper, G. M. , Shendure, J. , & Kircher, M. (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47, D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuser, A. J. J. , Ploeg, A. T. , Chien, Y.‐H. , Llerena, J. , Abbott, M.‐A. , Clemens, P. R. , Kimonis, V. E. , Leslie, N. , Maruti, S. S. , Sanson, B.‐J. , Araujo, R. , Periquet, M. , Toscano, A. , Kishnani, P. S. , & on behalf of the Pompe Registry Sites (2019). GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry. Human Mutation, 40, 2146–2164. 10.1002/humu.23878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, C. R. , Elliott, S. , Buroker, N. , Thomas, L. I. , Keutzer, J. , Glass, M. , Gelb, M. H. , & Turecek, F. (2013). Identification of infants at risk for developing Fabry, Pompe, or mucopolysaccharidosis‐I from newborn blood spots by tandem mass spectrometry. Journal of Pediatrics, 163(2), 498–503. 10.1016/j.jpeds.2013.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semplicini, C. , Letard, P. , De Antonio, M. , Taouagh, N. , Perniconi, B. , Bouhour, F. , Echaniz‐Laguna, A. , Orlikowski, D. , Sacconi, S. , Salort‐Campana, E. , Solé, G. , Zagnoli, F. , Hamroun, D. , Froissart, R. , Caillaud, C. , & Laforêt, P. (2018). Late‐onset Pompe disease in France: Molecular features and epidemiology from a nationwide study. Journal of Inherited Metabolic Disease, 41, 937–946. 10.1007/s10545-018-0243-7 [DOI] [PubMed] [Google Scholar]
- Sherry, S. T. , Ward, M. H. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E. M. , & Sirotkin, K. (2001). DbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29, 308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turaça, L. T. , Faria, D. O. S. , de Kyosen, S. O. , Teixeira, V. D. , Motta, F. L. , Pessoa, J. G. , Rodrigues e Silva, M. , Almeida, S. S. , de D'Almeida, V. , Munoz Rojas, M. V. , Martins, A. M. , & Pesquero, J. B. (2015). Novel GAA mutations in patients with Pompe disease. Gene, 561, 124–131. 10.1016/j.gene.2015.02.023 [DOI] [PubMed] [Google Scholar]
- van der Ploeg, A. T. , & Reuser, A. J. (2008). Pompe's disease. Lancet, 372, 1342–1353. 10.1016/S0140-6736(08)61555-X [DOI] [PubMed] [Google Scholar]
- van Gelder, C.M. , Hoogeveen‐Westerveld, M. , Kroos, M. A. , Plug, I. , van der Ploeg, A. T. , & Reuser, A. J. (2015). Enzyme therapy and immune response in relation to CRIM status: The Dutch experience in classic infantile Pompe disease. Journal of Inherited Metabolic Disease, 8(2), 305–314. 10.1007/s10545-014-9707-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann, J. , Karg, E. , Turi, S. , Legnini, E. , Wittmann, G. , Giese, A. K. , Lukas, J. , Gölnitz, U. , Klingenhäger, M. , Bodamer, O. , Mühl, A. , & Rolfs, A. (2012). Newborn screening for lysosomal storage disorders in Hungary. JIMD Reports, 6, 117–125. 10.1007/8904_2012_130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. F. , Liu, H. C. , Hsu, T. R. , Tsai, F. C. , Chiang, S. F. , Chiang, C. C. , Ho, H. C. , Lai, C. J. , Yang, T. F. , Chuang, S. Y. , Lin, C. Y. , & Niu, D. M. (2014). A large‐scale nationwide newborn screening program for Pompe disease in Taiwan: Towards effective diagnosis and treatment. American Journal of Medical Genetics, Part A, 164, 54–61. 10.1002/ajmg.a.36197 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The data described in this study is available upon request from the corresponding authors, and new variants have been added to the Pompe disease GAA variant database (http://www.pompevariantdatabase.nl/) and LOVD (http://gaa.lovd.nl/).