

Abstract
The N‐heterocyclic silylene [{Fe(η5‐C5H4‐NDipp)2}Si] (1DippSi, Dipp=2,6‐diisopropylphenyl) shows an excellent combination of pronounced thermal stability and high reactivity towards small molecules. It reacts readily with CO2 and N2O, respectively affording (1DippSiO2)2C and (1DippSiO)2 as follow‐up products of the silanone 1DippSiO. Its reactions with H2O, NH3, and FcPH2 (Fc=ferrocenyl) furnish the respective oxidative addition products 1DippSi(H)X (X=OH, NH2, PHFc). Its reaction with H3BNH3 unexpectedly results in B−H, instead of N−H, bond activation, affording 1DippSi(H)(BH2NH3). DFT results suggest that dramatically different mechanisms are operative for these H−X insertions.
Keywords: carbene homologues, insertion, metallocenes, silicon, subvalent compounds
A stable cyclic diaminosilylene featuring a ferrocene‐based backbone is reported. The dicoordinate SiII atom is part of a six‐membered FeC2N2Si ring and shows a comparatively large bond angle of ca. 107°. This N‐heterocyclic silylene can activate strong bonds (C=O, N=O, B−H, N−H, P−H, O−H) under ambient conditions.
The N‐heterocyclic silylene (NHSi) A [1] (Figure 1) is a heavier NHC [2] analogue and represents the first stable compound containing divalent and dicoordinate silicon. [3] Backbone‐saturated congeners are significantly more reactive. For example, whereas 1,3‐bis(2,6‐diisopropylphenyl)imidazolin‐2‐ylidene (IPr) is inert towards PH3, the backbone‐saturated congener SIPr readily inserts into a P−H bond, [4] and silylene B undergoes self‐insertion into an Si−N bond during its tetramerisation. [5] The first isolable dialkylsilylene C [6] exhibits a more pronounced ambiphilicity than diaminosilylenes and rearranges to a SiIV compound. [7]
Figure 1.
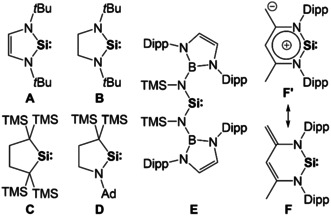
Silylenes A–F (Ad=1‐adamantyl, Dipp=2,6‐diisopropylphenyl, TMS=trimethylsilyl).
The rapid development of carbene chemistry has led to acyclic diaminocarbenes (ADACs), [8] ring‐expanded NHCs (reNHCs) with ring sizes >5 [9] and cyclic (alkyl)(amino)carbenes (CAACs), [10] which are all closely related to standard NHCs, but exhibit a more pronounced ambiphilicity, and hence higher reactivity. [11] While more than a dozen silicon analogues of standard NHCs have been isolated,[ 3 , 12 ] only a single example each has been reported for stable silicon analogues of CAACs, [13] ADACs [14] and reNHCs, [15] viz. silylenes D–F (Figure 1). The ambivalent reactivity of reNHSi F was rationalised by a significant contribution of N‐ylidic canonical structures summarised as F′. We here report on the reNHSi [{Fe(η5‐C5H4‐NDipp)2}Si] (1DippSi), which contains a six‐membered FeC2N2Si ring. 1DippSi is an analogue of our stable ferrocene‐based NHCs, whose ambiphilicity allowed for small‐molecule activation reactions unprecedented for diaminocarbenes.[ 16 , 17 ]
Our attempts to obtain reNHSis of the type 1RSi by reduction of corresponding SiIV dihalides 1RSiX2 (X=Cl, Br) or by α‐elimination of HCl from 1RSi(H)Cl were unsuccessful. [18] An alternative approach, which was introduced for the acyclic diaminosilylene (ADASi) E, is the reaction of [SiCl2(IPr)] [19] with the corresponding lithium amide. [14] This SiII precursor turned out to be the key to success. Its reaction with 1MesLi2 in C6D6 afforded the silylene 1MesSi together with IPr (Scheme 1). Although too unstable for isolation, 1MesSi was sufficiently persistent at room temperature for detecting its 29Si NMR signal (δ=121.5 ppm), which is significantly downfield‐shifted with respect to reNHSi F (δ=88.4 ppm) [15] and NHSi A (78.3 ppm). [1] The signal of the SiII atom in ADASi E was observed at even lower field (δ=204.6 ppm). [14] Trapping of 1MesSi with (PhSe)2 at room temperature in benzene solution afforded 1MesSi(SePh)2; details are provided in the Supporting Information (SI). The bulkier homologue 1DippSi, obtained from [SiCl2(IPr)] and 1DippLi2 in toluene at room temperature, is sufficiently stable for isolation (Scheme 1). IPr and 1DippSi could not be separated by crystallisation or sublimation. It was possible to remove IPr from ADASi E by stirring a hexane solution of the mixture at room temperature under an atmosphere of CO2, which led to the precipitation of IPr(CO2). [14] This method was not successful in our case, because, in contrast to E, 1DippSi reacts swiftly with CO2 under the same mild conditions, affording the orthocarbonate (1DippSiO2)2C (Scheme 2; see the SI). The primary products are most likely CO and the silanone 1DippSiO, [20] which subsequently undergoes a cycloaddition with CO2 in a 2:1 ratio. When generated by reaction of 1DippSi with N2O in benzene at room temperature, this silanone forms the expected dimer (1DippSiO)2 (Scheme 2; see the SI). [21] An analogous stepwise reaction with CO2 was first reported for decamethylsilicocene (Cp*2Si). [22] Dialkylsilylene C [23] as well as IPr=N‐Si‐OSitBu3 and IPr=N‐Si‐Si(SiMe3)3, an acyclic (imino)(siloxy)‐ and (imino)(silyl)silylene, [24] are the only examples containing dicoordinate SiII in this context to date. [25] We found that IPr is easily removed by complexation with ZnCl2, which is inert towards 1DippSi. In contrast to 1DippSi, [ZnCl2(IPr)] [26] is insoluble in hexane.
Scheme 1.
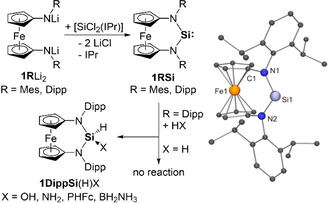
Synthesis of 1MesSi (persistent, Mes=mesityl) and 1DippSi (stable) and reactions of the latter with H2O, NH3, FcPH2 (Fc=ferrocenyl), and H3BNH3 under ambient conditions in benzene or toluene. Selected bond lengths [Å] and angles [°] for 1DippSi: Si1‐N1 1.7327(12), Si1‐N2 1.7344(12), N1‐Si1‐N2 106.58(6); sum of angles (Σ∡) at N1 359.9, at N2 360.0.
Scheme 2.
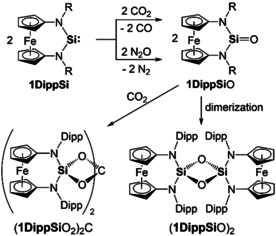
Reactions of 1DippSi with CO2 and N2O under ambient conditions in benzene, respectively affording (1DippSiO2)2C and (1DippSiO)2 via the silanone 1DippSiO as assumed intermediate.
The 29Si NMR signal of 1DippSi is located at δ=115.7 ppm, upfield‐shifted by 6 ppm with respect to 1MesSi. 1DippSi was structurally characterised by X‐ray diffraction (Scheme 1). The Si bond angle (106.6°) lies in between the values determined for reNHSi F (99.3°) [15] and ADASi E (110.9°) [14] and is close to that reported for a heterocyclic silylene with a six‐membered ring containing an NSiIIBP unit. [27] Silylenes whose dicoordinate SiII atom is part of a five‐membered ring exhibit more acute Si bond angles close to 90°.[ 3 , 6 , 12 , 13 , 28 ]
Similar to 1MesSi, 1DippSi undergoes an oxidative addition with (PhSe)2 in benzene solution at room temperature to afford 1DippSi(SePh)2 (see the SI). We next addressed the oxidative addition of strong H−X bonds of different polarities, which is of fundamental importance for chemical synthesis and catalysis. [29] While 1DippSi is inert towards H2 under ambient conditions, it reacted readily with H2O, NH3, and FcPH2, affording the corresponding derivatives of the type 1DippSi(H)X (Scheme 1, Figure 2; X=OH, NH2, PHFc; see the SI). The reaction of H2O with stable dicoordinate SiII compounds to the corresponding hydroxysilane has been reported only for the metallasilylene [Cp*(CO)3Cr‐Si‐SIPr]+[30] as well as for A [31] and F. [32] The hydroxysilanes A(H)OH and F(H)OH were not observed, but their intermediacy was merely inferred from the products isolated. In contrast, the analogous NH3 addition product F(H)NH2 was obtained in high yield from the reaction of F with NH3. [33] F is the exception to the rule that five‐ and six‐membered NHSis cannot be employed for NH3 activation, although they are more Lewis acidic and have a smaller singlet‐triplet gap compared to the corresponding NHCs. [34] Apart from [Cp*(CO)3Cr‐Si‐SIPr]+, [30] Dipp(Me3Si)N‐Si‐Si(SiMe3)3 [35] and IPr=N‐Si‐OSitBu3 [24a] we are not aware of any other stable silylene to undergo an oxidative addition of NH3. The reaction of NH3 with 1DippSi is remarkable because NH3 activation is a challenging target even for transition metal complexes [36] —the potential of low‐valent main‐group element compounds in this context was uncovered only recently.[ 25 , 37 ] The reaction of 1DippSi with FcPH2 afforded the oxidative addition product 1DippSi(H)(PHFc). In view of the ability of 1DippSi for N‐H activation, the activation of a P−H bond, which is weaker than an N−H bond by ca. 100 kJ mol−1, is not unexpected; [38] reNHSi F is also capable of P‐H activation. [39] We next addressed the reaction of 1DippSi with H3BNH3 (Scheme 1), [40] expecting the formation of 1DippSiH2, most likely by transfer of a protic and a hydridic H atom [41] to the divalent atom, as was observed for 1,3‐di‐tert‐butylimidazolin‐2‐ylidene [33] and reNHSi F. [42] Instead, the reaction furnished 1DippSi(H)(BH2NH3) (Figure 2), although the B−H bond is stronger than the N−H bond of H3BNH3. [43] First B−H bond activation reactions with SiII compounds were reported only recently, [44] and the reaction of the (silyl)(vinyl)silylene MeIPr=CH‐Si‐Si(SiMe3)3 (MeIPr=1,3‐bis(2,6‐diisopropylphenyl)‐4,5‐dimethylimidazolin‐2‐ylidene) with pinacolborane (HBPin), which affords MeIPr=CH‐Si(H)(Bpin)‐Si(SiMe3)3, is the only example involving dicoordinate SiII. [45]
Figure 2.
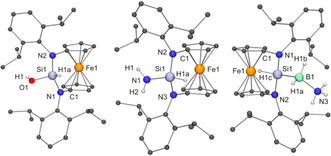
Molecular structures of 1DippSi(H)OH (left; selected bond lengths [Å] and angles [°]: Si1‐N1 1.7197(16), Si1‐N2 1.7199(16), Si1‐O1 1.6071(16), N1‐Si1‐N2 111.49(8); Σ∡ at N1 359.9, at N2 359.4), 1DippSi(H)NH2 (middle; selected bond lengths [Å] and angles [°]: Si1‐N1 1.691(4), Si1‐N2 1.721(3), Si1‐N3 1.730(3), N2‐Si1‐N3 111.72(14); Σ∡ at N2 358.5, at N3 358.0) and 1DippSi(H)(BH2NH3) (right; selected bond lengths [Å] and angles [°]: Si1‐N1 1.7571(14), Si1‐N2 1.7694(14), Si1‐B1 2.008(2), N3‐B1 1.600(3), N1‐Si‐N2 107.84(7), Si1‐B1‐N1 116.41(13); Σ∡ at N1 358.3, at N2 356.6).
We performed a DFT study on the electronic characteristics and the reactivity of 1, the full molecular model of 1DippSi. [46] At the PBEh‐3c level of DFT employed, the HOMO comprises the expected silylene lone pair together with significant contributions of the ferrocene moiety and the LUMO is dominated by the silylene p‐orbital, with a substantial HOMO–LUMO energy separation of ΔE H/L=6.4 eV. The unexpectedly low computed singlet‐triplet energy difference of ΔE S/T=0.4 eV does not correlate with this value because the lowest triplet state arises from a local excitation within the ferrocene moiety and does not involve the silylene p‐orbital (Figure 3). [47]
Figure 3.
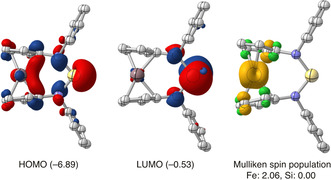
Frontier molecular orbitals and triplet spin‐density distribution computed for 1 (i. e. the full molecular model of 1DippSi; orbital energies in eV, isocontour surfaces at ±0.05 a0 −3/2 for orbitals and ±0.005 a0 −3 for the spin density, α spin: yellow, β spin: green; iPr groups and H atoms not shown).
Surprisingly, direct oxidative addition of H2O and NH3 to 1 is precluded by high kinetic barriers for both substrates (35 and 42 kcal mol−1, respectively), and two distinct alternative pathways were identified instead. The lowest‐energy pathway for NH3 activation commences with the formation of adduct 2 (Scheme 3, top). Proton transfer is facilitated by a second NH3 molecule acting as a proton shuttle and the experimentally observed product 3 is formed in a strongly exergonic step with a moderate overall barrier of 20 kcal mol−1. H2O, in turn, does not form a datively bonded adduct with 1, but directly adds across an Si−N bond via TS2 to form hydroxysilylene 4 in an exergonic step (Scheme 3, bottom). From there on out, silanone 5 is formed through a water‐assisted proton transfer; [48] the experimentally observed product 6 results in a strongly exergonic step after passage of a minute barrier in TS4.
Scheme 3.
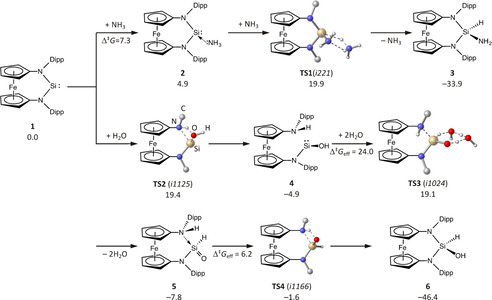
Computed lowest‐energy reaction pathway for the formal H‐X oxidative addition to 1 with NH3 (top) and H2O (bottom, ΔG 298 in kcal mol−1). Bonds formed or broken in transition states are dashed, unreactive H atoms are omitted and the orientation of the Dipp substituents in the transition state is indicated by showing the respective Cipso atom only.
The quantum‐chemical evaluation discloses a concerted dehydrogenation of H3BNH3 by 1 as initial step along the lowest‐energy pathway for ammonia‐borane activation (Scheme 4). Alternative direct insertion of 1 into an N−H or B−H bond is precluded by high kinetic barriers (53 and 35 kcal mol−1, respectively; see the SI). Whereas the resulting silane 7 forms as an unreactive side product, [49] H2BNH2 is a highly reactive species that has been thoroughly studied in the thermal and catalytic dehydrogenation of H3BNH3 and is known to polymerize below −150 °C. [50]
Scheme 4.
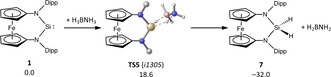
Computed reaction path for the formation of 7 from 1 and H3BNH3; ΔG 298 in kcal mol−1.
Obviously, B‐H insertion of 1 in H2BNH2 competes efficiently with the polymerization of the latter, leading to the formation of 8 (Scheme 5). With a low barrier of 12 kcal mol−1 8 can dehydrogenate a second equivalent of H3BNH3 through intermediate 9 yielding the experimentally observed product 10 while regenerating H2BNH2. After initial formation of H2BNH2 from 1 and H3BNH3 the follow‐up reaction cascade involving B‐H insertion by 1 and subsequent dehydrogenation of H3BNH3 is kinetically favoured over the formation of 7.
Scheme 5.
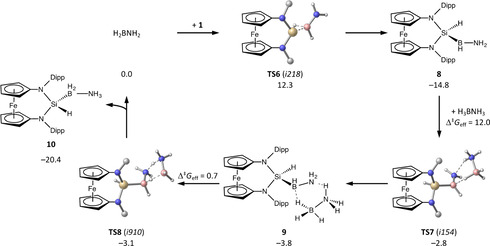
Computed reaction path for the formation of 10 from 1, H2BNH2and H3BNH3 (ΔG 298 in kcal mol−1 relative to the separated reactants, activation barriers relative to the preceding minimum).
In conclusion, we have described the synthesis and reactivity of the new stable reNHSi 1DippSi. 1DippSi reacts readily with N2O and CO2, which is in contrast to the inertness of F, the only other stable cyclic diaminosilylene featuring a ring‐expanded structure known to date. [25a] Studies on the reactivity of 1DippSi towards H‐X bonds of different strengths and polarities show parallels to previous reactivity studies on other silylenes. The reactions with NH3 and H2O both give the H‐X insertion products. Mechanistically, however, they differ significantly. More particularly, the lowest‐energy path of the reaction with H2O involves the N‐Si cooperative activation of an O−H bond. For H3BNH3 the reaction mechanism consists of two key elementary steps, the first one being the dehydrogenation of H3BNH3 to H2BNH2, which subsequently catalyses the conversion of 1DippSi to 1DippSi(H)(BH2NH3) with H3BNH3. In contrast to H3BNH3, H2BNH2 has a vacant p‐orbital, which enables insertion of the silylene in a B−H bond in the second step. This silylborane can in turn dehydrogenate a second equivalent of H3BNH3 to give the final product 1DippSi(H)(BH2NH3).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Quantum‐chemical were performed at the Center for Scientific Computing (CSC) Frankfurt on the Goethe and Fuchs high‐performance compute clusters. Open access funding enabled and organized by Projekt DEAL.
N. Weyer, M. Heinz, J. I. Schweizer, C. Bruhn, M. C. Holthausen, U. Siemeling, Angew. Chem. Int. Ed. 2021, 60, 2624.
Dedicated to Professor Peter Jutzi on the occasion of his 82nd birthday
Contributor Information
Prof. Dr. Max C. Holthausen, Email: max.holthausen@chemie.uni-frankfurt.de.
Prof. Dr. Ulrich Siemeling, Email: siemeling@uni-kassel.de.
References
- 1. Denk M., Lennon R., Hayashi R., West R., Belyakov A. V., Verne H. P., Haaland A., Wagner M., Metzler N., J. Am. Chem. Soc. 1994, 116, 2691–2692. [Google Scholar]
- 2. Hopkinson M. N., Richter C., Schedler M., Glorius F., Nature 2014, 510, 485–496. [DOI] [PubMed] [Google Scholar]
- 3. Asay M., Jones C., Driess M., Chem. Rev. 2011, 111, 354–396. [DOI] [PubMed] [Google Scholar]
- 4. Bispinghoff M., Tondreau A. M., Grützmacher H., Faradji C. A., Pringle P. G., Dalton Trans. 2016, 45, 5999–6003. [DOI] [PubMed] [Google Scholar]
- 5. Schmedake T. A., Haaf M., Apeloig Y., Müller T., Bukalov S., West R., J. Am. Chem. Soc. 1999, 121, 9479–9480. [Google Scholar]
- 6. Kira M., Ishida S., Iwamoto T., Kabuto C., J. Am. Chem. Soc. 1999, 121, 9722–9723. [Google Scholar]
- 7. Kira M., Chem. Commun. 2010, 46, 2893–2903. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Boyarskiy V. P., Luzyanin K. V., Kukushkin V. Y., Coord. Chem. Rev. 2012, 256, 2029–2056; [Google Scholar]
- 8b. Slaughter L. M., ACS Catal. 2012, 2, 1802–1816; [Google Scholar]
- 8c. Vignolle J., Cattoën X., Bourissou D., Chem. Rev. 2009, 109, 3333–3384. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Yong X., Thurston R., Ho C.-Y., Synthesis 2019, 51, 2058–2080; [Google Scholar]
- 9b. Li J., Shen W.-x., Li X.-r., Curr. Org. Chem. 2013, 16, 2879–2891. [Google Scholar]
- 10.
- 10a. Melaimi M., Jazzar R., Soleilhavoup M., Bertrand G., Angew. Chem. Int. Ed. 2017, 56, 10046–10068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10180–10203; [Google Scholar]
- 10b. Soleilhavoup M., Bertrand G., Acc. Chem. Res. 2015, 48, 256–266. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Munz D., Organometallics 2018, 37, 275–289; [Google Scholar]
- 11b. Martin D., Canac Y., Lavallo V., Bertrand G., J. Am. Chem. Soc. 2014, 136, 5023–5030. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Zhu L., Zhang J., Cui C., Inorg. Chem. 2019, 58, 12007–12010; [DOI] [PubMed] [Google Scholar]
- 12b. Fedushkin I. L., Lukoyanov A. N., Khvoinova N. M., Cherkasov A. V., Russ. Chem. Bull. 2013, 62, 2454–2461; [Google Scholar]
- 12c. Zark P., Schäfer A., Mitra A., Haase D., Saak W., West R., Müller T., J. Organomet. Chem. 2010, 695, 398–408. [Google Scholar]
- 13. Kosai T., Ishida S., Iwamoto T., Angew. Chem. Int. Ed. 2016, 55, 15554–15558; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15783–15787. [Google Scholar]
- 14. Hadlington T. J., Abdalla J. A. B., Tirfoin R., Aldridge S., Jones C., Chem. Commun. 2016, 52, 1717–1720. [DOI] [PubMed] [Google Scholar]
- 15. Driess M., Yao S., Brym M., van Wüllen C., Lentz D., J. Am. Chem. Soc. 2006, 128, 9628–9629. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Petrov A. R., Derheim A., Oetzel J., Leibold M., Bruhn C., Scheerer S., Oßwald S., Winter R. F., Siemeling U., Inorg. Chem. 2015, 54, 6657–6670; [DOI] [PubMed] [Google Scholar]
- 16b. Siemeling U., Färber C., Bruhn C., Leibold M., Selent D., Baumann W., von Hopffgarten M., Goedecke C., Frenking G., Chem. Sci. 2010, 1, 697–704. [Google Scholar]
- 17.
- 17a. Guthardt R., Oetzel J., Schweizer J. I., Bruhn C., Langer R., Maurer M., Vícha J., Shestakova P., Holthausen M. C., Siemeling U., Angew. Chem. Int. Ed. 2019, 58, 1387–1391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1401–1405; [Google Scholar]
- 17b. Oetzel J., Weyer N., Bruhn C., Leibold M., Gerke B., Pöttgen R., Maier M., Winter R. F., Holthausen M. C., Siemeling U., Chem. Eur. J. 2017, 23, 1187–1199. [DOI] [PubMed] [Google Scholar]
- 18. Oetzel J., Bruhn C., Siemeling U., Z. Anorg. Allg. Chem. 2018, 644, 935–944. [Google Scholar]
- 19. Ghadwal R. S., Roesky H. W., Merkel S., Henn J., Stalke D., Angew. Chem. Int. Ed. 2009, 48, 5683–5686; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5793–5796. [Google Scholar]
- 20.Recent examples for oxygenations of stable low-valent Si compounds with CO2:
- 20a. Protchenko A. V., Vasko P., Cao Huan Do D., Hicks J., Fuentes M. Á., Jones C., Aldridge S., Angew. Chem. Int. Ed. 2019, 58, 1808–1812; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1822–1826; [Google Scholar]
- 20b. Burchert A., Yao S., Müller R., Schattenberg C., Xiong Y., Kaupp M., Driess M., Angew. Chem. Int. Ed. 2017, 56, 1894–1897; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1920–1923; [Google Scholar]
- 20c. Wang Y., Chen M., Xie Y., Wei P., H. F. Schaefer III , Robinson G. H., J. Am. Chem. Soc. 2015, 137, 8396–8399; [DOI] [PubMed] [Google Scholar]
- 20d. Mück F. M., Baus J. A., Nutz M., Burschka C., Poater J., Bickelhaupt F. M., Tacke R., Chem. Eur. J. 2015, 21, 16665–16672. [DOI] [PubMed] [Google Scholar]
- 21. Li W., Hill N. J., Tomasik A. C., Bikzhanova G., West R., Organometallics 2006, 25, 3802–3805. [Google Scholar]
- 22. Jutzi P., Eikenberg D., Möhrke A., Neumann B., Stammler H.-G., Organometallics 1996, 15, 753–759. [Google Scholar]
- 23. Liu X., Xiao X.-Q., Xu Z., Yang X., Li Z., Dong Z., Yan C., Lai G., Kira M., Organometallics 2014, 33, 5434–5439. [Google Scholar]
- 24.
- 24a. Reiter D., Frisch P., Wendel D., Hörmann F., Inoue S., Dalton Trans. 2020, 49, 7060–7068; [DOI] [PubMed] [Google Scholar]
- 24b. Wendel D., Porzelt A., Herz F. A. D., Sarkar D., Jandl C., Inoue S., Rieger B., J. Am. Chem. Soc. 2017, 139, 8134–8137. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Shan C., Yao S., Driess M., Chem. Soc. Rev. 2020, 49, 6733–6754; [DOI] [PubMed] [Google Scholar]
- 25b. Fujimori S., Inoue S., Eur. J. Inorg. Chem. 2020, 3131–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jacquet O., Frogneux X., Das Neves Gomes C., Cantat T., Chem. Sci. 2013, 4, 2127–2131. [Google Scholar]
- 27. Rosas-Sánchez A., Alvarado-Beltran I., Baceiredo A., Saffon-Merceron N., Massou S., Branchadell V., Kato T., Angew. Chem. Int. Ed. 2017, 56, 10549–10554; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10685–10690. [Google Scholar]
- 28.
- 28a. Kobayashi R., Ishida S., Iwamoto T., Angew. Chem. Int. Ed. 2019, 58, 9425–9428; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9525–9528; [Google Scholar]
- 28b. Abe T., Tanaka R., Ishida S., Kira M., Iwamoto T., J. Am. Chem. Soc. 2012, 134, 20029–20032. [DOI] [PubMed] [Google Scholar]
- 29. Chu T., Nikonov G. I., Chem. Rev. 2018, 118, 3608–3680. [DOI] [PubMed] [Google Scholar]
- 30. Filippou A. C., Baars B., Chernov O., Lebedev Yu. N., Schnakenburg G., Angew. Chem. Int. Ed. 2014, 53, 565–570; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 576–581. [Google Scholar]
- 31. Haaf M., Schmiedel A., Schmedake T. A., Powell D. R., Millevolte A. J., Denk M., West R., J. Am. Chem. Soc. 1998, 120, 12714–12719. [Google Scholar]
- 32. Yao S., Brym M., van Wüllen C., Driess M., Angew. Chem. Int. Ed. 2007, 46, 4159–4162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 4237–4240. [Google Scholar]
- 33. Jana A., Schulzke C., Roesky H. W., J. Am. Chem. Soc. 2009, 131, 4600–4601. [DOI] [PubMed] [Google Scholar]
- 34. Metzler A., Inoue S., Präsang C., Driess M., J. Am. Chem. Soc. 2010, 132, 3038–3046. [DOI] [PubMed] [Google Scholar]
- 35. Protchenko A. V., Bates J. I., Saleh L. M. A., Blake M. P., Schwarz A. D., Kolychev E. L., Thompson A. L., Jones C., Mountford P., Aldridge S., J. Am. Chem. Soc. 2016, 138, 4555–4564. [DOI] [PubMed] [Google Scholar]
- 36. Hoover J., Science 2016, 354, 707–708. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Weetman C., Inoue S., ChemCatChem 2018, 10, 4213–4228; [Google Scholar]
- 37b. Yadav S., Saha S., Sen S. S., ChemCatChem 2016, 8, 486–501; [Google Scholar]
- 37c. Power P. P., Nature 2010, 463, 171–177. [DOI] [PubMed] [Google Scholar]
- 38.E−H bond dissociation energies for EH3 and MeEH2 are 450 and 425 kJ mol−1, respectively, for E=N, but 351 and 322 kJ mol−1, respectively, for E=P; see: Luo Y. R., Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007. [Google Scholar]
- 39. Präsang C., Stoelzel M., Inoue S., Meltzer A., Driess M., Angew. Chem. Int. Ed. 2010, 49, 10002–10005; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 10199–10202. [Google Scholar]
- 40.
- 40a. Demirci U. B., Int. J. Hydrogen Energy 2017, 42, 9978–10013; [Google Scholar]
- 40b. Staubitz A., Robertson A. P. M., Manners I., Chem. Rev. 2010, 110, 4079–4124. [DOI] [PubMed] [Google Scholar]
- 41. Boom D. H. A., Jupp A. R., Slootweg J. C., Chem. Eur. J. 2019, 25, 9133–9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stoelzel M., Präsang C., Inoue S., Enthaler S., Driess M., Angew. Chem. Int. Ed. 2012, 51, 399–403; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 411–415. [Google Scholar]
- 43.
- 43a. Matus M. H., Grant D. J., Tho Nguyen M., Dixon D. A., J. Chem. Phys. C 2009, 113, 16553–16560; [Google Scholar]
- 43b. Rablen P. R., Hartwig J. F., J. Am. Chem. Soc. 1996, 118, 4648–4653. [Google Scholar]
- 44.
- 44a. Swamy V. S. V. S. N., Raj K. V., Vanka K., Sen S. S., Roesky H. W., Chem. Commun. 2019, 55, 3536–3539; [DOI] [PubMed] [Google Scholar]
- 44b. Khoo S., Shan Y.-L., Yang M.-C., Li Y., Su M.-D., So C.-W., Inorg. Chem. 2018, 57, 5879–5887. [DOI] [PubMed] [Google Scholar]
- 45. Roy M. M. D., Ferguson M. J., McDonald R., Zhou Y., Rivard E., Chem. Sci. 2019, 10, 6476–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.All calculations were performed with the PBEh-3c method implemented in ORCA 4.1.2. For a benchmark against F12 coupled cluster theory and further technical details see the SI.
- 47.As it is becoming commonplace to tentatively correlate ΔE H/L and/or ΔE S/T of a silylene with its reactivity (see, for example, Ref. [25]), it is important to realize that both parameters exhibit a substantial method dependence (see Figure S46 in the SI), which is not commonly acknowledged in the literature. For comparison, we thus provide as SI (Figure S45) electronic characteristics evaluated at the PBEh-3c level for several silylenes published by others.
- 48.In view of Singleton's criticism, the corresponding alternative reaction paths identified are provided as SI. See: Plata R. E., Singleton D. A., J. Am. Chem. Soc. 2015, 137, 3811–3826.25714789 [Google Scholar]
- 49.The presence of traces of this silane in the crude product is indicated by a low-intensity 29Si NMR signal at δ=−34.1 ppm (see Figure S44 in the SI), which was absent after recrystallization.
- 50. Kwon C. T., H. A. McGee, Jr. , Inorg. Chem. 1970, 9, 2458–2461. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary