
Figure 1.
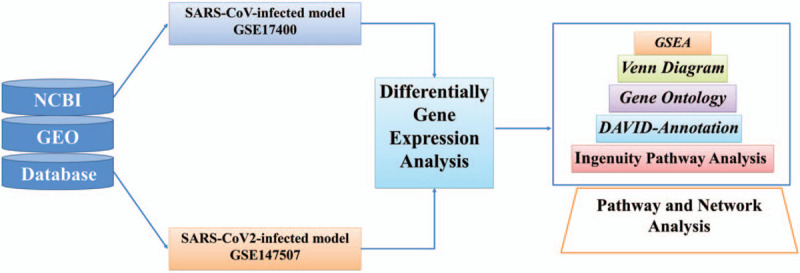
Schematic illustration of the study design. High-throughput data of severe acute respiratory syndrome coronavirus (SARS-CoV-)/SARS-CoV-2-infected human lung adenocarcinoma models were acquired from National Center for Biotechnology Information database with the accession number GSE17400 and GSE147507 datasets. Differentially expressed genes in the top 10% compared to mock-infected controls from (SARS-CoV) and SARS-CoV-2 were merged by the Venn diagram to find shared genes between these 2 groups. Furthermore, gene annotation and gene ontology were done using the Database for Annotation, Visualization, and Integrated Discovery database. Finally, gene set enrichment analysis, and Ingenuity pathway analysis were used for enrichment and downstream pathways analyses. DAVID = the Database for Annotation, Visualization, and Integrated Discovery, GEO = Gene Expression Omnibus, GO = gene ontology, GSEA = gene set enrichment analysis, IPA = Ingenuity pathway analysis, NCBI = National Center for Biotechnology Information.