
FIG 1.
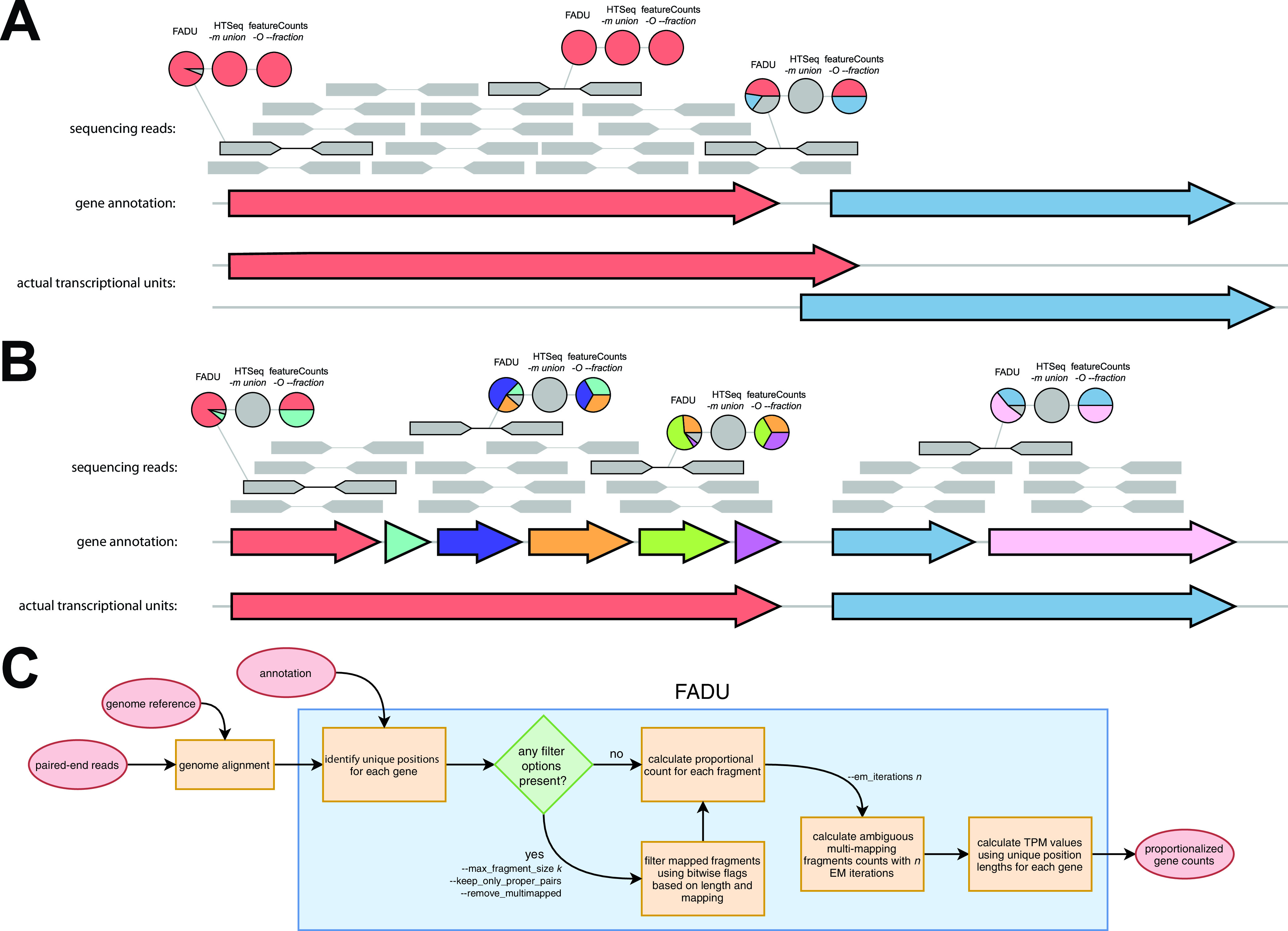
Implementation of FADU. (A) The workflow of FADU uses a BAM file and a GFF annotation file to identify proportional read counts for prokaryotic RNA-Seq analyses. (B) The implementation of FADU differs from those of other similar genome alignment-based quantification tools primarily in the quantification of ambiguous multigene fragments. The three sets of pie charts above a paired-end fragment display how the counts from the fragment are proportionally assigned to its overlapping genes. In the case of two overlapping genes, FADU accounts for only the unique portions of each gene and assigns a proportional count based on the length that the fragment overlaps the feature. (C) In the case of operons, FADU will assign proportional counts to the different genes based on the overlap between the mapping coordinates of the fragment and any overlapping genic features.