

Abstract
Soluble low-molecular-weight oligomers formed during the early stage of amyloid aggregation are considered the major toxic species in amyloidosis. The structure-function relationship between oligomeric assemblies and the cytotoxicity in amyloid diseases are still elusive due to the heterogeneous and transient nature of these aggregation intermediates. To uncover the structural characteristics of toxic oligomeric intermediates, we compared the self-assembly dynamics and structures of SOD128–38, a cytotoxic fragment of the superoxide dismutase 1 (SOD1) associated with the amyotrophic lateral sclerosis, with its two non-toxic mutants G33V and G33W using molecular dynamics simulations. Single-point glycine substitutions in SOD128–38 have been reported to abolish the amyloid toxicity. Our simulation results showed that the toxic SOD128–38 and its non-toxic mutants followed different aggregation pathways featuring distinct aggregation intermediates. Specifically, wild-type SOD128–38 initially self-assembled into random coil rich oligomers, among which fibrillar aggregates comprised of well-defined curved single-layer β-Sheets were nucleated via coil-to-sheet conversions and the formation of β-barrels as intermediates. In contrast, the non-toxic G33V/G33W mutants readily assembled into small β-sheet rich oligomers and then coagulated with each other into cross-β fibrils formed by two-layer β-sheets without forming β-barrels as the intermediates. The direct observation of β-barrel oligomers during the assembly of toxic SOD128–38 fragments but not the non-toxic glycine-substitution mutants strongly support β-barrels as the toxic oligomers in amyloidosis, probably via interactions with cell membrane and forming amyloid pores. With well-defined structures, the β-barrel might serve as the novel therapeutic targets against amyloid-related diseases.
Graphical Abstract
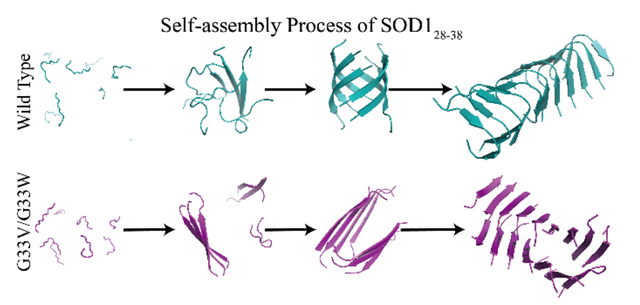
The self-assembly dynamics of SOD128–38 and its G33V/G33W mutants. The wild-type SOD128–38 initially nucleated into unstructured oligomers, followed by a conformational conversion into β-sheets and the formation of β-barrel intermediates before the emergence of nanofibrils comprised of curved single-layer β-sheets. The non-toxic G33V/G33W mutants aggregated via a different aggregation pathway, where the peptides first assembled into small β-sheets and these β-sheets further coagulated with each other into cross-β nanofibrils without forming β-barrels as intermediates.
Introduction
The self-assembly of proteins into amyloid fibrils has attracted great attention due to its association with not only a long list of human degenerative diseases1–3 (i.e., pathological amyloids) but also physiological functions in an organism4, 5 (i.e., functional amyloids) and potential applications as functional materials in nanotechnology6, 7 (i.e., artificial amyloids). For example, the pathological aggregation of amyloid beta8 (Aβ) and tau9, alpha-synuclein10 (αS), human islet amyloid polypeptide11 (hIAPP) and superoxide dismutase 112 (SOD1) are associated with Alzheimer's disease, Parkinson's disease, type 2 diabetes and amyotrophic lateral sclerosis (ALS), respectively. Despite differences in sequences and native structures of the aggregating proteins (e.g., Aβ, αS, hIAPP and SOD1), amyloid fibrils9, 13–16 share similar cross-β core structures with β-strands within each protofilament aligned perpendicular to the fibril axis. Increasing experimental evidences demonstrated that the soluble low-molecular-weight oligomers formed by diseases-related amyloid proteins during the early aggregation stage are more toxic than their mature fibrils17–19. Therefore, characterizations of their assembly dynamics and oligomeric intermediates during the early aggregation stage are of significant importance both for understanding the cytotoxicity mechanism and designing therapeutic approaches for the treatment of amyloid diseases. However, these oligomers are usually dynamic, transient and heterogeneous in sizes and structures, determining their structure–function relationships is extremely challenging1, 17–19.
Many experiments in vitro have demonstrated that various amyloid peptides could induce membrane leakage and disruption by forming pore-like structures in the membrane20–22. Consistent with these observations and the general structure-function principle in biology23, the toxic oligomers of different amyloid proteins may have well-defined three-dimensional structures with common features in order to perform the pathological functions. The formation of β-barrels, first structurally-determined for a slow-aggregating αB crystallin fragment (i.e., αBC90–100) by x-ray crystallography, was postulated as the potentially cytotoxic oligomers of amyloid aggregation24. The formation of β-barrels in amyloid aggregation was supported by experimental studies of both the full-length and fragments of other amyloid proteins22, 25–28. For instance, recent nuclear magnetic resonance spectroscopy and native mass spectrometry studies demonstrated that Aβ42 could form specific β-barrel structures in the lipid mimicking environment22, 26. Similar β-barrel oligomers formed by Aβ40 was also supported by another recent top-down hydrogen exchange mass spectrometry experiment27. Commentary to experimental studies, the stability of β-barrel oligomers in both aqueous solution and lipid membrane environment was also investigated through molecular dynamics (MD) simulation studies29, 30. Using atomistic discrete molecular dynamics (DMD) simulations – a unique type of MD algorithms, we have investigated the aggregation dynamics of a set of amyloid fragments derived from different amyloid proteins including Aβ, IAPP and αS and revealed that the β-barrel oligomers were common intermediates towards the formation of cross-β nano-fibrils31–34. In DMD simulations, interatomic interaction potentials are described by discrete stepwise instead of continuous functions, enabling efficient sampling of conformational spaces of biomolecules (Methods). Spontaneous formation of β-barrels by short amyloid fragments were also observed in other prior computational studies using MD35, 36, replica exchange molecular dynamics37–40, and Monte Carlo41 simulations. Moreover, a similar β-barrel formation was also observed in the early aggregation stage of full-length hIAPP in silico42. However, since the isolation of β-barrel intermediates from highly heterogeneous and dynamic aggregation species is experimentally challenging, pinpointing the structure-function relationships between β-barrel oligomers and amyloid cytotoxicity in amyloidosis remains to be fully established.
A recent experimental study reported that an 11-residue fragment derived from the ALS-related SOD1 (28KVKVWGSIKGL38, denoted as SOD128–38) is necessary and sufficient for mediating the toxicity of SOD116. Single-point glycine substituting mutants G33V/G33W (denoted as SOD128–38G33V and SOD128–38G33W) abolished the cytotoxicity but didn’t attenuate fibril formation16. With small sizes, the capability of forming amyloid fibrils but contrasting cytotoxicities, SOD128–38 and its non-toxic gly-substitution mutants are the ideal system for computational simulations to identify the toxic oligomers and investigate whether the formation of β-barrel intermediates is related to amyloid cytotoxicity. In this study, we applied atomistic DMD, a predictive and computationally efficient MD approach43-45, to investigate the assembly dynamics of SOD128–38, SOD128–38G33V and SOD128–38G33W with the number of aggregating peptides ranging from 4 to 16. Our simulation results showed that all three types of peptides could spontaneously assemble into well-defined nano-fibrils from isolated monomers, but the aggregation kinetics, pathways, and final fibril confirmations were dramatically different. Briefly, the toxic SOD128–38 initially formed oligomers with random coil and bend dominant structures, followed by a conformational transition into stable β-sheets and the formation of β-barrel intermediates before converting into ordered nano-fibril structures comprised of curved single-layer β-sheets. The critical nucleus size of SOD128–38 from unstructured monomers to stable β-sheets was ~4–6. In contrast, both SOD128–38G33V and SOD128–38G33W displayed significantly higher aggregation propensities. SOD128–38G33V/SOD128–38G33W could readily self-assemble into small single-layer β-sheets and then further associated with each other into cross-β nanofibrils formed by multi-layer β-sheets. The observation of wild-type SOD128-38 forming twisted single-layer β-sheet structures in DMD simulations is consistent with prior X-ray crystallography experiments, revealing that the SOD128-38 mainly assembled into highly twisted single β-sheet layer structure16, 46. The results that both toxic SOD128-38 and its non-toxic mutants formed cross-β fibrils in both experiments and simulations are also consistent with the consensus that mature fibrils are nontoxic or significantly less toxic than oligomers1, 17-19. Direct observation of the correlation between the formation of β-barrel intermediates and the cytotoxicity in SOD128–38 and two mutants, along with previous experimental22, 24, 26, 27, 38 and computational29-34, 39, 40, 42 evidences of observing β-barrel intermediates in the aggregation of toxic amyloid peptides, support β-barrels as the toxic oligomers in amyloidosis. These β-barrels may interact with cell membranes and form “amyloid-pores” as observed in many experiments20-22. With well-defined three-dimensional structures, the β-barrels might be served as a novel therapeutic target against the amyloid-related diseases.
Materials and methods
Molecular systems setup.
In this study, we systematically investigated the self-assembly dynamics and structures of SOD128–38 and its G33V/G33W mutant peptides (denoted as SOD128–38G33V/SOD128–38G33W) by using all-atom DMD simulations with implicit solvent. Following previous experimental studies16, 46 the sequence of wild-type SOD28–38 was 28KVKVWGSIKGL38. Both N- and C-termini were treated neutrally charged for all three types of peptides. To capture the aggregation dynamics and oligomer structures with different sizes, seven molecular systems with the even number of peptides ranging from 4 to 16 for each type of fragments were set up. For each system, twenty independent simulations were performed starting with different initial configurations (i.e., coordinates and velocities). Each independent simulation lasted 1000 ns. All peptides started with a fully extended conformation and were initially placed randomly in the simulation box with a minimum inter-molecular distance of 1.5 nm. In all cases, the same peptide concentration of ~15 mM was maintained by changing the size of simulation box. The periodic boundary was used. The details of all the simulations were summarized in Table 1.
Table 1.
The details of molecular system for each type of peptides in our DMD simulations, including the number of peptides (Npeptide), the corresponding dimension of the cubic simulation box, the number of DMD runs (Nrun), the length of each DMD simulations, and the accumulative total simulation time
| Npeptide | 4 | 6 | 8 | 10 | 12 | 14 | 16 |
| Dimension, nm | 7.6 | 8.8 | 9.7 | 10.4 | 11.1 | 11.7 | 12.2 |
| Nrun | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Time, μs | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Total time,μs | 20.0 | 20.0 | 20.0 | 20.0 | 20.0 | 20.0 | 20.0 |
Details of DMD simulations.
All the simulations were performed in the canonical NVT ensemble using the all-atom discrete molecular dynamics43–45 (DMD) simulations at 300K. DMD is a unique type of molecular dynamics algorithm with significantly enhanced sampling efficiency, widely used by various groups in studying protein folding43, 44, 47, 48, amyloid aggregation31–33, 42, and nanoparticle–protein49, 50 interactions. The major difference between DMD and traditional MD approaches is in the form of the interatomic interaction potential functions, where continuous potential functions in traditional MD are replaced by step functions. The dynamics of the system is dictated by a series of collision events at which two atoms meet at an energy step and change their velocities according to conservation laws. By iteratively updating only the two colliding atoms, predicting their new collisions with corresponding neighbors, and finding the next collision via quick sort algorithms, the sampling efficiency of DMD is significantly enhanced without frequent calculations of forces and accelerations (e.g., every ~1–2 fs) in traditional MD simulations. In our DMD simulations, the step function potentials are adapted from the CHARMM-based Medusa force field43, 51. Both bonded interactions (i.e., covalent bonds, bond angles, and dihedrals) and non-bonded interactions (i.e., van der Waals, solvation, hydrogen bond, and electrostatic terms) were considered in DMD. The bonded termed were parameterized based on statistical analysis of protein structures from protein data bank (PDB). The non-bonded parameters were adopted from the CHARMM force field52. The water molecules were implicitly modeled using the EEF1 implicit solvation model developed by Lazaridis and Karplus53. A reaction-like algorithm was used to model hydrogen bond43. The electrostatic interactions were screened using the Debye-Hückel approximation with screening length set to 10 Å, which corresponds to ~100 mM of NaCl under physiological conditions. The DMD software is available to academic researchers at Molecules In Action (www.moleculesinaction.com). The units of mass, time, length, and energy used in our united-atom with implicit water model were 1 Da, ~50 femtosecond, 1 Å, and 1 kcal/mol, respectively.
Computational Analysis.
The secondary structures were calculated using the dictionary secondary structure of protein (DSSP) program54. A hydrogen bond was defined if the distance between the backbone N and O atoms was ≤3.5 Å and the angle N–H...O ≥ 120°32. Two chains were considered to form a β-sheet when two or more consecutive residues in each chain adopted the β-strand conformation and these residues were connected by at least two backbone hydrogen bonds55. The size of a β-sheet was corresponded to by the number of strands in a β-sheet layer. If two peptides were connected by at least one intermolecular heavy atom contact, the minimum inter-molecular distance within 0.55 nm, they belonged to the same oligomer. The oligomer size was referred to the total number of peptides in an oligomer. If multiple β-sheets were connect by at least one heavy atom contact pair they were defined as a β-sheet oligomer and the total number of β-strand was defined as the β-sheet oligomer size. If every β-strand was connected by two neighboring β-strands through no less than two hydrogen bonds and formed a closed single β-sheet layer, it was treated as a β-barrel oligomer32, 33, 42. A network-based algorithm was used to identify β-barrel oligomers along the simulation trajectories. The potential of mean force (PMF) was calculated according to
| (1) |
where kB was the Boltzmann constant, T corresponded to the simulation temperature 300 K, P(x, y) was the probability distribution of two selected reaction coordinates, x and y.
Results and Discussions
To investigate the self-assembly dynamics and structures of SOD128–38, SOD128–38G33V and SOD128–38G33W, seven molecular systems for each type of peptides were studied with even number of peptides ranging from 4 to 16. For each molecular system, twenty independent implicit solvent DMD simulations each lasted 1 μs were performed at the room temperature by starting from different initial states including randomized positions, orientations and velocities (Methods). In all cases, the same concentration of peptides at ~15 mM was maintained by adjusting the simulation box sizes. Here, the high peptide concentration was arbitrarily chosen to increase the aggregation rates without affecting the structures of stable aggregates in silico56. To avoid the potential biases from the initial states, only the last 200 ns simulation data of each trajectory was used for equilibrium conformational analysis.
Glycine substitutions enhanced the β-sheet propensity of SOD128–38 aggregation.
We first analyzed the secondary structure properties of the assemblies formed by each type of peptides with different number of aggregating peptides (Fig. 1a). With four SOD128–38, the peptide system was predominantly random coil with the content as high as ~68%, the β-sheet probability was only ~12%. As the number of peptides increased to six, the averaged content of random coil decreased to ~56%, and β-sheet increased to ~30%. When the number of simulated peptides increased beyond six, the overall secondary compositions became saturated with the contents of random coil, β-sheet, bend and turn fluctuating around ~46%, ~45%, ~6% and ~3%, respectively (Fig. 1a). No helices were observed. The averaged secondary structure propensities per residue was also analyzed (Fig. 1d, g, j, m). Except for the two terminal residues, most residues of SOD128–38 predominantly formed β-sheets with over 60% propensities when the number of simulated peptides larger than six (Fig. 1g). Residue G33 displayed a high propensity (>20%) to adopt the bend conformation (Fig. 1j). Residues L37 and G38 also have ~10–20% probabilities to form bend and turn structures (Fig. 1j&m). Compared to SOD128–38, SOD128–38G33V and SOD128–38G33W displayed significantly higher β-sheet propensities with the average probability of ~65% in all simulated systems without the size dependence in the aggregation of wild-type (Fig. 1b&c). The coil content was less than 35%, while the propensities for other structures was less than 1%. The analyses of secondary structure propensities per residue in SOD128–38G33V (Fig. 1 e, h, k, n) and SOD128–38G33W (Fig. 1 f, i, l, o) demonstrated that the substitution of G33 to hydrophobic valine and tryptophan significantly enhanced β-sheet formation not only near the mutated G33 but also the rest of sequences, with all other secondary structures including coil, turn and bend significantly inhibited. For instance, residues 30–36 in SOD128–38G33V/SOD128–38G33W showed 90–95% propensities in β-sheet conformation, much higher than those in SOD128–38. The bend and turn structures formed by residue 33 were completely inhibited in SOD128–38G33V/SOD128–38G33W systems. Overall, the single-point mutations substituting the flexible glycine 33 with hydrophobic valine and tryptophan significantly enhanced the β-sheet formation of SOD128–38 assemblies.
Fig. 1.
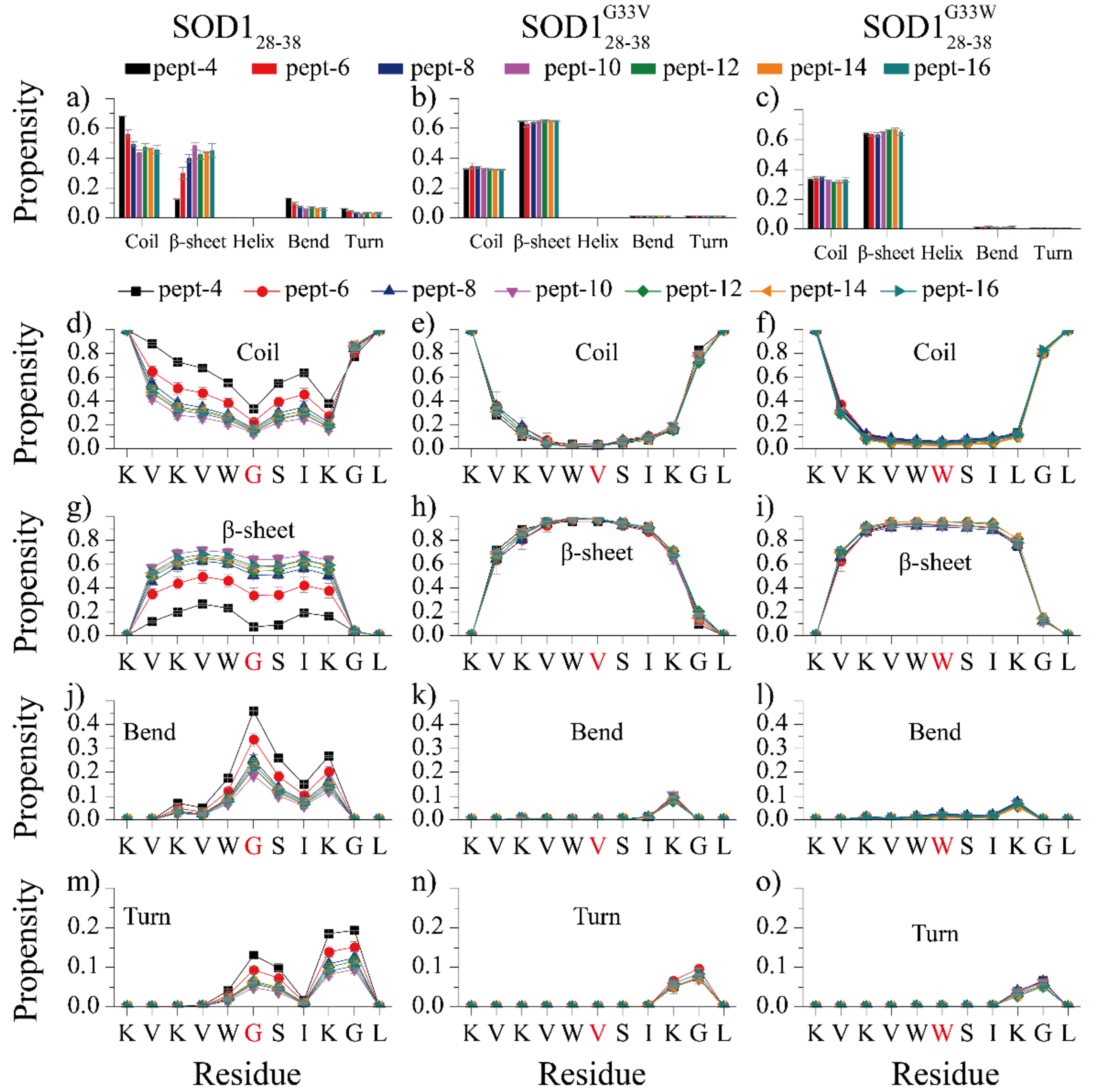
Secondary structure analyses of SOD128–38, SOD128–38G33V and SOD128–38G33W self-assemblies, (a-c) The average secondary structure contents in terms of coil, β-sheet, helix, bend and turn for SOD128–38, SOD128–38G33V and SOD128–38G33W aggregation with different number of peptides. The propensity of each residue adopted coil (d-f), β-sheet (g-i), bend (j-k) and turn (m-o) for each type of peptides with different system sizes. The error bars of secondary structure propensities correspond to the standard deviations of means from 20 independent simulations. Our results suggest that substituting flexible glycine at residue position 33 (highlighted in red in panels d-o) with hydrophobic valine and tryptophan significantly increased the β-sheet propensity in the aggregates.
Glycine substitutions altered the fibril structure of SOD128–38.
To investigate the effects of single-point glycine substitutions on the assembly structures of SOD128–38, we did conformational analysis of the equilibrium assemblies of SOD128–38 and its mutants. For each type of peptides, we calculated the mass-weighted probability distributions of oligomers, β-sheet oligomers and β-sheets with different sizes in each molecular system (Fig. 2a-i). An oligomer was defined as a cluster of peptides connected by inter-molecular heavy atom contacts. If two β-sheets were connected by at least one inter-molecular heavy atom pair, they belonged to the same β-sheet oligomer. The oligomer, β-sheet oligomer and β-sheet sizes denoted the total number of composite peptides. SOD128–38 oligomers were dynamic, with large size oligomers co-existing with monomers (Fig. 2a). In the four-peptide system, SOD128–38 peptides mainly stayed as monomers with weak populations to various aggregates from dimer to tetramer. As the system size increased to six, SOD128–38 oligomers and β-sheet oligomers with the size around 5–6 became the dominant species, where SOD128–38 mainly assembled into 5- and 6-strand β-sheets (Fig. 2 a, d, g). When the system size increased beyond six, SOD128–38 mainly assembled into single β-sheet oligomers with the size distributions of β-sheet oligomers and β-sheets similar to that of oligomers (Fig. 2 a, d, g). Our results also indicated that the critical nucleation size for SOD128–38 amyloid aggregation was between 4 to 6.
Fig. 2.
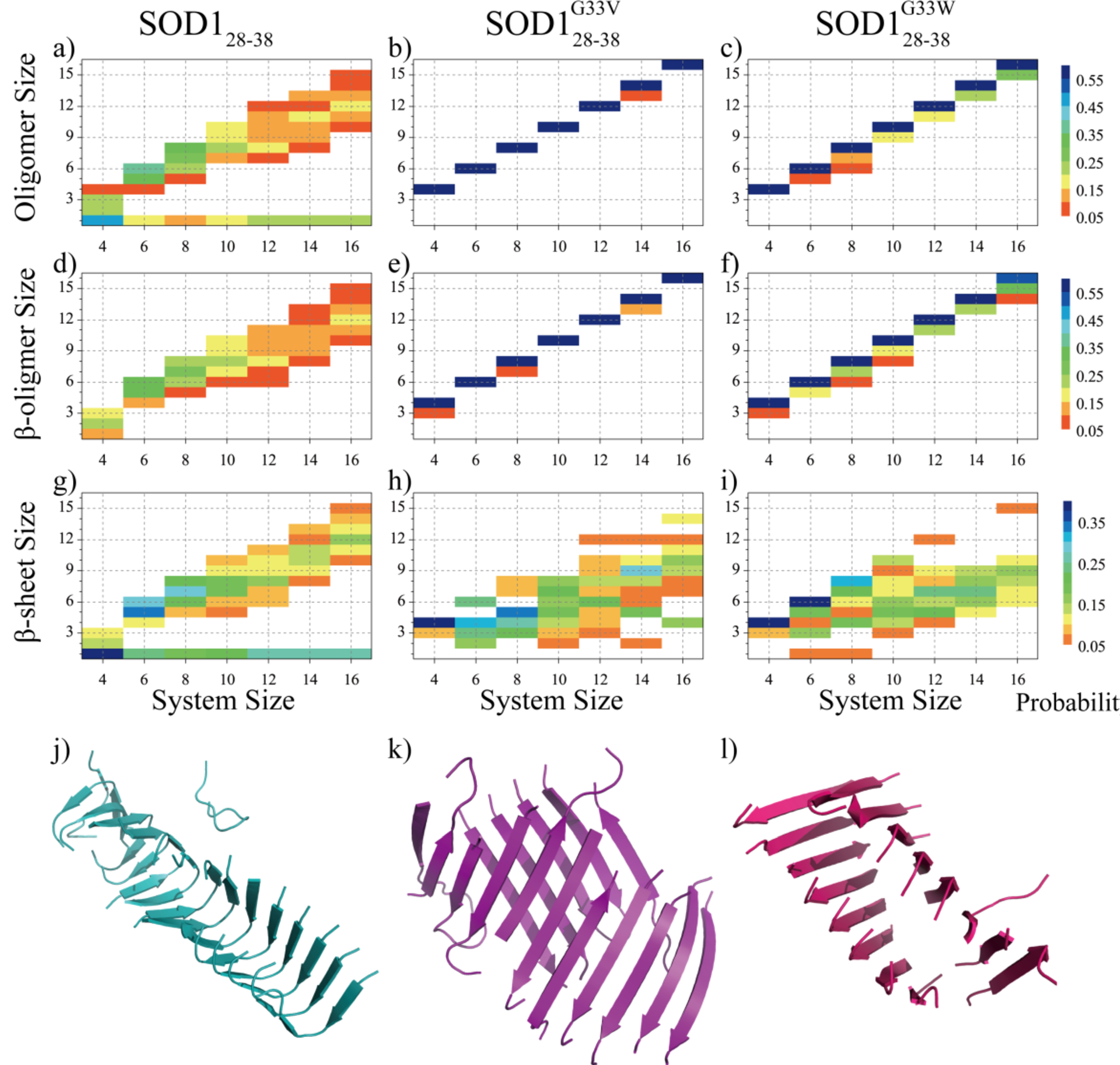
Conformational analyses of the SOD128–38, SOD128–38G33V and SOD128–38G33W assemblies. The probability distribution of the oligomer size (a-c), β-sheet oligomer size (d-f) and β-sheet size (g-i) for the SOD128–38, SOD128–38G33V and SOD128–38G33W self-assemblies in each molecular system. To avoid bias from the initial state, only the conformations of the last 200 ns of all independent simulations were included in the analysis. For each peptide type, representative aggregate structures in simulations of 16 peptides were also presented (j-k). SOD128–38, SOD128–38G33V and SOD128–38G33W peptides were colored in cyan, purple and pink, respectively.
In contrast, SOD128–38G33V and SOD128–38G33W preferred to aggregate into a single β-sheet rich oligomer in all simulated molecular systems with the most populated oligomers and β-sheet oligomers comprised of all peptides in simulations (Fig. 2b, c, e, f). Even in the smallest four-peptide systems, the mutants predominantly self-assembled into four-strand β-sheet layers indicating that the critical nucleation size was less than four. Both single- and two-layer β-sheets were observed in the six-peptide system as indicated by the population of β-sheet sizes ranging from 2 to 6, with the latter corresponding to a single β-sheet oligomer. As the system size increased, the final assemblies of SOD128–38G33V and SOD128–38G33W mainly featured two-layer β-sheet formation, as the most populated β-sheet sizes were roughly equivalent to the half of system sizes (Fig. 2h,i). Snapshots of representative aggregates (Fig. 2j, k, l) as well as final assembled structures from all independent simulations (Fig. S1–3) of the largest 16-peptide systems confirmed that SOD128–38 mainly formed single-layer β-sheet nanofibrils with each strand featuring a β-turn-β conformation, while SOD128–38G33V/SOD128–38G33W predominantly assembled into two-layer mated β-sheet fibrils. The turn in wild-type SOD128–38 was around the flexible glycine known as the breaker of ordered secondary structures. Mutating G33 with hydrophobic valine and tryptophan rendered the peptides to form a single straight β-strand, and thus alter the fibril structures with two-layer β-sheets to bury the hydrophobic residues on one-side of the β-strands.
Glycine substitution mutations promoted the inter-molecular interactions between central residues of SOD128–38.
To characterize the interactions stabilizing the β-rich assembly structures, we calculated the inter-peptide contact frequency maps between either the backbone or sidechain atoms of different residues (Fig. 3a-f). The backbone contact frequencies showed that SOD128–38 formed both parallel and anti-parallel β-sheets, but the parallel alignment pattern was more frequency (Fig. 3a). The side-chain contact frequencies revealed that the SOD128–38 assemblies were mainly stabilized by interactions among hydrophobic residues (Fig. 3d). The ratio of the hydrogen bonds in anti-parallel or parallel alignment of β-strands further conformed that the parallel alignment β-strands had higher probability than the anti-parallel alignment β-strands (Fig. 3g). In the SOD128–38G33V/SOD128–38G33W assemblies, the anti-parallel alignments of β-strands was enhanced (Fig. 3b&c), where the hydrogen bonds in anti-parallel and parallel alignment of β-strands were around 1:1 ratio (Fig. 3h&i). The side-chain residue-pairwise contact probability showed the G33V/G33W mutation decreased the V29-L38, and L38-L38 pairwise contact propensity, and the interaction between central hydrophobic residues were significantly enhanced. This was because the SOD128–38G33V/SOD128–38G33W mainly assembled into tightly mated two-layer β-sheet, and the SOD128–38 aggregated into single twisted β-sheet.
Fig. 3.
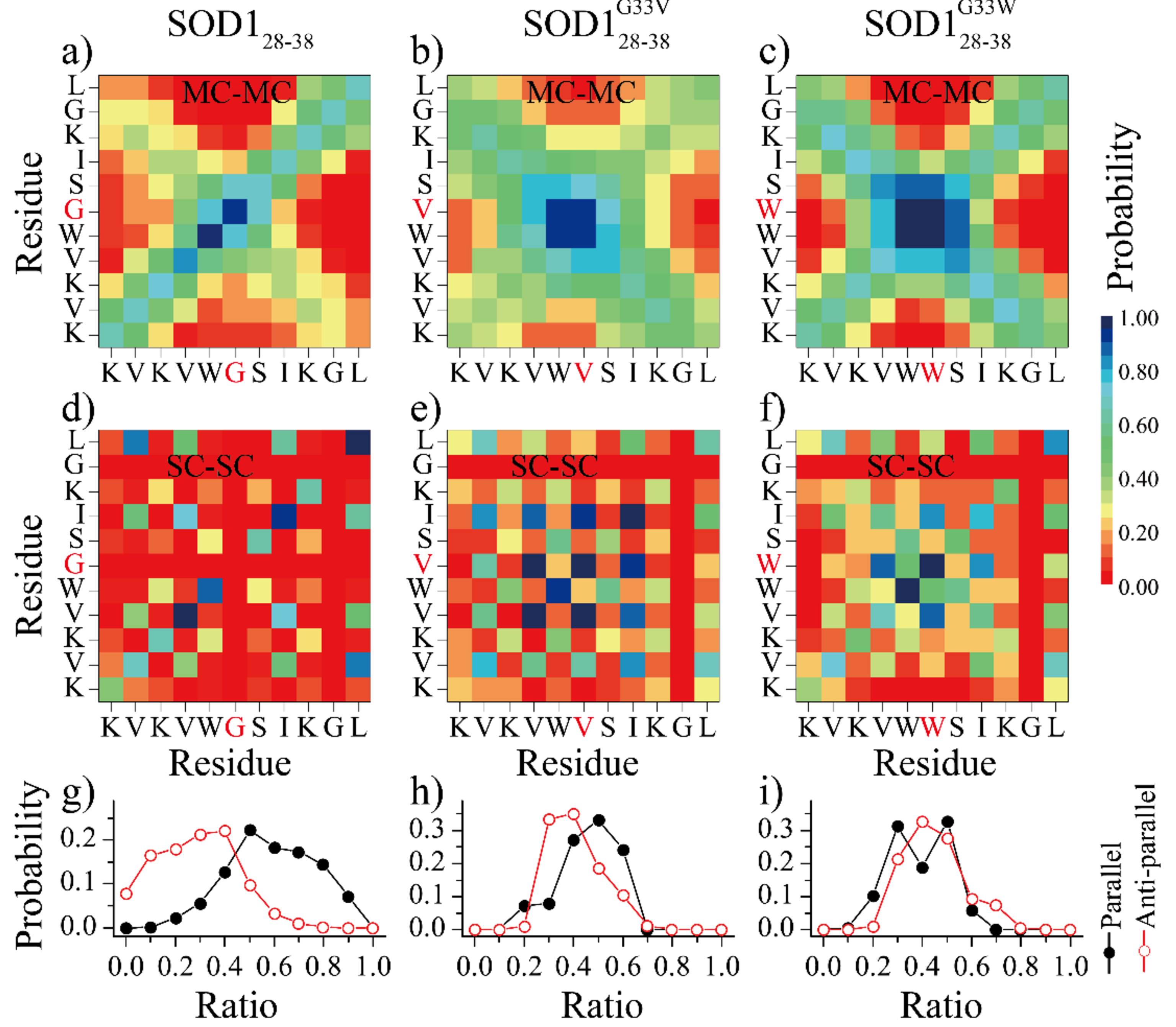
The inter–peptide interaction analysis. The residue-pairwise inter-molecular contact frequency maps were computed both between main-chain atoms (MC-MC) and between side-chain atoms (SC-SC) in simulations of sixteen peptides. Only the last 200ns of each 1000ns independent simulations was used for analysis.
Glycine substitutions changed the aggregation pathways of SOD128–38 and inhibited the formation of β-barrels as intermediates.
To examine details of conformational dynamics of fibrilization, we first analyzed simulation trajectories of eight peptides, with the system size larger than the critical nucleus size of ~4–6 for the wild type (Fig. 2). in detail for each type of peptide. For each type of peptides, we randomly picked a trajectory, where the time evolution of the largest oligomer, the largest β-sheet oligomer, the largest β-sheet, and the mass-weighted average β-sheet sizes, together with the β-barrel size were calculated (Fig. 4a). Following our previous studies, if a β-sheet had a closed form with each β-strand having at least two neighboring β-strands through at least two hydrogen bonds, we defined it as a β-barrel31–34, 42. The β-barrel size corresponded to the total number of composite peptides. As shown in Fig. 4a, the wild-type SOD128–38 first aggregated into small unstable oligomers with coil structures abundant where the largest oligomer size was larger than the largest β-sheet oligomer size. These initial oligomers were highly dynamic with frequent association and dissociation before assembling into pentamers (the first 400 ns in Fig. 4a). When the oligomer size increased up to five with addition of monomers, most of the peptides converted into a single β-sheet structures (e.g., the largest oligomer size was approximately the same to the largest β-sheet and average β-sheet sizes) and then subsequently formed β-barrel pentamer (e.g., snapshots at ~440 and 480 ns in Fig. 4a). A conformational conversion of β-barrel from pentamer to hexamer was also observed (e.g., ~500 to 800 ns in Fig. 4a) via dissociation and re-association. The inter-conversion between β-barrel and β-sheet layer was also detected (e.g., ~800 to 1000 ns in Fig. 4a). Simulations of other molecular systems with number of peptides displayed similar aggregation dynamics (Fig. S4). Overall, the self-assembly process of SOD128–38 was very dynamic with the frequent conformational conversion and the β-barrel intermediates abundant.
Fig. 4.
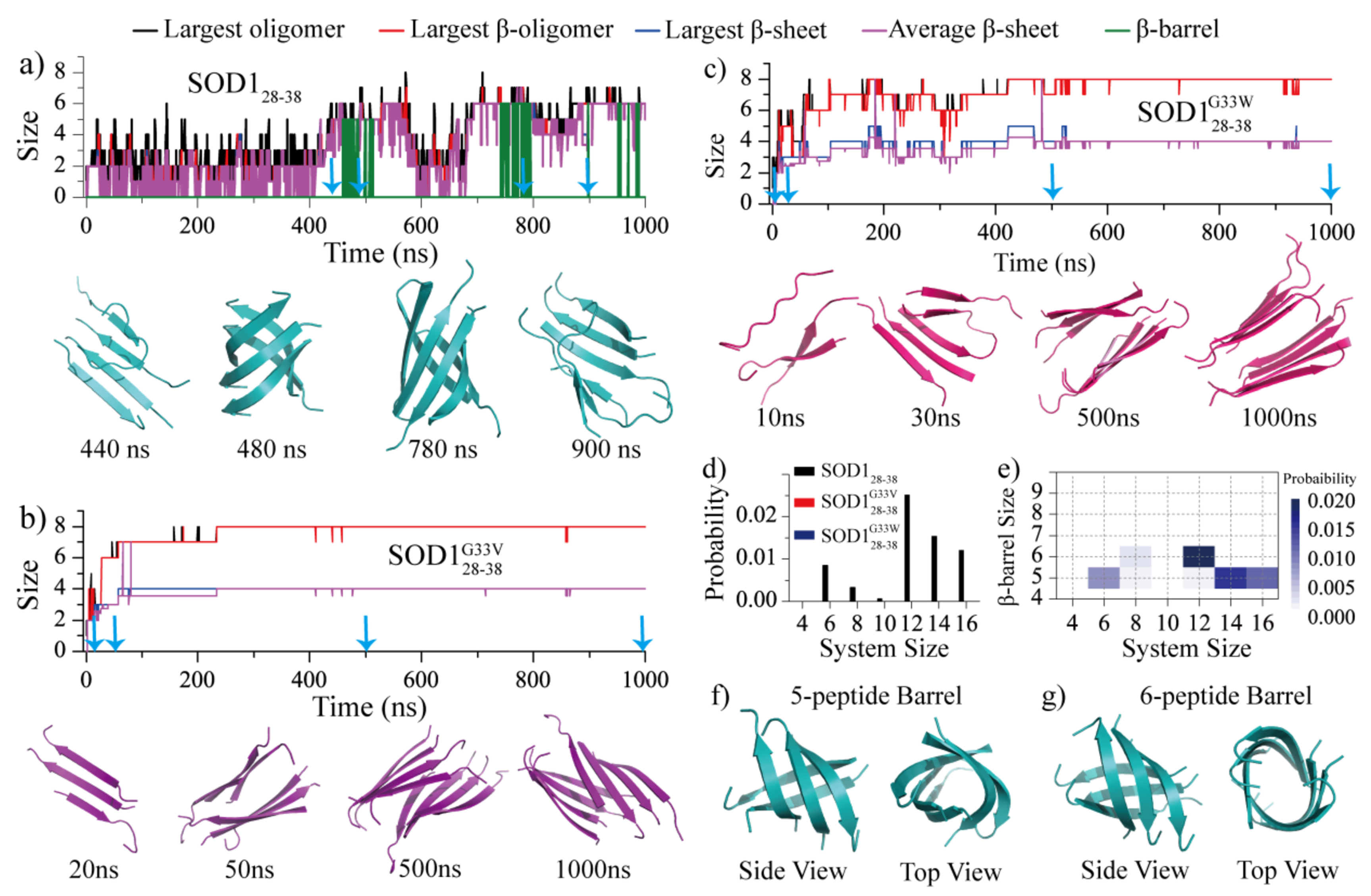
The self-assembly dynamics and conformations analysis. The self-assembly dynamics of eight SOD128–38 a), SOD128–38G33V b), and SOD128–38G33W c) peptides monitored by the time evolution of the largest oligomer size (black), largest β-sheet oligomer size (red), largest β-sheet size (blue), averaged β-sheet layer size (purple) and β-barrel size (green). The oligomer structures along the simulation trajectory as indicated by cyan arrows were present at the bottom, d) The probability of observing the formation of β-barrels in each molecular system for all three peptide types, e) The probability distribution of β-barrels with different sizes was calculated as a function of the number of simulated peptides for SOD128–38. f-g) The representative structure of SOD128–38 β-barrel pentamer and hexamer. SOD128–38, SOD128–38G33V and SOD128–38G33W were colored in cyan, purple and pink, respectively.
Compared to the SOD128–38, SOD128–38G33V and SOD128–38G33W peptides featured drastically different assembly dynamics (Fig. 4b&c). The glycine-substitution mutants displayed significantly higher aggregation propensity, where they readily self-assembled into small size β-sheets (e.g., ~20 ns in Fig. 4b, ~10 ns in Fig. 4c). There were much less fluctuations in terms of the average β-sheet size as the β-sheet oligomers grew, indicating that these small β-sheets acted as building blocks to form large-size β-sheet oligomers (e.g., ~50 ns in Fig. 4b, ~30 ns in Fig. 4c). Eventually, these small β-sheets coagulated with each other and re-adjusted themselves into well-defined two-layer β-sheet assemblies (e.g., ~50 and 500 ns in Fig. 4b, ~30 and 500 ns in Fig. 4c). No obvious conformational fluctuations were observed once the two-layer assemblies formed, suggesting that the aggregates of SOD128–38G33V/SOD128–38G33W were very stable, which was markedly different from that for SOD128–38. Besides, we didn’t observe any β-barrel formations during the assembly process of mutants in the selected trajectories.
The probability for each type of peptides to form β-barrel averaged over all independent simulations (during the whole 1000 ns trajectories) in every simulated system was also calculated (Fig. 4d and Table S1). Indeed, the β-barrel were only observed for the wild-type SOD128–38 when the system size was larger than 4 (Fig. S4). The mass-weighted size distribution of β-barrel formed by SOD128–38 was also analyzed for each system (Fig. 4e), SOD128–38 mainly assembled into five- and six-peptide β-barrel (shown in Fig. 4f&g). Prior cell viability assay showed that SOD128–38 was toxic to primary motor neurons, but the single-point mutation of G33V/G33W was non-toxic to motor neurons16. Along with the G33V/G33W mutation of SOD128–38 completely inhibited its ability to form β-barrel assemblies, our results suggested that the β-barrel might be the potential toxic oligomer of SOD128–38. The relationship between the propensity of forming β-barrel intermediates and amyloid cytotoxicity was also supported by prior hIAPP segments studies, where the β-barrel intermediates widely observed in toxic fragment hIAPP19–29 assemblies, and were not detected in the non-toxic hIAPP15–25 aggregates31,57. Such β-barrel formed by other amyloid fragments24, 31–34, 38–41 (e.g Aβ16–22, hIAPP8–20, αS68–78, hIAPP22–29, Aβ25–35, αB90–100) and full-length proteins22, 26, 29, 30, 42 (Aβ, hIAPP) were also reported by other computational and experimental studies. With well-defined three-dimensional structures and compatible to the “amyloid-pore” hypothesis, the directly observed the correlation between β-barrel oligomers formation and cytotoxicity revealed that the β-barrel may play a key important role for the cytotoxicity of amyloid aggregation.
The self-assembly free energy landscapes
To better understand the effects of glycine substitutions on the SOD128–38 self-assembly dynamics, we further computed the effective aggregation free energy landscape along with the aggregation dynamics for simulations with 16 peptides (Fig. 5). The free energy landscape estimated as the potential of mean force (PMF) was computed as a function of the oligomer size and the average number of residues forming β-sheet structures per peptide (Nβ-sheet) by using all the 1000-ns trajectories of twenty independent simulations to capture the early assembly process. There were two well-defined energy basins for the assembly free energy landscape of SOD128–38 around (1,0) and (13,7), corresponding to the isolated monomers and the β-sheet rich nano-fibrils, respectively. The oligomers with sizes less than 4 featured low β-sheet content (highlighted as regions 1 & 2 in the PMF and corresponding snapshots in Fig. 5a). When the oligomer size larger than 4, the dominant conformations of SOD128–38 assemblies were β-sheet rich, where the average β-sheet residues per chain increase up to 7 (labeled as 3 in Fig. 5a). Hence, the critical nucleus size for SOD128–38 aggregation was around 5. The assembly dynamics showed that the β-barrel intermediates were formed during the conformational conversion. The final β-sheet abundant assemblies (labeled as 4 in Fig. 5a) saturated around the oligomer size of ~13 with ~1–3 fluctuations in size, indicating the dynamic assembly equilibrium with monomers in the solution.
Fig. 5.
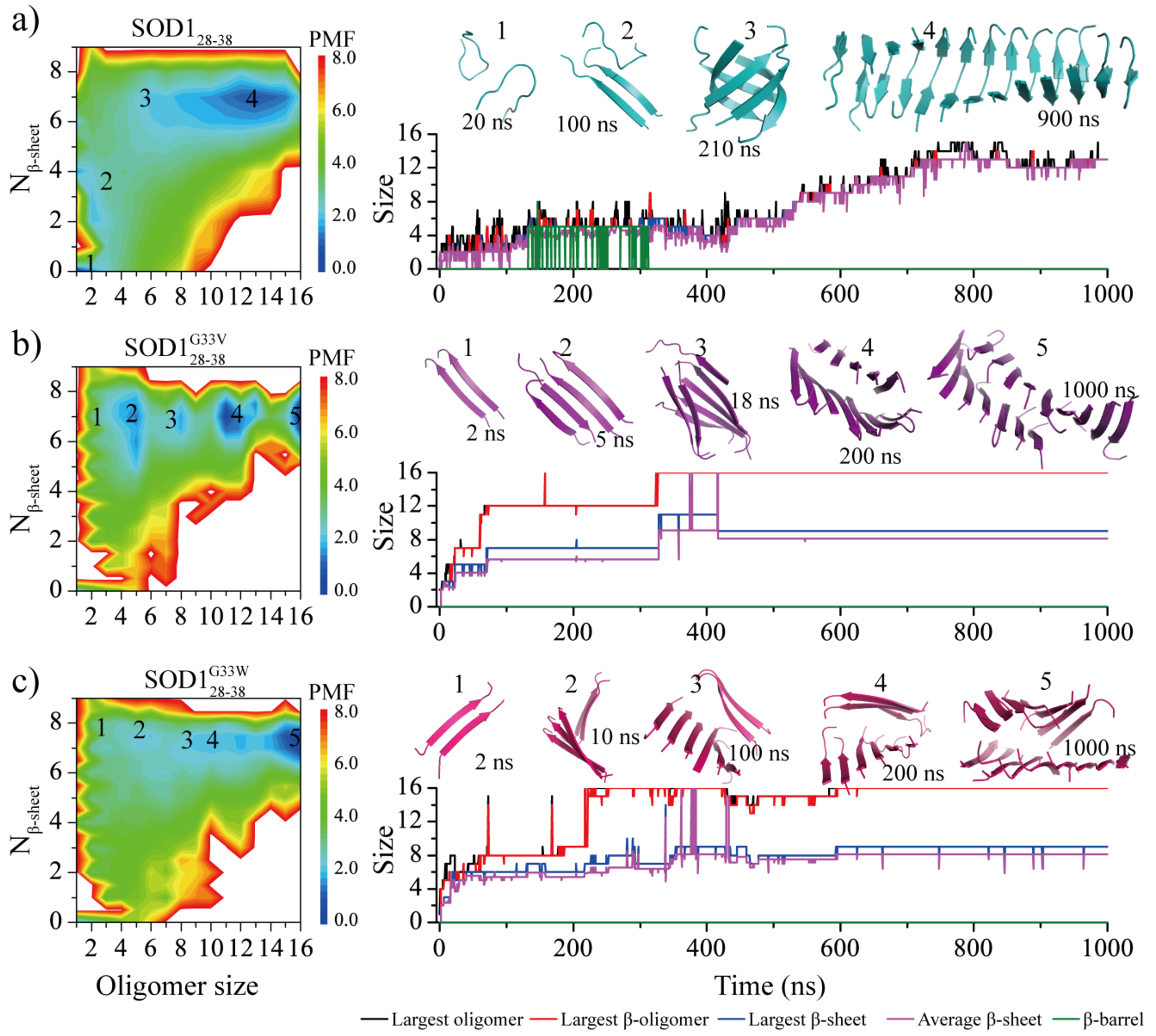
The self-assembly free energy landscape and aggregation dynamics analysis for sixteen-peptide simulations. For each of three peptide types - a) SOD128–38, b) SOD128–38G33V and c) SOD128–38G33W, the free energy landscape as a function of the oligomer size and the average number of residues adopting β-sheet conformation per chain (Nβ-sheet) are presented on the left. To capture all the conformation of assemblies during the aggregation process, the whole 1000 ns trajectory of 20 independent DMD runs were included in the analysis. A representative trajectory from isolated monomers into final nano-fibrils via all the intermediates corresponding to labeled basins in the PMF is also shown on the right. The size of the largest oligomer (black), largest β-sheet oligomer (red), largest β-sheet layer (blue), averaged β-sheet layer (purple) and β-barrel (green) are plotted as a function of simulation time. The snapshots along with the growth of assemblies are also shown in the inset on the right and their corresponding states in the free energy landscape are also labeled. SOD128–38, SOD128–38G33V and SOD128–38G33W peptides are colored in cyan, purple and pink, respectively.
The self-assembly free energy landscape of SOD128–38G33V/SOD128–38G33W mutants (Fig. 5b&c) were also drastically different from wild-type SOD128–38. The isolated monomers became unfavorable with high free energy. Small oligomers including dimers, trimers and tetramers were ready high in the β-sheet content, where the average β-sheet residues per chain were larger than 7 (e.g., regions 1&2 the PMF and corresponding snapshots in Fig. 5b&c), indicating that the critical nucleation size of SOD128–38G33V/SOD128–38G33W mutants should be around 2. The assembly dynamics along a simulation trajectory directly showed that SOD128–38G33V/SOD128–38G33W readily aggregated into small-size β-sheets and these small β-sheets subsequently assembled into large size β-sheet oligomer (e.g., regions and snapshots 3&4 in Fig. 5b&c) without forming β-barrel intermediates. All of the 16 SOD128–38G33V/SOD128–38G33W peptides assembled into a single ordered two-layer mated β-sheet nanofibrils (snapshots 5 in Fig. 4b&c) located at the lowest free energy basin (16, 8). There was no much fluctuation when the SOD128–38G33V/SOD128–38G33W during last the 400 ns, indicating the final two-layer β-sheet structure was very stable. Our results demonstrated the G33V/G33W single-point mutations of SOD128–38 enhanced the self-assembly and β-sheet propensity of SOD128–38, inhibited the β-barrel formation, and changed the final assemblies from single twisted β-sheet to tightly mated two-layer β-sheet.
The correlation between the formation of β-barrels and amyloid toxicity.
Eisenberg and coworkers recently showed that two different fragments of IAPP, IAPP19–29 and IAPP15–25, also featured different toxicities and fibril morphologies57. Toxic IAPP19–29 formed mated β-sheets, but the non-toxic IAPP15–25 formed unmated β-sheets. Toxic SOD128–38, on the other hand, aggregated into the “corkscrew” structure comprised of a unmated highly curved single-layer β-sheet16, 46. These observations suggest that amyloid cytotoxicity is independent of the final fibril structures, consistent with the toxic oligomer hypothesis1, 17–19. Interestingly, an early computational study showed that the toxic IAPP19–29 formed β-barrels as aggregation intermediates but the non-toxic IAPP15–25 did not, consistent with our computational results on the aggregation of SOD128–38 and two glycine-substitution mutants. Similarly, β-barrel intermediates were also observed in the aggregation of other toxic amyloid peptide fragments such as αS68–78 (also known as NACore)33, hIAPP22–2931 and Aβ16–2231, 34 as well as full-length amyloid peptides such as Aβ22, 26, 29, 30 and hIAPP42. The correlation between amyloid cytotoxicity and the formation of β-barrels as aggregation intermediates strongly supports β-barrel intermediates as the toxic oligomers in amyloidosis. Further questions of how amyloid proteins and peptides spontaneously form amyloid pores in the membrane environment and cause membrane damages remain to be uncovered both computationally and experimentally.
Conclusion
We systematically studied the self-assembly dynamics and structures of SOD128–38 and its G33V/G33W mutants from isolated monomers to well-defined β-sheet nano-fibrils in silico. Our simulation results demonstrated that the toxic SOD128–38 peptides first assembled into small unstructured oligomers, followed by coil-to-sheet conformational conversions in oligomers with at least 4–6 peptides and the formation of β-barrels as intermediates. The final twisted single-layer β-sheets of SOD128–38 was consistent with the prior X-ray crystallography study where the so-called “corkscrew” structure was also comprised of a twisted single-lay β-sheet16, 46. The non-toxic G33V and G33W mutants, on the other hand, displayed completely different assembly dynamics. The glycine-substituting mutant peptides first assembled into small β-sheets with even the dimeric assemblies dominant with β-sheet formations. These small β-sheets coagulated with each other and re-adjusted themselves into well-defined steric zipper two-layer β-sheets without the population of β-barrel intermediates. These results are also consistent with prior experiments demonstrated that the glycine-substitution mutations attenuated cytotoxicity but didn’t inhibited fibril formation. The observation that both toxic SOD128–38 and its non-toxic mutants formed cross-β fibrils in both experiments and simulations supports the consensus that mature fibrils are nontoxic or significantly less toxic than oligomers1, 17–19. Our systematic computational study of SOD128–38 and the two mutants as well as previous experimental22, 24, 26, 27, 38 and computational29–34, 39, 40, 42 evidences of observing β-barrel intermediates in the aggregation of toxic amyloid peptide support β-barrels as the toxic oligomers in amyloidosis. These β-barrels may interact with cell membranes and form “amyloid-pores” as observed in many experiments20–22. With well-defined three-dimensional structures, the β-barrels might be served as a novel therapeutic target against the amyloid-related diseases.
Supplementary Material
Acknowledgement
This work was supported in part by the National Natural Science Foundation of China under the Grant No. 11904189 (Sun), K.C. Wong Magna Fund in Ningbo University, China (Sun), NSF CBET-1553945 (Ding), and NIH R35GM119691 (Ding). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSFC, NIH and NSF.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Iadanza MG; Jackson MP; Hewitt EW; Ranson NA; Radford SE, A new era for understanding amyloid structures and disease. Nature reviews. Molecular cell biology 2018, 19, 755–773. [DOI] [PubMed] [Google Scholar]
- 2.Ke PC; Pilkington EH; Sun Y; Javed I; Kakinen A; Peng G; Ding F; Davis TP, Mitigation of Amyloidosis with Nanomaterials. Advanced materials 2020, 32, e1901690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ke PC; Sani MA; Ding F; Kakinen A; Javed I; Separovic F; Davis TP; Mezzenga R, Implications of peptide assemblies in amyloid diseases. Chemical Society reviews 2017, 46, 6492–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rouse SL; Matthews SJ; Dueholm MS, Ecology and Biogenesis of Functional Amyloids in Pseudomonas. Journal of molecular biology 2018, 430, 3685–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fowler DM; Koulov AV; Alory-Jost C; Marks MS; Balch WE; Kelly JW, Functional amyloid formation within mammalian tissue. PLoS biology 2006, 4, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu XZ; Liao MR; Gong HN; Zhang L; Cox H; Waigh TA; Lu JR, Recent advances in short peptide self-assembly: from rational design to novel applications. Curr Opin Colloid In 2020, 45, 1–13. [Google Scholar]
- 7.Guo C; Arnon ZA; Qi R; Zhang Q; Adler-Abramovich L; Gazit E; Wei G, Expanding the Nanoarchitectural Diversity Through Aromatic Di- and Tri-Peptide Coassembly: Nanostructures and Molecular Mechanisms. ACS nano 2016, 10, 8316–24. [DOI] [PubMed] [Google Scholar]
- 8.Murphy MP; LeVine H 3rd, Alzheimer's disease and the amyloid-beta peptide. Journal of Alzheimer's disease: JAD 2010, 19, 311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fitzpatrick AWP; Falcon B; He S; Murzin AG; Murshudov G; Garringer HJ; Crowther RA; Ghetti B; Goedert M; Scheres SHW, Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 2017, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefanis L, alpha-Synuclein in Parkinson's disease. Cold Spring Harbor perspectives in medicine 2012, 2, a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westermark P; Andersson A; Westermark GT, Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiological reviews 2011, 91, 795–826. [DOI] [PubMed] [Google Scholar]
- 12.Sangwan S; Eisenberg DS, Perspective on SOD1 mediated toxicity in Amyotrophic Lateral Sclerosis. Postepy biochemii 2016, 62, 362–369. [PubMed] [Google Scholar]
- 13.Kajava AV; Aebi U; Steven AC, The parallel superpleated beta-structure as a model for amyloid fibrils of human amylin. Journal of molecular biology 2005, 348, 247–52. [DOI] [PubMed] [Google Scholar]
- 14.Xiao Y; Ma B; McElheny D; Parthasarathy S; Long F; Hoshi M; Nussinov R; Ishii Y, Abeta(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nature structural & molecular biology 2015, 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y; Zhao C; Luo F; Liu Z; Gui X; Luo Z; Zhang X; Li D; Liu C; Li X, Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell research 2018, 28, 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sangwan S; Zhao A; Adams KL; Jayson CK; Sawaya MR; Guenther EL; Pan AC; Ngo J; Moore DM; Soriaga AB; Do TD; Goldschmidt L; Nelson R; Bowers MT; Koehler CM; Shaw DE; Novitch BG; Eisenberg DS, Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS). Proceedings of the National Academy of Sciences of the United States of America 2017, 114, 8770–8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glabe CG, Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiology of aging 2006, 27, 570–5. [DOI] [PubMed] [Google Scholar]
- 18.Glabe CG; Kayed R, Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006, 66, S74–8. [DOI] [PubMed] [Google Scholar]
- 19.Simoneau S; Rezaei H; Sales N; Kaiser-Schulz G; Lefebvre-Roque M; Vidal C; Fournier JG; Comte J; Wopfner F; Grosclaude J; Schatzl H; Lasmezas CI, In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS pathogens 2007, 3, e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Last NB; Rhoades E; Miranker AD, Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proceedings of the National Academy of Sciences of the United States of America 2011, 108, 9460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Scala C; Yahi N; Boutemeur S; Flores A; Rodriguez L; Chahinian H; Fantini J, Common molecular mechanism of amyloid pore formation by Alzheimer's beta-amyloid peptide and alpha-synuclein. Scientific reports 2016, 6, 28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serra-Batiste M; Ninot-Pedrosa M; Bayoumi M; Gairi M; Maglia G; Carulla N, Abeta42 assembles into specific beta-barrel pore-forming oligomers in membrane-mimicking environments. Proceedings of the National Academy of Sciences of the United States of America 2016, 113, 10866–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naqvi AAT; Mohammad T; Hasan GM; Hassan MI, Advancements in Docking and Molecular Dynamics Simulations Towards Ligand-receptor Interactions and Structure-function Relationships. Current topics in medicinal chemistry 2018, 18, 1755–1768. [DOI] [PubMed] [Google Scholar]
- 24.Laganowsky A; Liu C; Sawaya MR; Whitelegge JP; Park J; Zhao M; Pensalfini A; Soriaga AB; Landau M; Teng PK; Cascio D; Glabe C; Eisenberg D, Atomic view of a toxic amyloid small oligomer. Science 2012, 335, 1228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kandel N; Zheng T; Huo Q; Tatulian SA, Membrane Binding and Pore Formation by a Cytotoxic Fragment of Amyloid beta Peptide. The journal of physical chemistry. B 2017, 121, 10293–10305. [DOI] [PubMed] [Google Scholar]
- 26.Osterlund N; Moons R; Ilag LL; Sobott F; Graslund A, Native Ion Mobility-Mass Spectrometry Reveals the Formation of beta-Barrel Shaped Amyloid-beta Hexamers in a Membrane-Mimicking Environment. Journal of the American Chemical Society 2019, 141, 10440–10450. [DOI] [PubMed] [Google Scholar]
- 27.Pan J; Han J; Borchers CH; Konermann L, Structure and dynamics of small soluble Abeta(1–40) oligomers studied by top-down hydrogen exchange mass spectrometry. Biochemistry 2012, 51, 3694–703. [DOI] [PubMed] [Google Scholar]
- 28.Kandel N; Matos JO; Tatulian SA, Structure of amyloid beta25–35 in lipid environment and cholesterol-dependent membrane pore formation. Scientific reports 2019, 9, 2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen PH; Campanera JM; Ngo ST; Loquet A; Derreumaux P, Tetrameric Abeta40 and Abeta42 beta-Barrel Structures by Extensive Atomistic Simulations. II. In Aqueous Solution. The journal of physical chemistry. B 2019, 123, 6750–6756. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen PH; Campanera JM; Ngo ST; Loquet A; Derreumaux P, Tetrameric Abeta40 and Abeta42 beta-Barrel Structures by Extensive Atomistic Simulations. I. In a Bilayer Mimicking a Neuronal Membrane. The journal of physical chemistry. B 2019, 123, 3643–3648. [DOI] [PubMed] [Google Scholar]
- 31.Sun Y; Ge X; Xing Y; Wang B; Ding F, beta-barrel Oligomers as Common Intermediates of Peptides Self-Assembling into Cross-beta Aggregates. Scientific reports 2018, 8, 10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y; Kakinen A; Xing Y; Faridi P; Nandakumar A; Purcell AW; Davis TP; Ke PC; Ding F, Amyloid Self-Assembly of hIAPP8–20 via the Accumulation of Helical Oligomers, alpha-Helix to beta-Sheet Transition, and Formation of beta-Barrel Intermediates. Small 2019, 15, e1805166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y; Kakinen A; Zhang C; Yang Y; Faridi A; Davis TP; Cao W; Ke PC; Ding F, Amphiphilic surface chemistry of fullerenols is necessary for inhibiting the amyloid aggregation of alpha-synuclein NACore. Nanoscale 2019, 11, 11933–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ge X; Sun Y; Ding F, Structures and dynamics of beta-barrel oligomer intermediates of amyloid-beta16–22 aggregation. Biochimica et biophysica acta. Biomembranes 2018, 1860, 1687–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei G; Song W; Derreumaux P; Mousseau N, Self -assembly of amyloid-forming peptides by molecular dynamics simulations. Frontiers in bioscience: a journal and virtual library 2008, 13, 5681–92. [DOI] [PubMed] [Google Scholar]
- 36.Wei G; Mousseau N; Derreumaux P, Sampling the self -assembly pathways of KFFE hexamers. Biophysical journal 2004, 87, 3648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Simone A; Derreumaux P, Low molecular weight oligomers of amyloid peptides display beta-barrel conformations: a replica exchange molecular dynamics study in explicit solvent. The Journal of chemical physics 2010, 132, 165103. [DOI] [PubMed] [Google Scholar]
- 38.Do TD; LaPointe NE; Nelson R; Krotee P; Hayden EY; Ulrich B; Quan S; Feinstein SC; Teplow DB; Eisenberg D; Shea JE; Bowers MT, Amyloid beta-Protein C-Terminal Fragments: Formation of Cylindrins and beta-Barrels. Journal of the American Chemical Society 2016, 138, 549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie L; Luo Y; Wei G, Abeta(16–22) peptides can assemble into ordered beta-barrels and bilayer beta-sheets, while substitution of phenylalanine 19 by tryptophan increases the population of disordered aggregates. The journal of physical chemistry. B 2013, 117, 10149–60. [DOI] [PubMed] [Google Scholar]
- 40.Zou Y; Sun Y; Zhu Y; Ma B; Nussinov R; Zhang Q, Critical Nucleus Structure and Aggregation Mechanism of the C-terminal Fragment of Copper-Zinc Superoxide Dismutase Protein. ACS chemical neuroscience 2016, 7, 286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irback A; Mitternacht S, Spontaneous beta-barrel formation: an all-atom Monte Carlo study of Abeta16–22 oligomerization. Proteins 2008, 71, 207–14. [DOI] [PubMed] [Google Scholar]
- 42.Sun Y; Kakinen A; Xing Y; Pilkington EH; Davis TP; Ke PC; Ding F, Nucleation of beta-rich oligomers and beta-barrels in the early aggregation of human islet amyloid polypeptide. Biochimica et biophysica acta. Molecular basis of disease 2019, 1865, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding F; Tsao D; Nie H; Dokholyan NV, Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 2008, 16, 1010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urbanc B; Borreguero JM; Cruz L; Stanley HE, Ab initio discrete molecular dynamics approach to protein folding and aggregation. Methods in enzymology 2006, 412, 314–38. [DOI] [PubMed] [Google Scholar]
- 45.Shirvanyants D; Ding F; Tsao D; Ramachandran S; Dokholyan NV, Discrete molecular dynamics: an efficient and versatile simulation method for fine protein characterization. The journal of physical chemistry. B 2012, 116, 8375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sangwan S; Sawaya MR; Murray KA; Hughes MP; Eisenberg DS, Atomic structures of corkscrew-forming segments of SOD1 reveal varied oligomer conformations. Protein science: a publication of the Protein Society 2018, 27, 1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dokholyan NV; Buldyrev SV; Stanley HE; Shakhnovich EI, Discrete molecular dynamics studies of the folding of a protein-like model. Folding & design 1998, 3, 577–87. [DOI] [PubMed] [Google Scholar]
- 48.Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH, Conformational ensemble of native alpha-synuclein in solution as determined by short-distance crosslinking constraint-guided discrete molecular dynamics simulations. PLoS computational biology 2019, 15, e1006859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang M; Sun Y; Cao X; Peng G; Javed I; Kakinen A; Davis TP; Lin S; Liu J; Ding F; Ke PC, Graphene quantum dots against human IAPP aggregation and toxicity in vivo. Nanoscale 2018, 10, 19995–20006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faridi A; Sun Y; Mortimer M; Aranha RR; Nandakumar A; Li Y; Javed I; Kakinen A; Fan Q; Purcell AW; Davis TP; Ding F; Faridi P; Ke PC, Graphene quantum dots rescue protein dysregulation of pancreatic beta-cells exposed to human islet amyloid polypeptide. Nano research 2019, 12, 2827–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding F; Dokholyan NV, Emergence of protein fold families through rational design. PLoS computational biology 2006, 2, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karplus BRBREBBDODJSSSM, Charmm-a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. Journal of computational chemistry 1983, 4, 187–217. [Google Scholar]
- 53.Lazaridis T; Karplus M, Effective energy function for proteins in solution. Proteins 1999, 35, 133–52. [DOI] [PubMed] [Google Scholar]
- 54.Kabsch W; Sander C, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–637. [DOI] [PubMed] [Google Scholar]
- 55.Sun Y; Qian Z; Guo C; Wei G, Amphiphilic Peptides A6K and V6K Display Distinct Oligomeric Structures and Self-Assembly Dynamics: A Combined All-Atom and Coarse-Grained Simulation Study. Biomacromolecules 2015, 16, 2940–9. [DOI] [PubMed] [Google Scholar]
- 56.Matthes D; Gapsys V; de Groot BL, Driving forces and structural determinants of steric zipper peptide oligomer formation elucidated by atomistic simulations. Journal of molecular biology 2012, 421, 390–416. [DOI] [PubMed] [Google Scholar]
- 57.Krotee P; Rodriguez JA; Sawaya MR; Cascio D; Reyes FE; Shi D; Hattne J; Nannenga BL; Oskarsson ME; Philipp S; Griner S; Jiang L; Glabe CG; Westermark GT; Gonen T; Eisenberg DS, Atomic structures of fibrillar segments of hIAPP suggest tightly mated beta-sheets are important for cytotoxicity. eLife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.