

Abstract
Electrostatic interactions between phosphate anions and Toll-like receptor 4 / Myeloid differentiation factor-2 (TLR4/MD-2) protein complexes of human, murine, equine and canine species were computed. Such knowledge can provide mechanistic information about recognising LPS-like ligands, since anionic phosphate groups belong to the structural features of LPS with their diphosphorylated diglucosamine backbone. Sequence composition analyses, electrostatic interaction potentials and docked energies as well as molecular dynamics studies evaluated the phosphate interactions within the triangular LPS binding site (wedge). According to electrostatic analyses, human, horse and dog wedges possess phosphate-binding sites with indistinct positive and negative charge distributions, but the murine wedge shows a unique strong negative net charge at the site where antagonists bind in other species (Pan). Docking of a phosphate mono-anion (probe) confirmed its repulsion at this Pan site, but the Pag site of the murine wedge attracted the probe. It is occupied by phosphate groups of agonists in other species (Pag). Molecular dynamics trajectories show a variable degree of random walk across the wedges, that is, not following electrostatic preferences (neither Pag nor Pan). In summary, two opposing electrostatic patterns exist –murine versus human, equine and canine species – all of which reflect the potential dual activity mode of under-acylated ligands such as lipid IVA.
Keywords: Innate immune system, endotoxicity, molecular modelling, structure–activity relationships, phosphate, lipid IVA, TLR4, MD-2
Introduction
Gram-negative bacterial LPSs (also known as ‘endotoxin’) as well as lipid A (LA), which constitutes the lipophilic scaffold (or backbone) of LPS, activate the innate immune response through TLR4 and MD-2.1–4 Species-dependent activity has been summarised previously.5,6 LPS of Escherichia coli shows endotoxic activities throughout all species. Therefore, its absence is required for biological medical products (biopharmaceutical drugs).7 Lipid IVA (L4a), a substructure of LA, has been made available as precursor of LPS biosynthesis by extraction from natural sources as well as by total synthesis.1 Elimination of bacterial contaminants is another way to reduce endotoxicity risks.8 Structure–activity relationships (SARs) of different endotoxins were reviewed in 2010.9 More recently, a systematic panel of LPS derivatives was tested for endotoxicity to unravel SARs more systematically.10 Other synthetic LPS-like agents with modified diglucosamine backbone shed light on the geometrical requirements for endotoxicity upon binding to the TLR4/MD-2 complex.11,12
Target complex
LPS (Lig) binding to the TLR4/MD-2 complex triggers the dimerization of the extracellular TLR4/MD-2/Lig complex (per se a heterotrimer) to form an m-shaped dimer (TLR4/MD-2/Lig)2. This is thought to be the structural prerequisite for the species-dependent transmembrane-signalling of LPS-like ligands (agonists).13,14 In contrast, antagonistic L4a or Eritoran has not yet been crystallised as dimer.15,16
In site-directed mutagenesis studies, two murine residues –mLys367B and mArg434B – were replaced by human hGlu369B and hAsn436B, inferring human non-responsiveness to the murine cell system and vice versa.17 Yet, this view is complemented by another tenet challenging the extracellular contributions to receptor dimerization to form a (TLR4/MD-2/Lig)2 complex by experimentally determining another source of complex formation in the ‘intrinsic dimerization propensity of transmembrane and/or cytoplasmic domain(s) of TLR4’.18 The underpinnings of the present docking study have already been documented elsewhere.17 Here, we combined our computed findings with reported experimental data to assess the preferred locations of anionic phosphate groups on LPS-like binders.6,17,19,20
A much shorter stretch of counter TLR4* constitutes the wedge border to the left (Figure 1). This finding allows its N-terminal part to be cut away. The intersection points between the three wedge sides create edges which reflect the three contact zones between TLR4 and TLR4*, or TLR4 and MD-2, as well as TLR4* and MD-2 (Figure 1). Finally, the two diglucosamine phosphate ester groups show activity-related binding locations. They always reside in two of the three edges: either PagPan with Pag for agonists, or PagPan with Pan for antagonists.5,6 Of note, the binding locations and mechanisms for non-LPS-like ligands vary according to the nature of those structures.21,22
Figure 1.

Molecular model of dimeric canine TLR4/myeloid differentiation factor-2 (MD-2) receptor complex (left), and schematic representation of the ‘wedge’ (Δ).5,6 It was created from the 3D template of a human crystal receptor complex with liganded LPS (PDB code 3FXI).14 Of note, only one wedge is displayed based on the blue MD-2. The other wedge would reside on the green-coloured MD-2. The homodimer shows a symmetric ‘m’-shape and is composed of two functional TLR4/MD-2 units (dark and light blue or grey and green). Horseshoe-shaped TLR4 belongs to the LRR class of proteins (dark blue or grey). Globular MD-2 belongs to the ‘all β’ (only beta strands) class of proteins with a large lipophilic groove for the binding of fatty-acid chains of LPS-like ligands.
Probe design and expected molecular behaviour
The current literature attests that electrostatic interactions produce agonistic or antagonistic activities upon LPS-like ligand binding.20 The findings included that TLR4 and MD-2 were necessary to account for species-dependent ligand recognition.17,19,20,23 Ester phosphorylation of the diglucosamine backbone constitutes a pharmacophore element of LPS-like ligands and modulate endotoxin signalling. Phosphate groups are signal enhancers according to SAR analysis (Figure 2). LA is the immunoreactive portion of LPS. Both have been reported as full (or strong) agonists. For these reasons, the phosphate mono-anion was selected as interaction probe to evaluate the electrostatic properties within the empty space in TLR4/MD-2 complexes (wedges).
Figure 2.
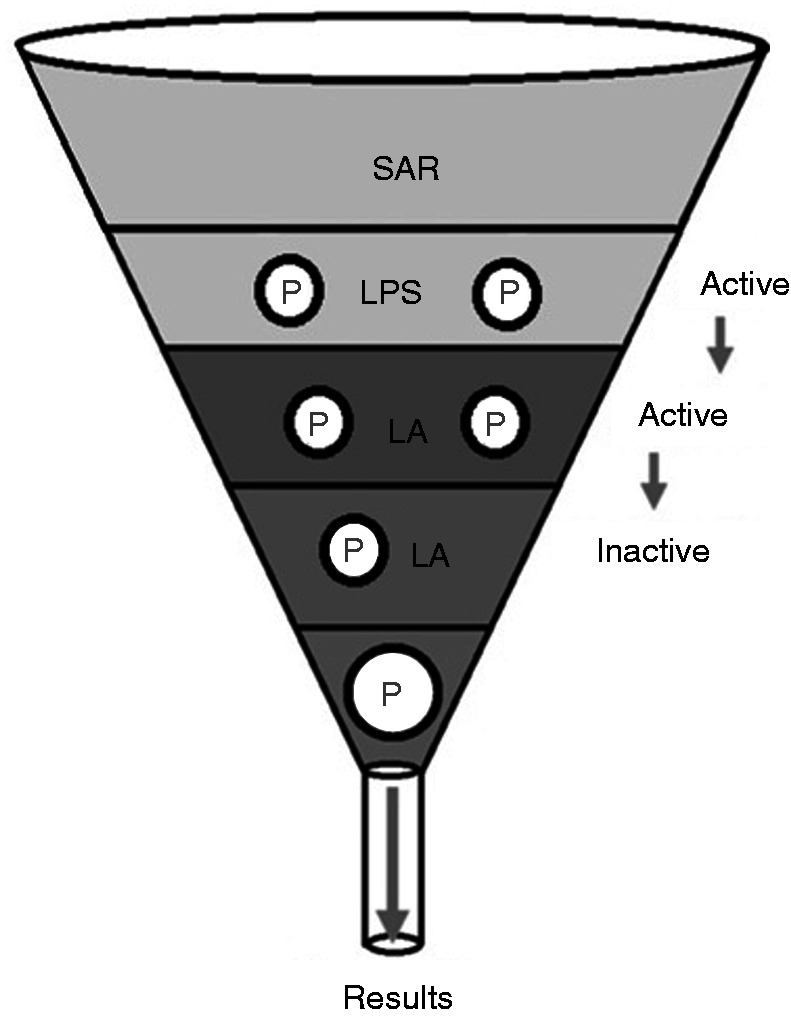
Structure–activity relationships of the immunoreactive portion of LPS and its substructure, lipid A (LA). Two Ps represent a diphosphorylated congener, while one P denotes a monophosphoryl derivative. The single P symbolises the H2PO4– probe in the present study.
Electronic (here measured as electrostatic forces of phosphate probes) and steric features of LPS-like molecules contribute synergistically to the ligand recognition by the receptor complex. This non-additive effect of variable phosphate and fatty-acid attachments to the diglucosamine scaffold was systematically explored.10 It can also be observed with LA of Capnocytophaga canimorsus with its penta-acylated diglucosamine-monophosphate backbone. It is at least 100 times less endotoxic than diphosphorylated LA from E. coli.3,5 Monophosphorylated LA from Salmonella enterica showed extremely weak endotoxic potency. A particular case is the dual mode of action for L4a. It constitutes a tetra-acylated antagonist in human species but acts as an endotoxin in murine cells.15,19,20,23 Moreover, related tetra-acylated LA monophosphates act as antagonists while pentacyl congeners in human cells do not.24
Particular attention was drawn to the presence of a protruding acyl chain at the dimerization interface (fatty acid labelled ‘FA 1’, ‘a’ or sometimes ‘γ’).14,25 The steric and conformational constraints have been studied by Zamyatina et al. for the diglucosamine backbone as well as by Marszalek et al. for disaccharide analogs.11,26 Precisely, the presence of both phosphate groups on LA has been found to contribute to the dimerization of the ‘monomeric’ TLR4/MD-2/LPS receptor unit into a dimeric (TLR4/MD-2/LPS)2 complex, a prerequisite of endotoxicity (agonism).14 A recent survey describes four decades of outstanding research in the hands of renowned scientists concerning the characterisation and endotoxin activities of LPS and its congeners, in particular the immunological activities of LA.27
Species dependency versus ligand dependency
The wedge concepts were based on hitherto known crystal structures and therein considered to be ‘natural’ (i.e. reasonable or plausible; Table 1). From a certain view point, the triangle is contoured to the left by the second or counter TLR4* (chain b, grey line in Figure 1), the TLR4 to the right (chain a, dark blue line in Figure 1) and, attached to the latter, the MD-2 at the bottom (chain c, horizontal light blue line in Figure 1).5 The diglucosamine scaffold of agonistic L4a is more solvent exposed compared to antagonistic L4a or Eritoran (Figure 1).14–16,28–30 Antagonistic L4a is buried deeply in the hydrophobic pocket of MD-2 and inverted (flipped) with regard to its agonist pose.5,6,19,23
Table 1.
Observed biological activities correlated to crystal structures with LPS or its derivatives.
| Ligand/species | H | M | e | c |
|---|---|---|---|---|
| LPS | Ag (TLR4/MD-2/LPS)2 ‘m’ ‘Δ’ 3FXI14, 4G8A27 | Ag (TLR4/MD-2/LPS)2 ‘m’ ‘Δ’ 3VQ228 | Ag– | Ag– |
| LA | Ag– | Ag (TLR4/MD-2/LA)2 ‘m’ ‘Δ’ 5iJD29 | Ag– | Ag– |
| L4a | An (–/MD-2/L4a)1 ‘u’ ‘–’ 2E5930 | Ag (TLR4/MD-2/L4a)2 ‘m’ ‘Δ’ 3VQI28 | (Ag)– | (An)– |
| Eri | An (TLR4/MD-2/Er)1 ‘>’ 2Z6515 | An– | An– | An– |
Ag: agonist; An: antagonist, partial or weak activity (Ag/An); ‘m’: m-shaped dimer; ‘u’: u-shaped MD-2; Δ: a complete wedge; >: an incomplete or open wedge without TLR4*; –: only MD-2 is present; LA: lipid A; L4a: lipid IVA; Eri: Eritoran.6
In human cells, L4a acts as antagonist, and therefore a monomeric hTLR4/hMD-2/L4a complex is regarded as ‘natural’ while a constructed (hTLR4/hMD-2/L4a)2 dimer is considered ‘non-natural’ or ill-assembled. In contrast, L4a is an agonist in murine cells, and therefore a dimeric (mTLR4/mMD-2/L4a)2 is considered ‘natural’. This observation implied that phosphate alone could distinguish between the (four) species. Each species would accommodate two phosphates into an agonistic constellation (PagPan and Pag) or antagonistic constellation (PagPan and Pan) in full correspondence to its endotoxic activity. Assuming such species-dependent behaviour driven by phosphates alone would clearly contradict the mere fact that certain diphosphorylated LPS-like ligands only act as either agonists or antagonists regardless of the species (species independency). The dual activity of under-acylated precursor L4a also hints at the oversimplification of this assumption (Table 2).
Table 2.
Correlation between endotoxic effects, ligand structure and tested organisms. Ligand dependency combined with species independency for LPS or LA as opposed to species dependency and ligand independency for L4a.
| Endotoxic activity | Observed for | In cells of | Dependency on |
|---|---|---|---|
| Dual | L4a | h, m | Species |
| Single | LPS, LA, Eritoran | h, m, e, c | Ligands |
| Agonism | LPS, LA | h, m, e, c | Ligands |
| Antagonism | Eritoran | h, m, e, c | Ligands |
Methods
Canine TLR4/MD-2 receptor generation by homology modelling
The TLR4/MD-2 sequences of all four studied species are homologous, with identity scores higher than 60%. The canine TLR4/MD-2 receptor complex has not been solved by X-ray (Table 1). With identity scores of 73% and 71% for TLR4 and MD-2, respectively, it was generated like the horse model.6,31
Preparing the wedge for molecular modelling
Available crystal structures were gathered, and their relevance was assessed by biochemical reasoning (Table 1).32 The literature lists all the experimentally determined TLR4/MD-2 complexes.5 In 2016, a new structure was released with PDB code 5IJD.29 As a most valuable asset, it confirmed the observation that agonists constitute dimeric complexes whereas antagonist ligands do not. At all times, the template (3FXI) served as reference of superposition (MagicFit, Matchmaker).32–34
Instead of complete dimeric complexes (shorthand notations: (TLR4/MD-2)2 or TLR4/MD-2/TLR4*/MD-2*), truncated dimeric complexes were prepared for human, mouse, horse and dog (h, m, e, c short_TLR4*/TLR4/MD-2). To this end, the template’s MD-2* (chain d in 3FXI) was removed using Swiss PDB Viewer (Figure 3).33 After truncation, our four shorter TLR4* models provided for the following sequence segments: hSer312b to hCys627b, mSer310b to mPhe621b, eSer312b to ePhe624b and cSer311b to cCys627b. Having three huge proteins engaged in binding the shorter TLR4* models saved simulation resources (software and hardware).
Figure 3.

Wedge model preparation for molecular simulations. First, the counter MD-2* was removed from the structure (left to centre), leaving only one complete wedge. Then, the N-terminal part of the TLR4* was cut out because it does not contribute to LPS-like ligand binding (Figure 2). The model, although with a shorter TLR4*, constitutes a complete interaction unit (wedge) with all amino acids for LPS-like ligand binding.
Assigning atom charges to study the electrostatic interactions
Modelling tools under academic licences were used for loading atomic charges as required by each molecular application. Prior to docking under AD4,35 we calculated Gasteiger charges for all amino acids and the phosphate probe.36 For simulations in the Tripos force field (FF)37 under Sybyl,38 we loaded Kollman-united charges for the amino acids. For interaction potential (IAP) studies, the Tripos atom types were converted into MMFF96 atom types, and the atomic charges came from the MMFF96 FF39 under MOE.40 For the molecular dynamics (MD) simulations, the phosphate model was retrieved at the PRODRG repository (Figure 4).41 Under physiological pH and equilibrium conditions, speciation leads to a 1:1 mixture of mono- and di-anionic phosphate groups (H2PO4–↔H++HPO42–, pKa 7.21).42 The mono-anionic form was used for simulations as a trade-off between electrostatic attractions through anions and the potential formation of hydrogen bond networks. The inorganic phosphate ion (Pi) shows two hydroxyl groups P–O–H with H-donor functions, as well as one negatively charged O=P–O– group with two H-acceptor functions. The built-in partial charges of PRODRG follow the inductive and mesomeric effects under ideal vacuum or homogenous solvent conditions. Here, however, the effective atomic charges had to reflect the polarisation effects at the very moment of non-covalent asymmetric binding between cationic arginine or lysine and the Pi probe. It also constitutes a strong Lewis base, thereby inducing asymmetric charge redistribution on the functional groups of mono-cationic side chains (Figure 4). The a priori charge assignments under FF applications cannot reflect the increased dipolar moment by charge–charge inductions upon close contact to functional side chains. Thus, a subtle change was introduced on one oxygen atom, keeping the molecular net charge at –1: the charge-modified ion yielded a dipole moment of 8.29 Debye compared to the standard value of 8.16 Debye for H2PO4– (Figure 4).
Figure 4.
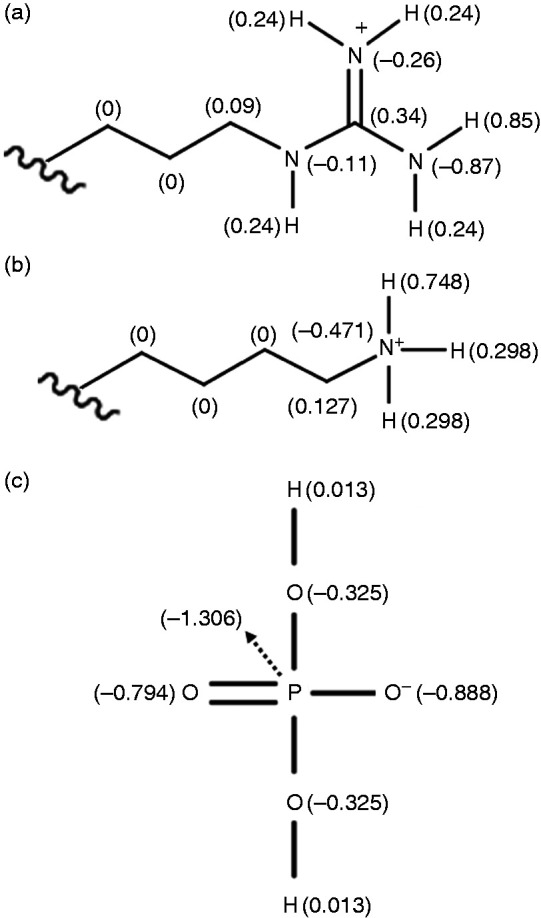
Assignment of partial charges for MD simulations: (a) arginine in the wedge; (b) lysine in the wedge; (c) phosphate mono-anion as molecular probe (H2PO4–). Panels (a) and (b) were derived from standard force-field parameters (Arg: H = 0.85, N = –0.87; Lys: H = 0.748, N = –0.471 in GROMOS force field as implemented in GROMACS43,44), while panel (c) was slightly modified from the phosphate model of PRODRG.41
Sequence analyses and electrostatic IAPs
To locate charge differences, IAP maps were generated employing licensed MOE software.40 This method permits the visualisation of attractive and repulsive interactions between a Pi probe and the molecular surface. To this end, the electrostatic fields were colour coded: blue for positive and red for negative charges, and white for uncharged hydrophobic propensities. The Surfaces and Maps menu of MOE was applied to display electrostatic properties of the side chains in the wedges based on MMFF94 FF partial charges.39
Binding site evaluation and binding energy estimations
The Tripos FF was applied for numerical determination of such electrostatic properties in form of pure Coulomb energies (CE; Kcal/mol) with the default cut-off value for non-bonded interactions (8 Å).37,38 Standard geometry optimisation without electrostatics was applied first until convergence was reached (criterion: local minima of potential energies). Then, the electrostatic equation term was activated for a one-run self-consistent field test (1SCF) to explore the charge attraction and repulsion forces experienced in the presence of the phosphate mono-anion. 1SCF was crucial to create physical systems of atoms with molecular uniformity (controlling all degrees of freedom) and thereby avoiding the introduction of rotational and translational noise, all of which allows direct comparison of the energy values on all levels of study: (a) the same site (either Pag, Pan or PagPan) for species-dependency tests, and (b) the same species (one of h, m, e or c) for site-specificity tests. As a direct result, the electrostatic term quantities were extracted from the total CE of the FF equations: CEliganded–CEunliganded=CEligand. Of note, prior to calculation, the bond types had to be adopted to suit the Tripos atom type parameterisations: P–O–H and P–O(–) as single bonds, and P=O as double bond.37
Auto-dock v4.2 and MGL tools were applied for Pi docking into the three crystallographic observed binding regions of the wedge (PagPan, Pag or Pan).35 To this end, a box size of 40 Å×40 Å×40 Å was chosen to give the phosphate the opportunity to explore the space around the three binding regions and to move away.5 The grid-projected electrostatic potentials were visualised in the box to demonstrate that in an overall positive attraction field governs mouse Pag, in contrast to its Pan site. This situation is inverted in human Pag and Pan sites (Figures 9–12). The phosphate mono-anion was placed at Pag, Pan or PagPan of the complete wedge (Δ) as its start position.5 Certain docking set-up parameters adopted non-standard values to further local search at the sites: tran0 with the initial coordinates; quaternion0 1 0 0 0 for initial ligand orientation; dihe0 0. 0. for two rotatable bonds with initial dihedrals; torsdof 2 for torsional degrees of freedom of two rotatable bonds at P–O–H substructure; axisangle0 0 0 0 0 0 0 for initial docking orientation at the start position; tstep 0.05 for very small translational steps (Å) for exhaustive docking at the start position; and qstep and dstep 0.05 for very small bond rotations (torsional steps) around the start conformation. The docking simulations were carried out merely to observe whether the mono-anionic phosphate probe would stay at its crystallographic defined binding site (as it would do as part of LPS or LA) or move far away and eventually towards another site (as it would do as part of L4a): from start position Pag (Pan) towards Pan (Pag, respectively). Under these premises, docking evaluated the probe’s preferential binding sites (Pag vs. Pan for LA vs. L4a).
Figure 9.

Visualisation of the +1 charge attraction fields from residues in the murine wedge calculated by AD4 (for a 40 Å×40 Å×40 Å box). TLR4 (green ribbons, right), counter TLR4* (red ribbons, left) and MD-2 (light blue ribbons, bottom) contour the broadly triangular cavity. The three sites are marked with a H2PO4– mono-anion: Pan (VDW volume, left), PagPan (sticks, right) and Pag (half space-filled, top, midsection). The murine Pag site is under the influence of the positive net charge (q = +1) which visualises the strong attraction field (grey to black wire frames). The mono-anionic probe prefers the position marked by the phosphate model at Pag. The field lines (wire frames) were drawn only for q = +1, and lines for 0 < q < +1 would expand to reach the probe model at Pag, but such lines would hide the molecular structures from display.
Figure 10.

Visualisation of the –1 charge attraction fields from residues in the murine wedge calculated by AD4 (for a 40 Å×40 Å×40 Å box). TLR4 (green ribbons, right), counter TLR4* (red ribbons, left) and MD-2 (light blue ribbons, bottom) contour the broadly triangular cavity. The three sites are marked with a H2PO4– mono-anion: Pan (VDW volume, left), PagPan (sticks, right) and Pag (half space-filled, top). The murine Pan site is almost completely engulfed in the negatively charged (q = –1) attraction fields (grey to black wire frames). The mono-anionic probe (q = –1) cannot approach and will make its way to the Pag site (top, midsection) which is completely free of negative electrostatic fields, that is, no wire frames contouring it. Compared to Figure 9, Pag lies way off the field lines (wire frames) on the top right corner of the box (top, almost centre).
Figure 11.

Visualisation of the +1 charge attraction fields from residues in the human wedge calculated by AD4 (for a 40 Å×40 Å×40 Å box). TLR4 (green ribbons, right), counter TLR4* (red ribbons, left) and MD-2 (light blue ribbons, bottom) contour the broadly triangular cavity. The three sites are marked with a H2PO4– mono-anion: Pan (VDW volume, left), PagPan (sticks, right) and Pag (half space-filled, top). A complete human wedge (Δ) exercises less attraction to phosphate anions than the murine one (Figure 9). The Pag site (top) interacts favourably with a negative probe, since it is under the influence of the positive net charge (q=+1), that is, the field lines (wire frames) reach and overlap in parts with Pag (top, midsection) on the top right corner of the box.
Figure 12.

Visualisation of the –1 charge attraction fields from residues in human wedge calculated by AD4 (for a 40 Å×40 Å×40 Å box). TLR4 (green ribbons, right) counter TLR4* (red ribbons, left) and MD-2 (light blue ribbons, bottom) contour the approximately triangular cavity. The three sites are marked with a H2PO4– mono-anion: Pan (VDW volume, left), PagPan (sticks, right) and Pag (half space-filled, top). In the presence of TLR4*, there is a strong repulsive force at Pan. Hence, a complete human wedge (Δ) is likely to exist only for agonistic LPS-like ligands occupying Pag and PagPan with their dephosphorylated diglucosamine backbone. The triangular shape of the wedge is formed by the three phosphate interaction sites: (a) Pag, on the top right corner of the box; (b) PagPan way down, buried in the cleft to its right, almost centre; and (c) Pan, leftmost phosphate model, which lies halfway on the z-axis (third dimension in the flat picture) between PagPan at the bottom and Pag at the top.
MD simulations
Classical MD is a well-accepted computational tool to evaluate and observe the behaviour of proteins in water, and to analyse the interaction of these biochemical macromolecules with small molecules.43 The classical Newton’s equations of motion of the interacting particles are integrated to generate the time evolution of the system. Then, within the framework of the statistical mechanics, the properties of interest are calculated as time averages.44 The total potential energy of the system is modelled through bonded and non-bonded terms containing the intra- and intermolecular interactions: bond, angle and torsional terms as well as Van der Waals and Coulomb interactions are included. The potential energy function, fed by a specific set of parameters, constitutes the FF.45 Temperature and pressure conditions are set by thermostat and barostat algorithms designed to control the thermodynamic state of the system.43
Under the FF of Gromos v53a6 in the GROMACS 5.0 package,46,47 the multimeric molecular receptor complex TLR4*/TLR4/MD-2/2H2PO4– containing the wedge with a shortened TLR4* was placed in the centre of a cubic water box (SPCE model). The wedge with a shortened TLR4* was treated by steepest descent energy minimisation, followed by 200,000 steps of NVT and 300,000 steps of NPT, both at 310 K for equilibration. MD was run using NPT at 310 K for 100 ns (50,000,000 steps, dt=2 fs or 0.002 ps, leap-frog as numerical integrator). The resulting trajectories were analysed with VMD software.48
For the construction of new ligands that are not found in the Gromacs standards or to modify an existing one, the following steps have to be taken. First, the topology file is built in two ways, using a server such as Automatic Topology Builder49 or PRODRG.41 The latter was used to build the topology from the PDB or GRO structure input files and obtain the output in Gromos format that can be included in the topology files. Second, the connections of the atoms in the molecule were constructed by hand and a set of new parameters was proposed. This new set of parameters can be calculated with the help of quantum chemistry or through methodologies designed to develop new FF.50 The justification for the atomic partial charges loaded on the phosphate moieties were the increasing dipole moment of the H2PO4- anion such that with the charge loaded in GROMOS the average dipole moment is 8.16 Debye units, albeit with the charges that we loaded the average dipole moment was 8.29, a mere slight increase as reported.50 They showed that slight modifications of the dipole moment of the probe favours the fit between calculated and experimental data. Charge assignment for electrostatic interactions play a crucial role which also applies at protein interfaces. This adjustment was carried out at the beginning during trial runs.
Results and discussion
Determination of the phosphate-binding sites in the wedge
The molecular interaction between probe and target models took place in the wedge. The phosphate mono-anion start poses (Pag, Pan and PagPan) were created by extracted them from the available crystal structures (3FXI for Pag and PagPan; 2Z65 for Pan and PanPag). The murine Pag and PagPan sites (3VQ2, 5IJD) matched perfectly with the human sites (3FXI, 4G8a). Hence, they were readily merged into the two homology models of horse and dog. As a minimalist messenger, the Pi probe moved around in the wedge according to electrostatic repulsions or attractions.
Studying the differential amino acids for all four species
In order to establish a possible charge-position dependency, the net charges of the Pag and Pan sites were determined (Table 3). The canine charge distribution is identical to the human one, while the equine distribution with its overall positive electrostatic features resembles more the positively charged mouse and human systems. As a direct consequence, the four species form two clusters according to the overall charge distributions in the four wedges (Table 3, cf. h, e, c vs. m; two last columns): the total charge at the Pag site depends on the presence of MD-2, either alone in the monomeric, antagonistic wedge (chain c, >; last two columns in Table 3) or with TLR4* in the dimeric, agonistic wedge (chains b and c; Δ; last two columns in Table 3). The total charge at the Pag site depends on the presence of one TLR4 alone (chain a, >) or both TLR4 proteins (chains a and b; Δ). In the cited literature, the horse and dog systems have been recognised as partial (or weakly acting) agonist or antagonist, respectively. Since the PagPan site is more conserved among the species and always occupied by one phosphate group of the diglucosamine backbone of either agonists or antagonists (providing for acronym PagPan), there is no need to compare it (Table 3).
Table 3.
Differential charge contributions of the three proteins (TLR4, TLR4* and MD-2) at the Pag or Pan sites for all four species (human, mouse, horse and dog).
|
TLR4 (a) |
TLR4* (b) |
MD-2 (c) |
Net charges |
|||||
|---|---|---|---|---|---|---|---|---|
| Species | Pag | Pan | Pag | Pan | Pag | Pan | Pag | Pan |
| h | +2= Lys362a Lys341a |
– | +1=–1 + 1+1 Glu369b Lys388b Arg435b |
–1 Glu439b | – | +2=Arg90c Lys122c |
+2 a > L4a Eri |
+2 c > L4a, Eri |
| +3 a,b Δ LPS, LA |
+1 b,c Δ LPS, LA |
|||||||
| m | +2= Lys360a Lys341a |
– | +3=1 + 1+1 Lys367b Lys433b Arg434b |
–1 Glu437b | – | 0=1–1 Arg90c Glu122c |
+2 a > Eri |
0 c > Eri |
| +5 a,b Δ LPS, LA, L4a |
–1 b,c Δ LPS, LA, L4a |
|||||||
| e | +3= Lys363a Arg342a Arg385a |
– | +1=–1 + 1+1
Glu370b Lys389b Lys436b |
–1 Asp440b | – | +2= Arg90c Arg122c |
+3 a > Eri |
+2 c > Eri |
| +4 a,b Δ LPS, LA, (L4a) |
+1 b,c Δ LPS, LA, (L4a) |
|||||||
| c | +2= Lys362a Arg384a |
– | +1=–1 + 1+1 Asp369b Lys388b Lys435b |
–1 Asp439b | – | +2=Arg90c Arg122C |
+2 a > (L4a), Eri |
+2 c > (L4a), Eri |
| +3 a,b Δ LPS, LA |
+1 Δ LPS, LA |
|||||||
All side chains orient towards the monomeric (>) or dimeric (Δ) wedge. Last two columns show the net charges with the chains (a, b and c) to which the net charges belong. For each species, two cases exist (horizontal line) to distinguish the > or Δ wedges. The data were taken from the respective liganded crystal structures with their ligands (Eritoran, L4a, LA or LPS). (L4a) indicates dual effects of L4a.
As a direct result, two wedge types with different electrostatic properties were detected: (a) human, equine as well as canine wedges possess Pag and Pan sites with positive and negative point charges, but (b) only the murine wedge shows a very strong positive total charge at Pag and a unique strong negative net charge at Pan sites (–1). Each chain contributes differently in each species to the overall (net) charge (see left columns in Table 3). Chains a and b do not show any species-specificities (neither for Pag nor for Pan) because their residue composition is basically the same (no differential amino acids). Chain b, however, contributes at the Pag site in a species-specific way: for human, equine and canine organisms with an electrostatic balance of ‘+1=–1 + 1+1’ versus ‘+3=1 + 1+1’ for mice (column 4 in Table 3). The electrostatic features also reflect the fact that L4a acts as antagonist in human cells. Moreover, in horse and dog systems, L4a acts with very weak activities, that is, with difficulties to designate partial antagonism or agonism.3
Comparing published interaction models and present advances
The human, murine and equine wedge models were revised, and two more phosphate-interacting residues were added (underlined residues in Figure 5).6 The differential amino acids in the canine wedge are presented for the first time (Figure 5). The implementation of the wedge concept allowed the following three interpretations. (1) The only subunit that can contact both sites (Pag and Pan) is TLR4* in the dimerization interface. The PagPan sites of the four species show the same total charge (+2). Always present are two lysine residues (mLys263a+mLys360a; eLys264a+eLys363a; cLys264a+cLys362a) or a combination of lysine with arginine residues (hLys362a+hArg264a). (2) While the Pan site can be formed by residues of MD-2 alone (hLys58c, hArg90c, mAsn58c, mArg90c, eArg58c, eArg90c, cLys58c, cArg90c), the Pag site cannot exist without the interplay with residues from TLR4* (hLys388b, hGln436b, mSer386b, mArg434b, eLys389b, eGln437b, cLys388b, cGln436b). (3) The chains a and c (TLR4, MD-2) are too far away from Pan and Pag, so they cannot contribute with their side chains (Table 3).
Figure 5.

Wedge residues interacting with the locations of the dephosphorylated glucosamine backbone (P1-Gln1-Gln2-P2) for human, mouse, equine and canine organisms. The underlined amino acids had not yet been reported in our previous work.5,6
Mapping the electrostatic IAPs
IAP maps were generated for all four species. The four wedges were screened by H2PO4– as probe. The counter TLR4* is present in all murine crystal structures (Table 1 and Figure 6). In contrast, it is only present in human crystal structures if complexed to LPS or LA but not with co-crystallised antagonists such as L4a or Eritoran (Table 1 and Figure 7). In view of biological (function) units packed into crystal unit cells, the human MD-2 protein appears in a different conformation than in the complete wedge (Table 1 and Figure 8).5 It was assumed that it must have suffered from crystal package deformation. After structure alignments in space, five crystal complexes with agonist poses pinpointed the site of PagPan (3FXI, 4G8A, 3VQ1, 3VQ2 and 5IJD). Hence, PanPag (from 2E59) was deprecated. In earlier reports, the antagonist pose had already been characterised by two features: (a) the GlcN2 phosphate P2 occupies the Pan site, and (b) the diglucosamine backbone (GlcN1-GlcN2) is horizontally flipped with respect to the agonist pose.5,14
Figure 6.

Display of the murine wedge (3VQ2, 5IJD; Δ in Table 1). The surfaces of the amino acids are colour coded by their electrostatic properties (red: anionic/acidic; blue: cationic/basic; and white: non-polar/hydrophobic). The three cycles locate the sites which interact with the two phosphate groups on the diglucosamine scaffold of LPS-like ligands. Two combinations have been crystallographic observed: PagPan (yellow) and Pag (black) occupied by both phosphate groups of agonists; or PagPan and Pan (green) for phosphate groups of antagonists. It can be seen that the Pan site is a very unfavourable place for anions.
Figure 7.

Display of the human wedge (3FXI, 4G8A; Δ in Table 1) with the counter TLR4*. The surfaces of the amino acids are colour coded by their electrostatic properties (red: anionic/acidic; blue: cationic/basic; and white: non-polar/hydrophobic). The Pag site (black cycle) exercises a strong attraction (+3 net charges; Table 1) complying with its physiological function to signal LPS binding into the cell. The Pan site (green cycle) is less privileged with a mixed (blue and red) charge distribution. When the complex is liganded by LPS-like antagonists, the TLR4* is not present in all hitherto published crystal structures (Figure 6 for crystal data).
Figure 8.

Display of the incomplete human wedge (3FXI, 2ZG5; > in Table 1) without counter TLR4*. The surfaces of the amino acids are colour coded by their electrostatic properties (red: anionic/acidic; blue: cationic/basic; and white: non-polar/hydrophobic). The Pan and PagPan sites (green and yellow cycles) mark the locations of the phosphates on the diglucosamine backbone of LPS-like antagonists. When co-crystallised with antagonists, the liganded complexes are not dimeric, and the resulting wedge remains incomplete (missing TLR4*, > or – in Table 1). This is the case for L4a with its rare dual activity (Figure 6).
Simulating phosphate binding and electrostatic energies
In order to reflect L4a’s dual activity, we expected that the electrostatic energies could be ranked in a species-specific order to reflect two differential poses: (a) the antagonist pose in human receptor complex would mean that Pan >> Pag and Pan ≈ PagPan; and (b) the agonist pose in case of murine complexes with Pan >> Pag and Pan ≈ PagPan. The computed values reproduced the attraction order: –34 >> –22 and –34 ≈ –35, or –27 >> –14 and –27 ≈ –20, respectively (Table 4). As a most valuable asset, these estimated electrostatic quantities of the probe are in good keeping with the sequence composition analysis (Table 3). Both studies hint at an explanation of why the murine wedge is exceptional among the four species. It exclusively accommodates phosphate anions at its Pag site because it possesses two sites with opposite net charges at extreme values: (a) the highest positive net charge of all with +5 at Pag; and (b) the –1 negative charge at its Pan site. Of all sites from all four species, this site uniquely exercises electrostatic repulsion for incoming phosphate anions which is then trapped at Pag under the strongest attraction forces (+5).
Table 4.
Electrostatic energy estimations under the Tripos force field (Kcal/mol).
| Species | Pag | Pan | PagPan |
|---|---|---|---|
| h | –22 | >0 to –34 | –35 |
| m | –27 | >0 to –17 | –20 |
| e | –27 | >0 to –24 | –34 |
| c | –24 | >0 to –17 | –30 |
The negative values within the energy ranges reflect poses with hydrogen bonds. In the first and second row the differences between Pag and Pan are most significant.
For murine and equine wedges, Pag was found to be more attractive than Pan sites (Table 4). This finding reflects that upon binding, L4a acts as agonist. Sharp potential cliffs were observed for all Pan sites when the probe was displaced by distances of > 2 [A°]. These energy fluctuations were a direct consequence of the fact that all Pan sites are very narrow. Hence, small movements were either detrimental due to Van-der-Waals contacts or favourable thanks to hydrogen bonding to amino-acid side chains. At the Pag and PagPan sites, however, H-bond networking was not observed because of the larger distances to the surrounding wedge residues. Since two phosphates are separated by the diglucosamine backbone, the fact that the Pan sites are extremely sensitive to phosphate displacements (potential energy cliffs), while occupying the Pag site provides more spatial tolerance, may be linked to the fact that LPS-like antagonists have not yet been crystallised in dimeric forms, that is, without a counter TLR4* (incomplete wedge forms ‘>’or ‘–’). The strongest electrostatic forces between Pag and Pan consistently correlate with the dual activity of L4a: Pag with the highest values for agonistic murine and equine systems, Pan with the highest values for antagonistic human systems. The electrostatic behaviour of the canine system remains unclear – a fact that may relate to its unclear activity profile; its designation to partial antagonism is doubtful.3
Observing the probe movements by receptor docking simulations
Docking results showed ligand affinities in the range of one- to two-digit millimolarities at all three sites and all four species. The final solutions could be grouped into no more than three clusters of docked poses (criterion: RMSD < 0.5 Å). The most populated cluster was always identical with the best scoring solutions (80–90 out of 100). The results could be interpreted and kept in line with the results obtained by all other methods: the crystallographic-determined locations of the (two) phosphate groups on the diglucosamine scaffold of LPS-like ligands (Pag, Pan and PagPan) also qualified as favourable binding sites during docking, regardless of species. The phosphate only moved into receptor regions that were adjacent to one of these three sites (within 3 Å). This is a most convincing finding regarding the enormous space available and the huge contact surfaces on the complete wedges (Δ). In the human and mouse models, the outcome confirmed the previous results. In the murine model (Δ), the probe did not stay at its initial site (Pan), and the best-scoring final solutions were found near the Pag site (Table 5). In the human system, the Pan was the preferred site. Equine and canine results were inconclusive, albeit consistent in a way that hints again at their weak or doubtful activities.
Table 5.
Docking of H2PO4– into the three crystallographic observed phosphate-binding sites. The solutions (100 runs) were RMSD clustered.
| Species | Pag | Pan | PagPan |
|---|---|---|---|
| h | Staying nearby | Staying nearby | Staying nearby* |
| m | Staying nearby | Moving away (8 to 15) towards Pag (all) | Staying nearby* |
| e | Staying nearby | Moving away (8 to 15) towards Pag (1st cluster) | Staying nearby |
| C | Staying nearby | Moving away (8 to 15) towards Pag (2nd cluster) and PagPan (1st cluster) | Staying nearby |
The start positions (Pag and PagPan) were successfully re-docked, while major displacements occurred at Pan, given in Å.
*Also tested for PanPag site.
The electrostatic attraction and repulsion forces were simulated and visualised by docking simulation of the phosphate mono-anion probe in the wedge of the four species. The probe moved away from the site which binds antagonists (Pan) in the murine wedge due to a unique strong negative net charge. Instead, the probe finally attached to the murine Pag site (Table 5 and Figure 9). The analyses of the electrostatic distributions in the human, horse and dog wedges revealed that they can be grouped together. So, a need arose to contrast murine against human positive and negative charge distributions for their remarkable distinctions (Figures 9–12).
Observing the probe movements by MD simulations
The different MD trajectories for each mono-anionic phosphate (at Pag and PagPan) in human and murine wedges were displayed (Figure 13). The probe interactions for human and mouse were stable at average distance of about 2.5 Å during 100 ns of simulation time (rightmost inlays in Figure 13). All time frames (simulation steps) were superimposed to document the preferential positions (phosphate interaction zones) during 100 ns for the four species. Although the equine wedge has an arginine-rich Pan site, the latter is not visited by any phosphate mono-anion which corresponds to an agonist behaviour.
Figure 13.

Trajectories of phosphate mono-anion inside the murine (positive control, dimeric receptor, top) and human (negative control, monomeric receptor, bottom) wedge. Left: Trajectory for phosphate with start position at the Pag site. Centre: Trajectory for phosphate with the start position at the PagPan site. Right: Interaction distances plots (distance in Å on y-axes) in function of time (ns, x-axes) between phosphates and the wedge residues. Topmost plots: Phosphate interaction distances with murine wedge residues. Bottom most plots: Phosphate interaction distances with human wedge residues. Red line: trajectory during the first third. White line: trajectory during the second third. Blue line: trajectory during the last third. Each letter labels a species (h: human; m: mouse).
In the human wedge model, the mono-anionic phosphate interacts with the hLys388b residue at Pag (Figure 5). It belongs to chain b (TLR4*) which is absent in crustal structures of antagonistic ligands (negative control). Residue mLys341a interacted clearly with the Pi probe at the Pag site and the other with mLys360a at the PagPan site in mice as the positive control (Table 6).
Table 6.
Expected and calculated location (positional preferences) of two phosphates in the wedge, with anionic hGlu439b, mGlu437b, eAsp440b and cAsp439b in the model (short TLR4*/TLR4/MD-2).
| Species | Expected location | Computed location |
|---|---|---|
| Human (h) | Antagonist Negative control with TLR4* Pag > not Pan PagPan > PagPan |
Not antagonist Pag > Pag(Lys341a) Pag > Pag(Lys388b) PagPan > Pag(Lys341a) PagPan > PagPan(Lys362a) PagPan > PagPan(Asn383a) |
| Murine (m) | Agonist Positive control with TLR4* Pag > Pag PagPan > PagPan |
Agonist Pag > Pag(Lys341a) PagPan > PagPan(Lys263a) PagPan > PagPan(Lys360a) |
| Equine (e) | Agonist Positive control with TLR4* Pag > Pag PagPan > PagPan |
Agonist Pag > Pag(Arg385a) PagPan > PagPan(Lys363a) |
| Canine (c) | Antagonist Negative control with TLR4* Pag > not Pan PagPan > PagPan |
Not antagonist Pag > Pag(Lys388b) Pag > Pag(Gly363a) PagPan > Pag(Arg384a) PagPan > PagPan(Lys362a) |
Residues are labelled by the ID number of equine sequence PDB5 and by their chain letter a, b or c (a: TLR4; b: TLR4*; c: MD-2). Almost all interactions fulfil the expectation value (see 3rd column), except underlined interactions: opposed to expectation (see 2nd column).5 The ‘ > ’ is a short form for an arrow symbol ‘→’, from initial location Pag to new location Pan: Pag > Pan.
Interpretation of the simulation results
Since human, equine and canine systems act along a single mode of action, they all can be grouped together because their sites have positive net charges allowing the attraction of phosphates. In contrast, the murine wedge hosts a unique negative overall charge at Pan (Table 7). Particularly, its agonist position of phosphate (Pag) is strongly favoured because it exercises the strongest attraction among all three sites: Pag > PagPan > Pan, or Pag >> Pan. Therefore, it must be qualified as an exceptional binding site, albeit only attractive for L4a – but why not for other LPS-like ligands? The fact that ligands other than L4a do not get trapped into this position hints at new possibilities of explaining the exceptional case of L4a. Finally, it is safe to say that other structural features overcompensate the electrostatic forces, resulting in a species-dependent distinction between murine Pag and human Pan sites (last column in Table 7).
Table 7.
Correlation between endotoxic effects, tested organisms and ligand structures.
| Endotoxic activities | Observed for ligands | For species | Endotoxicity depends on | Receptor electrostatics | Ligand structures |
|---|---|---|---|---|---|
| Dual mode | L4a | m | Species | mPan = –1 mPag = +5 | None (L4a = L4a) |
| Single mode | LPS, LA, L4a, Eri | h, e, c | Ligands | h, e, c Pan > 0 h, e, c Pag > 0 |
Number of fatty acids |
| Agonism | LPS, LA | h, m, e, c | Ligands | p. but overc. | 6 acyl chains |
| Antagonism | Eri | h, m, e, c | Ligands | p. but overc. | 4 acyl chains |
Ligand dependency combined with species independency for LPS or LA as opposed to species dependency and ligand independency for L4a.
p. but overc.: present but overcompensated (by … entry in last column).
In view of the results, our reported differential amino acids5,6 were revised for the human and murine systems (Figure 14). We identified murine versus human residue pairs counteracting with bound LPS-like ligands due to opposing net charges – mArg266A versus hGly267A, mLys367B versus hGlu369B and mGlu122C versus h Lys122C – while the following pairs act in the same direction to stabilise the common phosphate position to all agonists and antagonists – mLys263A and hArg264A, mArg337A and hAsn339A, mGln339A and hLys341, and finally mLys360A and hLys362A. It makes sense that on the chain, a side of the wedge (TLR4) to which chain c with its LPS-binding pocket is attached to (MD-2), all amino acids work in the same direction to stabilise the phosphate binding common to LPS-like agonists and antagonists. In contrast, on the opposite side of the wedge, chain b (counter TLR4*), there are the differentiating residues, namely mLys367 versus hGlu369 in cooperativity with mGlu122C versus hLys122C on chain c at the bottom of the wedge (pocket entry to MD-2). This can explain why the dimerization mediated by the LPS-like ligands forms a dimeric receptor system in the case of agonists by ion interactions, while in the opposite case, antagonists fail to bridge the two TLR4 proteins. The second TLR4 with its attached MD-2 either leaves or never come in close contact to form the dimer which is believed to trigger the transmembrane signalling, thereby starting the endotoxic pathway downstream into the cells as part of the innate immune system. In addition, non-conserved methionine on chain b, mMet417B, is larger than other residues at the equivalent position and thereby could already cause steric hindrance for the non-substituted ligand L4a in the murine case.51 Besides the differences on the TLR4 proteins, MD-2 may contribute with its species-specific residues at position 122. From an evolutionary perspective, it is remarkable how the same and identical protein (TLR4) is taken twice, with one concave side (inward face of TLR4) and one convex side (outward face of counter TLR4*) to continue evolving to either keep the phosphates (PagPan) or distinguish between two phosphate-binding sites (Pag or Pan). This way, the very same gene product, its expression and its corresponding protein structure serve two opposing biological functions, all of which makes heritage less error prone and saves up metabolic energies for the living cells.
Figure 14.

The wedge with the schematic representation of the ligand-interacting differential amino acids which could explain the lack of endotoxic responsiveness in the case of human cell lines regarding the murine cell system. When the site ‘Pan__Pan’ is occupied, only chain a (green line) and chain c (yellow line) are bound to the antagonistic ligand. This binding mode with an inverted ligand scaffold is seen in all hitherto known crystal structures alike (ANiX). The agonist pose (‘Pag__Pag’) also links the a-chain to the b-chain which is a prerequisite for receptor dimerization (of which chains b and d form part; d is another MD-2 protein).
The following five observations underscore the usefulness of the wedge models:
Comparing Pag to Pan for all four organisms, the Pag sites are always equally or more positively charged than the Pan sites.
For a given species, the Pag site of a dimeric wedge (chains a and b; Δ) is always more positively charged than the monomeric wedge (chain a; >). This finding confers consistent activity relatedness to all known crystal complexes: dimers represent agonism, and monomers represent antagonism.
The strongest total net charge distribution can be ascribed to the dimeric mouse wedge what fairly reflects its unique agonistic effect on L4a. In addition, human, equine and murine receptor complexes need highly acetylated ligands (LA or LPS) to show an agonistic effect because they indiscriminately possess phosphate-attractive sites (positive Pag and Pan).
TLR4* and MD-2 (chains b and c) contribute to the phosphate preferences.
The four wedges belong to two electrostatic patterns: (a) for mouse, and (b) for human, equine and also canine organisms. The mouse possesses unique charge distribution between Pag and Pan sites which is not shared by the three others.
All computed findings – except for MD simulations – are in excellent keeping with the extant literature, also cited by Meng et al.20: ‘Bryant and coworkers demonstrated that under defined conditions, MD-2 and TLR4 were both required for the species-specific activation of L4a, partially reconciling the contradiction between the two theories’.52 In a docking study, Ohto et al. reported how lipids interact with the lipophilic deep cavities of a ‘MD-2–related lipid recognition family’ of proteins.53 Standard MD falls short of expectations in our case because the trajectories are subject to atomic-scale decisions, and if the attracting side chains are deflected during probe passage, no electrostatic influence is exercised as if no charged amino acid had been present at that particular location. Any effort to interpret the trajectory as a plain reflection of electrostatic forces must go astray. Yet, MD has offered opportunities in other areas to investigate complex molecular behaviour where conformational changes of ligands play a pivotal role in a time-dependent manner and therefore have already been applied to LPS-like ligands.4,12,54–58
Conclusions
Phosphate mono-anions interacted with amino acids of the TLR4/MD-2 receptor complexes of four species to identify intrinsic attraction or repulsion forces. Partial and total charge effects were systematically studied by the phosphate probes in the human, murine, equine and canine wedges. To exclude misinterpretation of the hitherto poorly understood concert of receptor species specificities and steric and electronic influences of LPS-like ligands with their variable bulk of fatty-acid substituents, the computer simulations were reduced to observe the electrostatic behaviour of the probe in the four wedges.
Our wedge concept proved its usefulness because all hitherto known agonist or antagonist crystal complexes contain either a dimeric receptor complex with complete wedge (Δ) or merely a TLR4/MD-2 monomer or MD-2 with incomplete wedges (> or –) for binding.
Results of sequence composition analyses for differential residues, IAP maps and electrostatic energy estimations coincide with docking results much more than with MD. MD fell short of expectation due to a larger portion of random walk of the probes during production runs of 100 ns. The identification of the driving electrostatic attraction/repulsion forces was hampered in that it remained impossible to know how large the degree of random walk contributions to the full trajectory was during MD simulations. All other computational results were consistent and coincided in that the human, equine and canine wedges belong to one group sharing general electrostatic attractions at all phosphate interaction sites. These sites had been identified based on crystal structures prior to modelling. In contrast, the murine system showed the strongest attraction at the phosphate-binding site for agonists (Pag). Conversely, it possesses a unique repulsive phosphate interaction site (Pan) which directs all hitherto known antagonistic LPS-like ligands upon binding to mTLR4/mMD-2.
The studied electrostatic forces alone cannot fully explain why the common binding position of LPS-like agonists (Pag) differs from that of antagonists (Pan) in general, and also why endotoxic activity of L4a is species dependent in particular. A more exhaustive view has to incorporate the fatty-acid substitution pattern, of which the under-acylation of L4a is a well-documented illustration leading to dual activity.
In the same systematic way as the present study did for electrostatics, future modelling studies could evaluate those steric effects of variations in acylation patterns (four, five or six fatty acids) in light of the crystallographic observed binding differences.
Acknowledgements
Lozano-Aponte is grateful for his Mexican CONACYT for PhD scholarship, CONACYT – Mexico City (2013-2017/360064. The authors thankfully acknowledge computer resources and technical expertise provided by Laboratorio Nacional de Supercómputo del Sureste de México and Dr Ignacio Martinez L. director at VIEP, BUAP.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
ORCID iDs
Jorge Lozano-Aponte https://orcid.org/0000-0003-1719-5011
Thomas Scior https://orcid.org/0000-0003-2196-2682
References
- 1.Alexander C, Rietschel ET. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res 2001; 7: 167–202. [PubMed] [Google Scholar]
- 2.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140: 805–820. [DOI] [PubMed] [Google Scholar]
- 3.Simon I, Buko L, Marco S, et al. The lipopolysaccharide from Capnocytophaga canimorsus reveals an unexpected role of the core-oligosaccharide in MD-2 binding. PLoS Pathog 2012; 8: e1002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molinaro A, Holst O, Di Lorenzo F, et al. Chemistry of lipid A: at the heart of innate immunity. Chem Eur J 2015; 21: 500–519. [DOI] [PubMed] [Google Scholar]
- 5.Scior T, Alexander C, Zaehringer U. Reviewing and identifying amino acids of human, murine, canine and equine TLR4/MD-2 receptor complexes conferring endotoxic innate immunity activation by LPS/lipid A, or antagonistic effects by Eritoran, in contrast to species-dependent modulation by lipid IVa. Comput Struct Biotechnol J 2013; 5: e201302012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scior T, Lozano-Aponte J, Figueroa-Vazquez V, et al. Three-dimensional mapping of differential amino acids of human, murine, canine and equine TLR4/MD-2 receptor complexes conferring endotoxic activation by lipid A, antagonism by Eritoran and species-dependent activities of lipid IVa in the mammalian LPS sensor system. Comput Struct Biotechnol J 2013; 7: e201305003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mamat U, Wilke K, Bramhill D, et al. Detoxifying Escherichia coli for endotoxin-free production of recombinant proteins. Microb Cell Fact 2015; 14: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandenburg K, Heinbockel L, Correa W, et al. Peptides with dual mode of action: killing bacteria and preventing endotoxin-induced sepsis. Biochim Biophys Acta 2016; 1858: 971–979. [DOI] [PubMed] [Google Scholar]
- 9.Brandenburg K, Schromm AB, Gutsmann T. Endotoxins: relationship between structure, function, and activity. Subcell Biochem 2010; 53: 53–67. [DOI] [PubMed] [Google Scholar]
- 10.Zariri A, Pupo E, van Riet E, et al. Modulating endotoxin activity by combinatorial bioengineering of meningococcal lipopolysaccharide. Sci Rep 2016; 6: 36575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garate JA, Stöckl J, Fernández-Alonso Mdel C, et al. Anti-endotoxic activity and structural basis for human MD-2·TLR4 antagonism of tetraacylated LA mimetics based on βGlcN(1↔1)αGlcN scaffold. Innate Immun 2015; 21: 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Artner D, Oblak A, Ittig S, et al. Conformationally constrained lipid A mimetics for exploration of structural basis of TLR4/MD-2 activation by lipopolysaccharide. ACS Chem Biol 2013; 8: 2423–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin MS, Kim SE, Heo JY, et al. Crystal structure of the TLR1–TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007; 130: 1071–1082. [DOI] [PubMed] [Google Scholar]
- 14.Park BS, Song DH, Kim HM, et al. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009; 458: 1191–1195. [DOI] [PubMed] [Google Scholar]
- 15.Ohto U, Fukase K, Miyake K, et al. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 2007; 316: 1632–1634. [DOI] [PubMed] [Google Scholar]
- 16.Kim HM, Park BS, Kim JI, et al. Crystal structure of the TLR4–MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007; 130: 906–917. [DOI] [PubMed] [Google Scholar]
- 17.Meng J, Gong M, Björkbacka H, et al. Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J Immunol 2011; 187: 3683–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panter G, Jerala R. The ectodomain of the Toll-like receptor 4 prevents constitutive receptor activation. J Biol Chem 2011; 286: 23334–23344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng J, Drolet JR, Monks BG, et al. MD-2 residues tyrosine 42, arginine 69, aspartic acid 122, and leucine 125 provide species specificity for lipid IVA. J Biol Chem 2010; 285: 27935–27943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng J, Lien E, Golenbock DT. MD-2-mediated ionic interactions between lipid A and TLR4 are essential for receptor activation. J Biol Chem 2010; 285: 8695–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang LD, Lin HJ, Huang PH, et al. Synthesis of serine-based glycolipids as potential TLR4 activators. Org Biomol Chem 2011; 9: 2492–2504. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Smith C, Yin H, et al. Targeting Toll-like receptors with small molecule agents. Chem Soc Rev 2013; 42: 4859–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akashi S, Nagai Y, Ogata H, et al. Human MD-2 confers on mouse Toll-like receptor 4 species-specific lipopolysaccharide recognition. Int Immunol 2001; 13: 1595–4599. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Gaekwad J, Wolfert MA, et al. Synthetic tetra-acylated derivatives of lipid A from Porphyromonas gingivalis are antagonists of human TLR4. Org Biomol Chem 2008; 6: 3371–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Resman N, Vasl J, Oblak A, et al. Essential roles of hydrophobic residues in both MD-2 and Toll-like receptor 4 in activation by endotoxin. J Biol Chem 2009; 284: 15052–15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, Lee G, Marszalek PE. Atomic cranks and levers control sugar ring conformations. J Phys Condens Matter 2005; 17: S1427–S1442. [Google Scholar]
- 27.Kawahara K. Synthetic chemistry with friendships that unveiled the long-lasting mystery of LA. Innate Immun 2019; 25: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohto U, Yamakawa N, Akashi-Takamura S, et al. Structural analyses of human Toll-like receptor 4 polymorphisms D299G and T399I. J Biol Chem 2012; 287: 40611–40617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohto U, Fukase K, Miyake K, et al. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc Natl Acad Sci U S A 2012; 109: 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Su L, Morin MD, et al. TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc Natl Acad Sci U S A 2016; 113: E884–E893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scior T, Wahab A. 2007. Structure prediction of proteins with very low homology: a comprehensive introduction and a case study on aminopeptidase In: Kaplan SP. (ed.) Drug design research perspectives. 1st ed New York: Nova Science Publishers, 2007, pp.275–306. [Google Scholar]
- 32.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000; 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johansson MU, Zoete V, Michielin O, et al. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinformatics 2012; 13: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem 2004; 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
- 35.Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009; 30: 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity – a rapid access to atomic charges. Tetrahedron 1980; 36: 3219–3228. [Google Scholar]
- 37.Clark M, Cramer RD, Van Opdenbosch N. Validation of the general purpose Tripos 5.2 force field. J Comp Chem 1989; 10: 982–1012. [Google Scholar]
- 38.Certara. SYBYL-X Suite, http://tripos.com (accessed 10 December 2019).
- 39.Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 1996; 17: 490–519. [Google Scholar]
- 40.Chemical Computing Group. Molecular Operating Environment 2016.07, https://www.chemcomp.com/MOE-Molecular_Modeling_and_Simulations.htm (accessed 10 December 2019).
- 41.Schüttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D Biol Crystallogr 2004; 60: 1355–1363. [DOI] [PubMed] [Google Scholar]
- 42.Bruice PY. Organic chemistry. 5th ed Upper Saddle River, NJ: Prentice Hall, 2006, Appendix II. [Google Scholar]
- 43.Allen MP, Tildesley DJ. Computer simulation of liquids. 2nd ed Oxford: Oxford University Press, 2017. [Google Scholar]
- 44.Hansen JP, McDonald IR. Theory of simple liquids: with applications to soft matter. Amsterdam: Elsevier, 2013. [Google Scholar]
- 45.Guvench O, MacKerell AD., Jr. Comparison of protein force fields for molecular dynamics simulations. Methods Mol Biol 2008; 443: 63–88. [DOI] [PubMed] [Google Scholar]
- 46.Pronk S, Páll S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013; 29: 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oostenbrink C, Villa A, Mark AE, et al. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem 2004; 25: 1656–1676. [DOI] [PubMed] [Google Scholar]
- 48.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996; 14: 33–38 [DOI] [PubMed] [Google Scholar]
- 49.Malde AK, Zuo L, Breeze M, et al. An Automated force field Topology Builder (ATB) and repository: version 1.0. J Chem Theory Comput 2011; 7: 4026–4037. [DOI] [PubMed] [Google Scholar]
- 50.Salas FJ, Méndez-Maldonado GA, Núñez-Rojas E, et al. Systematic procedure to parametrize force fields for molecular fluids. J Chem Theory Comput 2015; 11: 683–693. [DOI] [PubMed] [Google Scholar]
- 51.Resman N, Oblak A, Gioannini TL, et al. Tetraacylated LA and paclitaxel-selective activation of TLR4/MD-2 conferred through hydrophobic interactions. J Immunol 2014; 192: 1887–1895. [DOI] [PubMed] [Google Scholar]
- 52.Walsh C, Gangloff M, Monie T, et al. Elucidation of the MD-2/TLR4 interface required for signaling by lipid IVa. J Immunol 2008; 181: 1245–1254. [DOI] [PubMed] [Google Scholar]
- 53.Kondo Y, Ikeda K, Tokuda N, et al. TLR4-MD-2 complex is negatively regulated by an endogenous ligand, globotetraosylceramide. Proc Natl Acad Sci U S A 2013; 110: 4714–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pastor N, Amero C. Information flow and protein dynamics: the interplay between nuclear magnetic resonance spectroscopy and molecular dynamics simulations. Front Plant Sci 2015; 6: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan H, Wei K, Bao J, et al. In silico study on multidrug resistance conferred by I223R/H275Y double mutant neuraminidase. Mol Biosyst 2013; 9: 2764–2774. [DOI] [PubMed] [Google Scholar]
- 56.Muvva C, Singam ER, Raman SS, et al. Structure-based virtual screening of novel, high-affinity BRD4 inhibitors. Mol Biosyst 2014; 10: 2384–2397. [DOI] [PubMed] [Google Scholar]
- 57.Muthukumaran R, Sangeetha B, Amutha R. Conformational analysis on the wild type and mutated forms of human ORF1p: a molecular dynamics study. Mol Biosyst 2015; 11: 1987–1999. [DOI] [PubMed] [Google Scholar]
- 58.Plazinska A, Plazinski W. Stereoselective binding of agonists to the β2-adrenergic receptor: insights into molecular details and thermodynamics from molecular dynamics simulations. Mol Biosyst 2017; 13: 910–920. [DOI] [PubMed] [Google Scholar]