

Abstract
Objective:
People with diabetes are at a significantly higher risk of cardiovascular disease, in part, due to accelerated atherosclerosis. Diabetic subjects have increased number of platelets that are activated, more reactive, and respond sub-optimally to anti-platelet therapies. We hypothesized that reducing platelet numbers by inducing their premature apoptotic death would decrease atherosclerosis.
Approach and Results:
This was achieved by targeting the anti-apoptotic protein Bcl-xL (which is essential for platelet viability) via distinct genetic and pharmacological approaches. In the former, we transplanted bone marrow (BM) from mice carrying the Tyr15 to Cys loss of function allele of Bcl-x (known as Bcl-xPlt20) or wild-type littermate controls into atherosclerotic prone Ldlr+/− mice made diabetic with Streptozotocin (STZ) and fed a Western diet. Reduced Bcl-xL function in hematopoietic cells significantly decreased platelet numbers, exclusive of other hematological changes. This led to a significant reduction in atherosclerotic lesion formation in Bcl-xPlt20 BM transplanted Ldlr+/− mice. To assess the potential therapeutic relevance of reducing platelets in atherosclerosis, we next targeted Bcl-xL with a pharmacological strategy. This was achieved by low-dose administration of the BH-3 mimetic, ABT-737 tri-weekly, in diabetic Apoe−/− mice for the final 6 weeks of a 12-week study. ABT-737 normalized platelet numbers along with platelet and leukocyte activation to that of non-diabetic controls, significantly reducing atherosclerosis, whilst promoting a more stable plaque phenotype.
Conclusion:
These studies suggest that selectively reducing circulating platelets, by targeting Bcl-xL to promote platelet apoptosis, can reduce atherosclerosis and lower cardiovascular disease risk in diabetes.
Graphical Abstract
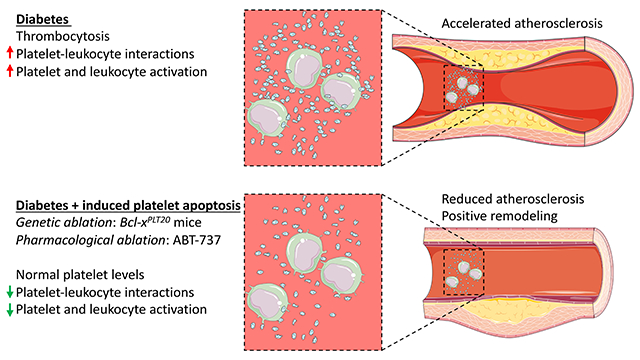
The prevalence and severity of atherosclerotic cardiovascular disease (CVD) remains the leading cause of mortality worldwide and is significantly increased in people with diabetes (Type 1 and Type 2)1–3,4, 5. We have previously shown that the hematopoietic compartment is overactive in the setting of diabetes, insulin resistance and obesity, resulting in the expansion and activation of myeloid cells (monocytes and neutrophils), which directly contribute to inflammation in the vessel wall6–8. There is also increased thrombopoiesis in people with diabetes, correlating with CVD severity9, 10. Platelets are generally associated with thrombotic CVD, but have also been causally implicated in the development and progression of atherosclerotic lesions11–14. Thus, it is not surprising that inhibiting platelet activation significantly reduces the risk of CV events15. However, this platelet focused approach appears to be less efficacious in people with diabetes16–18. This is likely due to enhanced thrombopoiesis, generating more reactive platelets19, 20,17, 19, 21–25, which appear to be more resistant to standard anti-platelet therapies18, 21, 26.
We have recently defined one of the mechanisms driving thrombopoiesis in diabetes, initiated by enhanced production of thrombopoietin (TPO) in the inflamed liver10. The primary responder to increased blood glucose levels appears to be the circulating neutrophil, which becomes activated and releases the sterile inflammatory mediator, S100A8/A96, 7, 27. S100A8/A9 then stimulates Kupffer cells, instructing the liver to produce TPO, enhancing thrombopoiesis and thereby accelerating atherosclerosis. In addition to diabetic models, we have also reported increased atherosclerosis burden as a result of enhanced thrombopoiesis, albeit driven by different mechanisms13, 28. Given that platelets play a causal role in promoting atherosclerosis, we reasoned that normalizing the increased platelet numbers seen in diabetes to that of non-diabetic controls would slow the progression of atherosclerosis.
Platelet production is a complicated process and tightly regulated by the well-established TPO-feedback loop, where platelets regulate their own abundance by acting as a sink for TPO, which is usually under constitutive expression in the liver. Given that platelet depletion by antibodies is a challenging and costly chronic intervention, we explored alternative options to reduce platelet numbers. Platelets have a pre-programmed life span based on their abundance of the anti-apoptotic protein Bcl-xL29. Inhibition of Bcl-xL causes platelet apoptosis and clearance through the reticuloendothelial system in the spleen and liver. Importantly, there are a number of small molecule inhibitors termed BH3 mimetics that target the pro-survival proteins including Bcl-xL. These mimetics have been developed for the treatment of hematological malignancies and solid tumors. One such mimetic, ABT-737 has been shown to target Bcl-xL and reduce platelet numbers, causing clearance predominantly in the liver30. Importantly, apoptosis induced by ABT-737 does not induce platelet activation, and in fact, appears to suppress it31, 32. We therefore designed experimental strategies, genetic and pharmacological, to specifically target Bcl-xL and tested the idea that inducing platelet apoptosis would reduce the abundance of circulating platelets and atherosclerotic lesion burden in diabetes.
Methods
Data available on request from the authors
Mice.
Animal experiments were approved by the Alfred Medical Research Education Precinct (AMREP) Animal Ethics Committee (application #1348) and conducted in accordance with the National Health and Medical Research Council (NHMRC) of Australia Guidelines for Animal Experimentation, along with the NIH Guide for the Care and Use of Laboratory Animals. Ldlr+/− mice, generated by crossing male Ldlr−/− mice (Jax mice, stock #002207) with female C57Bl/6J mice (Jax mice, stock #000664), and Apoe−/− mice (Jax mice, stock # 002052), were bred and housed in the Alfred Alliance’s Precinct Animal Centre (PAC) in a pathogen-free facility under controlled conditions, exposed to a 12:12 hour light dark cycle and fed a standard laboratory diet (Specialty Feeds, Glen Forrest, AUS). Bcl-xPlt20 mice on a C57Bl/6J background were bred and housed at the Walter and Eliza Hall Institute of Medical Research (WEHI) animal facility in accordance with the regulatory standards and approved by the WEHI animal ethics committee. Male mice were used in this study as, unlike females, they are susceptible to Streptozotocin-induced diabetes.
All mice were randomized into different groups and the endpoint measures were collected and analyzed in a blinded manner. Mice that failed to reconstitute their BM, become diabetic or developed skin lesions (which can be common in atherosclerotic mice) were excluded from the study and humanely euthanized. Primary outcomes for the studies were circulating platelet counts or atherosclerotic lesion size. Mice were euthanised by CO2.
Bone Marrow Transplantation (BMT).
As previously described, Ldlr+/− recipient mice were treated with 10mg/L neomycin and 10mg/L polymyxin for 2 weeks before and 6 weeks after the BMT procedure. Recipient mice were irradiated (from a cesium gamma source) with 2 doses of 5.5 Gy separated by at least 4h. BM (5x106 cells/recipient) from Bcl-xPlt20 or littermate control mice were transplanted into the Ldlr+/− recipient mice via tail vein injection followed by an 8-week reconstitution period.
Diets.
Ldlr+/− mice post BMT and induction of diabetes (Streptozotocin; STZ) were fed a Western type diet (WTD) (SF100-219, Specialty feeds, 21% fat, 0.15% cholesterol) for the remainder of the study (i.e. 10 weeks) in order to increase plasma cholesterol levels and induce atherosclerosis. Apoe−/− mice were fed a standard laboratory diet for the entire study.
Treatments.
To induce diabetes, STZ was administered to mice by i.p injection (50 mg/kg for 5 days) using the recommended protocol from the Animal Models of Diabetic Complications Consortium (AMDCC)33. Mice were considered diabetic if their non-fasted glucose level exceeded 17 mmol/L. ABT-737 was administered by i.p. injection tri-weekly at a dose of 30 mg/kg. ABT-737 was prepared as previously described29. Briefly, ABT-737 was solubilized in 10% DMSO and diluted in a solution consisting of 30% propylene glycol, 5% Tween 80 and 65% D5W, pH 4-5. The final concentration of DMSO was less than 1%.
Complete Blood Count.
Complete blood counts were measured in whole blood obtained from tail vein bleeding using an automated hematology analyzer (Sysmex XS-1000i; Kobe, Japan).
Blood glucose, plasma cholesterol.
Blood glucose was determined by a glucometer (Accu-chek, Roche). Plasma cholesterol was determined by a calorimetric enzymatic assay as per manufacturer’s instruction (Wako Diagnostics, Japan).
Quantification of reticulated platelets.
Reticulated platelets were measured as previously described13. In brief, undiluted EDTA-anticoagulated blood (5μL) within 30mins of collection was mixed with an anti-CD41-APC antibody (eBioscience,) and the fluorescent dye thiazole orange (final concentration 1μg/mL) and incubated at room temperature in the dark for 20 min. Reticulated platelets were identified as CD41+thiazole orangehi. Reticulated platelets numbers presented were determined by normalizing to complete blood count of platelets.
Hematopoietic stem and progenitor cells.
BM cells were obtained from femurs and tibias and stained with a cocktail of antibodies before analysis by flow cytometry as previously described13, 28, 34. Briefly, to identify lineage committed cells antibodies to CD45R, CD19, CD11b, CD3e, TER-119, CD2, CD8, CD4 and Ly6-C/G were used (eBioscience or Biolegend), with antibodies against Sca1 (Biolegend) and cKit (eBioscience) to identify progenitor cell populations and Hematopoietic Stem and Progenitor cells (HSPCs, aka LSKs) (Lineage−,Sca1+,cKit+) cells and with antibodies to CD16/CD32 (FcγRII/III) and CD34 (BD Biosciences) to separate Common Myeloid Progenitors (CMPs) (Lineage−, Sca1−, cKit+, CD34int, FcγRII/IIIint) and Megakaryocyte Erythroid Progenitors (MEPs) (Lineage−, Sca1−, cKit+, CD34lo, FcγRII/IIIlo).
Neutrophil and monocyte platelet aggregates.
Blood was collected from the tail vein into EDTA-lined tubes kept at room temperature. Red blood cells (RBCs) were lysed, and washed cells were then stained with antibodies against CD45 (Invitrogen), CD115 (eBioscience), Gr1 (Ly6-C/G; BD Biosciences), CD11b (eBioscience) and CD41 (eBioscience) at 1:400 dilution for 30 min on ice. The cells were carefully washed, resuspended in FACS buffer and run on an LSRII flow cytometer to detect leukocyte platelet interactions and leukocyte activation. Viable cells were selected on the basis of forward and side scatter characteristics, and then CD45+ leukocytes were selected. Ly6-Chi monocyte platelet aggregates were identified as CD115+Gr1hi (Ly6-Chi) and CD41+. Neutrophil-platelet aggregates were identified as CD115−Gr1+ (Ly6-G+) and CD41+. Platelet-dependent activation of Ly6-Chi monocytes and neutrophils was measured by CD11b expression (MFI). Monocyte and neutrophil numbers were assessed using flow cytometry as above without selecting based on CD41 and were normalized to complete blood counts of WBCs.
Atherosclerosis analysis:
Aortic arch plaque analysis (en face).
Lipid content in the aortic arch was measured by en face analysis. Dissected aortas were fixed in 4% (w/v) paraformaldehyde and, prior to staining, all fat and connective tissue was removed from the outer layers of the vessel. The aorta was cut longitudinally, stained with Oil Red O followed by washing and mounting on a silicone coated dish. Images of the aortas were captured on a SZX10 microscope (X0.63 objective) and quantification of ORO staining was performed off-line using Adobe Photoshop 2020.
Proximal aorta lesion analysis.
Hearts were dissected after the mouse was perfusion with saline and were frozen in optimal cutting temperature compound (OCT). Serial 6 μM sections of the aortic root were prepared. Six sections per stain from the beginning to the end of the plaque were used for quantification, with the single data point representing the average. H&E (lesion size), Oil Red O (lipid abundance), CD68 (macrophage content) and Picrosirius red (collagen) staining were performed as previously described10. In addition, immunohistochemistry staining was also performed to detect smooth muscle actin using anti-C anti-smooth muscle alpha-actin antibody at 1:100 dilution. Images were acquired on an Olympus BX43 microscope. Quantification of all images were performed using Adobe Photoshop 2020.
Statistical analysis
Data are presented as mean ± SEM and were analyzed using the two-tailed Student t-test or one-way ANOVA followed by Dunnet’s post hoc test. P<0.05 was considered significant. the normality and variance were not tested to determine whether the applied parametric tests were appropriate. All tests were performed using the Prism software (GraphPad Software, Inc., La Jolla, CA).
Results
Genetically induced platelet apoptosis to lower platelets does not alter traditional cardiovascular risk factors in diabetes
We have previously shown that enhanced platelet production in diabetes is in part responsible for accelerated atherogenesis in diabetes10. To directly test the role of platelets on enhanced atherogenesis in diabetes, we performed a BMT study using low-density lipoprotein receptor (LDLR) heterozygous (Ldlr+/−) mice reconstituted with BM from either mice with genetically induced loss-of-function in Bcl-XL (Bcl-xPLT20) or littermate controls (WT) (Figure 1A). The Bcl-xPLT20 mice have a mutation in the pro-survival protein Bcl-XL, which causes the platelets to undergo apoptosis prematurely, resulting in a significant reduction in steady-state platelet count 35. Ldlr+/− mice were employed for two main reasons; 1) The hematopoietic cells from the Bcl-xPLT20 mice express Apoe, thus if Apoe−/− recipient mice were used, the reintroduction of haematopoietic expressed Apoe would cause a normalisation of plasma cholesterol, preventing atherosclerosis. 2) Ldlr+/− mice recipients were used over Ldlr−/− mice, to allow us to better control plasma cholesterol levels. We have previously found that diabetic Ldlr−/− mice fed this diet develop sever hypercholesterolemia (3000-4000mg/dl) and die prematurely. Thus, the Ldlr+/− mice as reported below have more appropriate cholesterol levels. After 8 weeks of reconstitution, mice were made diabetic with STZ and fed a high fat (21%) high cholesterol (0.15%) diet for 10 weeks. Importantly, blood glucose and plasma cholesterol levels were not significantly different between the WT BMT and Bcl-xPLT20 BMT mice (Figure 1B, C). Consistent with the model, the number of total and mature circulating platelets were significantly reduced in the Bcl-xPLT20 BM recipient mice compared to the WT recipient mice, while the abundance of reticulated platelets was increased as expected (Figure 1D). Importantly, circulating leukocytes were similar between the groups (Figure 1E). We did observe an increase in the abundance of cells that constitute the thrombopoietic branch of hematopoiesis (i.e., HSPCs, CMPs and MEPs; Figure 1F). However, not reporting the absolute cell number may limit our ability to distinguish the cellularity in the BM. These data suggest that any downstream effects on atherosclerosis are not due to differences in the classical cardiovascular risk factors such as hyperglycemia, hypercholesterolemia and increased myeloid cell number, but are related to changes in platelet number and/or function.
Figure 1: Genetic loss of function in Bcl-xL decreases platelets in diabetic Ldlr+/− mice.
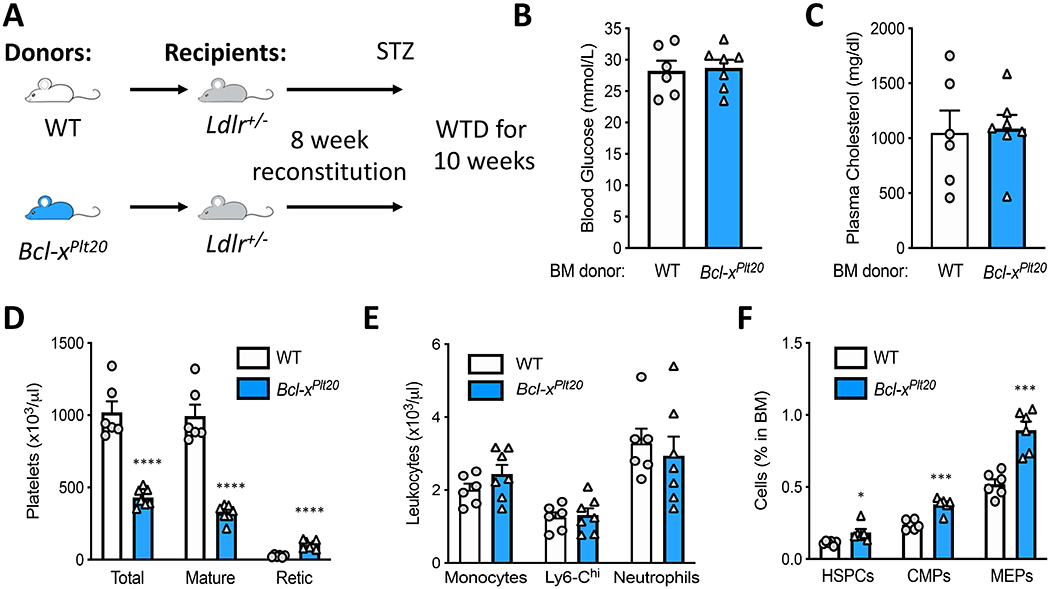
A) Experimental overview: Ldlr+/− mice were transplanted with BM from Bcl-xPLT20 mice or littermate controls (WT), made diabetic with STZ and fed a WTD. B) Blood glucose and C) plasma cholesterol levels at the end of the study. D) Circulating platelets, E) leukocytes and F) BM haematopoietic stem and progenitor cells were quantified by flow cytometry. Data are presented mean ± SEM and analyzed using a Student’s unpaired t-test, biological replicates of n=6 & 7/group; *p<0.05, ***p<0.001, ****p<0.0001.
Genetic induction of platelet apoptosis significantly decreases atherosclerosis in diabetic mice.
To determine the effect of platelet apoptosis in diabetic Bcl-xPLT20 BMT Ldlr+/− mice, we assessed atherosclerotic plaque size and composition. First, we quantified atherosclerotic burden in the aortic arch by Oil-Red-O (ORO) staining and en face analysis. We observed a striking decrease in lipid abundance in the diabetic Ldlr+/− mice that received Bcl-xPLT20 BM (Figure 2A). Next, we determined the size of atherosclerotic plaques in the aortic sinus. We observed a significant reduction in atherosclerotic plaque size in this region when platelets were reduced by genetically targeting the anti-apoptotic protein Bcl-XL (Figure 2B). Importantly, not only were lesions found to be smaller, but also contained less lipids and macrophages (Figure 2C & D). Moreover, we also observed a trend in increased collagen deposition in Bcl-xPLT20 BMT mice compared to WT BMT mice (Figure 2E). These data suggest that lowering platelet number by intrinsic platelet apoptosis not only slows the progression of atherosclerosis, but also allows favorable remodeling of plaques towards a more ‘stable’ phenotype (Figure 2).
Figure 2: Genetically induced platelet apoptosis decreases atherosclerosis in diabetic Ldlr+/− mice.
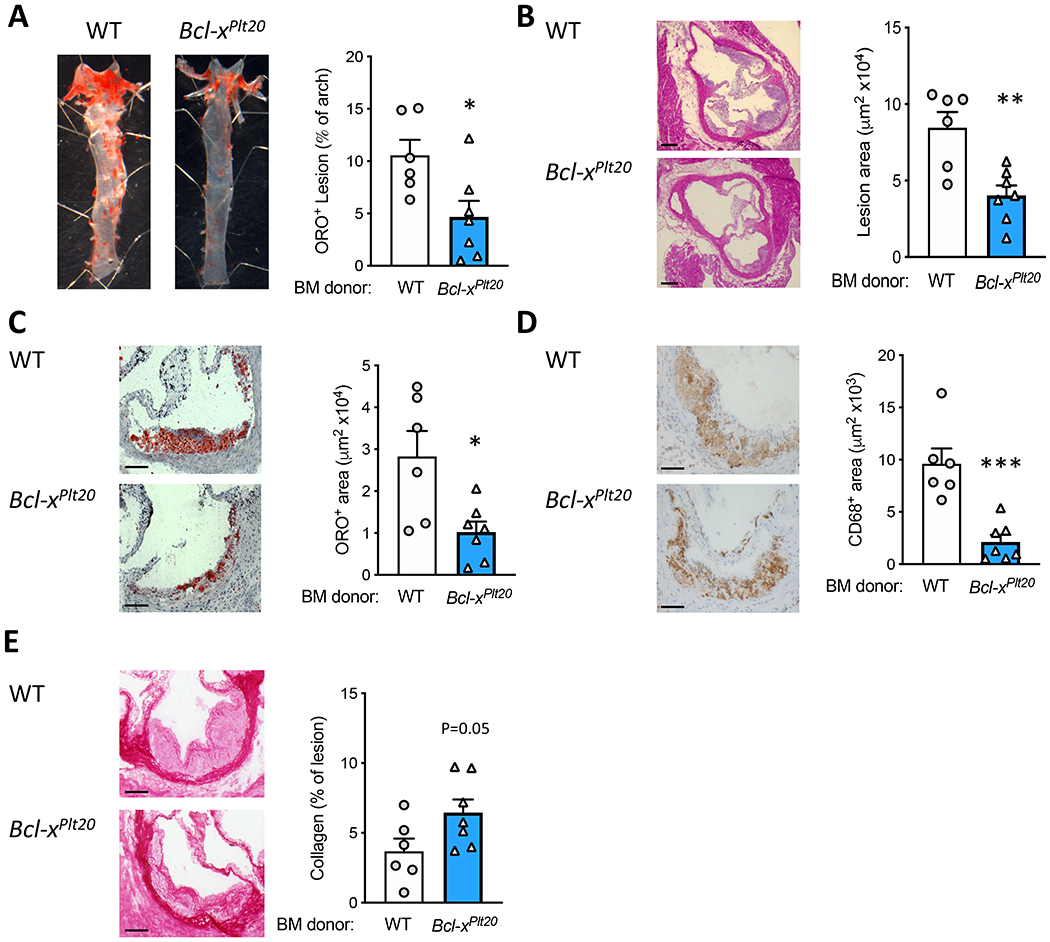
A) Atherosclerosis was quantified in the aortic arch by Oil-Red-O staining and en face analysis. In the aortic sinus, plaques were assessed by B) H&E (size; scale bars = 100 μm), C) Oil-Red-O (lipid), D) CD68+ area (macrophages) and E) picrosirius red (collagen), scale bars: 50 μM. Data are presented mean ± SEM and analyzed using a Student’s unpaired t-test, biological replicates of n=6 & 7/group; *p<0.05, **p<0.01, ****p<0.0001.
Intrinsic platelet apoptosis reduces platelet-leukocyte interactions and leukocyte activation
We have previously shown that diabetes causes enhanced platelet-leukocyte interactions and through these interactions, leukocytes become activated and express adhesion molecules which can facilitate their recruitment to the atherosclerotic plaque 10. Thus, we hypothesized that inducing platelet apoptosis would lower platelet-leukocyte interactions and consequently reduce leukocyte activation. We determined this by assessing platelet-leukocyte aggregates and cell-surface expression of CD11b, a marker of leukocyte activation. Consistent with our hypothesis, we observed significantly fewer platelet-Ly6-Chi monocyte and platelet-neutrophil aggregates in mice that received Bcl-xPLT20 BM (Figure 3A). Exploring further, we also noted a significant reduction in the key activation marker CD11b in mice that received Bcl-xPLT20 BM (Figure 3B). These data suggest that lowering platelet abundance by intrinsic apoptosis has a significant impact on atherosclerosis lesion burden, potentially by reducing leukocyte activation and could be targeted to achieve better CVD outcomes in diabetes.
Figure 3: Genetically induced platelet apoptosis results in fewer platelet-leukocyte interactions and reduced leukocyte activation.
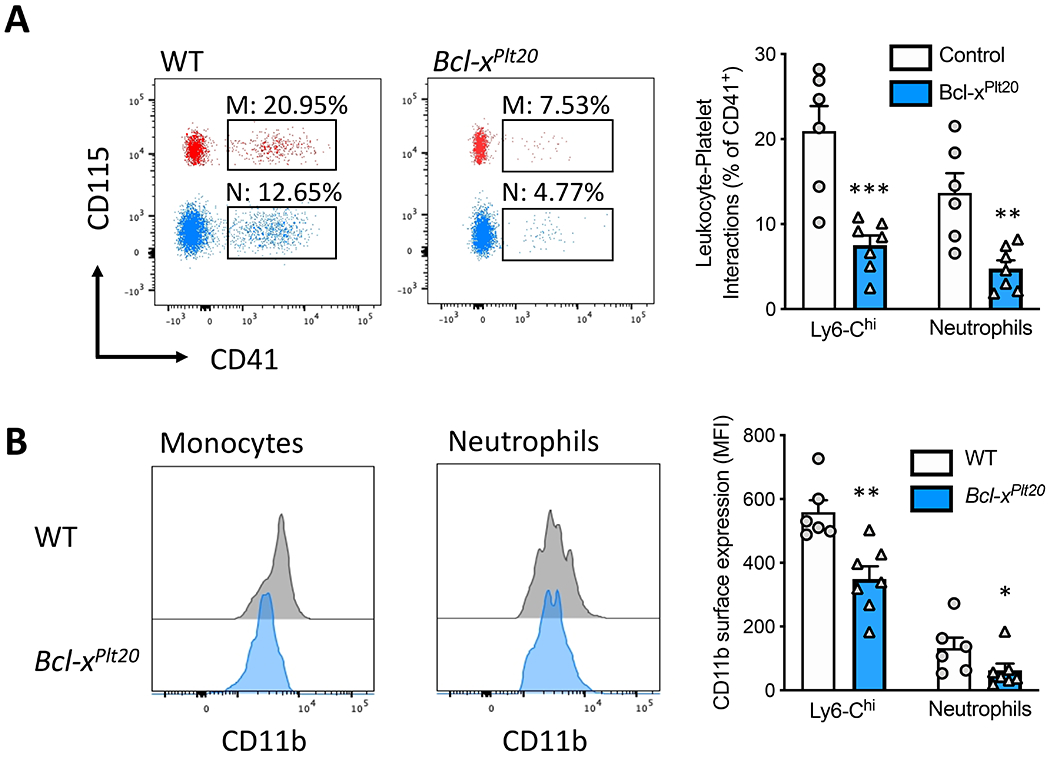
A) Platelet-leukocyte interactions were assessed by flow cytometry. Ly6-Chi monocyte (M) and neutrophil (N) populations, identified as per the methods, were overlayed on the same FACS plot for visualization purpose. B) Leukocyte activation (cell-surface CD11b expression) on the platelet-interacting cells was quantified by flow cytometry. Data are presented mean ± SEM and analyzed using a Student’s unpaired t-test, biological replicates of n=6 & 7/group; *p<0.05, **p<0.01, ****p<0.0001.
Inducing platelet apoptosis with low-dose administration of BH3 mimetic ABT-737 normalizes platelet counts in atherosclerotic-prone mice with diabetes
Given that genetic disruption of the platelet pro-survival gene Bcl-xL reduces atherosclerosis in diabetic mice through intrinsic platelet apoptosis, we next explored the utility of pharmacologically targeting this pathway through the use of the BH3 mimetic ABT-737. This drug, a precursor to ABT-236 (navitoclax), was originally developed as an intervention against hematological malignancies as it was shown to target Bcl-2 and Bcl-w, but was also found to induce thrombocytopenia due to its high affinity for Bcl-xL. We hypothesized that ABT-737 could be administered at a low-dose to normalize platelet counts in diabetic mice which could then presumably afford vascular protection. In a dosing-frequency study we determined that the optimal dose to normalize platelets without causing thrombocytopenia or rebound thrombocytosis was a tri-weekly administration of 30mg/kg ABT-737 (2.5-fold less than the 75mg/kg dose shown to cause an ~80% reduction in platelets after a single injection; Supplementary Figure I). With the vision to translate these studies for clinical application, we propose that the use of a drug to induce platelet apoptosis in people without either diabetes, increased platelet levels (i.e. essential thrombocythemia) or those at high-risk of an event is impractical. Therefore, we chose to only explore the use of this small molecule inhibitor as an intervention in the setting of diabetes only. Our goal was to normalize platelet levels to those seen in non-diabetic mice and as such we rationalised not to include a non-diabetic treated group.
Exploring the utility of ABT-737 as an intervention, we rendered standard laboratory diet fed Apoe−/− mice diabetic with STZ and then 6 weeks later treated mice with either the vehicle or ABT-737 (30mg/kg tri-weekly) for 6 additional weeks. In parallel, a non-diabetic group of standard laboratory diet fed Apoe−/− mice treated with vehicle was included to compare the effect of diabetes and interventional effects (Figure 4A). We chose to use the Apoe−/− mouse for this study as it is the preferred model of diabetic atherosclerosis, when a BMT is not required. The Apoe−/− mouse develops atherosclerotic plaques on a standard laboratory diet (unlike the Ldlr−/− mouse), allowing us to include a non-diabetic control group. As expected, blood glucose and plasma cholesterol levels were significantly higher in diabetic mice compared to non-diabetic Apoe−/− mice (Figure 4B,C). Importantly, pharmacologically induced platelet apoptosis with ABT-737 had no effect on blood glucose or plasma cholesterol levels, suggesting that any changes in atherosclerotic plaque development was independent of these two important drivers of disease (Figure 4B, C).
Figure 4: Low dose ABT-737 normalizes platelets in diabetic Apoe−/− mice.
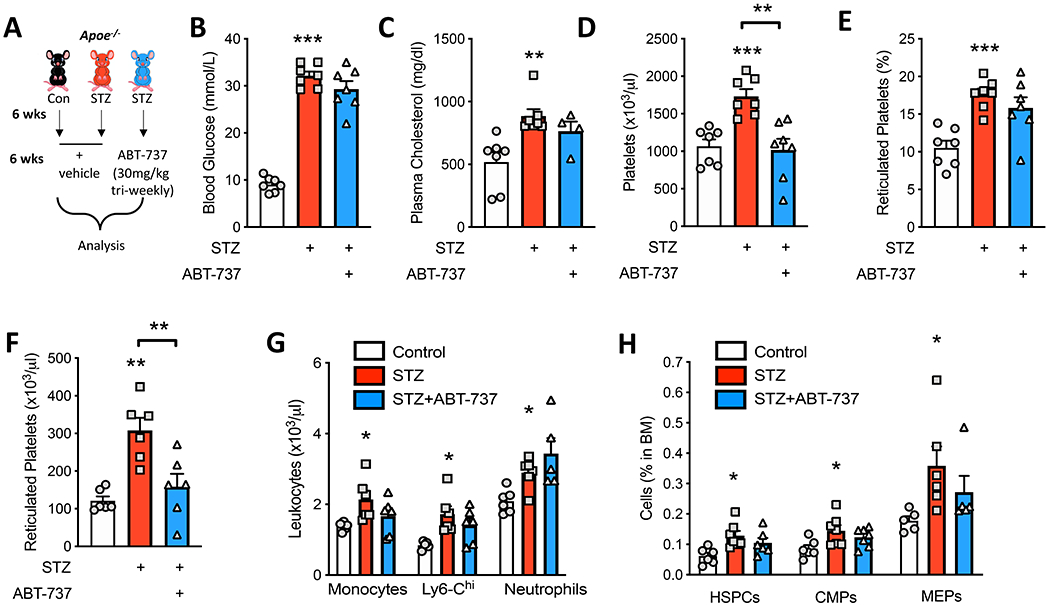
A) Experimental overview: Apoe−/− mice were made diabetic (STZ) or left as control and fed a standard laboratory diet for 12 weeks. After 6 weeks, a group of the diabetic mice were treated with ABT-737 (30mg/kg/tri-weekly i.p.) for additional 6 weeks. B) Blood glucose (n=7/group) and C) plasma cholesterol levels were measured at the end of the study (n=7, 7, 5/group). Circulating D) platelets, E) percentage and F) number of reticulated platelets (n=7/group), and G) leukocytes were measured by flow cytometry (n=5-6/group). H) BM Haematopoietic stem and progenitor cells were quantified by flow cytometry (n=6-7/group). Data are presented mean ± SEM and analyzed using a one-way ANOVA followed by a Dunnett’s multiple comparisons test, biological replicates n=5-7/group; *p<0.05, **p<0.01, ****p<0.0001 c.f. control, or as indicated.
We next explored the hematological parameters in these mice. Consistent with the mechanism of action of ABT-737 and the dosage and frequency selected, we observed a normalization of platelet numbers in the treated mice to non-diabetic levels (Figure 4D). While the percentage of the immature reticulated platelets was unchanged, we did observe a significant reduction in the overall number of these cells (Figure 4E, F). Importantly, the changes in hematological parameters appeared to be confined to platelets as blood monocytes and neutrophils, which are increased in the setting of diabetes6, were unchanged by ABT-737 treatment (Figure 4G). This was also reflected in the BM haematopoietic stem and progenitor cell populations, where diabetes promoted an expansion in myelopoiesis and thrombopoiesis, which was not influenced by ABT-737 (Figure 4H).
ABT-737 reduces atherosclerosis in diabetic mice.
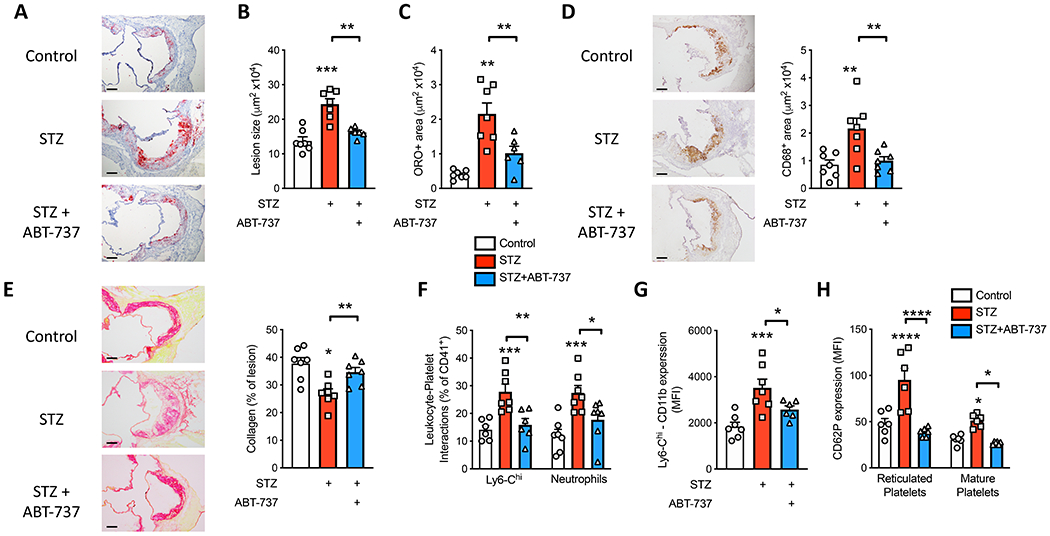
Our previous findings10, along with the data presented in Figure 2, suggest that platelets contribute to accelerated atherosclerotic plaque pathology observed in diabetes. Encouraged by the normalization of platelets in the diabetic mice treated with ABT-737, we hypothesized that atherosclerotic plaques would be smaller in these mice. Reiterating that ABT-737 was administered for the final 6 weeks of the study, we next assessed plaque burden in the aortic sinus. As expected, we observed significantly larger plaques in the diabetic, vehicle treated mice compared to the non-diabetic mice. Importantly ABT-737 treatment resulted in a significant reduction in plaque size, to the levels trending towards those seen in the non-diabetic mice (Figure 5A, B). This inhibition in plaque growth with ABT-737 was paralleled by a reduction in plaque lipid and macrophage content (Figure 5A, C, D). We also observed an increase in plaque collagen content in the ABT-737 treated mice (Figure 5E), indicative of positive remodelling to a more stable phenotype, which is consistent with our observations following genetic disruption of Bcl-xL (Figure 2). We also determined smooth muscle cell (SMC) content in the plaques. Consistent with previous reports36 we noted a significant increase in SMCs in the lesions from the diabetic mice compared to control mice, which was reversed in the lesions of mice treated with ABT-737 (Supplementary Figure II). We attempted to identify platelets within the atherosclerotic plaques without success (data not shown), thus we suggest that a limitation of these data is that we are unable to conclude whether platelets directly influenced atherosclerosis by playing a role within the atherosclerotic plaque.
Figure 5: ABT-737-induce platelet apoptosis in diabetic Apoe−/− mice and reduce atherosclerosis.
Atherosclerotic plaques were assessed in the aortic sinus. A) Representative images of lesion size and lipid composition with ORO staining; B) lesion size and C) lipid positive area. D) CD68+ area (macrophages) and E) picrosirius red (collagen), scale bars: 50 μM. F) Platelet-leukocyte interactions and G) Ly6-Chi monocyte activation, as assessed by CD11b levels on the platelet-interacting cells, were quantified by flow cytometry. H) CD62P (P-selectin) expression was assessed on reticulated and mature platelets by flow cytometry. Data are presented mean ± SEM and analyzed using a one-way ANOVA followed by a Dunnett’s multiple comparisons test, biological replicates, n=7, 7, 6/group; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 c.f. control, or as indicated.
In line with our hypothesis that reduced platelets would cause fewer interactions with leukocytes and prevent their activation, we also found that pharmacological induction of platelet apoptosis decreased the abundance of platelet-leukocyte interactions, specifically with Ly6-Chi monocytes and neutrophils (Figure 5F). This was accompanied by reduced Ly6-Chi monocyte activation (CD11b; Figure 5G). Interestingly, we also observed reduced P-selectin expression on mature and reticulated platelets, providing a potential mechanism for reduced platelet-leukocyte interactions via leukocyte PSGL-1, which would in turn prevent outside-in signaling to induce leukocyte CD11b expression (Figure 5H). These findings are consistent with the observations in our genetic studies and are likely to explain, at least in part, the reduction in atherosclerosis in these mice. Taken together, these two studies, with complimentary genetic and pharmacological approaches, reveal that targeting platelet apoptosis can reduce atherosclerotic complications that are highly prevalent in people with diabetes.
Discussion
Diabetes accelerates atherosclerotic CVD, the largest cause of morbidity and mortality in individuals with both type 1 and type 2 diabetes. While a number of mechanisms have been proposed to explain accelerated atherosclerosis in diabetes, it is apparent that enhanced leukocyte and platelet production along with the activation of these cells is a major contributor6, 10. Here we found that limiting platelet numbers by inducing platelet apoptosis in genetic and pharmacological studies attenuated the accelerated atherosclerotic phenotype in diabetic mice. This beneficial effect was not through changes in the abundance of monocytes and neutrophils, which are well-established to drive atherogenesis34, 37–40, but was instead associated with a decrease in the activation of these immune cells. This suggests that an approach targeting platelets ultimately is able to lead to reduced capacity of monocytes to be recruited to and infiltrate atherosclerotic plaques.
Platelets, particularly through their activation, are known contributors to atherosclerotic CVD11, 13. Even prior to the direct evidence, platelet activation has long been targeted to treat CVD, where anti-thrombotic drugs such as aspirin, clopidogrel, prasugrel and ticagrelor are often prescribed following an ischemic event to reduce the risk of clotting41. This reflects the longstanding view that platelets only become important during an ischemic event. Anti-platelet therapies decrease the risk of CVD, but these drugs are less efficacious in people with diabetes19–21. We have found that increased platelet numbers (i.e. thrombocytosis) can directly contribute to atherogenesis13, 28, and this is particularly relevant in the setting of diabetes10. Thus, it stands to reason that limiting platelet production may reduce the accelerated CVD seen in the context of diabetes. Indeed, targeting one of the upstream mediators that we identified to be responsible for enhanced thrombopoiesis, S100A8/A9, was effective at reducing platelet production and atherosclerosis in animal studies10. However, S100A8/A9 can also play other important roles in sterile inflammation and communicate with a number of different cells. This led us to question if more direct platelet focused interventions could be more appropriate, while allowing us to determine just how important platelets are in mediating the vascular pathology seen in diabetes.
Over a decade ago, it was discovered that the brief circulating life span of platelets is circumscribed by the induction of an intrinsic mitochondrial apoptosis program29. The two critical components of this program are pro-survival Bxl-xL and pro-apoptotic Bak. Loss of the former dramatically reduces platelet life span, loss of Bak results in almost doubling29. Given the abundance of platelets that are cleared from the circulation daily without apparently eliciting an inflammatory response through the reticuloendothelial system, we suggest that platelet apoptosis is an immune-silent event. With this knowledge, we hypothesized that promoting early apoptosis specifically in platelets would be an immunologically silent tactic to reduce platelet levels thereby limiting some of the vascular complications seen in diabetes.
We discovered that reducing platelets, through either genetic or pharmacological approaches to induce platelet apoptosis, significantly inhibits atherogenesis in diabetes. We and others have previously show that diabetes causes adverse lesion remodelling as plaques evolve, highlighted by increased lipid and macrophage accumulation and reduced collagen content, which is a phenotype indicative of an unstable lesion that is prone to plaque rupture6, 10. We did note increased SMCs in the diabetic mice, which was reduced when the diabetic mice were treated with ABT-737. While one may expect SMC abundance to go hand in hand with collagen staining, it has be previously noted that lesions from diabetic animals do indeed present with an increased abundance of SMCs, which is not directly influenced by the potential proliferative effects of glucose36. Perhaps the collagen levels and matrix deposition could have been decreased in the diabetic mice due to platelets interacting with endothelial cells to induce matrix metalloproteinase 9, influencing collagen levels42. Platelet-leukocyte interactions are known to activate monocytes and neutrophils, inducing the expression of the adhesion molecule CD11b (aka Mac-1), contributing to the recruitment of these cells to the atherosclerotic plaque, likely explaining increased macrophage and lipid abundance. This inflammatory occurrence is known to be more prominent in individuals with diabetes43–46, and is even suggested to be a more robust readout than the activation status of circulating platelets47. Further, ex vivo platelet aggregation, which is also enhanced individuals with diabetes, is not reduced with anti-platelet therapy48, which is also similar to the finding with platelet-leukocyte aggregates43. In models of increased platelets, leukocyte-platelet aggregates are indeed observed and appear to be important in driving atherosclerosis11, 13, 28. However, other pathways including the generation of platelet-derived microparticles49, 50 or altered proteomes (intracellular and surface)51 and RNA52 content may also play an important role in atherosclerosis in the setting diabetes. Nonetheless, our previous findings do indeed support this series of events10, 13, 28, and the data presented herein confirms the hypothesis that lowering platelets, and potentially their ability to become activated, through targeted inhibition of Bcl-xL31, 32, reverses this atherogenic pathway in the setting of diabetes. Taken together, it is likely that a combination of enhanced platelet production and altered platelet content/composition are important factors in driving the accelerated atherosclerotic phenotype observed in diabetes.
BH3 mimetics designed to induce cell death, have been almost exclusively trialled in the setting of cancers, both hematological and solid tumors. Given that diabetes and obesity, along with other cardiovascular risk factors induce a myeloproliferative state, albeit not as aggressive, it is therefore postulated that inducing apoptosis to control the abundance of myeloid cells could limit vascular disease. In this study we explored the Bcl-xL inhibitor ABT-737, which has been redesigned to the orally active compound ABT-263, also known as navitoclax. Given the potency of this drug at high concentrations, we optimized dosing in order to ‘normalize’ platelet levels in diabetic mice. Another advantage of targeting Bcl-xL with ABT-737 is that platelet reactivity is also reduced31, 32. Our data suggest that chronic administration of a low dose of ABT-737 is able to normalize platelet levels and their activation to limit atherosclerosis. Importantly, low dose ABT-263 was shown to have no effect on bleeding time, which is an important side effect to avoid in platelet-based therapies31. While ABT-236 does inhibit Bcl-xL, it also inhibits Bcl-2 and Bcl-w. Thus, we postulate that while being careful to avoid thrombocytopenia, low-dose administration of a specific Bcl-xL inhibitor could normalize platelet counts in people with diabetes, with the clinical potential to translate into a reduced risk of a major adverse cardiac event53–55. We propose that short-term studies in humans are warranted to determine if surrogate platelet-related cardiovascular markers such as platelet-leukocyte interactions or systemic inflammatory cytokine levels are reduced in individuals treated with such compounds that induce platelet apoptosis. Bearing in mind platelet that quality is also important, the development of specific Bcl-xL inhibitors to treat CVD will likely be in conjunction with standard anti-platelet therapies and perhaps drugs that alter platelet content. As the world of hematological malignancies (i.e. essential thrombocythemia, clonal hematopoiesis with indeterminant potential, etc.) and CVD become more and more intertwined37, 56, perhaps drugs traditionally targeted to treat leukemias could be repurposed at sub-toxic doses as interventions to limit CVD in high risk settings such as diabetes57.
Supplementary Material
Highlights:
Diabetes accelerates atherosclerosis, which is associated with increased platelet production.
Genetically induced platelet apoptosis, though mutation of Bcl-xL, dramatically lowered platelet numbers and inhibited atherosclerosis in diabetes.
Pharmacologically targeting Bcl-xL with the BH3 mimetic ABT-737 at a low dose normalised platelet counts and prevented the acceleration in atherosclerotic plaque development.
Reducing platelet numbers via the Bcl-xL pathway appeared to decrease atherosclerosis by preventing platelet-monocyte interactions and subsequently monocyte activation. However, Bcl-xL inhibition also decreased platelet activation, likely also contributing to the observed reduction in atherosclerosis.
Acknowledgements
We thank Keti Florides, Jason Corbin and Marion Lebois for outstanding assistance.
Sources of Funding
This work was supported by NHMRC grant (APP1106154) to AJM and NIH (R01HL137799 & R00HL122505) to PRN. NMJH is supported by the Dutch Heart foundation (2017T039), Dutch Diabetes foundation (2017.85.005) and the EFSD). AJM is supported by Career Development Fellowship from the NHMRC (APP1085752), a Future Leader Fellowship from the National Heart Foundation (100440), a Viertel Award from Diabetes Australia Research Trust and a Centenary Award from CSL. E.C.J is the recipient of a fellowship from the Lorenzo and Pamela Galli Charitable Trust.
Nonstandard abbreviations and acronyms:
- Bcl-xL
B-cell lymphoma-extra large
- Ldlr
Low-density lipoprotein receptor
- Apoe
Apolipoprotein E
- CVD
Cardiovascular disease
- TPO
Thrombopoietin
- BMT
Bone marrow transplant
- STZ
Streptozotocin
- BH3
B Cell Lymphoma-2 Homology Domain 3
Footnotes
Declarations
None.
References
- 1.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. The New England journal of medicine. 1998;339:229–234 [DOI] [PubMed] [Google Scholar]
- 2.Laakso M Cardiovascular disease in type 2 diabetes from population to man to mechanisms: The kelly west award lecture 2008. Diabetes care. 2010;33:442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah AD, Langenberg C, Rapsomaniki E, Denaxas S, Pujades-Rodriguez M, Gale CP, Deanfield J, Smeeth L, Timmis A, Hemingway H. Type 2 diabetes and incidence of cardiovascular diseases: A cohort study in 1.9 million people. The lancet. Diabetes & endocrinology. 2015;3:105–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luscher TF, Creager MA, Beckman JA, Cosentino F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part ii. Circulation. 2003;108:1655–1661 [DOI] [PubMed] [Google Scholar]
- 5.Pasterkamp G Methods of accelerated atherosclerosis in diabetic patients. Heart. 2013;99:743–749 [DOI] [PubMed] [Google Scholar]
- 6.Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flynn MC, Kraakman MJ, Tikellis C, Lee MK, Hanssen NM, Kammoun HL, Pickering R, Dragoljevic D, Al-Sharea A, Barrett TJ, et al. Transient intermittent hyperglycemia accelerates atherosclerosis by promoting myelopoiesis. Circ Res. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014;19:821–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sokunbi DO, Wadhwa NK, Suh H. Vascular disease outcome and thrombocytosis in diabetic and nondiabetic end-stage renal disease patients on peritoneal dialysis. Advances in peritoneal dialysis. Conference on Peritoneal Dialysis. 1994;10:77–80 [PubMed] [Google Scholar]
- 10.Kraakman MJ, Lee MK, Al-Sharea A, Dragoljevic D, Barrett TJ, Montenont E, Basu D, Heywood S, Kammoun HL, Flynn M, et al. Neutrophil-derived s100 calcium-binding proteins a8/a9 promote reticulated thrombocytosis and atherogenesis in diabetes. J Clin Invest. 2017;127:2133–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein e. Nature medicine. 2003;9:61–67 [DOI] [PubMed] [Google Scholar]
- 12.Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nature medicine. 2009;15:97–103 [DOI] [PubMed] [Google Scholar]
- 13.Murphy AJ, Bijl N, Yvan-Charvet L, Welch CB, Bhagwat N, Reheman A, Wang Y, Shaw JA, Levine RL, Ni H, et al. Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nature medicine. 2013;19:586–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin JF, Kristensen SD, Mathur A, Grove EL, Choudry FA. The causal role of megakaryocyte-platelet hyperactivity in acute coronary syndromes. Nature reviews. Cardiology. 2012;9:658–670 [DOI] [PubMed] [Google Scholar]
- 15.Collaboration AT. Collaborative overview of randomised trials of antiplatelet therapy--i: Prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. Antiplatelet trialists’ collaboration. Bmj. 1994;308:81–106 [PMC free article] [PubMed] [Google Scholar]
- 16.Angiolillo DJ. Antiplatelet therapy in diabetes: Efficacy and limitations of current treatment strategies and future directions. Diabetes care. 2009;32:531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guthikonda S, Alviar CL, Vaduganathan M, Arikan M, Tellez A, DeLao T, Granada JF, Dong JF, Kleiman NS, Lev EI. Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel in patients with stable coronary artery disease. J Am Coll Cardiol. 2008;52:743–749 [DOI] [PubMed] [Google Scholar]
- 18.Neergaard-Petersen S, Hvas AM, Kristensen SD, Grove EL. Platelets and antiplatelet therapy in patients with coronary artery disease and diabetes. Semin Thromb Hemost. 2016;42:234–241 [DOI] [PubMed] [Google Scholar]
- 19.Papanas N, Symeonidis G, Maltezos E, Mavridis G, Karavageli E, Vosnakidis T, Lakasas G. Mean platelet volume in patients with type 2 diabetes mellitus. Platelets. 2004;15:475–478 [DOI] [PubMed] [Google Scholar]
- 20.Muscari A, De Pascalis S, Cenni A, Ludovico C, Castaldini N, Antonelli S, Bianchi G, Magalotti D, Zoli M. Determinants of mean platelet volume (mpv) in an elderly population: Relevance of body fat, blood glucose and ischaemic electrocardiographic changes. Thrombosis and haemostasis. 2008;99:1079–1084 [DOI] [PubMed] [Google Scholar]
- 21.Guthikonda S, Lev EI, Patel R, DeLao T, Bergeron AL, Dong JF, Kleiman NS. Reticulated platelets and uninhibited cox-1 and cox-2 decrease the antiplatelet effects of aspirin. J Thromb Haemost. 2007;5:490–496 [DOI] [PubMed] [Google Scholar]
- 22.Hekimsoy Z, Payzin B, Ornek T, Kandogan G. Mean platelet volume in type 2 diabetic patients. Journal of diabetes and its complications. 2004;18:173–176 [DOI] [PubMed] [Google Scholar]
- 23.Shah B, Valdes V, Nardi MA, Hu L, Schrem E, Berger JS. Mean platelet volume reproducibility and association with platelet activity and anti-platelet therapy. Platelets. 2014;25:188–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tschoepe D, Roesen P, Esser J, Schwippert B, Nieuwenhuis HK, Kehrel B, Gries FA. Large platelets circulate in an activated state in diabetes mellitus. Semin Thromb Hemost. 1991;17:433–438 [DOI] [PubMed] [Google Scholar]
- 25.Vaduganathan M, Alviar CL, Arikan ME, Tellez A, Guthikonda S, DeLao T, Granada JF, Kleiman NS, Ballantyne CM, Lev EI. Platelet reactivity and response to aspirin in subjects with the metabolic syndrome. American heart journal. 2008;156:1002 e1001–1002 e1007 [DOI] [PubMed] [Google Scholar]
- 26.Karpatkin S Heterogeneity of human platelets. Ii. Functional evidence suggestive of young and old platelets. J Clin Invest. 1969;48:1083–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, Quaife-Ryan GA, Al-Sharea A, Pernes G, Dragoljevic D, et al. Neutrophil-derived s100a8/a9 amplify granulopoiesis after myocardial infarction. Circulation. 2020;141:1080–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy AJ, Sarrazy V, Wang N, Bijl N, Abramowicz S, Westerterp M, Welch CB, Schuetz JD, Yvan-Charvet L. Deficiency of atp-binding cassette transporter b6 in megakaryocyte progenitors accelerates atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:751–758 [DOI] [PubMed] [Google Scholar]
- 29.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–1186 [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–951 [DOI] [PubMed] [Google Scholar]
- 31.Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, Josefsson EC, Alwis I, Ono A, Willcox A, et al. Bcl-xl-inhibitory bh3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118:1663–1674 [DOI] [PubMed] [Google Scholar]
- 32.Vogler M, Hamali HA, Sun XM, Bampton ET, Dinsdale D, Snowden RT, Dyer MJ, Goodall AH, Cohen GM. Bcl2/bcl-x(l) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood. 2011;117:7145–7154 [DOI] [PubMed] [Google Scholar]
- 33.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, et al. Nadph oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127:1888–1902 [DOI] [PubMed] [Google Scholar]
- 34.Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, et al. Apoe regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebois M, Dowling MR, Gangatirkar P, Hodgkin PD, Kile BT, Alexander WS, Josefsson EC. Regulation of platelet lifespan in the presence and absence of thrombopoietin signaling. J Thromb Haemost. 2016;14:1882–1887 [DOI] [PubMed] [Google Scholar]
- 36.Suzuki LA, Poot M, Gerrity RG, Bornfeldt KE. Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: Lack of direct growth-promoting effects of high glucose levels. Diabetes. 2001;50:851–860 [DOI] [PubMed] [Google Scholar]
- 37.Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J. 2016;37:1113–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, van Rooijen N, et al. Extramedullary hematopoiesis generates ly-6c(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, et al. Monocyte subsets differentially employ ccr2, ccr5, and cx3cr1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angiolillo DJ, Goodman SG, Bhatt DL, Eikelboom JW, Price MJ, Moliterno DJ, Cannon CP, Tanguay JF, Granger CB, Mauri L, et al. Antithrombotic therapy in patients with atrial fibrillation undergoing percutaneous coronary intervention: A north american perspective-2016 update. Circ Cardiovasc Interv. 2016;9 [DOI] [PubMed] [Google Scholar]
- 42.Zeng M, Luo Y, Xu C, Li R, Chen N, Deng X, Fang D, Wang L, Wu J, Luo M. Platelet-endothelial cell interactions modulate smooth muscle cell phenotype in an in vitro model of type 2 diabetes mellitus. Am J Physiol Cell Physiol. 2019;316:C186–C197 [DOI] [PubMed] [Google Scholar]
- 43.Allen N, Barrett TJ, Guo Y, Nardi M, Ramkhelawon B, Rockman CB, Hochman JS, Berger JS. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis. 2019;282:11–18 [DOI] [PubMed] [Google Scholar]
- 44.Davison GM, Nkambule BB, Mkandla Z, Hon GM, Kengne AP, Erasmus RT, Matsha TE. Platelet, monocyte and neutrophil activation and glucose tolerance in south african mixed ancestry individuals. Sci Rep. 2017;7:40329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elalamy I, Chakroun T, Gerotziafas GT, Petropoulou A, Robert F, Karroum A, Elgrably F, Samama MM, Hatmi M. Circulating platelet-leukocyte aggregates: A marker of microvascular injury in diabetic patients. Thromb Res. 2008;121:843–848 [DOI] [PubMed] [Google Scholar]
- 46.Zahran AM, El-Badawy O, Mohamad IL, Tamer DM, Abdel-Aziz SM, Elsayh KI. Platelet activation and platelet-leukocyte aggregates in type i diabetes mellitus. Clin Appl Thromb Hemost. 2018;24:230S–239S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu H, Li N, Yngen M, Ostenson CG, Wallen NH, Hjemdahl P. Enhanced leukocyte-platelet cross-talk in type 1 diabetes mellitus: Relationship to microangiopathy. J Thromb Haemost. 2004;2:58–64 [DOI] [PubMed] [Google Scholar]
- 48.Tatarunas V, Kupstyte-Kristapone N, Zvikas V, Jakstas V, Zaliunas R, Lesauskaite V. Factors associated with platelet reactivity during dual antiplatelet therapy in patients with diabetes after acute coronary syndrome. Sci Rep. 2020;10:3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.French SL, Butov KR, Allaeys I, Canas J, Morad G, Davenport P, Laroche A, Trubina NM, Italiano JE, Moses MA, et al. Platelet-derived extracellular vesicles infiltrate and modify the bone marrow during inflammation. Blood Adv. 2020;4:3011–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: A transcellular delivery system for rantes promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. 2005;25:1512–1518 [DOI] [PubMed] [Google Scholar]
- 51.Velez P, Izquierdo I, Rosa I, Garcia A. A 2d-dige-based proteomic analysis reveals differences in the platelet releasate composition when comparing thrombin and collagen stimulations. Sci Rep. 2015;5:8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freedman JE, Larson MG, Tanriverdi K, O’Donnell CJ, Morin K, Hakanson AS, Vasan RS, Johnson AD, Iafrati MD, Benjamin EJ. Relation of platelet and leukocyte inflammatory transcripts to body mass index in the framingham heart study. Circulation. 2010;122:119–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koehler MF, Bergeron P, Choo EF, Lau K, Ndubaku C, Dudley D, Gibbons P, Sleebs BE, Rye CS, Nikolakopoulos G, et al. Structure-guided rescaffolding of selective antagonists of bcl-xl. ACS Med Chem Lett. 2014;5:662–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM, Deshayes K, Fairbrother WJ, et al. Structure-guided design of a selective bcl-x(l) inhibitor. Nat Chem Biol. 2013;9:390–397 [DOI] [PubMed] [Google Scholar]
- 55.Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, Boghaert ER, Catron ND, Chen J, Colman PM, et al. Discovery of a potent and selective bcl-xl inhibitor with in vivo activity. ACS Med Chem Lett. 2014;5:1088–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee MKS, Dragoljevic D, Bertuzzo Veiga C, Wang N, Yvan-Charvet L, Murphy AJ. Interplay between clonal hematopoiesis of indeterminate potential and metabolism. Trends Endocrinol Metab. 2020;31:525–535 [DOI] [PubMed] [Google Scholar]
- 57.Tang Y, Liu W, Wang W, Fidler T, Woods B, Levine RL, Tall AR, Wang N. Inhibition of jak2 suppresses myelopoiesis and atherosclerosis in apoe(−/−) mice. Cardiovasc Drugs Ther. 2020;34:145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.