

Abstract
There is a strong correlation between some pathogens and certain cancer types. One example is Helicobacter pylori and gastric cancer. Exactly how they contribute to host tumorigenesis is, however, a mystery. Pathogens often interact with the host through proteins. To subvert defense, they may mimic host proteins at the sequence, structure, motif, or interface levels. Interface similarity permits pathogen proteins to compete with those of the host for a target protein and thereby alter the host signaling. Detection of host–pathogen interactions (HPIs) and mapping the re-wired superorganism HPI network—with structural details—can provide unprecedented clues to the underlying mechanisms and help therapeutics. Here, we describe the first computational approach exploiting solely interface mimicry to model potential HPIs. Interface mimicry can identify more HPIs than sequence or complete structural similarity since it appears more common than the other mimicry types. We illustrate the usefulness of this concept by modeling HPIs of H. pylori to understand how they modulate host immunity, persist lifelong, and contribute to tumorigenesis. H. pylori proteins interfere with multiple host pathways as they target several host hub proteins. Our results help illuminate the structural basis of resistance to apoptosis, immune evasion, and loss of cell junctions seen in H. pylori-infected host cells.
Keywords: computational prediction of host–pathogen interactions, interface mimicry, protein–protein interaction, structural network, superorganism network
Introduction
Microbiota—commensals and pathobionts—are an essential part of the host, having important roles in physiological processes, including immunity and metabolism [1,2]. They interact with the host through proteins, metabolites, small molecules, and nucleic acids [3]. They employ effector proteins to modulate—inhibit or activate—host signaling pathways. In susceptible hosts, they may convert to pathogens (commensal-to-pathogen transition). Helicobacter pylori is the prevalent species in human gastric microbiota, which colonizes the gastric epithelium. It is the most widespread bacterial infection worldwide and can persist lifelong [4]. More than half of the human population is infected with H. pylori [5]. It causes persistent chronic inflammation and increases gastritis and gastric cancer risk [6]. Every infected individual develops gastritis which lasts decades, but only a small population develops cancer [6]. The risk of gastric cancer depends on strain-specific bacterial virulence factors and their host–pathogen interactions (HPIs) [7]. There are various H. pylori strains due to intra-genomic diversification (e.g., point mutations and recombination) and inter-genomic recombination [7]. This genetic variability leads to different sets of virulence factors and H. pylori strains with varying degrees of pathogenicity [8]. Elucidation of the pathogenic players and their HPIs that increase gastric cancer risk is crucial to provide molecular insights into the pathogenesis mechanisms and effective therapeutics.
To subvert host pathways and evade immune surveillance, pathogens mimic host proteins at different levels: sequence, structure, motif, and interface [3]. Interface mimicry seems the most common type and is frequent within intra- [9–11] and inter-species [12,13], indicating that pathogens target more host proteins by hijacking host interfaces. Interface mimicry, where proteins with different global structures can interact in similar ways, was suggested already over two decades ago [14–16]. This similarity in endogenous (within-host/intra-species) and exogenous (host–pathogen/inter-species) protein–protein interfaces permits pathogen proteins to compete with the host’s [13], alter physiological signaling, and cause persistent infections as well as cancer. Identification of the crosstalk between host and pathogens and of the targeted host pathways will delineate virulence strategies and insight into some of the underlying principles of infections. Horizontal gene transfer and convergent evolution allowed pathogens to adopt interface mimicking strategy [3]. In horizontal gene transfer, pathogens attain host genes and shape them over time to acquire new functionality. However, in convergent evolution, pathogens, and their hosts evolved independently toward similar interface architectures [17,18].
HPI data including structural details are far from complete, and experimental characterization of the large-scale inter-species interactions is challenging [19]. Computational tools are increasingly exploited in enriching the HPI data, uncovering their complexed (bound) structures, and complementing experiments. Although modeling of intra-species protein–protein interactions (PPIs) is a well-established field, prediction of inter-species interactions is relatively new. Still, numerous efforts aim to develop computational methods to detect HPIs [19], most of which rely on sequence homology [20–28]. Homologybased approaches give accurate results only if the sequence similarity is high. However, not all pathogenic proteins have homologs in human. For instance, VacA, a secreted effector from H. pylori, does not possess sequence similarity to any other known bacterial or eukaryotic proteins [5], but it modulates signaling through several host pathways [29]. Therefore, it is impossible to identify its HPIs with sequence-based methods, emphasizing the importance of involvement of protein 3D structures in predicting HPIs. Sequence homology alone is not always sufficient to detect targets of pathogenic proteins. There are also comparative homology-based approaches that consider structure [27,28,30–36]; conserved interactions—so called “interologs” (interacting homologs) [37,38]—of the pathogenic and host proteins; and transcriptome data [39]. Structure-based approaches often exploit global structural similarity rather than interface mimicry [31,36]. Several studies computationally identified large-scale HPIs and built interspecies protein interaction networks for viruses and bacteria [21,24,26,31–33,40–44].
One method uses interface information together with sequence homology and gene expression, but in this method, host and pathogenic proteins are predicted to interact only if they satisfy a minimum of 80% sequence identity over at least 50% of template host protein complexes [30]. To the best of our knowledge, none of the approaches utilize solely interface structures to predict HPIs. Here, we report a novel computational approach that identifies putative HPIs and their 3D structures as complexes based only on interface similarity (only local structural similarity is required, no need for sequence similarity) (Fig. 1). This approach reveals not only targets of pathogenic proteins but also the host endogenous PPIs, which may be disrupted by these potential HPIs. It has been suggested that the available interface structures are already diverse enough to represent most endogenous interactions [52–55], and hence, success of template-based methods for prediction of intra-species PPIs is very high [10]. Since exogenous interfaces mimic endogenous ones, available endogenous and exogenous interface structures may also cover majority of the HPI space.
Fig. 1.
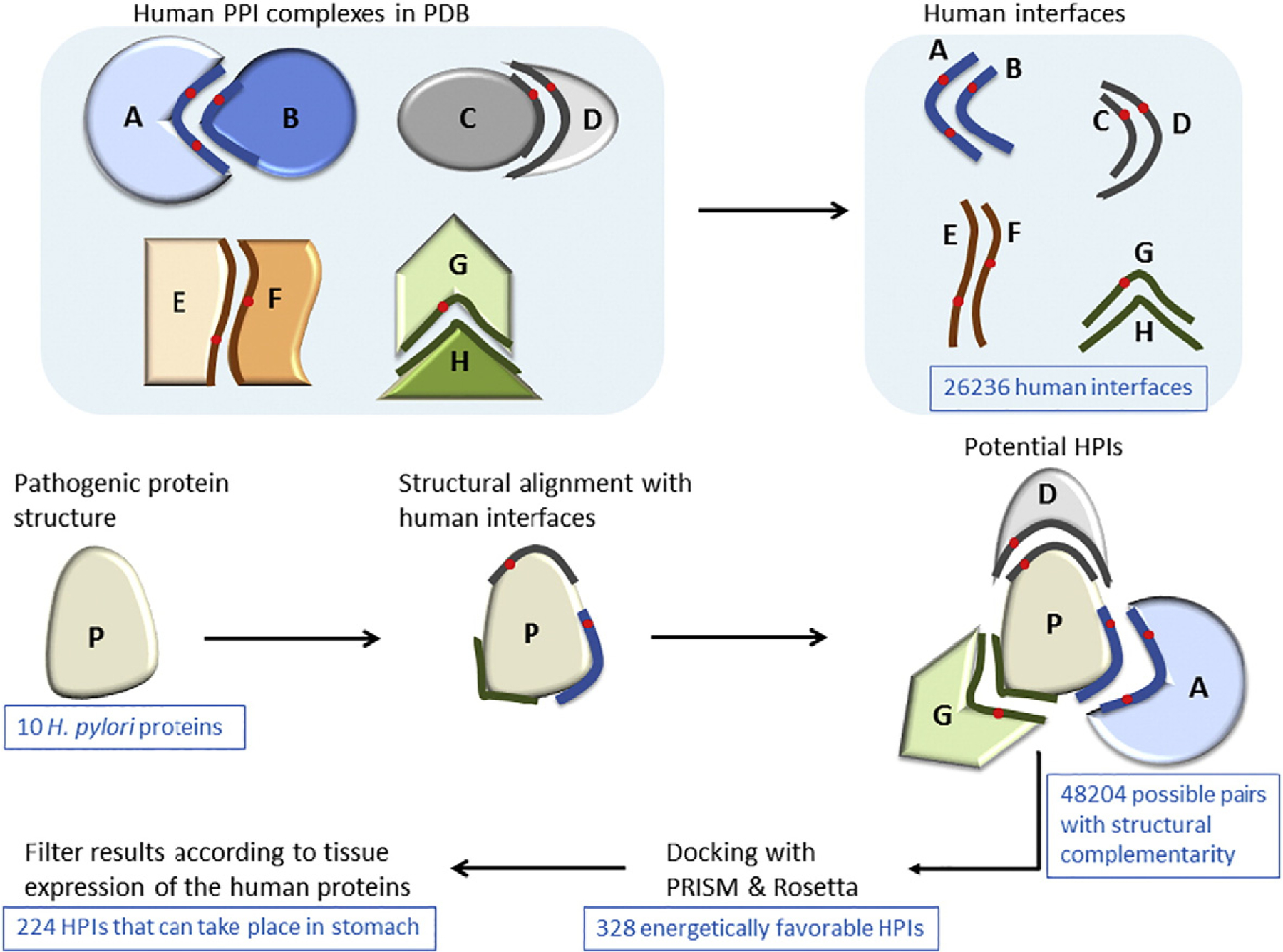
Workflow of our interface-based HPI modeling/prediction approach. We first extract human interfaces from the PDB. Then, we obtain the structures of the H. pylori proteins from the PDB. For docking, we need two target proteins. However, for a given pathogenic protein, we do not know the potential partner proteins in the human. Before docking, we need to identify the potential partners. To do so, we structurally align the pathogenic proteins with all human interfaces. If the pathogenic protein is aligned with the B-face of the interface, it can interact with the complementary A-face. Now, that we have potential partners of the pathogenic protein, we can perform docking for these pairs with PRISM [45–48] and Rosetta (local refinement) [49–51]. We employ these docking methods to select the energetically favorable HPIs, since structural complementarity may not necessarily confer electrochemical complementarity. Finally, we filter our energetically favorable HPI results according to tissue expression of the human proteins, checking whether the human partners of these pathogenic proteins are expressed in the same tissue where the pathogen is found. We further evaluate the HPI models based on the percent match of the interface residues with the template interface and probability of the template interface being a real biological interface.
To illustrate the utility of this concept, we applied our interface-based approach to H. pylori. We modeled its HPIs and built its structural superorganism network, where every pairwise interaction has a 3D structure as a complex. Integrated metaorganism networks that consider host and microbiota/pathogens—as a whole—are vital for an in-depth understanding the virulence strategies of pathogens and how they evade immune surveillance [1]. Our results indicate that H. pylori proteins potentially target hub proteins in the human PPI network and show that HPI models can unravel human pathways targeted by pathogenic proteins. Potential targets of H. pylori proteins are enriched in several cancer pathways, such as viral carcinogenesis, chemical carcinogenesis, and pancreatic cancer.
Results
H. pylori can live and replicate both outside and inside host cells, suggesting that it may be a facultative intracellular bacteria [56]. It has a type-IV secretion system through which it injects its virulence factors—secreted effectors—directly into host cytoplasm. Although H. pylori and host cells physically encounter one another, it does not guarantee that all their proteins are accessible to interact with each other. Only outer-membrane proteins (OMPs) and secreted effectors of H. pylori can interact with the host proteins. About 1% of the H. pylori genome encodes for OMPs, also known as adhesins, which facilitate the adherence of bacteria to epithelial cells [24] to establish persistent colonization [4]. The presence of adhesins increases the risk of gastric cancer, thus serving as biomarkers for H. pylori-associated gastric cancer [57]. Using our interface-based approach, we analyzed 10 H. pylori OMPs and secreted effectors (Table 1) including SabA, BabA, CagA, VacA, and gGT, respectively.
Table 1.
Secreted effectors and OMPs of H. pylori that we analyzed with our interface-based computational approach
| H. pylori protein | PDB | OMP or secreted effector | Known targets in human | Complex structures for the known HPIs in PDB | Number of HPIs modeled |
|---|---|---|---|---|---|
| CagA (HP0547) | 4dvyP | Secreted | TP53B2 [58] MARK2 (PAR1B) [59] CRK [60] E-Cadherin (CADH1) [61,62] MET (HGFR) [61] ZO1 [63] JAM1 [63] PLCγ [64] GRB2 [65] CSK [66] SHP-2 (PTN11) [5,67] SHP-1 (PTN6) [68] PI3K (P85A) [68] GRB7 [68] RASA1 (Ras-GAP1) [68] TAK1 [69] |
4irv:AE 3iec:AE |
69 |
| VacA | 2qv3A | Secreted | LFA1 (ITGAL) [70] CD18 (ITGB2) [71] RPTP-α [72] RPTP-β [72] LRP1 [73] Fibronectin [74] |
15 | |
| BabA (HP1243) | 4zh0A | OMP | Lewis(b) antigen [75] MUC5B [76] |
16 | |
| SabA (HP0725) | 4o5jA | OMP | Lewis(x) [77] MUC5B [76] |
18 | |
| gGT (HP1118) | 2nqoA 2nqoB |
Secreted | No known HPIs | 25 | |
| Cagα (HP0525) | 2pt7A | OMP | No known HPIs | 26 | |
| HP1286 | 3hpeA | Secreted | No known HPIs | 12 | |
| HP0231 | 3tdgA | Secreted | No known HPIs | 28 | |
| HP0721 | 2xrhA | Secreted | No known HPIs | 9 | |
| HP0827 | 2ki2A | Secreted | No known HPIs | 6 |
Our results revealed several potential HPIs for the 10 H. pylori OMPs and secreted effectors. All our HPI models have 3D structures as complexes. As can be seen in Fig. 1, the 328 HPI complexes have structural and electrochemical complementarity. Of the 328 targets (host proteins), 224 are expressed in the stomach according to Human Protein Atlas, where H. pylori resides [78,79]. Their details and the human PPIs that they may disrupt are given in Tables S1 and S2, respectively. Forty-nine of the targets are known not to be expressed in stomach and there are no expression data for the remaining 55 targets. We considered the 49 targets as false positives. Although theoretically they can interact with H. pylori proteins, these interactions are not possible since they cannot encounter H. pylori proteins. So, rough calculation of our false positive rate is about 18% (49/(49 + 224)). It is important to note that the 224 HPIs which can take place in the stomach may also have false positives, but due to limited experimental HPI data, we cannot calculate the exact false-positive rate.
Below, we provide examples of how novel HPI candidates may elucidate the roles of H. pylori in modulation of host signaling and contribution to malignant transformation, and how they can explain some phenotypes, such as hummingbird, occurring in infected individuals. Then, we present our structural superorganism network between H. pylori and human, with some hub proteins targeted by H. pylori proteins. Finally, to validate our predictions, we calculate the stability and dynamics of some of the HPI models by molecular dynamics (MD) simulation.
Examples of potential HPIs
CagA and VacA are widely studied H. pylori secreted toxins. They have pleiotropic effects, targeting multiple host pathways [5]. Our results also confirm this showing that they have various targets in the host. Other H. pylori proteins also have several putative HPIs that can potentially abolish several endogenous host PPIs due to competition. Below, we present some of the intriguing targets of H. pylori proteins.
We found that OMPs and secreted effectors mimic the interfaces between cytokines and their receptors, such as interferon alpha/beta receptor 2—interferon alpha 2 (INAR1–IFNA2) and INAR2–IFNA2 (Fig. 2). Cytokine and chemokine signaling plays important roles in T-cell recruitment to the infected host tissue to clear the pathogens and in regulation of their activation and differentiation [80]. Blockage of these pathways by bacterial proteins may underlie the lifelong persistence of H. pylori infection since it may prevent the cytokine signaling. This could also allow malignant precursor cells in the stomach to evade the host immune system, which is one of the hallmarks of cancer and may explain how this pathogen contributes to initiation of gastric tumor. As opposed to antagonistic effects, these bacterial proteins could also act as ligands, activate receptors, and induce inflammatory responses. Excess inflammation predisposes individuals to cancer [81].
Fig. 2.
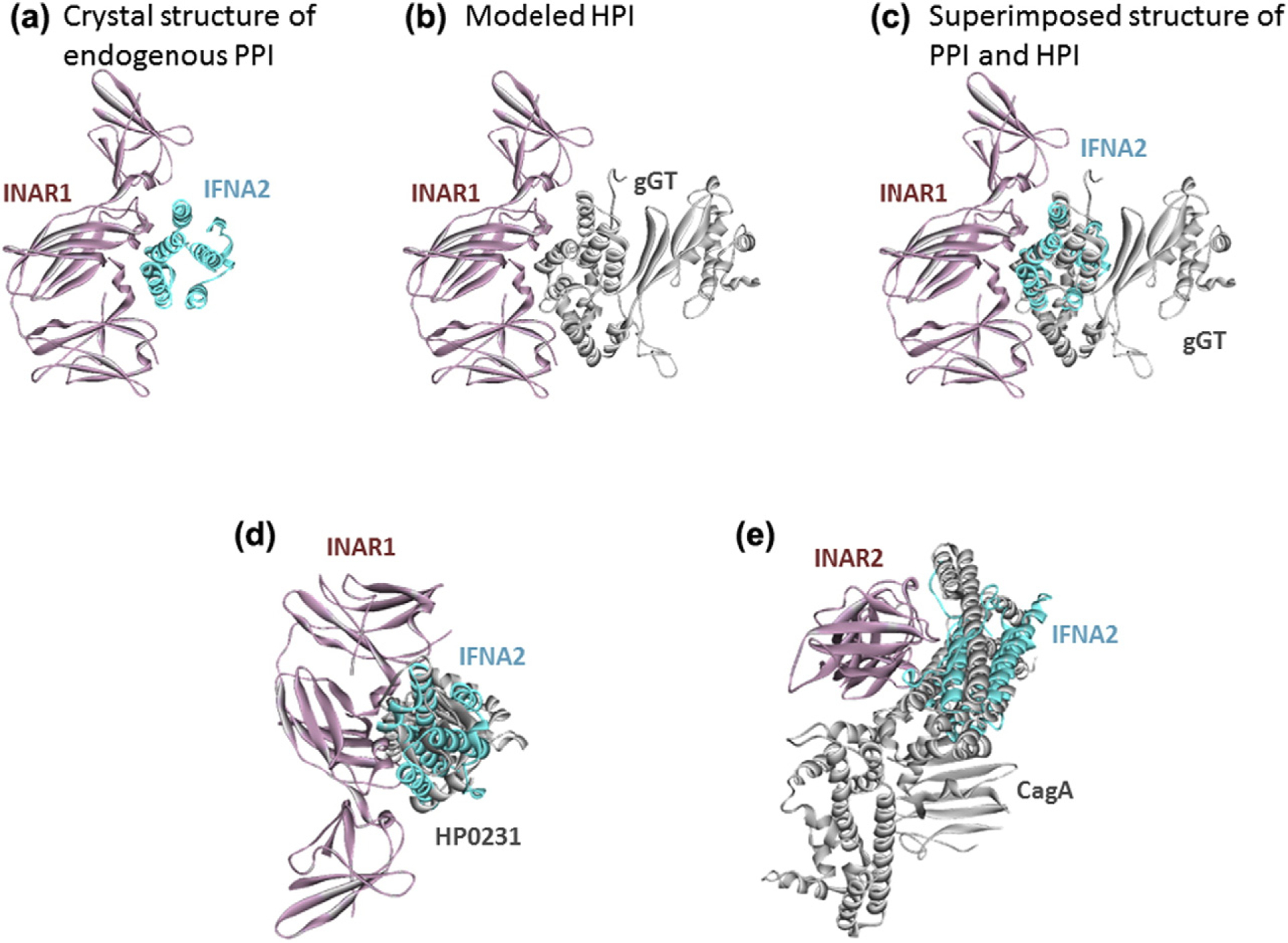
H. pylori proteins mimic the interfaces between cytokines and their receptors. Thus, they may block the cytokine signaling, which may prevent T-cell recruitment to infected tissue and lifelong persistent infection. (a) Endogenous human PPI between INAR1 and IFNA2. (b) Our HPI model between INAR1 and gGT. (c) Superimposed view of PPI and HPI shows that gGT almost perfectly mimics the interface on IFNA2 to bind to INAR1. Panels d and e also show the superimposed structures of endogenous human PPIs and modeled HPIs. Cyan and pink proteins are human cytokines and cytokine receptors, respectively. Gray proteins are H. pylori proteins. Gray proteins bind to pink proteins by hijacking the interface on cyan proteins (only the interface is similar, not the global structure). Thus, they may block the pink–cyan protein interactions.
Our HPI models also include interactions with cell cycle regulators, such as Cyclin-A2 and cyclindependent kinase-5 (Fig. 3a, b). It is known that H. pylori induces cell cycle arrest at the G1 phase of the cell cycle in T cells [82]. These HPIs may provide mechanistic insight into inhibition of T-cell proliferation and immune evasion.
Fig. 3.
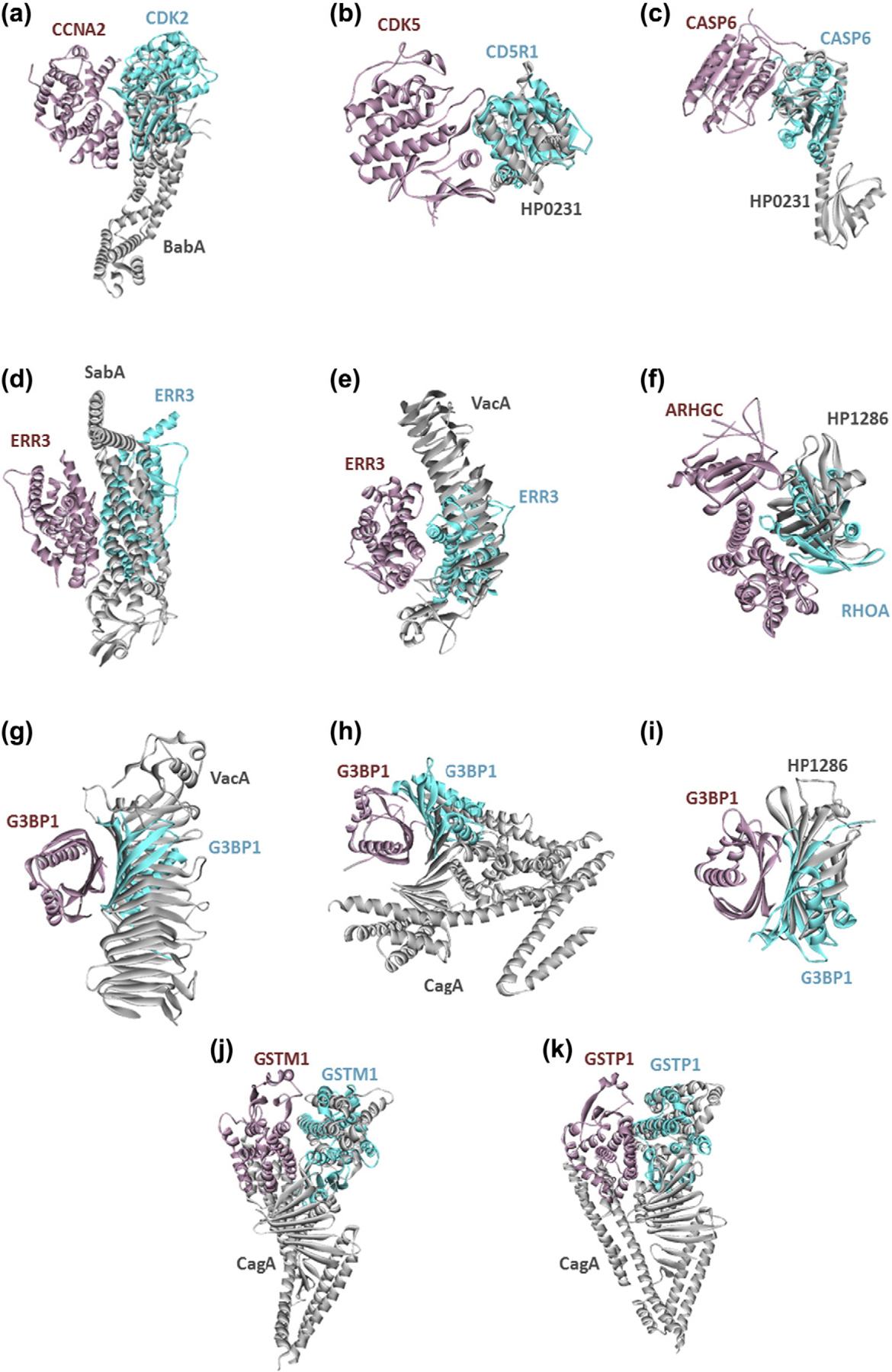
H. pylori proteins targeting the cell-cycle regulators (a and b), apoptosis regulators (c), estrogen receptors (d and e), Ras and Rho GTPase family members (f, g, h, and i), and glutathione metabolism (j and k). Figures show the superimposed structures of endogenous human PPIs and modeled HPIs. Pink and cyan proteins are human and gray ones are H. pylori proteins. Gray proteins bind to pink proteins by hijacking the interface on cyan proteins (only the interface is similar, not the global structure). Thus, they may block the pink–cyan protein interactions.
Cell death pathways are also targeted by H. pylori proteins. For instance, HP0231 of H. pylori may abolish the homodimerization of CASP6, which is crucial for propagation to apoptosis (Fig. 3c). Short-term exposure to H. pylori induces apoptosis in host cells. However, chronic exposure makes host cells resistant to apoptosis (not only to H. pylori-induced apoptosis but also to radiation- and chemotherapy-induced apoptosis) [58]. These novel HPIs may help us to understand the molecular details of resistance to apoptosis seen in H. pylori-infected cells, which is another hallmark of cancer.
Interestingly, we found that estrogen receptors, such as ERR3, are putative targets of H. pylori, where the pathogenic proteins interfere with the dimerization of these receptors (Fig. 3d, e). Gastric cancer is more frequent in men than in women. Female hormones reduce gastric cancer risk. Estrogen protects against H. pylori-induced gastric cancer in mice [83,84], but how is not known. If estrogen binding to dimeric receptors hinders HPIs with the receptors, this may explain why estrogen protects against gastric cancer.
Other intriguing interactions among our HPI models include Ras and Rho GTPase family members, such as ARHGC (Rho guanine nucleotide exchange factor C) and G3BP1 (Ras GTPase activating protein-binding protein 1) (Fig. 3f, g, h, i). These proteins mediate several signaling pathways, including cytoskeletal reorganization in the host cell. Our HPI candidates may contribute to the morphological changes—hummingbird phenotype [85]—that is observed in H. pylori-infected host epithelial cells and linked with elevated cell motility and metastasis [86].
Remarkably, H. pylori proteins target glutathione metabolism, including GSTM1 and GSTP1 proteins (Fig. 3j, k). It was reported that H. pylori infection lowers the glutathione levels in gastric mucosa [87] and oral glutathione supplement administration reduces the gastric pathologies [88]. These HPIs may provide clues for why glutathione levels are low in infected individuals.
In addition to mimicking endogenous interfaces, H. pylori proteins mimic exogenous interfaces (Table S3). For example, CagA–TP53BP2 complex already has a resolved crystal structure and we can recover this complex through our approach (Fig. 4a). We found that CagA can also mimic outer capsid protein sigma-1 of mammalian orthoreovirus and bind to human JAM1 (Fig. 4b). CagA may also bind to human Beclin-1, by mimicking the anti-apoptotic v-Bcl2 of Murid herpesvirus (Fig. 4c). So, although CagA has no sequence or global structural similarity to these pathogenic proteins, it may still mimic their interfaces and bind to the same host proteins in a similar fashion that other pathogens target them.
Fig. 4.
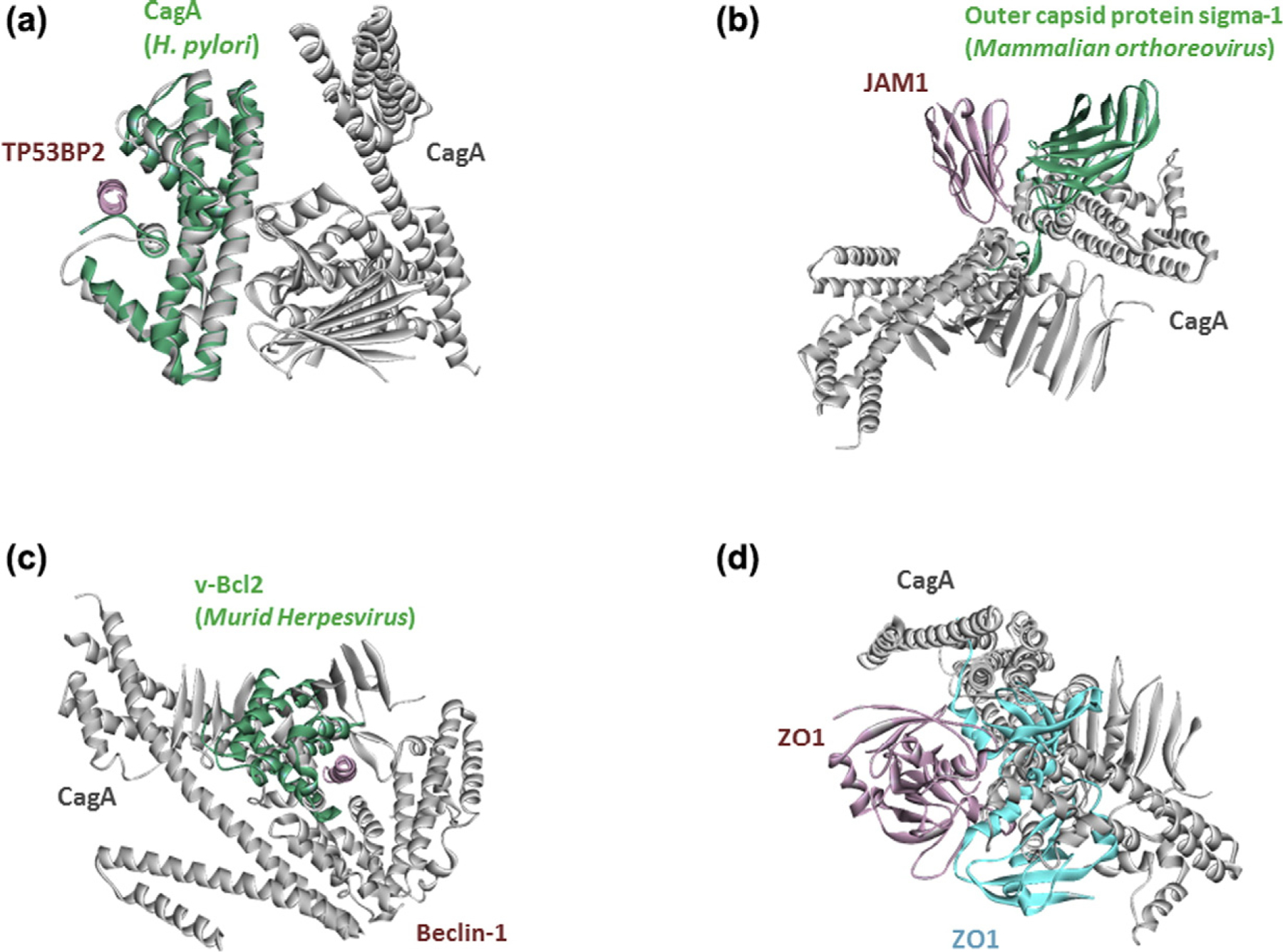
H. pylori proteins mimic not only host interactions, but also HPIs (a, b, and c). Our interface-based approach uncovered a known HPI for CagA (d). It is known that CagA interacts with ZO1, but their complex structure is not available. Figures show the superimposed structures of our HPI models for H. pylori with the known exogenous or endogenous human PPIs. Pink and cyan proteins are from human, greens are proteins from bacteria or virus, and gray proteins are H. pylori proteins. Gray proteins bind to pink proteins by hijacking the interfaces on green and cyan proteins (only the interface is similar, not the global structure).
Recovery of known HPIs
Among the known HPIs of H. pylori, only two structures with known targets are available: TP53BP2 (4irvAE.pdb) [58] and MARK2 (3iecAE.pdb) [59]. The availability of the structures of complexes is important because these show how the proteins interact, whether they overlap with other protein interactions, and how mutations can affect these interactions. They are also useful in drug design [89].
In addition to novel HPIs, we also recovered some of the known HPIs of H. pylori, such as CagA interactions with TP53BP2 (p53 binding protein 2) (Fig. 4a), JAM1 (junctional adhesion molecule A) (Fig. 4b), and ZO1 (zonula occluden 1) (Fig. 4d). It is important to note that our models for these known HPIs have Rosetta interface score very close to −5 but not lower than −5 (this situation changes depending on whether you ignore or consider disulfide bonds). Thus, these models are not as favorable as the examples above. To test their stability, we also performed MD simulations.
Although the CagA–ZO1 and CagA–JAM1 interactions were known to take place in the host cell, it was not known how they interact or whether they abolish any endogenous human PPIs. JAM1 and ZO1 are important in tight junctions [63], and H. pylori is known to disrupt the epithelial integrity by dysregulating them [90]. Loss of cell adherence may lead to epithelial-to-mesenchymal transition, facilitating metastasis [91]. We found that both proteins bind to the same site on CagA. Our model shows that CagA binding can interfere with ZO1 homodimerization which is necessary for tight junctions (Fig. 4d) [92]. The template interface for CagA– JAM1 interaction is another exogenous interaction between a capsid protein from mammalian orthoreovirus and JAM1. The viral capsid protein is known to disrupt the JAM1-homodimer structure [93], whose structure is not available currently. If our CagA–JAM1 model is correct, CagA binding to JAM1 can also abolish its dimerization. Since the JAM1 homodimer structure is unavailable, we are unable to confirm that our HPI model interferes with the JAM1 homodimer. Together, these results may illustrate the molecular mechanisms of how H. pylori infection leads to disruption of tight junctions and epithelial integrity that is seen in H. pylori infected-tissues.
Still, our results did not reveal all known HPIs of CagA. The reason for this may be that these known human targets do not have structures in PDB, or they do not have interfaces in their PDB structures. Also, the available structures may not cover the full-length proteins: structures of the interacting domains may be missing.
MD simulations of some HPI models
To assess the stability and dynamics of four HPI models (three recovered known HPIs with TP53BP2, ZO1, and JAM1, and one novel HPI model—gGT–INAR1) and compare them with those of their corresponding template PPIs, we performed explicitsolvent MD simulations for 100 ns. In all simulation systems, no immediate dissociation of the proteins was observed.
For CagA–TP53BP2, we simulated only the template PPI (4irv:AE.pdb), since our HPI model is almost identical to the template. This HPI complex was very stable, with a maximum RMSD of 2.5 Å (with respect to the initial conformation) throughout the trajectory. Structures of the initial and the final conformations and plots of RMSD values are given in Fig. 5.
Fig. 5.
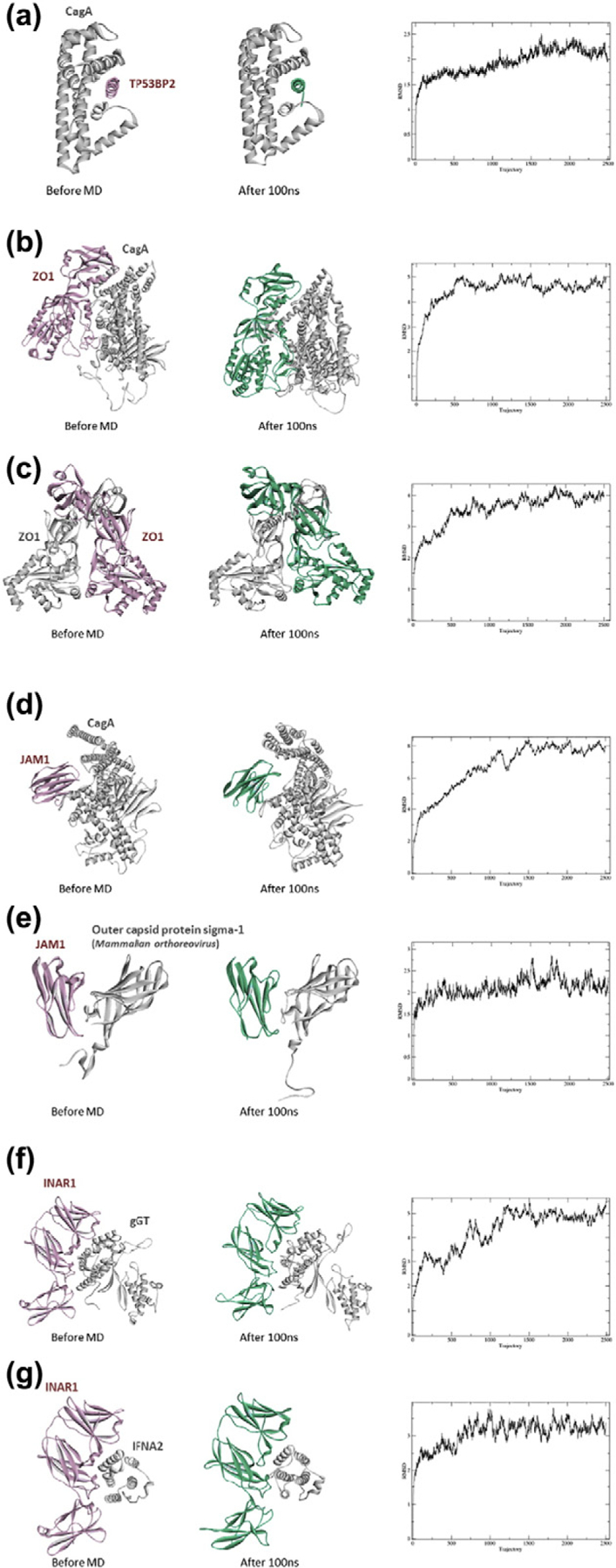
MD results for four HPI models and their corresponding template PPIs. The first figure in each panel shows the initial conformation of the HPI/PPI, and the second figure shows the conformation after 100-ns simulation. The last figure shows the RMSD values with respect to the initial structures.
CagA–ZO1 complex and the template endogenous PPI (ZO1-homodimer) are stable throughout the simulations. Our CagA–ZO1 model has about 4.5 Å and template ZO1-homodier 4.5-Å maximum RMSD (Fig. 5). Although CagA–ZO1 complex stays intact, we lost the original interface on CagA that we predicted in the first place. ZO1 slides from the edge toward the middle region of TP53BP2-binding region of CagA.
CagA–JAM1 HPI model, as well as the template exogenous PPI (JAM1–outer capsid protein sigma-1 of mammalian orthoreovirus) are stable, with maximum RMSDs of 8 and 3 Å, respectively (Fig. 5). Here, the initial interface between CagA and JAM1 is still preserved at the end of the simulation.
Lastly, the gGT–INAR1 model HPI and the template endogenous PPI (INAR1–IFNA2) also do not show any dissociation and their maximum RMSDs are about 5 and 4 Å, respectively (Fig. 5). Again, the initial interface of the HPI is preserved.
Overall, these MD results indicate that our HPI candidates are feasible models. Three out of four simulated HPI models (CagA–TP53BP2, CagA– JAM1, and gGT–INAR1) show that they are stable complexes and preserved the initial interface. Only one (CagA–ZO1) lost the initial interface, although the complex stayed intact.
We also computed the binding free energies (ΔGb), of all HPIs and PPIs by generalized Born with a simple switching (GBSW) and entropy calculations. Table 2 shows that all four HPIs are favorable, with negative ΔGb, and the template PPIs have higher affinity (lower ΔGb) than the HPI models, which suggests that the pathogenic proteins cannot out-compete their endogenous competitors to bind to their targets. However, if pathogenic proteins are more abundant, then they may out-compete their counterparts.
Table 2.
Binding free energies of the simulated HPI models and template PPIs
| Systems | 〈ΔGgas〉 (kcal/mol) |
〈ΔGsol〉 (kcal/mol) |
−TΔS (kcal/mol) |
〈ΔGb〉 (kcal/mol) |
|---|---|---|---|---|
| CagA–TP53BP2 (template) | −462.4 ± 230.3 | 366 ± 182.2 | 28.8 | −67.5 ± 49 |
| CagA–ZO1 (HPI model) | −1731.3 ± 216.4 | 1581 ± 217.8 | 37.1 | −113 ± 13.8 |
| ZO1-ZO1 (template) | −336.4 ± 149.7 | 136 ± 134.4 | 36.1 | −164.1 ± 22.7 |
| CagA–JAM1 (HPI model) | −245.9 ± 90.5 | 198.6 ± 83.5 | 33.8 | −13.4 ± 10.8 |
| JAM1–viral capsid protein (template) | −240.8 ± 169.6 | 177.6 ± 133.5 | 32.4 | −30.7 ± 37.7 |
| gGT–INAR1 (HPI model) | −529.4 ± 78 | 434.3 ± 69.3 | 35.6 | −59.4 ± 15 |
| INAR1–IFNA2 (template) | −505.5 ± 63.3 | 387 ± 59.1 | 33.6 | −84.8 ± 9.4 |
Angle brackets denote an average along the MD trajectory.
Structural superorganism network for H. pylori and human
Genome-wide mapping of HPIs has a potential to reveal systematic network trends, where bacteria and viruses favor targeting host hub proteins [13,94,95]. Structural networks provide higher spatial resolution and allows in-depth analysis compared to binary PPI networks. Our structural inter-species network (Fig. 6) contains 224 HPIs (exogenous interactions) and 3366 host PPIs (endogenous interactions). Here our HPI models serve as the exogenous and the template PPIs serve as the endogenous interactions. In our network, all pairwise interactions, endogenous and exogenous, have structures as complexes. All endogenous interactions are from the template interfaces (crystal structures, experimental data), whereas HPIs are our models. Figure 6a illustrates that H. pylori proteins target the highly connected part of the network. The majority of the targets of individual H. pylori proteins are distinct, but there are human proteins that are common targets to more than one H. pylori protein (Fig. 6b). Thus, multiple H. pylori proteins can target the same pathway.
Fig. 6.
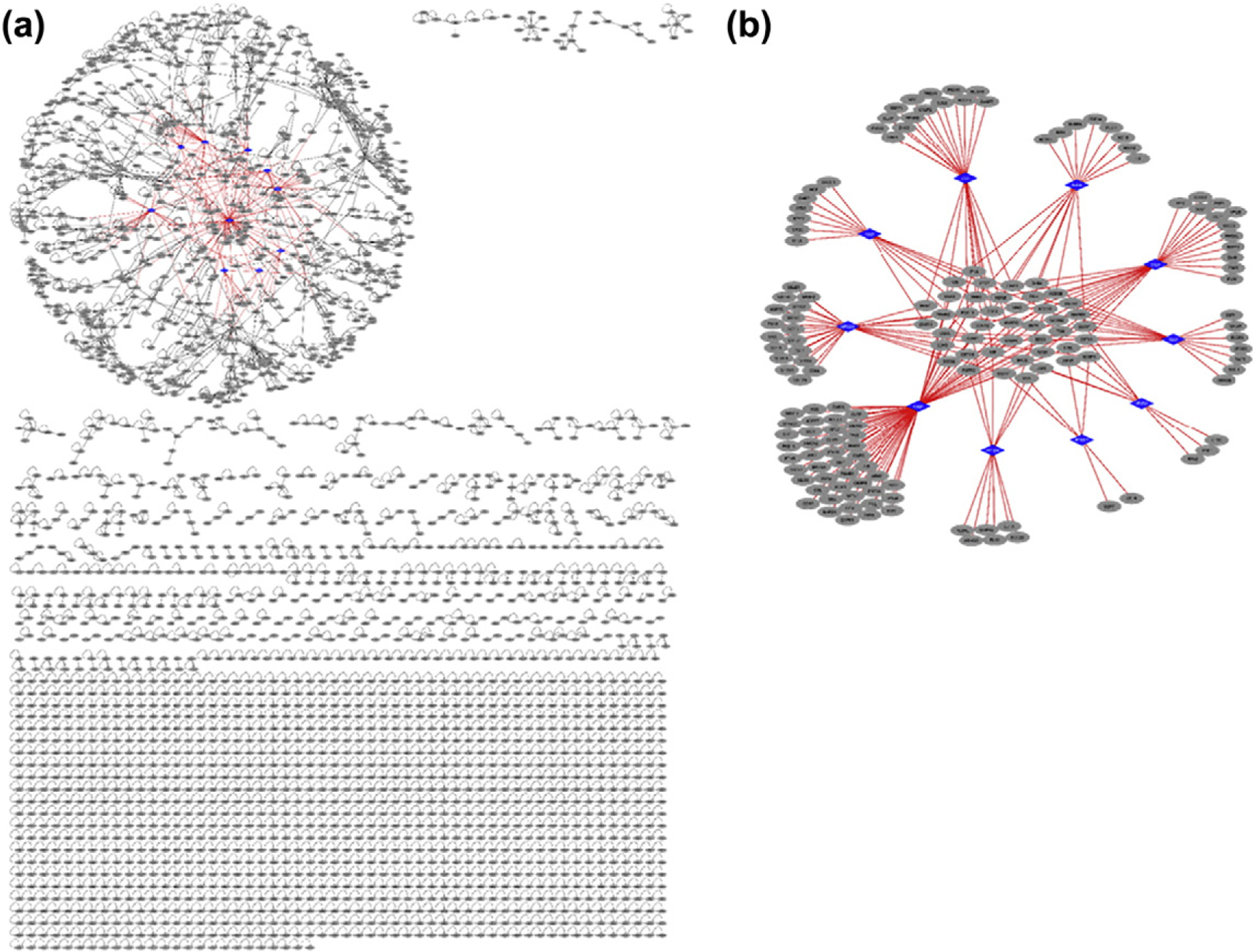
Structural inter-species interaction network for H. pylori. All pairwise interactions have structures as complexes. Endogenous human interactions (black edges) are obtained from crystal structures (our template interface set), where human proteins are shown as gray circular nodes. Exogenous interactions (red edges) are our HPI models for 10 H. pylori proteins and are shown as red edges. (a) H. pylori proteins (blue diamond-shaped nodes) target the highly connected part (hair-ball) of the human PPI. (b) Structural HPI network without the endogenous human interactions. Most of the targets of individual H. pylori proteins are distinct, but some are shared across different H. pylori proteins. Thus, multiple H. pylori proteins target the same pathway.
We analyzed the topological features of our structural superorganism network to better understand the properties of host proteins that interact with H. pylori proteins. We found that H. pylori proteins target hubs (proteins with high degree/connectivity) in the human structural PPI network, such as Cyclin-A2, INAR2, ARGHC, and ERR3. Hub proteins play a central role in many cellular functions, ensuring the cross-talk between pathways. Thus, it is a good strategy for pathogens to attack these proteins. Functional annotation shows that host proteins targeted by H. pylori are enriched in 48 KEGG pathways according to analysis by DAVID [96,97] (Table S4). Among the highly enriched, there are pathways in cancer, chemical carcinogenesis, viral carcinogenesis, renal cell carcinoma, and cytokine signaling.
We also constructed the structural superorganism network for all known HPIs in the PDB for bacterial, viral, and yeast species (Fig. 7). Here, both the endogenous and exogenous interactions are supported by solved 3D structures of complexes in the PDB, not our models. Unlike H. pylori, which targets only the highly connected part of the human PPI network, other species target the less connected parts as well. While some human proteins are targeted by several pathogenic proteins, others are targeted by only one.
Fig. 7.
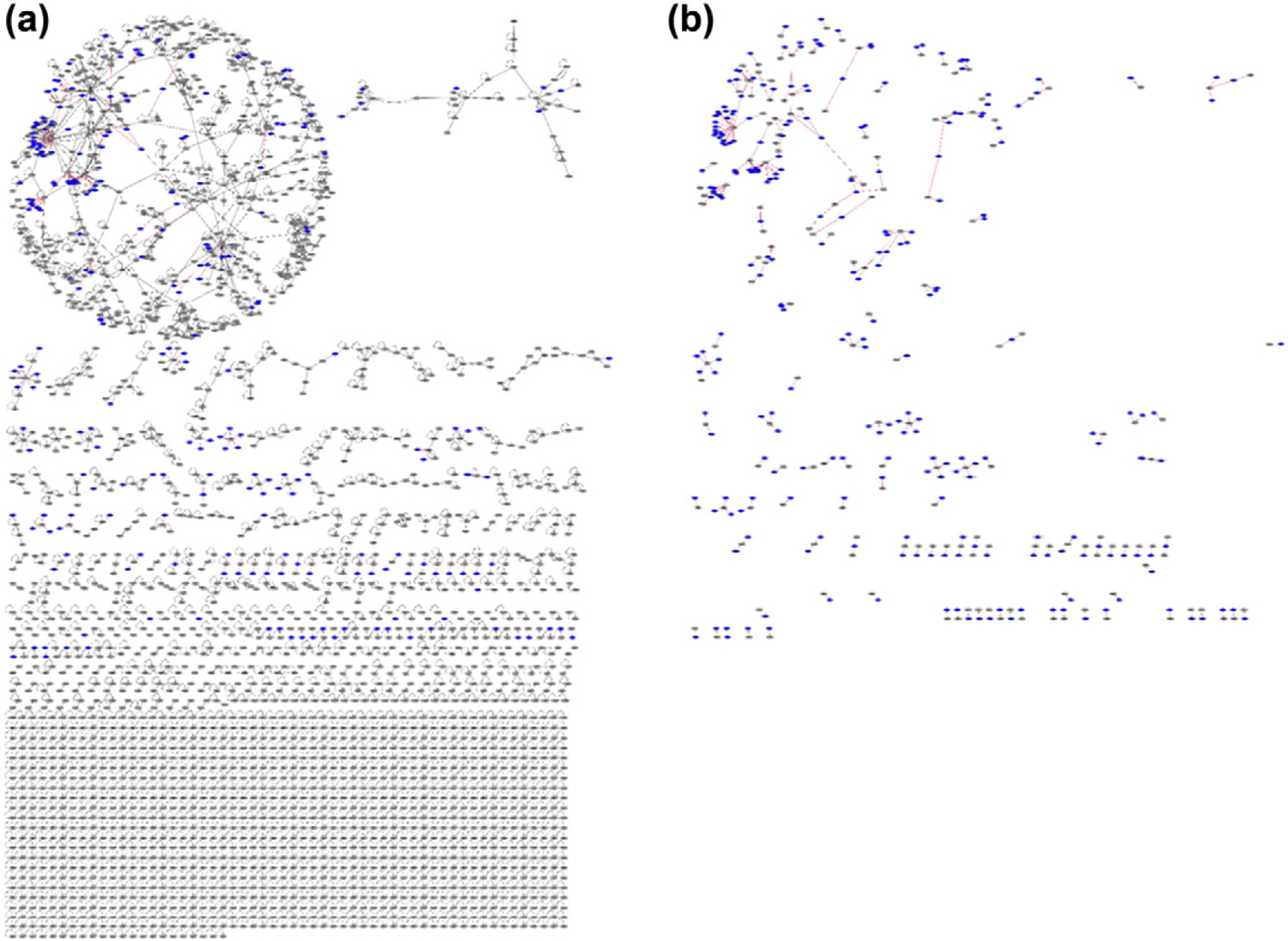
Structural interspecies network with all available HPI data for several bacterial, viral, and yeast species, and PPI data in PDB. (a) Combined HPIs and PPIs that are available in PDB. All endogenous (black edges) and exogenous interactions (red edges) have structures as complexes in PDB. There are 299 HPIs with proteins from bacterial, viral, and yeast species and 3366 endogenous interactions. Unlike our H. pylori HPI models, proteins from other bacterial, viral, and yeast species target both highly connected and less-connected part of the network. (b) All HPIs available in PDB. Some pathogenic proteins target the same host protein, whereas others have distinct targets. Some human proteins, such as 1A02, UBC, 2B11, and DRA, are hubs that are targeted by several non-human proteins.
Discussion
Very little is known about the pathogenic mechanisms of infectious diseases and pathogen-driven cancers on the molecular level. Molecular details of HPIs can help discern the roles of pathogenic virulence factors in the modulation of the host signaling and pathogenesis of pathogen-driven diseases, including cancer. HPI data may allow for development of more potent drugs [98]. Although computational methods to enrich experimental HPI data were developed, most of them rely on sequence homology, which restricts the application of these tools since not all pathogenic proteins have homologs in human. As interface similarity seems more frequent than global structural similarity, interfacebased methods hold promise to enrich HPI data. Here, we present the first interface-based HPI prediction approach which solely depends on the local structural (binding site) similarity of human interfaces with pathogenic proteins. With this approach, we show not only which proteins can interact but also how they interact. The concept behind the approach is that interface architectures are conserved in nature, regardless of the entire sequence. This concept mimics folding of single-chain proteins [99,100]. Proteins with different sequences can still adopt similar folds. Of note, those folds common in single chains resemble the ones at interfaces, with the only difference being the absence of chain connectivity in interfaces. If a pathogenic protein mimics the interface between two host proteins, it may potentially disrupt that endogenous interaction due to competition. To illustrate the usefulness of this approach, we applied our method to the most prevalent microbial species in the stomach, H. pylori, which is associated with gastric cancer. We identified several potential HPIs that could be relevant to disease phenotype. We found that H. pylori proteins may interfere with the functioning of host cell adhesion, cytokine signaling, cell cycle regulation, and apoptosis pathways. Our results can provide molecular insights into resistance to apoptosis, immune evasion, and loss of cell junctions that are seen in H. pylori-infected host cells. Our structural superorganism network further indicates that H. pylori proteins target the highly connected part of the network, indicating the requirement of interference with multiple host pathways.
The endogenous PPI data for H. pylori were constructed before by experimental techniques such as yeast-2-hybrid [101,102] and also by computational methods [41]. Its exogenous HPIs were predicted by sequence-based computational techniques [24]. They identified 833 interactions between 623 H. pylori proteins and 6559 human proteins [24]. Out of these, there are only 2 H. pylori proteins (HP0231 and HP1286), but no predicted HPIs in common with this study. The human proteins that they found to interact with these 2 H. pylori proteins either have no PDB structures or interfaces. This shows that sequence-based methods and interface-based methods can complement each other. Both are useful to enrich the HPI data. Sequence-based methods do not cover pathogenic proteins with no or limited sequence similarity to endogenous host proteins, and interface-based methods are limited by the number of available crystal structures and interfaces.
We performed MD simulations to validate the stability of our models, and we observed that all of the HPI complexes that we simulated were stable with three out of four of the HPIs preserving the modeled interfaces.
Models should be tested by experiments. The success of prediction methods depends on two factors, coverage and accuracy [30]. Coverage indicates the portion of all interactions covered by the method. The performance of interface-based methods depends on the availability of the interface structures in the PDB [103]. Thanks to the rapid increase in the number of resolved 3D structures of endogenous and exogenous interactions in recent years [104], the performance and the coverage of interface-based methods is expected to improve. Available interface structures were, however, projected to already be diverse enough to cover most endogenous interactions [52–55]. They may also cover the majority of the HPI space due to interface mimicry of exogenous interfaces of the endogenous ones. As to accuracy, due to the scarcity of experimental and computational HPI data for H. pylori, we were unable to calculate the exact false-positive and false-negative rates. A rough estimation of our false-positive rate is about 18%. We tried to minimize the error rates by calculating the percent-match of the HPI models with the corresponding template PPI and incorporating the probability of template interfaces being real biological interfaces. We further validated the stabilities of some of the HPI models by explicit solvent simulations and found that three out of four preserved the initial interface.
It is important to also note objective caveats, including that the coverage of endogenous human PPIs is low [105], disordered proteins are underrepresented in the PDB [104,106] and that most pathogenic proteins lack crystal structures, which may be somewhat alleviated by developments in ab initio modeling [107] for almost any microbial proteins. Notably, earlier interface-based modeling of endogenous PPIs generated results that are in agreement with available experimental mutagenesis data [108] and they are better at handling the protein complexes which undergo conformational change upon binding [109]. Computational screening of big data can provide possible leads to experiments guiding functional characterization, while avoiding millions of possible binary combinations of host and pathogenic proteins.
Our approach is based on the reasonable assumption that pathogenic proteins may alter host signaling. However, other modes of modulation of the host responses, such as through metabolites and small molecules, cannot be underestimated. Interaction of a particular pathogen with other microbial species in the microbiota also affects the overall response. Also, proteins often assemble into multi-protein complexes. Modeling only pairwise interactions between host and pathogenic proteins may not be sufficient. In addition, protein post-translational modification states may change the interactions.
In conclusion, large-scale characterization of the HPIs is vital for understanding of pathogenesis strategies and forecasting the impacts of pathogenic proteins in biological events. Despite the limitations of computational approaches, proteome-wide modeling of HPIs necessitates such tools. Completing the integrated inter-species interactome will help advance insight into virulence strategies, infection mechanisms, and better therapeutics.
Methods
Modeling HPIs
Here, we report a novel computational approach that utilizes interface mimicry to predict HPIs and determine the structures of their complexes. Figure 1 displays the workflow of our approach. Normally, docking algorithms require structures of two target proteins as inputs to dock to each other. However, in this case, we know only the pathogenic proteins (one of the targets), but not their interactors in the host (second target). Thus, before docking, we need to identify their potential partners in the host. To do that, we first generate all known human interfaces (both endogenous and exogenous) in the piface interface database, as described in Ref. [9]. The piface database was released in 2013. We have 26,236 human interfaces in our template set. Each interface is composed of two chains (partners/sides). Then, we structurally align all human interfaces with the H. pylori proteins by MultiProt [110]. The “match” thresholds for structural alignment are taken according to the PRISM algorithm [45–48]: at least 15 matching residues and 1 matching hotspot residue with a maximum RMSD of 2 Å. If the pathogenic protein is aligned with one side of the interface, the pathogenic protein can interact with the complementary side. Thus, the pathogenic protein can compete with the first side of interface to bind to second side and it can disrupt the endogenous pairwise interaction in the template PPI that it is aligned (Fig. 1). Shape complementarity (local structural complementarity here) does not always confer chemical complementarity. For instance, 10 H. pylori proteins aligned with 48,204 interfaces, but only 328 of them are energetically favorable. Therefore, after determination of the putative partners in human, we perform docking with two programs; PRISM [45–48] and Rosetta (local refinement) [49–51]. We consider HPIs as energetically favorable only if their Rosetta interface scores (I_sc) are below −5 and total energy scores below zero. We also calculated Rosetta I_sc for the template interfaces (endogenous PPIs) and compared them with those of HPIs to infer which one has higher affinity, that is, whether the pathogenic protein will out-compete the endogenous partner to bind to a host protein. For some template PPIs, Rosetta gives extremely low, non-realistic interface scores, as low as −5000 due to intermolecular disulfide bonds. To correct this, we calculated Rosetta interface scores with both considering and ignoring the disulfide bonds. We took the HPIs that are common to both Rosetta scorings as favorable interactions. The values given in Tables S1 and S3 are the results of Rosetta where we ignored disulfide bonds. It is important to note that Rosetta I_sc does not reflect/correspond the real binding free energy and have units. It only indicates whether an interaction is favorable or not.
Finally, we filtered our results according to tissue expression, checking whether the host proteins (targets of pathogenic proteins) are expressed in the same tissue where pathogen is found. We obtained the protein tissue expression data from the Human Protein Atlas, which covers 19,709 human proteins and 7106 human PDBs [78,79].
To further explore the likelihoods of our HPI models, we calculated the “percent-match” of the interfaces by dividing the number of interface residues that are aligned with the pathogenic protein by the number of interface residues in the endogenous template PPI. The greater the percent match, we consider that the better is the structural alignment with the template. We assigned weights based on the size of endogenous template interface with larger interfaces having higher weights. If the template interface is small, with less than 30 residues (n < 30), the weight is 0.5; if 30 < n < 50, the weight is 1; if 50 < n < 80, the weight is 1.5; and if n > 80 (very large interface), the weight is 2. We assigned weight 0.5 to coiled-coil interfaces regardless of the interface size because they can be aligned with almost any α-helices in pathogenic proteins and thus give unreliable models. Score1 given in Table S1 is obtained by the multiplication of the percent match of the interfaces and the corresponding weights.
We checked whether the template interfaces are real biological interfaces or crystal artifacts. We utilized the Evolutionary Protein–Protein Interface Classifier [111], which gives the probability of a particular interface to be biological. Score2 in Table S1 is obtained by multiplication of Score1 by the probability of being a biological interface. The higher the Score2, the more confidence we have that the HPI model would occur, as they are better mimics of real biological endogenous interfaces.
Constructing the structural superorganism network
We constructed the structural superorganism network between human and H. pylori. We took all available human interfaces (the template set) as endogenous pairwise interactions and our models of HPIs as exogenous interactions. Hence, all the interactions in our network have structures as complexes. There are 3366 human PPIs and 224 HPIs in this network. Our 26,236 interfaces map to 3366 distinct human PPIs. The topological features of the superorganism network that we generated are calculated by the NetworkAnalyzer [112] application in Cytoscape [113]. To compare H. pylori with other pathogens, we also built the structural interspecies network for all known HPIs in PDB. There are 299 HPIs in PDB for human with different bacterial, yeast, and viral species.
Functional annotation of human proteins predicted to interact with H. pylori proteins is performed by DAVID [96,97].
Stability and dynamics of the modeled HPIs by MD simulations
The stability of the complexes may serve as a means of validation of the modeled HPIs. MD simulations also provide dynamics information not shown in crystal structures. We performed simulations for four HPI models and their corresponding template human PPIs (Table 3). We utilized the CHARMM-GUI server [114] and CHARMM36 force field [115] with TIP3 explicit solvent and NaCl to construct the initial conformations and relax the system. Following pre-equilibrium, a 100-ns production run was performed with NAMD 2.10 [116] on a Biowulf cluster at the National Institutes of Health. The particle mesh Ewald method was used to calculate electrostatic and van der Waals interactions. The simulations were executed in isothermal– isobaric (NPT) ensemble with periodic boundary conditions. The temperature was kept at 310 K with Langevin temperature control, and pressure was maintained at 1 atm with Nosé–Hoover Langevin piston pressure control.
Table 3.
Details of the simulated systems
| Simulation system | Model or template | PDB |
|---|---|---|
| CagA–TP53BP2 | Template endogenous PPI | 4irvAE |
| CagA–ZO1 | HPI model | 4dvyP and 3tswD |
| ZO1–ZO1 | Template endogenous PPI | 3tswBD |
| CagA–JAM1 | HPI model | 4dvyP and 3eoyI |
| JAM1–viral capsid protein | Template exogenous PPI | 3eoyCI |
| gGT–INAR1 | HPI model | 2nqoA and 3se3A |
| INAR1–IFNA2 | Template endogenous PPI | 3se3AB |
Missing residues in the crystal structures of ZO1, CagA, and INAR1 were modeled by SWISS-MODEL [117]. Since the CagA–TP53BP2 interaction complex in the PDB and our predicted HPI are almost identical, we only simulated the template PPI.
To compare the stability of the systems throughout the trajectory, the RMSDs with respect to the initial structures of systems were calculated for each HPI model and their respective template PPIs.
To calculate the binding free energy of the simulated systems, GBSW [118] and entropy calculations were carried out with the CHARMM program [119]. We included only the protein atoms in GBSW and only the backbone atoms in the entropy calculations. The average binding free energy, ΔGb is calculated as the sum of gas phase, solvation energy, and the entropic contributions,
| (1) |
where 〈 〉 denotes an average along the MD trajectory (100 ns). The change in binding free energy due to complex formation (either HPIs or PPIs) is calculated by the following equation:
| (2) |
Supplementary Material
Acknowledgment
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health (NIH), under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This study utilized the high-performance computational capabilities of the Biowulf PC/Linux cluster at the NIH, Bethesda, MD (http://biowulf.nih.gov).
Abbreviations used:
- HPIs
host–pathogen interactions
- PPIs
protein–protein interactions
- OMPs
outer-membrane proteins
- MD
molecular dynamics
- INAR1–IFNA2
interferon alpha/beta receptor 2—interferon alpha 2
- GBSW
generalized Born with a simple switching
Footnotes
Conflict of Interest Statement: The authors declare that there are no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2017.10.023.
References
- [1].Guven-Maiorov E, Tsai CJ, Nussinov R, Structural host–microbiota interaction networks, PLoS Comput. Biol 13 (2017), e1005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vieira SM, Pagovich OE, Kriegel MA, Diet, microbiota and autoimmune diseases, Lupus 23 (2014) 518–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Guven-Maiorov E, Tsai CJ, Nussinov R, Pathogen mimicry of host protein–protein interfaces modulates immunity, Semin. Cell Dev. Biol 58 (2016) 136–145. [DOI] [PubMed] [Google Scholar]
- [4].Posselt G, Backert S, Wessler S, The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis, Cell Commun. Signal 11 (2013) 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jones KR, Whitmire JM, Merrell DS, A tale of two toxins: Helicobacter pylori CagA and VacA modulate host pathways that impact disease, Front. Microbiol 1 (2010) 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Polk DB, Peek RM Jr., Helicobacter pylori: gastric cancer and beyond, Nat. Rev. Cancer 10 (2010) 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dorer MS, Talarico S, Salama NR, Helicobacter pylori’s unconventional role in health and disease, PLoS Pathog 5 (2009), e1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yamaoka Y, Graham DY, Helicobacter pylori virulence and cancer pathogenesis, Future Oncol 10 (2014) 1487–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cukuroglu E, Gursoy A, Nussinov R, Keskin O, Nonredundant unique interface structures as templates for modeling protein interactions, PLoS One 9 (2014), e86738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Muratcioglu S, Guven-Maiorov E, Keskin O, Gursoy A, Advances in template-based protein docking by utilizing interfaces towards completing structural interactome, Curr. Opin. Struct. Biol 35 (2015) 87–92. [DOI] [PubMed] [Google Scholar]
- [11].Keskin O, Nussinov R, Similar binding sites and different partners: implications to shared proteins in cellular pathways, Structure 15 (2007) 341–354. [DOI] [PubMed] [Google Scholar]
- [12].Franzosa EA, Garamszegi S, Xia Y, Toward a three-dimensional view of protein networks between species, Front. Microbiol 3 (2012) 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Franzosa EA, Xia Y, Structural principles within the human–virus protein–protein interaction network, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 10538–10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tsai CJ, Lin SL, Wolfson HJ, Nussinov RA, Dataset of protein–protein interfaces generated with a sequence-order-independent comparison technique, J. Mol. Biol 260 (1996) 604–620. [DOI] [PubMed] [Google Scholar]
- [15].Tsai CJ, Lin SL, Wolfson HJ, Nussinov R, Protein–protein interfaces: architectures and interactions in protein–protein interfaces and in protein cores. Their similarities and differences, Crit. Rev. Biochem. Mol. Biol 31 (1996) 127–152. [DOI] [PubMed] [Google Scholar]
- [16].Keskin O, Nussinov R, Favorable scaffolds: proteins with different sequence, structure and function may associate in similar ways, Protein Eng. Des. Sel 18 (2005) 11–24. [DOI] [PubMed] [Google Scholar]
- [17].Hagai T, Azia A, Babu MM, Andino R, Use of host-like peptide motifs in viral proteins is a prevalent strategy in host-virus interactions, Cell Rep 7 (2014) 1729–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Stebbins CE, Galan JE, Structural mimicry in bacterial virulence, Nature 412 (2001) 701–705. [DOI] [PubMed] [Google Scholar]
- [19].Nourani E, Khunjush F, Durmus S, Computational approaches for prediction of pathogen–host protein–protein interactions, Front. Microbiol 6 (2015) 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Petrenko P, Doxey AC, mimicMe: a web server for prediction and analysis of host-like proteins in microbial pathogens, Bioinformatics 31 (2015) 590–592. [DOI] [PubMed] [Google Scholar]
- [21].Huo T, Liu W, Guo Y, Yang C, Lin J, Rao Z, Prediction of host–pathogen protein interactions between Mycobacterium tuberculosis and Homo sapiens using sequence motifs, BMC Bioinformatics 16 (2015) 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Krishnadev O, Srinivasan N, Prediction of protein–protein interactions between human host and a pathogen and its application to three pathogenic bacteria, Int. J. Biol. Macromol 48 (2011) 613–619. [DOI] [PubMed] [Google Scholar]
- [23].Dyer MD, Murali TM, Sobral BW, Computational prediction of host–pathogen protein–protein interactions, Bioinformatics 23 (2007) i159–66. [DOI] [PubMed] [Google Scholar]
- [24].Tyagi N, Krishnadev O, Srinivasan N, Prediction of protein–protein interactions between Helicobacter pylori and a human host, Mol. BioSyst 5 (2009) 1630–1635. [DOI] [PubMed] [Google Scholar]
- [25].Doxey AC, McConkey BJ, Prediction of molecular mimicry candidates in human pathogenic bacteria, Virulence 4 (2013) 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Evans P, Dampier W, Ungar L, Tozeren A, Prediction of HIV-1 virus–host protein interactions using virus and host sequence motifs, BMC Med. Genet 2 (2009) 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mahajan G, Mande SC, Using structural knowledge in the protein data bank to inform the search for potential host–microbe protein interactions in sequence space: application to Mycobacterium tuberculosis, BMC Bioinformatics 18 (2017) 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mariano R, Wuchty S, Structure-based prediction of host–pathogen protein interactions, Curr. Opin. Struct. Biol 44 (2017) 119–124. [DOI] [PubMed] [Google Scholar]
- [29].Manente L, Perna A, Buommino E, Altucci L, Lucariello A, Citro G, et al. , The Helicobacter pylori’s protein VacA has direct effects on the regulation of cell cycle and apoptosis in gastric epithelial cells, J. Cell. Physiol 214 (2008) 582–587. [DOI] [PubMed] [Google Scholar]
- [30].Davis FP, Barkan DT, Eswar N, McKerrow JH, Sali A, Host pathogen protein interactions predicted by comparative modeling, Protein Sci 16 (2007) 2585–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Doolittle JM, Gomez SM, Structural similarity-based predictions of protein interactions between HIV-1 and Homo sapiens, Virol. J 7 (2010) 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Doolittle JM, Gomez SM, Mapping protein interactions between Dengue virus and its human and insect hosts, PLoS Negl. Trop. Dis 5 (2011), e954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].de Chassey B, Meyniel-Schicklin L, Aublin-Gex A, Navratil V, Chantier T, Andre P, et al. , Structure homology and interaction redundancy for discovering virus-host protein interactions, EMBO Rep 14 (2013) 938–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Drayman N, Glick Y, Ben-nun-shaul O, Zer H, Zlotnick A, Gerber D, et al. , Pathogens use structural mimicry of native host ligands as a mechanism for host receptor engagement, Cell Host Microbe 14 (2013) 63–73. [DOI] [PubMed] [Google Scholar]
- [35].Aloy P, Bottcher B, Ceulemans H, Leutwein C, Mellwig C, Fischer S, et al. , Structure-based assembly of protein complexes in yeast, Science 303 (2004) 2026–2029. [DOI] [PubMed] [Google Scholar]
- [36].Rajasekharan S, Rana J, Gulati S, Sharma SK, Gupta V, Gupta S, Predicting the host protein interactors of Chandipura virus using a structural similarity-based approach, Pathog Dis 69 (2013) 29–35. [DOI] [PubMed] [Google Scholar]
- [37].Lee SA, Chan CH, Tsai CH, Lai JM, Wang FS, Kao CY, et al. , Ortholog-based protein–protein interaction prediction and its application to inter-species interactions, BMC Bioinformatics 9 (Suppl. 12) (2008) S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Krishnadev O, Srinivasan NA, Data integration approach to predict host–pathogen protein–protein interactions: application to recognize protein interactions between human and a malarial parasite, In Silico Biol 8 (2008) 235–250. [PubMed] [Google Scholar]
- [39].Schulze S, Henkel SG, Driesch D, Guthke R, Linde J, Computational prediction of molecular pathogen–host interactions based on dual transcriptome data, Front. Microbiol 6 (2015) 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, et al. , A physical and regulatory map of host–influenza interactions reveals pathways in H1N1 infection, Cell 139 (2009) 1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xu Q, Xiang EW, Yang Q, Transferring network topological knowledge for predicting protein–protein interactions, Proteomics 11 (2011) 3818–3825. [DOI] [PubMed] [Google Scholar]
- [42].Uetz P, Dong YA, Zeretzke C, Atzler C, Baiker A, Berger B, et al. , Herpesviral protein networks and their interaction with the human proteome, Science 311 (2006) 239–242. [DOI] [PubMed] [Google Scholar]
- [43].Remmele CW, Luther CH, Balkenhol J, Dandekar T, Muller T, Dittrich MT, Integrated inference and evaluation of host–fungi interaction networks, Front. Microbiol 6 (2015) 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang M, Su S, Bhatnagar RK, Hassett DJ, Lu LJ, Prediction and analysis of the protein interactome in Pseudomonas aeruginosa to enable network-based drug target selection, PLoS One 7 (2012), e41202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tuncbag N, Gursoy A, Nussinov R, Keskin O, Predicting protein–protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM, Nat. Protoc 6 (2011) 1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Keskin O, Nussinov R, Gursoy A, PRISM: protein–protein interaction prediction by structural matching, Methods Mol. Biol 484 (2008) 505–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Baspinar A, Cukuroglu E, Nussinov R, Keskin O, Gursoy A, PRISM: a Web server and repository for prediction of protein–protein interactions and modeling their 3D complexes, Nucleic Acids Res 42 (2014) W285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ogmen U, Keskin O, Aytuna AS, Nussinov R, Gursoy A, PRISM: protein interactions by structural matching, Nucleic Acids Res 33 (2005) W331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, et al. , Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations, J. Mol. Biol 331 (2003) 281–299. [DOI] [PubMed] [Google Scholar]
- [50].Wang C, Schueler-Furman O, Baker D, Improved sidechain modeling for protein–protein docking, Protein Sci 14 (2005) 1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang C, Bradley P, Baker D, Protein–protein docking with backbone flexibility, J. Mol. Biol 373 (2007) 503–519. [DOI] [PubMed] [Google Scholar]
- [52].Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, et al. , Structure-based prediction of protein–protein interactions on a genome-wide scale, Nature 490 (2012) 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhang QC, Petrey D, Norel R, Honig BH, Protein interface conservation across structure space, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 10896–10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gao M, Skolnick J, Structural space of protein–protein interfaces is degenerate, close to complete, and highly connected, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 22517–22522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kundrotas PJ, Zhu Z, Janin J, Vakser IA, Templates are available to model nearly all complexes of structurally characterized proteins, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 9438–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Dubois A, Boren T, Helicobacter pylori is invasive and it may be a facultative intracellular organism, Cell. Microbiol 9 (2007) 1108–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Su YL, Huang HL, Huang BS, Chen PC, Chen CS, Wang HL, et al. , Combination of OipA, BabA, and SabA as candidate biomarkers for predicting Helicobacter pylori-related gastric cancer, Sci Rep 6 (2016), 36442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nesic D, Buti L, Lu X, Stebbins CE, Structure of the Helicobacter pylori CagA oncoprotein bound to the human tumor suppressor ASPP2, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 1562–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nesic D, Miller MC, Quinkert ZT, Stein M, Chait BT, Stebbins CE, Helicobacter pylori CagA inhibits PAR1–MARK family kinases by mimicking host substrates, Nat. Struct. Mol. Biol 17 (2010) 130–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Suzuki M, Mimuro H, Suzuki T, Park M, Yamamoto T, Sasakawa C, Interaction of CagA with Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion, J. Exp. Med 202 (2005) 1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Oliveira MJ, Costa AM, Costa AC, Ferreira RM, Sampaio P, Machado JC, et al. , CagA associates with c-Met, E-cadherin, and p120-catenin in a multiproteic complex that suppresses Helicobacter pylori-induced cell-invasive phenotype, J. Infect. Dis 200 (2009) 745–755. [DOI] [PubMed] [Google Scholar]
- [62].Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. , Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells, Oncogene 26 (2007) 4617–4626. [DOI] [PubMed] [Google Scholar]
- [63].Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S, Disruption of the epithelial apical–junctional complex by Helicobacter pylori CagA, Science 300 (2003) 1430–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Churin Y, Al-Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M, Helicobacter pylori CagA protein targets the c-met receptor and enhances the motogenic response, J. Cell Biol 161 (2003) 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C, Grb2 is a key mediator of Helicobacter pylori CagA protein activities, Mol. Cell 10 (2002) 745–755. [DOI] [PubMed] [Google Scholar]
- [66].Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M, Attenuation of Helicobacter pylori CagA × SHP-2 signaling by interaction between CagA and Cterminal Src kinase, J. Biol. Chem 278 (2003) 3664–3670. [DOI] [PubMed] [Google Scholar]
- [67].Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, et al. , SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein, Science 295 (2002) 683–686. [DOI] [PubMed] [Google Scholar]
- [68].Selbach M, Paul FE, Brandt S, Guye P, Daumke O, Backert S, et al. , Host cell interactome of tyrosine-phosphorylated bacterial proteins, Cell Host Microbe 5 (2009) 397–403. [DOI] [PubMed] [Google Scholar]
- [69].Lamb A, Yang XD, Tsang YH, Li JD, Higashi H, Hatakeyama M, et al. , Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination, EMBO Rep 10 (2009) 1242–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Salama NR, Hartung ML, Muller A, Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori, Nat. Rev. Microbiol 11 (2013) 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sewald X, Gebert-Vogl B, Prassl S, Barwig I, Weiss E, Fabbri M, et al. , Integrin subunit CD18 is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin, Cell Host Microbe 3 (2008) 20–29. [DOI] [PubMed] [Google Scholar]
- [72].Yahiro K, Wada A, Nakayama M, Kimura T, Ogushi K, Niidome T, et al. , Protein-tyrosine phosphatase alpha, RPTP alpha, is a Helicobacter pylori VacA receptor, J. Biol. Chem 278 (2003) 19183–19189. [DOI] [PubMed] [Google Scholar]
- [73].Yahiro K, Satoh M, Nakano M, Hisatsune J, Isomoto H, Sap J, et al. , Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA, J. Biol. Chem 287 (2012) 31104–31115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hennig EE, Godlewski MM, Butruk E, Ostrowski J, Helicobacter pylori VacA cytotoxin interacts with fibronectin and alters HeLa cell adhesion and cytoskeletal organization in vitro, FEMS Immunol. Med. Microbiol 44 (2005) 143–150. [DOI] [PubMed] [Google Scholar]
- [75].Hage N, Howard T, Phillips C, Brassington C, Overman R, Debreczeni J, et al. , Structural basis of Lewis(b) antigen binding by the Helicobacter pylori adhesin BabA, Sci. Adv 1 (2015), e1500315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Walz A, Odenbreit S, Mahdavi J, Boren T, Ruhl S, Identification and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays, Glycobiology 15 (2005) 700–708. [DOI] [PubMed] [Google Scholar]
- [77].Sheu BS, Odenbreit S, Hung KH, Liu CP, Sheu SM, Yang HB, et al. , Interaction between host gastric SialylLewis X and H. pylori SabA enhances H. pylori density in patients lacking gastric Lewis B antigen, Am. J. Gastroenterol 101 (2006) 36–44. [DOI] [PubMed] [Google Scholar]
- [78].Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, et al. , A human protein atlas for normal and cancer tissues based on antibody proteomics, Mol. Cell. Proteomics 4 (2005) 1920–1932. [DOI] [PubMed] [Google Scholar]
- [79].Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. , Proteomics. Tissue-based map of the human proteome, Science 347 (2015), 1260419. [DOI] [PubMed] [Google Scholar]
- [80].Luther SA, Cyster JG, Chemokines as regulators of T cell differentiation, Nat. Immunol 2 (2001) 102–107. [DOI] [PubMed] [Google Scholar]
- [81].Trinchieri G, Cancer and inflammation: an old intuition with rapidly evolving new concepts, Annu. Rev. Immunol 30 (2012) 677–706. [DOI] [PubMed] [Google Scholar]
- [82].Schmees C, Prinz C, Treptau T, Rad R, Hengst L, Voland P, et al. , Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase, Gastroenterology 132 (2007) 1820–1833. [DOI] [PubMed] [Google Scholar]
- [83].Ohtani M, Garcia A, Rogers AB, Ge Z, Taylor NS, Xu S, et al. , Protective role of 17 beta-estradiol against the development of Helicobacter pylori-induced gastric cancer in INS-GAS mice, Carcinogenesis 28 (2007) 2597–2604. [DOI] [PubMed] [Google Scholar]
- [84].Ohtani M, Ge Z, Garcia A, Rogers AB, Muthupalani S, Taylor NS, et al. , 17 Beta-estradiol suppresses Helicobacter pylori-induced gastric pathology in male hypergastrinemic INS-GAS mice, Carcinogenesis 32 (2011) 1244–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chang CC, Kuo WS, Chen YC, Perng CL, Lin HJ, Ou YH, Fragmentation of CagA reduces hummingbird phenotype induction by Helicobacter pylori, PLoS One 11 (2016), e0150061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Hatakeyama M, Higashi H, Helicobacter pylori CagA: a new paradigm for bacterial carcinogenesis, Cancer Sci 96 (2005) 835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Shirin H, Pinto JT, Liu LU, Merzianu M, Sordillo EM, Moss SF, Helicobacter pylori decreases gastric mucosal glutathione, Cancer Lett 164 (2001) 127–133. [DOI] [PubMed] [Google Scholar]
- [88].De Bruyne E, Ducatelle R, Foss D, Sanchez M, Joosten M, Zhang G, et al. , Oral glutathione supplementation drastically reduces Helicobacter-induced gastric pathologies, Sci Rep 6 (2016), 20169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Guven Maiorov E, Keskin O, Gursoy A, Nussinov R, The structural network of inflammation and cancer: merits and challenges, Semin. Cancer Biol 23 (2013) 243–251. [DOI] [PubMed] [Google Scholar]
- [90].Caron TJ, Scott KE, Fox JG, Hagen SJ, Tight junction disruption: Helicobacter pylori and dysregulation of the gastric mucosal barrier, World J. Gastroenterol 21 (2015) 11411–11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Bourzac KM, Botham CM, Guillemin K, Helicobacter pylori CagA induces AGS cell elongation through a cell retraction defect that is independent of Cdc42, Rac1, and Arp2/3, Infect. Immun 75 (2007) 1203–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Fanning AS, Lye MF, Anderson JM, Lavie A, Domain swapping within PDZ2 is responsible for dimerization of ZO proteins, J. Biol. Chem 282 (2007) 37710–37716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T, Structure of reovirus sigma1 in complex with its receptor junctional adhesion molecule-A, PLoS Pathog 4 (2008), e1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Dyer MD, Neff C, Dufford M, Rivera CG, Shattuck D, Bassaganya-Riera J, et al. , The human-bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis, PLoS One 5 (2010), e12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Durmus Tekir S, Cakir T, Ulgen KO, Infection strategies of bacterial and viral pathogens through pathogen–human protein–protein interactions, Front. Microbiol 3 (2012) 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].da W. Huang, Sherman BT, Lempicki RA, Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists, Nucleic Acids Res 37 (2009) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Huang da W, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, Nat. Protoc 4 (2009) 44–57. [DOI] [PubMed] [Google Scholar]
- [98].Mondal SI, Mahmud Z, Elahi M, Akter A, Jewel NA, Muzahidul Islam M, et al. , Study of intra-inter species protein–protein interactions for potential drug targets identification and subsequent drug design for Escherichia coli O104:H4 C277–11, In Silico Pharmacol 5 (2016) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Tsai CJ, Xu D, Nussinov R, Structural motifs at protein– protein interfaces: protein cores versus two-state and three-state model complexes, Protein Sci 6 (1997) 1793–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Tsai CJ, Lin SL, Wolfson HJ, Nussinov R, Studies of protein–protein interfaces: a statistical analysis of the hydrophobic effect, Protein Sci 6 (1997) 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, et al. , The protein–protein interaction map of Helicobacter pylori, Nature 409 (2001) 211–215. [DOI] [PubMed] [Google Scholar]
- [102].Hauser R, Ceol A, Rajagopala SV, Mosca R, Siszler G, Wermke N, et al. , A second-generation protein–protein interaction network of Helicobacter pylori, Mol. Cell. Proteomics 13 (2014) 1318–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Negroni J, Mosca R, Aloy P, Assessing the applicability of template-based protein docking in the twilight zone, Structure 22 (2014) 1356–1362. [DOI] [PubMed] [Google Scholar]
- [104].Franzosa EA, Xia Y, Structural models for host–pathogen protein–protein interactions: assessing coverage and bias, Pac. Symp. Biocomput (2012) 287–298. [PubMed]
- [105].Anishchenko I, Kundrotas PJ, Vakser IA, Structural quality of unrefined models in protein docking, Proteins 85 (2017) 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Janin J, Sternberg MJ, Protein flexibility, not disorder, is intrinsic to molecular recognition, F1000 Biol. Rep 5 (2013) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ovchinnikov S, Park H, Varghese N, Huang PS, Pavlopoulos GA, Kim DE, et al. , Protein structure determination using metagenome sequence data, Science 355 (2017) 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Guven-Maiorov E, Keskin O, Gursoy A, VanWaes C, Chen Z, Tsai CJ, et al. , The architecture of the TIR domain signalosome in the Toll-like receptor-4 signaling pathway, Sci Rep (2015) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Vreven T, Hwang H, Pierce BG, Weng Z, Evaluating template-based and template-free protein–protein complex structure prediction, Brief. Bioinform 15 (2014) 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Shatsky M, Nussinov R, Wolfson HJ, A method for simultaneous alignment of multiple protein structures, Proteins 56 (2004) 143–156. [DOI] [PubMed] [Google Scholar]
- [111].Duarte JM, Srebniak A, Scharer MA, Capitani G, Protein interface classification by evolutionary analysis, BMC Bioinformatics 13 (2012) 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Yang H, Ke Y, Wang J, Tan Y, Myeni SK, Li D, et al. , Insight into bacterial virulence mechanisms against host immune response via the Yersinia pestis-human protein–protein interaction network, Infect. Immun 79 (2011) 4413–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. , Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res 13 (2003) 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jo S, Kim T, Iyer VG, Im W, CHARMM-GUI: a Webbased graphical user interface for CHARMM, J. Comput. Chem 29 (2008) 1859–1865. [DOI] [PubMed] [Google Scholar]
- [115].Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, et al. , Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types, J. Phys. Chem. B 114 (2010) 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. , Scalable molecular dynamics with NAMD, J. Comput. Chem 26 (2005) 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. , SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information, Nucleic Acids Res 42 (2014) W252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Im W, Lee MS, Brooks III CL, Generalized born model with a simple smoothing function, J. Comput. Chem 24 (2003) 1691–1702. [DOI] [PubMed] [Google Scholar]
- [119].Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M, Charmm—a program for macromolecular energy, minimization, and dynamics calculations, J. Comput. Chem 4 (1983) 187–217. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.