

Abstract
Inborn errors of immunity (IEI) are a heterogeneous group of disorders, mainly resulting from mutations in genes associated with immunoregulation and immune host defense. These disorders are characterized by different combinations of recurrent infections, autoimmunity, inflammatory manifestations, lymphoproliferation, and malignancy. Interestingly, it has been increasingly observed that common allergic symptoms also can represent the expression of an underlying immunodeficiency and/or immune dysregulation.
Very high IgE levels, peripheral or organ-specific hypereosinophilia, usually combined with a variety of atopic symptoms, may sometimes be the epiphenomenon of a monogenic disease. Therefore, allergists should be aware that severe and/or therapy-resistant atopic disorders might be the main clinical phenotype of some IEI. This could pave the way to target therapies, leading to better quality of life and improved survival in affected patients.
Keywords: Inborn errors of immunity, Primary immunodeficiency, Atopy, Atopic phenotypes, Allergy
Introduction
Inborn errors of immunity (IEI) are a group of mostly monogenic disorders arising from mutations in genes responsible for immune host defense and immunoregulation.1,2 Typical clinical features include recurrent infections, autoimmunity, inflammatory manifestations, lymphoproliferation, and malignancy.1 Interestingly, recent evidence suggests that also common allergic symptoms may represent the expression of an underlying immunodeficiency and/or immune dysregulation.3 The recognition of IEI in the context of an allergic phenotype is crucial to ensure prompt diagnosis and appropriate treatment aimed to modulate pathophysiological mechanisms and improve clinical symptoms. Indeed, clinical management and expected outcomes are profoundly different from the ones reported for typical allergic conditions. Also, the correct diagnosis could pave the way for targeted therapies.4
The article presents a practical approach to diagnose and manage IEI presenting with atopic phenotypes. We will discuss known monogenic disorders leading to IEI with severe atopic phenotypes in humans. Moreover, we will focus on the red flags that need to be considered to suspect these conditions and on differential diagnosis.
Atopic phenotypes as clinical manifestations of inborn errors of immunity
Relying on the complex interplay between activation and regulation, the immune system has a fundamental role in protecting the host from pathogenic infections while discriminating between self- and non-self antigens.5,6 In this context, allergy, defined as an immune-mediated hypersensitivity reaction, represents an exaggerated immune response against specific non-self antigens, known as allergens. Frequent allergic manifestations include eczema, allergic rhinitis, asthma, and food allergy, and classic testing used to investigate allergic diseases often shows increased serum immunoglobulin (Ig) E and peripheral blood eosinophilia. It is now clear that in some IEI, allergic symptoms may dominate the clinical presentation.3,7,8 In particular, the allergic triad defined by increased IgE, eosinophilia, and eczema is shared by different IEI that may be misdiagnosed as common allergic diseases.3 Also, different and more complex atopic phenotypes have been recently described. Interestingly, the number of newly identified genes associated with IEI has exponentially increased over the last decade. In addition to identifying novel IEI-related genes, it is now clear that distinct clinical phenotypes may be sustained by gain-of-function (GOF) or loss-of-function (LOF) mutations in the same gene. Moreover, different activity degrees of mutant proteins due to hypomorphic and hypermorphic mutations may also cause IEI phenotypic variability.5 In this setting, referring to monogenic disorders leading to a predominant allergic inflammation, Milner et al proposed the term “primary atopic disorders”.9,10 The study of these conditions has provided fundamental insights into human immunity and the pathogenesis of allergic diseases.11 The main pathways implicated in the development of atopy range from focal defects in immune cells and epithelial barrier function to global changes in metabolism. In particular, they include impaired T-cell receptor (TCR) signaling and cytoskeletal remodeling, TCR restriction, altered cytokine signaling, tolerance failure, cellular metabolic disturbance, mast cell dysregulation, and skin barrier disruption.12 A significant goal of investigating heritable single-gene disorders that lead to severe clinical allergic diseases is to unveil fundamental pathways responsible for hypersensitivity that could be targeted to provide novel therapeutic strategies for patients with allergic diseases, syndromic and non-syndromic alike.9
Individual inborn errors of immunity with atopic phenotypes
Focusing on IEI associated with atopic phenotypes, the broad spectrum of clinical and immunological features associated with individual IEI makes it challenging to define a universal classification. According to the predominant clinical and laboratory characteristics, they can be generally classified into six different phenotypes:
-
[1]
Hyper-IgE syndromes (HIES);
-
[2]
Omenn syndrome (OS);
-
[3]
Wiskott-Aldrich syndrome (WAS) and WAS-like conditions;
-
[4]
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-like conditions;
-
[5]
CBM-opathies due to mutations in genes encoding for Caspase recruitment domain (CARD) proteins – B-cell CLL/lymphoma 10 (BCL10) – MALT1 paracaspase (MALT1), altogether known as CBM complexes;
-
[6]
a miscellanea of other IEI presenting with allergic manifestations.
The literature review has been performed employing EMBASE, Pubmed, Scopus, and Web of Science databases, retrieving all publications on IEI with atopic phenotypes. The search strategy was performed using a free-text search (keywords: inborn errors of immunity, primary immunodeficiency, atopy, atopic phenotypes, allergy) and thesaurus descriptors search (MeSH and Emtree), adapted for all the selected databases. We searched all articles published up to August 2020. The inclusion criteria for eligible articles were the following: publication in peer-reviewed journals and the English language. Articles were excluded by title, abstract, or full text for irrelevance to the analyzed topic. Lastly, to identify further studies that met the inclusion criteria, the references of the selected articles were also reviewed.
Patients suffering from IEI with atopic phenotypes usually present with peculiar associated clinical manifestations and laboratory findings that need to be carefully analyzed in order to identify the underlying disease. In addition, it is fundamental to assess the presence or absence of a positive family history for primary immunodeficiencies and/or consanguinity, as well as the presence of pre- and perinatal factors that may have influenced the early development of the immune system, including maternal infection during pregnancy.
Table 1 and Table 2 summarize the common features of IEI with atopic phenotypes and the red flags that clinicians should consider in the diagnostic work-up, respectively. Table 3 shows an overview of each IEI analyzed in the text, highlighting the distinguishing features from classical allergic disorders. Fig. 1 depicts a proposal for a diagnostic algorithm for the identification of IEI with atopic phenotypes.
Table 1.
Common features of inborn errors of immunity with atopic phenotypes
| Early-onset atopic disease, usually at birth or in the first months of life |
| Severe atopic disease, usually not responsive to standard therapy (e.g. severe and recalcitrant eczema) |
| High levels of Th2 biomarkers (e.g. increased total serum IgE, eosinophilia) |
| Presence of other affected family members (inheritance pattern, including family history for primary immunodeficiencies and/or familial severe atopic diathesis), family history of consanguinity |
| Associated clinical featuresa |
| Associated immunological abnormalitiesa |
| Efficacy of targeted therapies |
See Table 2, Red flags
Table 2.
Red flags to suspect inborn errors of immunity with atopic phenotypes.
| Serum total IgE >2000 kU/L, especially in the first 3 months of life |
| Neonatal erythroderma |
| Congenital ichthyosis |
| AD + Serum total IgE >2000 kU/L + recurrent skin and pulmonary infections ± skeletal abnormalities ± neurodevelopmental delay |
| Atopic diathesis + recurrent/severe infections (especially due to opportunistic pathogens and Herpesviridae, including CMV, EBV, HHV-6) |
| AD + autoimmunity ± recurrent infections |
| Atopic diathesis + lymphopenia |
| Atopic diathesis + cytopenias (neutropenia/thrombocytopenia/anemia) |
| AD + diarrhea + endocrinopathy ± failure to thrive |
| AD + diarrhea + bleeding ± failure to thrive |
| EGID + severe eosinophilia (>1500 cells/mm3) ± atopic diathesis |
AD, atopic dermatitis; CMV, cytomegalovirus; EBV, Epstein-Barr virus; EGID, eosinophilic gastrointestinal disease; HHV-6, Human herpesvirus 6; IgE, immunoglobulin E
Table 3.
Inborn errors of immunity with atopic phenotypes.
| Disease | Genetic defect | Inheritance | Main Features | Distinguishing features from common allergic disorders |
|---|---|---|---|---|
| Hyper-IgE syndromes (HIES) | ||||
| AD-HIES STAT3 deficiency (Job syndrome) | STAT3 | AD LOF | Eczema, skin abscesses, CMC, recurrent pneumonias leading to pneumatocoeles, and skeletal and connective tissue abnormalities | Early-onset eczema; peculiar thickened texture of the facial skin, retroauricular fissures, and severe folliculitis of the axillae and groin; cold abscesses; distinctive facial, and skeletal features, low frequency of allergy |
| DOCK8 deficiency | DOCK8 | AR | Severe eczema, severe allergies, immunodeficiency with increased susceptibility to bacterial and viral infections, autoimmunity, and increased risk for malignancies | Severe eczema associated with warts, severe skin and sinopulmonary infections |
| ZNF341 deficiency | ZNF341 | AR | Phenocopy of AD-HIES | Same as AD-HIES |
| IL6 signal transducer (IL6ST) deficiency | IL6ST | AR or AD LOF | Largely overlapping with AD-HIES: eczema, recurrent skin and pulmonary infections, craniosynostosis, neurodevelopmental delay | Severe eczema, recurrent cutaneous and pulmonary infections, distinctive skeletal features |
| IL6 receptor deficiency | IL6R | AR | Partially overlapping with AD-HIES: no skeletal abnormalities | Recurrent pyogenic infections, cold abscesses |
| ERBIN deficiency | ERBB2IP | AD LOF | Eczema, eosinophilic esophagitis, skeletal and connective tissue abnormalities like STAT3-HIES | Skeletal and connective tissue abnormalities |
| Loeys-Dietz syndrome (TGFBR deficiency) | TGFBR1 TGFBR2 | AD | Marfan-like syndrome, high prevalence of allergic diseases | Skeletal and connective tissue abnormalities |
| PGM3 deficiency | PGM3 | AR | Skeletal dysplasia, immunodeficiency and tendency to bone marrow failure, severe atopy, neurodevelopmental delay; some patients display renal, intestinal, and heart defects. | Complex syndromic phenotype associated with atopy |
| Comel-Netherton syndrome | SPINK5 | AR | Congenital ichthyosis, bamboo hair, atopic diathesis; increased bacterial infections; enteropathy, failure to thrive | Congenital ichthyosis |
| TYK2 deficiency | TYK2 | AR | Susceptibility to intracellular bacteria (mycobacteria, Salmonella) and viruses; dermatitis | Peculiar susceptibility to infections |
| Omenn syndrome | ||||
| OS is associated with multiple genetic abnormalities |
RAG1, RAG2, IL2RG, IL7R, LIG4, ADA, DCLRE1C, RMRP, CHD7, ZAP70, 22q11del and more |
AR, XL | Erythroderma, lymphadenopathy, eosinophilia, and combined immunodeficiency | Erythroderma or neonatal eczematous rash; immunodeficiency |
| Wiskott-Aldrich syndrome (WAS) and WAS-like conditions | ||||
| Wiskott-Aldrich syndrome | WAS | XL | Thrombocytopenia, recurrent infections, eczema, bloody diarrhea, haematological malignancies, autoimmune manifestations | Eczema associated with thrombocytopenia and recurrent infections |
| WIP deficiency | WIPF1 | AR | Thrombocytopenia with or without small platelets, recurrent infections, eczema, bloody diarrhea | WAS-like phenotype |
| ARPC1B deficiency | ARPC1B | AR | Mild thrombocytopenia, recurrent infections, autoimmunity; dermatitis | WAS-like phenotype |
| NOCARH | CDC42 | AD | Neonatal-onset cytopenia, autoinflammation, rash, and episodes of hemophagocytic lymphohistiocytosis; wide phenotypic heterogeneity | Autoinflammation, cytopenia, episodes of HLH |
| Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-like conditions | ||||
| IPEX | FOXP3 | XL | Autoimmune enteropathy, early onset diabetes, thyroiditis, hemolytic anemia, thrombocytopenia, severe early-onset dermatitis, recurrent severe infections, elevated IgE and IgA | Severe early-onset dermatitis associated with multiorgan autoimmunity |
| CD25 deficiency | IL2RA | AR | IPEX-like syndrome; chronic viral, fungal, and bacterial infections | IPEX-like syndrome |
| STAT5b deficiency | STAT5B | AR or AD LOF |
Growth-hormone insensitive dwarfism; dysmorphic features; eczema; prominent autoimmunity Growth-failure; eczema (no immune defects compared to AR STAT5b deficiency) |
IPEX-like syndrome, dwarfism, dysmorphic features |
| STAT1 GOF | STAT1 | AD GOF | CMC, infections, autoimmunity (thyroiditis, diabetes, cytopenias), enteropathy | CMC, autoimmunity |
| ITCH deficiency | ITCH | AR | Autoimmunity, failure to thrive, developmental delay, dysmorphic facial features | Autoimmunity, dysmorphic facial features |
| CBM-opathies | ||||
| CADINS | CARD11 | AD LOF | Atopic disease, respiratory tract infections and cutaneous viral infections Increased IgE, eosinophilia, Th-2 skewed immune response |
Severe atopic disease associated with susceptibility to infections and immune dysregulation |
| CARD14 deficiency | CARD14 | AD LOF | Atopic disease, recurrent pyogenic and viral skin infections and respiratory tract infections | See CARD11 |
| MALT1 deficiency | MALT1 | AR | Recurrent infections of the skin and of the respiratory and gastrointestinal tracts, failure to thrive, periodontal disease and inflammatory gastrointestinal disease | Recurrent infections and inflammatory gastrointestinal disease |
| Other IEI presenting with atopic phenotypes | ||||
| Selective IgA deficiency (SIgAD) | Unknown | Unknown | Frequently asymptomatic. Susceptibility to infections, autoimmunity and allergy Serum IgA levels (<0.07 g/L), normal serum IgG and IgM on at least two determinations |
Isolated IgA deficiency |
| RLTPR deficiency | CARMIL2 | AR | Recurrent infections, EBV lymphoproliferation and other malignancy, atopy | Infections, atopy, malignancies |
| JAK1 GOF | JAK1 | AD GOF | Eosinophilia, hepatosplenomegaly, eosinophilic enteritis, poor growth, viral infections | Hypereosinophilic syndrome |
| MyD88 deficiency | MYD88 | AR | Bacterial infections (pyogens), high IgE levels | Peculiar susceptibility to pyogenic infections |
| EDA-ID due to IKBKG (NEMO) deficiency | IKBKG (NEMO) | XL | Anhidrotic ectodermal dysplasia; susceptibility to infections (bacteria, mycobacteria, viruses, fungi) | Peculiar phenotype of anhidrotic ectodermal dysplasia |
| NFKB1 deficiency | NFKB1 | AD | Recurrent respiratory infections, EBV proliferation, autoimmunity | Susceptibility to infections, autoimmunity (cytopenias, alopecia, thyroiditis) |
| NFKB2 deficiency | NFKB2 | AD | Recurrent respiratory infections, autoimmunity | Susceptibility to infections, autoimmunity (alopecia and endocrinopathies) |
| Hypereosinophilic syndrome due to somatic mutations in STAT5b | STAT5B (GOF) – somatic mutations | – | Eosinophilia, atopic dermatitis, urticarial rash, diarrhea | Hypereosinophilic syndrome |
AR, autosomal recessive; AD, autosomal dominant; CADINS, CARD11-associated atopy with dominant interference of NF-kB signaling; CMC, chronic mucocutaneous candidiasis; EDA-ID, Anhidrotic Ectodermal Dysplasia with ImmunoDeficiency; GOF, gain of function; HLH, hemophagocytic lymphohistiocytosis; LOF, loss of function; XL, X-linked
Fig. 1.

Proposal for a diagnostic algorithm for the identification of IEI with atopic phenotypes
Hyper-IgE syndromes
IgE antibodies play a central role in the pathogenesis of atopic diseases and in host immunity against parasitic infections. Serum IgE levels in non-atopic subjects are usually very low (0–200 IU/mL)13,14 but vary significantly according to age and ethnicity.15,16 Atopic patients have elevated antigen-specific and total serum IgE levels (1000–10,000 IU/mL).17 It is now well established that different IEI can manifest with elevated serum IgE as a sign of immune dysregulation.7,18 Classically, and up to recent years, markedly elevated serum IgE levels have been the hallmark of HIES. Moreover, many other IEI, including WAS, IPEX, Omenn syndrome, and atypical DiGeorge syndrome are characterized by an increase in serum IgE (see described below).
Focusing on HIES, the prototypic syndrome is caused by dominant-negative germline mutations in Signal transducer and activator of transcription 3 (STAT3), resulting in an autosomal dominant Hyper-IgE (AD-HIES or STAT3-HIES) syndrome, formerly known as Job syndrome, characterized by eczema, skin abscesses [Fig. 2], recurrent pneumonia leading to pneumatocoeles, and skeletal and connective tissue abnormalities, such as bone fragility, scoliosis, and decidual teeth retention.14,19,20 Other reported manifestations include an increased incidence of both Hodgkin and non-Hodgkin lymphomas;21,22 vascular abnormalities as aneurysms, dilation, and tortuosity of middle-sized arteries such as coronary and cerebral arteries;23 gastrointestinal disease as dysmotility, gastro-esophageal reflux, and eosinophilic esophagitis.24 The most typical laboratory finding is an elevated serum IgE level (often higher than 2000 IU/mL). Eosinophilia can be observed at the complete blood count (CBC). Immunoglobulin levels are usually normal, but specific antibody responses to encapsulated bacteria can be impaired.25 Lymphocyte phenotyping often reveals diminished memory T and B cells and very low IL-17 producing T cells.25 The National Institutes of Health (NIH)-scoring system has been developed and validated to support clinicians in the recognition and diagnosis of STAT3-HIES.13 Compared to atopic dermatitis (AD), skin findings in STAT3-HIES are characterized by the peculiar thickened texture of the facial skin, retro auricular fissures, and severe folliculitis of the axillae and groin; these skin manifestations appear very early in life (first month) and may sometimes be already present at birth.26 The possible presence of chronic mucocutaneous candidiasis (CMC) in patients with STAT3-HIES is another distinguishing feature from AD.27 Also, STAT3-HIES manifests with poor clinical and biological inflammation, predisposing to the development of cold abscesses of the skin and lungs. Paradoxically, despite extremely high total serum IgE levels, specific IgE values and skin prick testing are often negative, and STAT3-HIES patients tend to present with lower lifetime frequency and severity of food allergy than AD patients.28,29 The discordance between total IgE and allergic symptoms is at least partially explained by the essential role of STAT3 signaling in mast cell degranulation.29 Prophylactic therapy with anti-staphylococcal and antifungal agents and topical antiseptics are fundamental to reduce the risk of cutaneous and sinopulmonary bacterial infections.25 The role of hematopoietic stem cell transplantation (HSCT) in treating STAT3-HIES is still under investigation, with encouraging reports on improvement in immunologic and nonimmunologic features of the underlying disease.30
Fig. 2.

Upper eyelid abscess in a patient with STAT3-HIES
When severe eczema is associated with recurrent viral infections, a combined immunodeficiency (CID) syndrome should also be considered. Dedicator of Cytokinesis 8 (DOCK8) deficiency is an autosomal recessive CID presenting with severe eczema [Fig. 3], severe allergies, immunodeficiency with increased susceptibility to bacterial, fungal, and viral infections, autoimmunity, neurological manifestations, cerebral vascular malformations and increased risk for malignancies.31,32 Although some clinical features overlap with STAT3-HIES, including severe eczema, skin, and sinopulmonary infections, elevated IgE, and eosinophilia, DOCK8 deficiency mainly differs for (i) susceptibility to cutaneous viral infections such as human papillomavirus (HPV) causing diffuse warts, disseminated molluscum contagiosum, herpes simplex viruses; (ii) a higher frequency of allergic manifestations including atopic dermatitis, food allergies, asthma, and eosinophilic esophagitis; (iii) the risk of malignancies that can be secondary to poor control of viruses such as HPV-associated squamous cell carcinomas or not associated with viral infections as rapidly progressive T-cell lymphoma; (iv) no predisposition to develop pneumatoceles, fractures, scoliosis or to retain teeth.25,33, 34, 35 At present, HSCT is the only curative option for DOCK8 deficiency and is recommended at the early stages of the disease.36 Interestingly, not all disease-related manifestations responded equally well to transplantation: infections and eczema resolved quicker than food allergies.
Fig. 3.

Severe eczema in a patient with DOCK8 deficiency
A novel autosomal recessive (AR) form of HIES was described in 2018; it is due to biallelic mutations in Zinc Finger Protein 341 (ZNF341), a transcription factor that regulates the transcription of STAT3, thereby also regulating its expression and activity.37,38
Moreover, Schwerd et al reported that severely hypomorphic mutations of the Interleukin 6 Signal Transducer (IL6ST) gene are also responsible for a severe AR form of HIES.39 Interestingly, the IL6ST gene encodes for the gp130 co-receptor of IL-6 family cytokines that include IL-6, IL-11, IL-27, and transduce the signal via STAT3. Recently, heterozygous, dominant-negative mutations in IL6ST have been described as the second genetic etiology of autosomal dominant HIES.40
Although only a few cases have been described carrying these recently identified mutations, it seems that the clinical phenotypes of the patients with these genetic etiologies of HIES mostly, but not entirely, overlap.5 In contrast, patients with autosomal recessive Interleukin 6 Receptor (IL-6R) deficiency,41 although presenting with similar clinical features, do not display skeletal phenotypes.
The significant overlap between allergic and connective tissue features has been better-understood thanks to Lyons et al., who, in 2017, reported a family with a loss-of-function (LOF) mutation in ERBB2IP, which encodes for the ERBB2-interacting protein (ERBIN).42 ERBIN deficiency presents with elevated IgE, recurrent respiratory infections, eosinophilic esophagitis, joint hypermobility, and vascular abnormalities; these patients do not manifest mucosal susceptibility to candida and T- and B-cell memory impairment as observed in STAT3-HIES. Of note, it is now known that ERBIN is fundamental for STAT3-mediated downregulation of Transforming Growth Factor Beta (TGF-β) signaling. Loss of ERBIN induces T-regulatory cell proliferation and Th2 polarization, recapitulating the allergic and connective tissue phenotypes of STAT3-HIES.42
The same molecular pathway is involved in the pathogenesis of Loeys-Dietz syndrome due to autosomal dominant mutations in the TGF-β receptor pathway.43 Affected individuals present with a Marfan-like syndrome, familial thoracic aortic aneurysms, and high prevalence of allergic manifestations, including eczema, food allergy, asthma, allergic rhinitis, and eosinophilic gastrointestinal disease.
Among HIES, complex and widespread clinical manifestations are reported in patients with Phosphoglucomutase 3 (PMG3) deficiency.44 The enzyme PMG3 is involved in multiple glycosylation pathways, and PMG3 mutations cause an AR disease characterized by severe skeletal dysplasia, severe atopy, and autoimmunity along with immunodeficiency and tendency to bone marrow failure, often associated with neurodevelopmental delay; moreover, some patients display renal, intestinal, and heart defects.
Although previously not considered among primary immunodeficiencies, Comèl-Netherton syndrome is now included in the IUIS classification of IEI.1,45,46 It is a congenital ichthyosis syndrome caused by AR mutations in the serine protease inhibitor gene Kazal-type 5 (SPINK5), which plays a pivotal role in maintaining skin barrier integrity.47 Comèl-Netherton syndrome is characterized by an early-onset generalized rash that evolves into severe ichthyosis with typical bamboo hair (trichorrhexis invaginata).45 Along with skin disease, these patients present with enteropathy and recurrent bacterial infections.46 In particular, Renner et al reported impaired vaccine responses, particularly to polysaccharide vaccines.46 Comèl-Netherton syndrome is also classified among the inherited skin disorders sharing pathogenetic pathways with atopic conditions48 together with ichthyosis vulgaris caused by null mutations in Filaggrin (FLG),49 the inflammatory peeling skin syndrome due to mutations in Corneodesmosin (CDSN),50 the severe skin dermatitis, multiple allergies and metabolic wasting (SAM) syndrome due to bi-allelic mutations in DSG1, encoding the desmosomal cadherin desmoglein 1 (DSG1),51 or in DSP, encoding another desmosomal protein, desmoplakin.52 Interestingly, a functional role for DSG1 and its dysregulation in the pathophysiology of eosinophilic esophagitis has been reported.52,53 Moreover, a favorable response to the treatment with ustekinumab, a monoclonal antibody targeting IL-12 and IL-23 as well as downstream IL-17 pathways, has been recently described in patients with DSP mutations54,55
Further data are needed to evaluate if these other barrier defects may be associated with immunodeficiency.12
Tyrosine kinase 2 (TYK2) deficiency was formerly defined in a patient suffering from an autosomal recessive form of HIES.56 Minegishi et al described a 22-year-old Japanese male patient who displayed the characteristic features of HIES associated with susceptibility to various pathogens, including mycobacteria and herpes simplex virus.56 More recently, the comprehensive immunological investigation of other TYK2-deficient patients has revealed a wider spectrum of disease, including phenotypes with mycobacterial and viral infections without hyper-IgE syndrome.57, 58, 59, 60
According to the 2019 IUIS classification of IEI, also heterozygous dominant-negative mutations in Caspase Recruitment Domain Family Member 11 (CARD11) cause a Hyper-IgE syndrome. However, considering the specific molecular pathway involved in the disease, it will be discussed among CMB-opathies (see below).
Omenn syndrome
Omenn syndrome (OS) was first described in 1965, in infants who presented with generalized erythroderma, lymphadenopathy, eosinophilia, and CID.61 Although this condition has been initially associated with mutations in Recombination activating gene 1 and 2 (RAG1 and RAG2),62,63 genetic alterations in other genes have also been reported,64, 65, 66, 67, 68, 69, 70, 71, 72 including the ones responsible for ARTEMIS deficiency, ADA deficiency, Cartilage Hair Hypoplasia, CHARGE syndrome, EXTL3 deficiency and atypical complete DiGeorge syndrome. Moreover, leaky severe combined immunodeficiency (SCID) caused by hypomorphic mutations in the common γ-chain (IL-2 receptor γ), IL-7 receptor α, ZAP70, and DNA ligase 4 may present with an OS phenotype. It is now clear that OS is not an isolated form of CID and is not caused by a single genetic defect.73 Instead, it is an exaggerated inflammatory condition that can be caused by different genetic alterations that significantly reduce, but do not abrogate, T cell development, resulting in an oligoclonal expansion of CD4+ T cells.
With regards to the atopic manifestations of OS, the disease usually presents at birth with generalized erythroderma, defined as skin inflammation affecting more than 90% of the body surface.74 Differential diagnosis of a newborn with erythroderma includes infections, inborn errors of metabolism, ichthyoses and inflammatory skin disorders, drug hypersensitivity reactions, and congenital immunodeficiencies.26 Of note, although the initial cutaneous manifestation of OS is most commonly described as erythroderma, it may present with a neonatal eczematous rash.75 Early recognition of OS is fundamental to allow for early HSCT that is the only curative treatment for this otherwise fatal disease.76 The diagnostic work-up should include an immunological evaluation with immune phenotype analysis and immunoglobulin dosage. Although IgGs are delivered to the infant through the placenta, this is not true for IgA and IgM. Thus, correct evaluation of all Ig isotypes should be performed and should always be confronted with age-matched values. Moreover, it is fundamental to consider the possibility of maternal engraftment that can confound the diagnostic process. Although basic flow cytometry evaluation (CD3, CD4, CD8, CD19, CD56, and HLA-DR expression) may be able to indicate a maternal engraftment by excessive expression of HLA-DR on patients' T cells — indicative in the context of CID suspicion of maternal origin — the variable number of tandem repeat (VNTR) analysis, also referred to as microsatellite analysis and/or in situ hybridization, represents the current gold standard to evaluate the maternal engraftment. VNTR probes give strong hybridization signals allowing for earlier detection of chimerism as well as detection of small numbers of cells.77
Among SCID, atopy and eosinophilia are frequently reported in Adenosine deaminase (ADA)-SCID. Allergic rhinitis and asthma, atopic dermatitis, urticaria and food allergy are the most common atopic manifestations identified in this population.78,79
Wiskott-Aldrich syndrome (WAS) and WAS-like conditions
Wiskott-Aldrich syndrome (WAS) is an X-linked IEI typically presenting with the triad of immunodeficiency, eczema, and thrombocytopenia with small platelets (mean platelet volume, MPV <6 fL). Recurrent and/or chronic infections, autoimmune manifestations, and increased susceptibility to malignancies, especially EBV-associated lymphoma, represent the main features of the syndrome.80 With an estimated incidence of 1 in 100 000 live male births, WAS is caused by mutations in the WAS gene, encoding the WAS protein (WASP), mainly involved in signal transduction and cytoskeleton remodeling. WASP plays a pivotal role in the immunological synapse formation and in the migration of myeloid and lymphoid cells in response to chemotactic signals.81 A broad spectrum of clinical phenotypes has been described in patients with WAS mutations. Of note, it is now known that hypomorphic mutations of WAS cause isolated X-linked thrombocytopenia (XLT),82 which may even be intermittent83; moreover, gain-of-function (GOF) mutations in the GTPase-binding domain of WASp are responsible for isolated X-linked congenital neutropenia.84
Typically, patients suffering from WAS present early in life with severe eczema, bloody diarrhea, and recurrent infections.80 Bacterial respiratory infections are common; patients are also at risk for chronic viral infections, particularly caused by herpesviruses, papillomavirus, and molluscum contagiosum. Autoimmunity usually manifests with hemolytic anemia, inflammatory bowel disease, arthritis, and IgA nephropathy. Increased risk of EBV-driven lymphoproliferative disease, lymphoma, and leukemia is reported. Progressive lymphopenia with impaired T-cell proliferation, altered NK-cytolytic function, decreased levels of IgM with increased IgA and IgE, impaired production of antibodies (especially to polysaccharide antigens), and reduced number of switched memory B cells represent the main immunological features.80 As for the atopic phenotype, eczema has been reported in 81% of patients with WAS [Fig. 4].85 Eczema may resemble classical AD but is usually more severe and widespread, is associated with petechiae and purpura due to the hemorrhagic diathesis, and typically present during the first year of life. Antimicrobial prophylaxis and immunoglobulin replacement, if required, represent the mainstays of supportive therapy, while HSCT and gene therapy represent the curative treatment.86,87
Fig. 4.

Severe eczema, petechiae and purpura in a patient with Wiskott-Aldrich syndrome
A WAS-like phenotype has been reported in patients with WIP deficiency and ARPC1B deficiency, in both cases associated with congenital thrombocytopenia.
WASP-interacting protein (WIP) is fundamental for WASP molecular stabilization88 and is part of the DOCK8-WIP-WASP complex that links the T-cell receptor (TCR) to the actin cytoskeleton.89 Mutations of the WIPF1 gene, causing WIP deficiency, are responsible for an IEI resembling WAS,90 with eczema being reported in most patients.91
ARPC1B deficiency is an AR form of CID associated with immune dysregulation and platelet abnormalities.92, 93, 94 The Actin-Related Protein Complex 1B (ARPC1B) is required for the assembly and maintenance of the ARP2/3 complex that plays a pivotal role in actin branching. ARPC1B-deficient patients present with clinical and laboratory features suggestive of WAS, including dermatitis, thrombocytopenia with bloody diarrhea, vasculitis, recurrent infections, autoimmune and atopic diathesis95,96; in addition, episodes of macrophage activation syndrome have been reported in these patients [Fig. 5].94,96
Fig. 5.
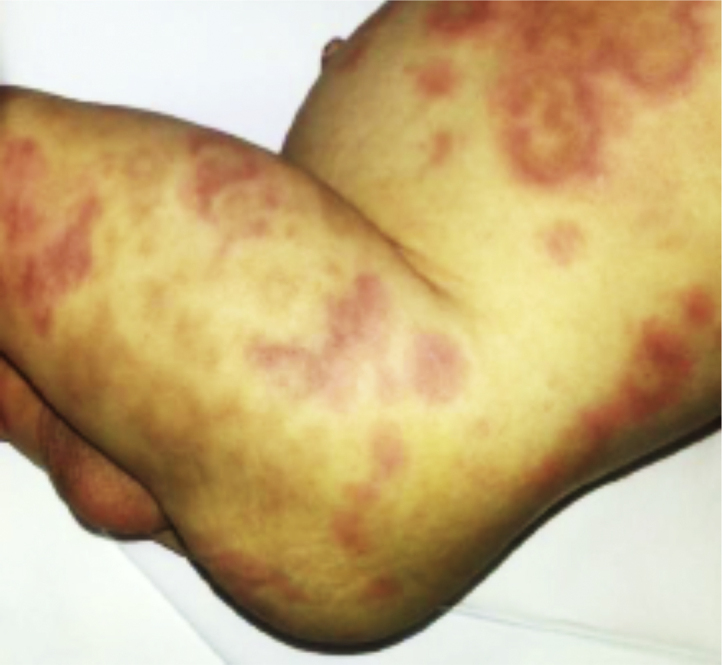
Cutaneous rash associated with macrophage activation syndrome (MAS) in a patient with ARPC1B deficiency (adapted from Brigida et al.96
A disease characterized by neonatal-onset cytopenia, autoinflammation, rash, and episodes of hemophagocytic lymphohistiocytosis (NOCARH) has been recently described in 4 unrelated patients carrying the same de novo heterozygous missense mutation in Cell division cycle 42 (CDC42) at p.Arg186Cys.97 CDC42 is a member of the Ras-homologous (Rho) GTPase family, functioning as a signaling node controlling a number of cellular processes, including proliferation, migration and adhesion.98,99 Interestingly, NOCARH differs considerably from the conditions previously associated with CDC42 mutations that showed a heterogeneous collection of neurodevelopmental phenotypes, including Takenouchi-Kosaki syndrome.100,101 More recently, additional reports further expanded the clinical spectrum of human diseases caused by inherited CDC42 mutations.102, 103, 104, 105, 106, 107, 108 Of note, He et al and Bekhouche et al reported on 2 patients with the p.Arg186Cys mutation, dysmorphism, and NOCARH along with elevation in total serum IgE.107,108
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-like conditions
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) is a X-linked recessive IEI that manifests in infancy with enteropathy, eczema, and severe autoimmune manifestations, including cytopenias, type 1 diabetes mellitus, autoimmune hepatitis, nephropathy, and myopathy.109,110 IPEX is caused by mutations in the FOXP3 gene, encoding for the Forkhead box protein 3, which is fundamental for regulatory T (Treg) cell function and immune tolerance.110 Immune abnormalities include lack of CD4+ CD25+ FOXP3+ Treg cells, eosinophilia, elevated serum IgE, and increased levels of autoantibodies.111,112 The most frequent atopic feature is severe eczematous dermatitis. However, other less common cutaneous manifestations may be present, including erythroderma, psoriasiform dermatitis, urticaria, pemphigoid nodularis, and alopecia universalis.113 Moreover, IPEX patients present with an increased incidence of food allergies.114 IPEX is usually fatal if not adequately treated. Medical management of IPEX with immunosuppressive agents such as tacrolimus and rapamycin may alleviate symptoms of the disease, but also expose patients to an increased risk of infections.115 Reports on HSCT have shown encouraging results, but it is fundamental to transplant before organ damage develops.115
As reported for WAS and WAS-like conditions, several IPEX-like syndromes have been described in the last years,116 including CD25 deficiency, STAT5b deficiency, and Itchy E3 Ubiquitin Protein Ligase (ITCH) deficiency [Table 3]. Moreover, gain-of-function mutations (GOF) in STAT1, generally associated with mucocutaneous candidiasis, may manifest as an IPEX-like phenotype. Even though the presentation of these diseases shares many features with IPEX, clinical manifestations specific of each disorder may support the differential diagnosis.116 For instance, CD25 deficiency also manifests with chronic viral, fungal, and bacterial infections,117 while STAT5b deficiency is also characterized by growth-hormone insensitive dwarfism.118 Regarding the atopic phenotypes, allergic dysregulation with eczema and food allergy have been variably reported in all these conditions, often associated with elevated IgE levels and evidence of overt Th2 skewing.116
CBM-opathies
Caspase recruitment domain (CARD) proteins – B-cell CLL/lymphoma 10 (BCL10) – MALT1 paracaspase (MALT1), altogether known as CBM complexes, play a key role as signal transducers, favoring inflammatory and immune responses associated to both cell surface and intracellular receptors.119,120
Diseases due to mutations in genes that are part of this complex (termed CBM-opathies) are extremely heterogeneous and present with a wide variety of clinical manifestations, ranging from CID to primarily atopic phenotypes.119,120 In particular, germline CBM-opathies typically manifest with early-onset, severe atopic diseases include those carrying germline mutations affecting CARD11, CARD14, and MALT1.119,120
While complete LOF mutations in CARD11 cause profound CID121,122 and heterozygous GOF mutations cause an immunodeficiency associated to B-cell lymphoproliferative disease and referred to as B-cell expansion with NF-kB and T-cell anergy (BENTA),123, 124, 125, 126, 127 heterozygous dominant-negative mutations are responsible for a distinctive clinical entity called CARD11-associated atopy with dominant interference of NF-kB signaling (CADINS).128, 129, 130, 131 The most typical clinical manifestations reported in patients with CADINS include atopic disease, respiratory tract infections, and cutaneous viral infections.129 Nearly 90% of patients with CADINS present atopic diseases, with AD and asthma being the most frequent, followed by allergic rhinoconjunctivitis, food allergy, and eosinophilic esophagitis.129 Partial clinical overlap with previously described IEI has been reported in some patients with CADINS: atopy and viral infections (DOCK8 deficiency), skeletal abnormalities as retained teeth (STAT3-HIES), failure to thrive, diarrhea, and severe atopic dermatitis (IPEX).120 Besides, similar to these conditions, increased IgE, eosinophilia, and Th-2 skewed immune response are frequently observed in CADINS. Immunological phenotype is characterized by normal absolute T- and NK-cell numbers with normal/low B-cell numbers; T-cell proliferation is impaired and hypogammaglobulinemia with altered specific antibody response has been reported.129 Antimicrobial prophylaxis and intravenous immunoglobulin can be considered depending on the patient's immune profile and infectious history.129 Therapies under investigation include biologics targeting allergic immune dysregulation, such as dupilumab (anti-IL4Rα) or mepolizumab (anti-IL-5)4 and glutamine that showed promising in vitro results in partially restoring T-cell proliferation.129
Recently, Peled et al reported that heterozygous dominant-negative LOF mutations in CARD14 cause severe atopic dermatitis.132 Of note, GOF CARD14 mutations were previously linked to psoriasis and pityriasis rubra pilaris.133,134 Patients with LOF mutations generally manifest with severe atopic dermatitis along with other atopic features, including markedly increased serum IgE levels, asthma, allergic rhinitis, and food allergies. Susceptibility to recurrent pyogenic and viral skin infections and respiratory tract infections is also commonly described in these patients.
Finally, patients carrying biallelic LOF mutations in MALT1 may present with atopic diseases, mainly dermatitis135, 136, 137, 138; however, most frequent clinical manifestations include recurrent infections of the skin and of the respiratory and gastrointestinal tracts, failure to thrive, periodontal disease and inflammatory gastrointestinal disease.
Other IEI presenting with allergic manifestations
Selective IgA deficiency (SIgAD) has a prevalence in Europe of nearly 1 in 600.139 However, the genetic causes underpinning SIgAD are known for a limited number of cases and a clinical/immunologic work-up followed by targeted gene mutation analysis has been proposed for an approach to IgA deficient patients.140 Although it is often asymptomatic, SIgA may present with recurrent respiratory infections and autoimmune diseases; moreover, allergic diseases may be the first and/or only clinical manifestation of this condition.139
Mutations in CARMIL2 (Capping Protein Regulator And Myosin 1 Linker 2), also known as RLTPR (RGD, leucine-rich repeat, tropomodulin and proline-rich-containing protein) affect the CD28-responsive pathway in T cells and the BCR-responsive pathway in B cells and have been reported in patients with cutaneous and pulmonary allergy, as well as a variety of bacterial and fungal infectious diseases, including invasive tuberculosis and mucocutaneous candidiasis.141
Janus kinase 1 (JAK1) GOF is responsible for severe atopic dermatitis and hypereosinophilic syndrome characterized by severe eosinophilia with eosinophilic infiltration of the liver and gastrointestinal tract, massive hepatosplenomegaly , autoimmune thyroid disease, and failure to thrive.142
Myeloid differentiation primary response protein 88 (MYD88) deficiency is responsible for a Mendelian predisposition to bacterial infections caused principally by pyogenic bacteria. MYD88 is a cytosolic protein recruited by IL-1 receptors (IL-1Rs) and toll-like receptors (TLRs) to trigger the activation of NF-kB pathway and inflammatory cytokine gene transcription. High IgE levels have been reported in patients with MYD88 deficiency, but their correlation with allergic manifestations need to be clearly defined.143,144
NF-kB is a ubiquitous transcription factor member of the Rel proto-oncogene family and regulates the expression of several genes involved in inflammatory and immune responses.145 Mutations in genes that affect nuclear factor kB (NF-kB)– dependent signaling are associated with a number of immunodeficiencies including anhidrotic ectodermodysplasia with immunodeficiency (EDA-ID, also known as NEMO deficiency), NFKB1 deficiency and NFKB2 deficiency, in addition to the already described CADINS.145, 146, 147 EDA-ID is characterized by hypotrichosis, hypodontia, hypohidrosis and typical facial features (protruding forehead, characteristic periorbital hyperpigmentation) which are usually associated with immunologic defects such as susceptibility to opportunistic infections, hypogammaglobulinemia, and impaired NK-cell activity.148 Heterozygous NFKB1 gene mutations cause common variable immunodeficiency (CVID)149,150 while NFKB2 gene defects have been shown to be associated with B cell dysregulation in patients with common variable immunodeficiency (CVID) or combined immunodeficiency (CID)151 Among phenocopies of IEI, somatic, GOF STAT5b mutation in a hematopoietic progenitor has been recently reported in 2 patients with a novel syndrome of nonclonal eosinophilia, atopic dermatitis, urticarial rash, and diarrhea.152
Conclusions
Human IEI represent an expanding universe.5,153 In the last 10 years, fundamental insights into the immunopathogenesis of allergic diseases derived from the studies on allergic phenotypes caused by discrete monogenic mutations. Improvement in genetic testing has led to more specific diagnosis and delineation of immune dysregulation syndromes characterized by the hyper IgE phenotype of eczema, recurrent infections, elevated serum IgE and/or hypereosinophilia.
IEI could be misrecognized because of the predominant clinical features of atopy. Without considering an underlying IEI, some individuals will remain undiagnosed, with a high risk of morbidity and mortality. An underlying IEI should be considered, especially in severe cases of atopic diseases with concurrent signs of autoimmunity and recurrent infections, unusual clinical course and lack of response to classical treatment strategies. Common features of IEI with atopic phenotypes and red flags to suspect IEI in the context of atopy should always be carefully considered for every patient. Once suspected, a comprehensive immunological evaluation is required, and genetic testing is essential to identify the specific genetic abnormality.
Integration of knowledge between allergists and immunologists is necessary to make a timely and correct diagnosis of IEI, predict the clinical course, and determine the indication for HSCT and targeted therapies.
Funding
This work was fully supported by the Italian Society of Pediatric Allergy and Immunology (SIAIP).
Consent for publication
All the authors give the consent for publication in the journal.
Ethics approval
Ethics approval was not required for this literature review. Informed consent was obtained from patients to publish the pictures reported in the four figures.
Author contributions
RC and FC conceived the review and the research method of bibliographic sources. All the authors performed the research, the analysis and the selection of the sources. RC wrote the first draft of the manuscript. All the authors critically revised the manuscript. GLM and FC supervised the project. All the authors accepted the final version of the manuscript.
Availability of data and materials
All the cited sources are available and reported in the reference list.
Declaration of competing interest
The authors report no competing interests. The authors have no conflict of interest to disclose with respect to this study.
Acknowledgements
This is a work of the Immunology Task Force of the Italian Society of Pediatric Allergy and Immunology (SIAIP). RC is supported by the Fellowship “Progressi in Biologia e Medicina” from Fondazione Ghislieri, Collegio Ghislieri, Pavia, Italy.
References
- 1.Tangye S.G., Al-Herz W., Bousfiha A. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee [published correction appears in J clin immunol. 2020 feb 22;:] J Clin Immunol. 2020;40(1):24–64. doi: 10.1007/s10875-019-00737-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delmonte O.M., Castagnoli R., Calzoni E., Notarangelo L.D. Inborn errors of immunity with immune dysregulation: from bench to bedside. Front Pediatr. 2019 Aug 27;7:353. doi: 10.3389/fped.2019.00353. PMID: 31508401; PMCID: PMC6718615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan S.K., Gelfand E.W. Primary immunodeficiency masquerading as allergic disease. Immunol Allergy Clin. 2015;35(4):767–778. doi: 10.1016/j.iac.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Castagnoli R., Licari A., Manti S., Chiappini E., Marseglia G.L. Type-2 inflammatory mediators as targets for precision medicine in children. Pediatr Allergy Immunol. 2020 Nov;31;26:17–19. doi: 10.1111/pai.13340. PMID: 33236434. [DOI] [PubMed] [Google Scholar]
- 5.Notarangelo L.D., Bacchetta R., Casanova J.L., Su H.C. Human inborn errors of immunity: an expanding universe. Sci Immunol. 2020;5(49) doi: 10.1126/sciimmunol.abb1662. eabb1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Herz W., Chou J., Delmonte O.M. Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population. Front Immunol. 2019;9:3146. doi: 10.3389/fimmu.2018.03146. Published 2019 Jan 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozcan E., Notarangelo L.D., Geha R.S. Primary immune deficiencies with aberrant IgE production. J Allergy Clin Immunol. 2008;122(6):1054–1064. doi: 10.1016/j.jaci.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 8.Sokol K., Milner J.D. The overlap between allergy and immunodeficiency. Curr Opin Pediatr. 2018;30(6):848–854. doi: 10.1097/MOP.0000000000000697. [DOI] [PubMed] [Google Scholar]
- 9.Milner J.D. Primary atopic disorders. Annu Rev Immunol. 2020;38:785–808. doi: 10.1146/annurev-immunol-042718-041553. [DOI] [PubMed] [Google Scholar]
- 10.Sacco K.A., Milner J.D. Gene-environment interactions in primary atopic disorders. Curr Opin Immunol. 2019;60:148–155. doi: 10.1016/j.coi.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Lyons J.J., Milner J.D. The clinical and mechanistic intersection of primary atopic disorders and inborn errors of growth and metabolism. Immunol Rev. 2019;287(1):135–144. doi: 10.1111/imr.12727. [DOI] [PubMed] [Google Scholar]
- 12.Lyons J.J., Milner J.D. Primary atopic disorders. J Exp Med. 2018;215(4):1009–1022. doi: 10.1084/jem.20172306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woellner C., Gertz E.M., Schaffer A.A. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. J Allergy Clin Immunol. 2010;125:424–432. doi: 10.1016/j.jaci.2009.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grimbacher B., Holland S.M., Gallin J.I. Hyper-IgE syndrome with recurrent infections - an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton R.G., Adkinson N.F. 23. Clinical laboratory assessment of IgE-dependent hypersensitivity. J Allergy Clin Immunol. 2003;111(2 Supplement 2):S687–S701. doi: 10.1067/mai.2003.123. [DOI] [PubMed] [Google Scholar]
- 16.Litonjua A.A., Celedón J.C., Hausmann J. Variation in total and specific IgE: effects of ethnicity and socioeconomic status. J Allergy Clin Immunol. 2005;115:751–757. doi: 10.1016/j.jaci.2004.12.1138. [DOI] [PubMed] [Google Scholar]
- 17.Grimbacher B., Belohradsky B.H., Holland S.M. Immunoglobulin E in primary immunodeficiency diseases. Allergy. 2002;57:995–1007. doi: 10.1034/j.1398-9995.2002.02168.x. [DOI] [PubMed] [Google Scholar]
- 18.Rael E.L., Marshall R.T., McClain J.J. The hyper-IgE syndromes: lessons in nature, from bench to bedside. World Allergy Organ J. 2012 Jul;5(7):79–87. doi: 10.1097/WOX.0b013e31825a73b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis S.D., Schaller J., Wedgwood R.J. Job's Syndrome. Recurrent, "cold", staphylococcal abscesses. Lancet. 1966;1(7445):1013–1015. doi: 10.1016/s01406736(66)90119-x. [DOI] [PubMed] [Google Scholar]
- 20.Minegishi Y., Saito M., Tsuchiya S. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 21.Kumanovics A., Perkins S.L., Gilbert H. Diffuse large B cell lymphoma in hyper-IgE syndrome due to STAT3 mutation. J Clin Immunol. 2010;30(6):886–893. doi: 10.1007/s10875-010-9452-z. [DOI] [PubMed] [Google Scholar]
- 22.Leonard G.D., Posadas E., Herrmann P.C. Non-Hodgkin’s lymphoma in Job's syndrome: a case report and literature review. Leuk Lymphoma. 2004;45(12):2521–2525. doi: 10.1080/10428190400004463. [DOI] [PubMed] [Google Scholar]
- 23.Freeman A.F., Avila E.M., Shaw P.A. Coronary artery abnormalities in Hyper-IgE syndrome. J Clin Immunol. 2011;31(3):338–345. doi: 10.1007/s10875-011-9515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arora M., Bagi P., Strongin A. Gastrointestinal manifestations of STAT3-deficient Hyper-IgE syndrome. J Clin Immunol. 2017;37(7):695–700. doi: 10.1007/s10875-017-0429-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergerson J.R.E., Freeman A.F. An update on syndromes with a hyper-IgE phenotype. Immunol Allergy Clin. 2019;39(1):49–61. doi: 10.1016/j.iac.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 26.Ponsford M.J., Klocperk A., Pulvirenti F. Hyper-IgE in the allergy clinic - when is it primary immunodeficiency? Allergy. 2018;73(11):2122–2136. doi: 10.1111/all.13578. [DOI] [PubMed] [Google Scholar]
- 27.Schimke L.F., Sawalle-Belohradsky J., Roesler J. Diagnostic approach to the hyper-IgE syndromes: immunologic and clinical key findings to differentiate hyper-IgE syndromes from atopic dermatitis [published correction appears in J Allergy Clin Immunol. 2010 Nov;126(5):1015] J Allergy Clin Immunol. 2010;126(3):611–617. doi: 10.1016/j.jaci.2010.06.029. e1. [DOI] [PubMed] [Google Scholar]
- 28.Boos A.C., Hagl B., Schlesinger A. Atopic dermatitis, STAT3-and DOCK8-hyper-IgE syndromes differ in IgE-based sensitization pat- tern. Allergy. 2014;69:943–963. doi: 10.1111/all.12416. [DOI] [PubMed] [Google Scholar]
- 29.Siegel A.M., Stone K.D., Cruse G. Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast cell degranulation. J Allergy Clin Immunol. 2013;132:1388–1396. doi: 10.1016/j.jaci.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castagnoli R., Delmonte O.M., Calzoni E., Notarangelo L.D. Hematopoietic stem cell transplantation in primary immunodeficiency diseases: current status and future perspectives. Front Pediatr. 2019;7:295. doi: 10.3389/fped.2019.00295. Published 2019 Aug 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q., Davis J.C., Lamborn I.T. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046–2055. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engelhardt K.R., McGhee S., Winkler S. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–1302. doi: 10.1016/j.jaci.2009.10.038. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aydin S.E., Kilic S.S., Aytekin C. DOCK8 deficiency: clinical and immunological phenotype and treatment options - a review of 136 patients. J Clin Immunol. 2015;35(2):189–198. doi: 10.1007/s10875-014-0126-0. [DOI] [PubMed] [Google Scholar]
- 34.Engelhardt K.R., Gertz M.E., Keles S. The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2015;136(2):402–412. doi: 10.1016/j.jaci.2014.12.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Herz W., Ragupathy R., Massaad M.J. Clinical, immunologic and genetic profiles of DOCK8-deficient patients in Kuwait. Clin Immunol. 2012;143(3):266–272. doi: 10.1016/j.clim.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aydin S.E., Freeman A.F., Al-Herz W. Hematopoietic stem cell transplantation as treatment for patients with DOCK8 deficiency. J Allergy Clin Immunol Pract. 2019;7:848–855. doi: 10.1016/j.jaip.2018.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Béziat V., Li J., Lin J.X. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. 2018;3(24) doi: 10.1126/sciimmunol.aat4956. eaat4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frey-Jakobs S., Hartberger J.M., Fliegauf M. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci Immunol. 2018;3(24) doi: 10.1126/sciimmunol.aat4941. eaat4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwerd T., Twigg S.R.F., Aschenbrenner D. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med. 2017;214(9):2547–2562. doi: 10.1084/jem.20161810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Béziat V., Tavernier S.J., Chen Y.H. Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med. 2020;217(6):e20191804. doi: 10.1084/jem.20191804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spencer S., Köstel Bal S., Egner W. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med. 2019;216(9):1986–1998. doi: 10.1084/jem.20190344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyons J.J., Liu Y., Ma C.A. ERBIN deficiency links STAT3 and TGF-beta pathway defects with atopy in humans. J Exp Med. 2017;214(3):669–680. doi: 10.1084/jem.20161435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frischmeyer-Guerrerio P.A., Guerrerio A.L., Oswald G. TGFβ receptor mutations impose a strong predisposition for human allergic disease. Sci Transl Med. 2013;5(195) doi: 10.1126/scitranslmed.3006448. 195ra194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y., Yu X., Ichikawa M. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. 2014;133(5):1400–1409. doi: 10.1016/j.jaci.2014.02.013. 1409.e1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Comel M. Ichthyosis linearis circumflexa. Dermatol. 1949;98:133–136. [PubMed] [Google Scholar]
- 46.Renner E.D., Hartl D., Rylaarsdam S. Comèl-Netherton syndrome – defined as primary immunodeficiency. J Allergy Clin Immunol. 2009;124:536–543. doi: 10.1016/j.jaci.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chavanas S., Bodemer C., Rochat A. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25(2):141–142. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 48.Taiber S., Samuelov L., Mohamad J. SAM syndrome is characterized by extensive phenotypic heterogeneity. Exp Dermatol. 2018;27(7):787–790. doi: 10.1111/exd.13551. [DOI] [PubMed] [Google Scholar]
- 49.Smith F.J., Irvine A.D., Terron-Kwiatkowski A. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38(3):337–342. doi: 10.1038/ng1743. [DOI] [PubMed] [Google Scholar]
- 50.Israeli S., Zamir H., Sarig O., Bergman R., Sprecher E. Inflammatory peeling skin syndrome caused by a mutation in CDSN encoding corneodesmosin. J Invest Dermatol. 2011;131(3):779–781. doi: 10.1038/jid.2010.363. [DOI] [PubMed] [Google Scholar]
- 51.Samuelov L., Sarig O., Harmon R.M. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. 2013;45(10):1244–1248. doi: 10.1038/ng.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McAleer M.A., Pohler E., Smith F.J. Severe dermatitis, multiple allergies, and metabolic wasting syndrome caused by a novel mutation in the N-terminal plakin domain of desmoplakin. J Allergy Clin Immunol. 2015;136(5):1268–1276. doi: 10.1016/j.jaci.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sherrill J.D., Kc K., Wu D. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol. 2014;7(3):718–729. doi: 10.1038/mi.2013.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paller A.S., Czarnowicki T., Renert-Yuval Y. The spectrum of manifestations in desmoplakin gene (DSP) spectrin repeat 6 domain mutations: immunophenotyping and response to ustekinumab. J Am Acad Dermatol. 2018;78:498–505. doi: 10.1016/j.jaad.2017.10.026. e2. [DOI] [PubMed] [Google Scholar]
- 55.Vakkilainen S., Puhakka L., Klemetti P. Novel DSP spectrin 6 region variant causes neonatal erythroderma, failure to thrive, severe herpes simplex infections and brain lesions. Acta Derm Venereol. 2019;99(9):789–796. doi: 10.2340/00015555-3203. [DOI] [PubMed] [Google Scholar]
- 56.Minegishi Y., Saito M., Morio T. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006 Nov;25(5):745–755. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 57.Nemoto M., Hattori H., Maeda N. Compound heterozygous TYK2 mutations underlie primary immunodeficiency with T-cell lymphopenia. Sci Rep. 2018 May 3;8(1):6956. doi: 10.1038/s41598-018-25260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kreins A.Y., Ciancanelli M.J., Okada S. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. 2015 Sep 21;212(10):1641–1662. doi: 10.1084/jem.20140280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kilic S.S., Hacimustafaoglu M., Boisson-Dupuis S. A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr. 2012 Jun;160(6):1055–1057. doi: 10.1016/j.jpeds.2012.01.056. Epub 2012 Mar 7. Erratum in: J Pediatr. 2012 Nov;161(5):974. Erratum in: J Pediatr. 2013 Mar;162(3):658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu P., Chen S., Wu B., Chen J., Lv G. A TYK2Gene mutation c.2395G>A leads to TYK2 deficiency: a case report and literature review. Front Pediatr. 2020 May 27;8:253. doi: 10.3389/fped.2020.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Omenn G.S. Familial reticulonendotheliosis with eosinophilia. N Engl J Med. 1965;273:427–432. doi: 10.1056/NEJM196508192730806. [DOI] [PubMed] [Google Scholar]
- 62.Villa A., Santagata S., Bozzi F. Partial V(D)J recombination activity leads to Omenn syndrome. Cell. 1998;93:885–896. doi: 10.1016/s0092-8674(00)81448-8. [DOI] [PubMed] [Google Scholar]
- 63.Villa A., Notarangelo L.D. RAG gene defects at the verge of immunodeficiency and immune dysregulation. Immunol Rev. 2019;287(1):73–90. doi: 10.1111/imr.12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roifman C.M., Gu Y., Cohen A. Mutations in the RNA component of RNase mito- chondrial RNA processing might cause Omenn syndrome. J Allergy Clin Immunol. 2006;117:897–903. doi: 10.1016/j.jaci.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 65.Ege M., Ma Y., Manfras B. Omenn syndrome due to Artemis mutations. Blood. 2005;105:4179–4186. doi: 10.1182/blood-2004-12-4861. [DOI] [PubMed] [Google Scholar]
- 66.Shibata F., Toma T., Wada T. Skin infiltration of CD56(bright) CD16(-) natural killer cells in a case of X-SCID with Omenn syndrome-like manifestations. Eur J Haematol. 2007;79:81–85. doi: 10.1111/j.1600-0609.2007.00874.x. [DOI] [PubMed] [Google Scholar]
- 67.Giliani S., Bonfim C., de Saint Basile G. Omenn syndrome in an infant with IL7RA gene mutation. J Pediatr. 2006;148:272–274. doi: 10.1016/j.jpeds.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 68.Roifman C.M., Zhang J., Atkinson A., Grunebaum E., Mandel K. Adenosine deaminase deficiency can present with features of Omenn syndrome. J Allergy Clin Immunol. 2008;121:1056–1058. doi: 10.1016/j.jaci.2007.12.1148. [DOI] [PubMed] [Google Scholar]
- 69.Gennery A.R., Slatter M.A., Rice J. Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell + severe combined immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol. 2008;153(1):75–80. doi: 10.1111/j.1365-2249.2008.03681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grunebaum E., Bates A., Roifman C.M. Omenn syndrome is associated with mutations in DNA ligase IV. J Allergy Clin Immunol. 2008;122(6):1219–1220. doi: 10.1016/j.jaci.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 71.Turul T., Tezcan I., Artac H. Clinical heterogeneity can hamper the diagnosis of patients with ZAP70 deficiency. Eur J Pediatr. 2009;168(1):87–93. doi: 10.1007/s00431-008-0718-x. [DOI] [PubMed] [Google Scholar]
- 72.Volpi S., Yamazaki Y., Brauer P.M. EXTL3 mutations cause skeletal dysplasia, immune deficiency, and developmental delay. J Exp Med. 2017 Mar 6;214(3):623–637. doi: 10.1084/jem.20161525. Epub 2017 Feb 1. PMID: 28148688; PMCID: PMC5339678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Villa A., Notarangelo L.D., Roifman C.M. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. 2008;122(6):1082–1086. doi: 10.1016/j.jaci.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 74.Hoeger P.H., Harper J.I. Neonatal erythroderma: differential diagnosis and management of the “red baby”. Arch Dis Child. 1998;79:186–191. doi: 10.1136/adc.79.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lehman H., Gordon C. The skin as a window into primary immune deficiency diseases: atopic dermatitis and chronic mucocutaneous candidiasis. J Allergy Clin Immunol Pract. 2019;7(3):788–798. doi: 10.1016/j.jaip.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 76.Mazzolari E., Moshous D., Forino C. Hematopoietic stem cell transplantation in Omenn syndrome: a single-center experience. Bone Marrow Transplant. 2005;36(2):107–114. doi: 10.1038/sj.bmt.1705017. [DOI] [PubMed] [Google Scholar]
- 77.Denianke K.S., Frieden I.J., Cowan M.J., Williams M.L., McCalmont T.H. Cutaneous manifestations of maternal engraftment in patients with severe combined immunodeficiency: a clinicopathologic study. Bone Marrow Transplant. 2001 Aug;28(3):227–233. doi: 10.1038/sj.bmt.1703128. PMID: 11535989. [DOI] [PubMed] [Google Scholar]
- 78.Williams K.W., Milner J.D., Freeman A.F. Eosinophilia associated with disorders of immune deficiency or immune dysregulation. Immunol Allergy Clin. 2015 Aug;35(3):523–544. doi: 10.1016/j.iac.2015.05.004. PMID: 26209898; PMCID: PMC4688016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lawrence M.G., Barber J.S., Sokolic R.A. Elevated IgE and atopy in patients treated for early-onset ADA-SCID. J Allergy Clin Immunol. 2013 Dec;132(6):1444–1446. doi: 10.1016/j.jaci.2013.05.040. Epub 2013 Jul 26. PMID: 23895897; PMCID: PMC3844080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Candotti F. Clinical manifestations and pathophysiological mechanisms of the wiskott-aldrich syndrome. J Clin Immunol. 2018;38:13–27. doi: 10.1007/s10875-017-0453-z. [DOI] [PubMed] [Google Scholar]
- 81.Blundell M.P., Worth A., Bouma G., Thrasher A.J. The Wiskott-Aldrich syndrome: the actin cytoskeleton and immune cell function. Dis Markers. 2010;29:157–175. doi: 10.1155/2010/781523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Villa A., Notarangelo L., Macchi P. X–linked thrombocytopenia and Wiskott–Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nat Genet. 1995;9:414–417. doi: 10.1038/ng0495-414. [DOI] [PubMed] [Google Scholar]
- 83.Notarangelo L.D., Mazza C., Giliani S. Missense mutations of the WASP gene cause intermittent X-linked thrombocytopenia. Blood. 2002;99:2268–2269. doi: 10.1182/blood.V99.6.2268. [DOI] [PubMed] [Google Scholar]
- 84.Devriendt K., Kim A.S., Mathijs G. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27:313–317. doi: 10.1038/85886. [DOI] [PubMed] [Google Scholar]
- 85.Sullivan K.E., Mullen C.A., Blaese R.M., Winkelstein J.A. A multi-institutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994;125:876–885. doi: 10.1016/s0022-3476(05)82002-5. [DOI] [PubMed] [Google Scholar]
- 86.Elfeky R.A., Furtado-Silva J.M., Chiesa R. One hundred percent survival after transplantation of 34 patients with Wiskott-Aldrich syndrome over 20 years. J Allergy Clin Immunol. 2018;142:1654–1656. doi: 10.1016/j.jaci.2018.06.042. e7. [DOI] [PubMed] [Google Scholar]
- 87.Ferrua F., Marangoni F., Aiuti A., Roncarolo M.G. Gene therapy for Wiskott-Aldrich syndrome: history, new vectors, future directions. J Allergy Clin Immunol. 2020;146(2):262–265. doi: 10.1016/j.jaci.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sasahara Y. WASP-WIP complex in the molecular pathogenesis of Wiskott-Aldrich syndrome. Pediatr Int. 2016;58:4–7. doi: 10.1111/ped.12819. [DOI] [PubMed] [Google Scholar]
- 89.Janssen E., Tohme M., Hedayat M. A DOCK8-WIP-WASp complex links T cell receptors to the actin cytoskeleton. J Clin Invest. 2016;126:3837–3851. doi: 10.1172/JCI85774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lanzi G., Moratto D., Vairo D. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med. 2012;209:29–34. doi: 10.1084/jem.20110896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schwinger W., Urban C., Ulreich R. The phenotype and treatment of WIP deficiency: literature synopsis and review of a patient with pre-transplant serial donor lymphocyte infusions to eliminate CMV. Front Immunol. 2018;9:2554. doi: 10.3389/fimmu.2018.02554. Published 2018 Nov 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kuijpers T.W., Tool A.T.J., van der Bijl I. Combined immunodeficiency with severe inflammation and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol. 2017;140:273–277. doi: 10.1016/j.jaci.2016.09.061. e10. [DOI] [PubMed] [Google Scholar]
- 93.Kahr W.H.A., Pluthero F.G., Elkadri A. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease.Nat Commun. 2017;8:14816. doi: 10.1038/ncomms14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Volpi S., Cicalese M.P., Tuijnenburg P. A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol. 2019;143:2296–2299. doi: 10.1016/j.jaci.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Somech R., Lev A., Lee Y.N. Disruption of thrombocyte and T lymphocyte development by a mutation in ARPC1B. J Immunol. 2017;199:4036–4045. doi: 10.4049/jimmunol.1700460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brigida I., Zoccolillo M., Cicalese M.P. T-cell defects in patients with ARPC1B germline mutations account for combined immunodeficiency. Blood. 2018;132:2362–2374. doi: 10.1182/blood-2018-07-863431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lam M.T., Coppola S., Krumbach O.H.F. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med. 2019 Dec 2;216(12):2778–2799. doi: 10.1084/jem.20190147. Epub 2019 Oct 10. PMID: 31601675; PMCID: PMC6888978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou Y., Johnson J.L., Cerione R.A., Erickson J.W. Prenylation and membrane localization of Cdc42 are essential for activation by DOCK7. Biochemistry. 2013 Jun 25;52(25):4354–4363. doi: 10.1021/bi301688g. Epub 2013 Jun 14. PMID: 23718289; PMCID: PMC3752685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baschieri F., Confalonieri S., Bertalot G. Spatial control of Cdc42 signalling by a GM130-RasGRF complex regulates polarity and tumorigenesis. Nat Commun. 2014 Sep 11;5:4839. doi: 10.1038/ncomms5839. PMID: 25208761; PMCID: PMC4449154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Takenouchi T., Kosaki R., Niizuma T., Hata K., Kosaki K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: yet another locus for thrombocytopenia and developmental delay. Am J Med Genet. 2015 Nov;167A(11):2822–2825. doi: 10.1002/ajmg.a.37275. Epub 2015 Aug 6. PMID: 26386261. [DOI] [PubMed] [Google Scholar]
- 101.Martinelli S., Krumbach O.H.F., Pantaleoni F., Coppola S., Amin E., Pannone L., Nouri K., Farina L., Dvorsky R., Lepri F., Buchholzer M., Konopatzki R., Walsh L., Payne K., Pierpont M.E., Vergano S.S., Langley K.G., Larsen D., Farwell K.D., Tang S., Mroske C., Gallotta I., Di Schiavi E., Della Monica M., Lugli L., Rossi C., Seri M., Cocchi G., Henderson L., Baskin B., Alders M., Mendoza-Londono R., Dupuis L., Nickerson D.A., Chong J.X., University of Washington Center for Mendelian Genomics. Meeks N., Brown K., Causey T., Cho M.T., Demuth S., Digilio M.C., Gelb B.D., Bamshad M.J., Zenker M., Ahmadian M.R., Hennekam R.C., Tartaglia M., Mirzaa G.M. Functional dysregulation of CDC42 causes diverse developmental phenotypes. Am J Hum Genet. 2018 Feb 1;vol. 102(2):309–320. doi: 10.1016/j.ajhg.2017.12.015. Epub 2018 Jan 25. PMID: 29394990; PMCID: PMC5985417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Su H.C., Orange J.S. The growing spectrum of human diseases caused by inherited CDC42 mutations. J Clin Immunol. 2020 May;40(4):551–553. doi: 10.1007/s10875-020-00785-8. PMID: 32417998; PMCID: PMC7335263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gernez Y., de Jesus A.A., Alsaleem H. Severe autoinflammation in 4 patients with C-terminal variants in cell division control protein 42 homolog (CDC42) successfully treated with IL-1 beta inhibition. J Allergy Clin Immunol. 2019;144(4):1122–1125. doi: 10.1016/j.jaci.2019.06.017. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bucciol G., Pillay B., Casas-Martin J. Systemic inflammation and myelofibrosis in a patient with Takenouchi-Kosaki syndrome due to CDC42 Tyr64Cys mutation. J Clin Immunol. 2020;40(4):567–570. doi: 10.1007/s10875-020-00742-5. Epub 2020 Jan 18. [DOI] [PubMed] [Google Scholar]
- 105.Verboon J.M., Mahmut D., Kim A.R. Infantile myelofibrosis and myeloproliferation with CDC42 dysfunction. J Clin Immunol. 2020;40(4) doi: 10.1007/s10875-020-00778-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Szczawinska-Poplonyk A., Ploski R., Bernatowska E., Pac M. A novel CDC42 mutation in an 11-year old child manifesting as syndromic immunodeficiency, autoinflammation, hemophagocytic lymphohistiocytosis, and malignancy: a case report. Front Immunol. 2020;11:318. doi: 10.3389/fimmu.2020.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.He T., Huang Y., Ling J., Yang J. A new patient with NOCARH syndrome due to CDC42 defect. J Clin Immunol. 2020;40(4) doi: 10.1007/s10875-020-00786-7. [DOI] [PubMed] [Google Scholar]
- 108.Bekhouche B., Tourville A., Ravichandran Y. A toxic palmitoylation of Cdc42 enhances NFkappaB signaling and drives a severe autoinflammatory syndrome. J Allergy Clin Immunol. 2020;146(5):1201–1204. doi: 10.1016/j.jaci.2020.03.020. Epub 2020 Apr 10. [DOI] [PubMed] [Google Scholar]
- 109.Powell B.R., Buist N.R., Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. 1982;100:731–737. doi: 10.1016/s0022-3476(82)80573-8. [DOI] [PubMed] [Google Scholar]
- 110.Cepika A.-M., Sato Y., Liu J.M.-H., Uyeda M.J., Bacchetta R., Roncarolo M.G. Tregopathies: monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol. 2018;142:1679–1695. doi: 10.1016/j.jaci.2018.10.026. [DOI] [PubMed] [Google Scholar]
- 111.Gambineri E., Torgerson T.R., Ochs H.D. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol. 2003;15:430–435. doi: 10.1097/00002281-200307000-00010. [DOI] [PubMed] [Google Scholar]
- 112.Barzaghi F., Passerini L., Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. 2012;3:211. doi: 10.3389/fimmu.2012.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McGinness J.L., Bivens M.M., Greer K.E., Patterson J.W., Saulsbury F.T. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) associated with pemphigoid nodularis: a case report and review of the literature. J Am Acad Dermatol. 2006;55:143–148. doi: 10.1016/j.jaad.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 114.Torgerson T.R., Linane A., Moes N. Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology. 2007;132:1705–1717. doi: 10.1053/j.gastro.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 115.Barzaghi F., Amaya Hernandez L.C., Neven B. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J Allergy Clin Immunol. 2018;141:1036. doi: 10.1016/j.jaci.2017.10.041. 1049.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Verbsky J.W., Chatila T.A. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr Opin Pediatr. 2013;25(6):708–714. doi: 10.1097/MOP.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Caudy A.A., Reddy S.T., Chatila T., Atkinson J.P., Verbsky J.W. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 + lymphocytes. J Allergy Clin Immunol. 2007;119:482–487. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 118.Kofoed E.M., Hwa V., Little B. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349:1139–1147. doi: 10.1056/NEJMoa022926. [PubMed: 13679528] [DOI] [PubMed] [Google Scholar]
- 119.Lu H.Y., Bauman B.M., Arjunaraja S. The CBM-opathies-A rapidly expanding spectrum of human inborn errors of immunity caused by mutations in the CARD11-BCL10-MALT1 complex. Front Immunol. 2018;9:2078. doi: 10.3389/fimmu.2018.02078. Published 2018 Sep. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lu H.Y., Biggs C.M., Blanchard-Rohner G., Fung S.Y., Sharma M., Turvey S.E. Germline CBM-opathies: from immunodeficiency to atopy. J Allergy Clin Immunol. 2019;143(5):1661–1673. doi: 10.1016/j.jaci.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 121.Stepensky P., Keller B., Buchta M. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol. 2013;131:477–485. doi: 10.1016/j.jaci.2012.11.050. e1. [DOI] [PubMed] [Google Scholar]
- 122.Greil J., Rausch T., Giese T. Wholeexome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol. 2013;131:1376–1383. doi: 10.1016/j.jaci.2013.02.012. e3. [DOI] [PubMed] [Google Scholar]
- 123.Snow A.L., Xiao W., Stinson J.R. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med. 2012;209:2247–2261. doi: 10.1084/jem.20120831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Buchbinder D., Stinson J.R., Nugent D.J. Mild B-cell lymphocytosis in patients with a CARD11 C49Y mutation. J Allergy Clin Immunol. 2015;136:819–821. doi: 10.1016/j.jaci.2015.03.008. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brohl A.S., Stinson J.R., Su H.C. Germline CARD11 mutation in a patient with severe congenital B cell lymphocytosis. J Clin Immunol. 2015;35:32–46. doi: 10.1007/s10875-014-0106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gupta M., Aluri J., Desai M. Clinical, immunological, and molecular findings in four cases of B cell expansion with NF-kB and T cell anergy disease for the first time from India. Front Immunol. 2018;9:1049. doi: 10.3389/fimmu.2018.01049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Outinen T., Syrjanen J., Rounioja S., Saarela J., Kaustio M., Helminen M. Constant B cell lymphocytosis since early age in a patient with CARD11 mutation: a 20- year follow-up. Clin Immunol. 2016;165:19–20. doi: 10.1016/j.clim.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 128.Ma C.A., Stinson J.R., Zhang Y. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet. 2017;49:1192–1201. doi: 10.1038/ng.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dorjbal B., Stinson J.R., Ma C.A. Hypomorphic caspase activation and recruitment domain 11 (CARD11) mutations associated with diverse immunologic phenotypes with or without atopic disease. J Allergy Clin Immunol. 2019;143:1482–1495. doi: 10.1016/j.jaci.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dadi H., Jones T.A., Merico D. Combined immunodeficiency and atopy caused by a dominant negative mutation in caspase activation and recruitment domain family member 11 (CARD11) J Allergy Clin Immunol. 2018;141:1818–1830. doi: 10.1016/j.jaci.2017.06.047. e2. [DOI] [PubMed] [Google Scholar]
- 131.Biggs C.M., Lu H.Y., Turvey S.E. Monogenic immune disorders and severe atopic disease. Nat Genet. 2017;49:1162–1163. doi: 10.1038/ng.3925. [DOI] [PubMed] [Google Scholar]
- 132.Peled A., Sarig O., Sun G. Loss-of-function mutations in caspase recruitment domain-containing protein 14 (CARD14) are associated with a severe variant of atopic dermatitis. J Allergy Clin Immunol. 2017;143:173–181. doi: 10.1016/j.jaci.2018.09.002. e10. [DOI] [PubMed] [Google Scholar]
- 133.Jordan C.T., Cao L., Roberson E.D. PSORS2 is due to mutations in CARD14. Am J Hum Genet. 2012;90:784–795. doi: 10.1016/j.ajhg.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fuchs-Telem D., Sarig O., van Steensel M.A. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am J Hum Genet. 2012;91:163–170. doi: 10.1016/j.ajhg.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jabara H.H., Ohsumi T., Chou J. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol. 2013;132:151–158. doi: 10.1016/j.jaci.2013.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McKinnon M.L., Rozmus J., Fung S.Y. Combined immunodeficiency associated with homozygous MALT1 mutations. J Allergy Clin Immunol. 2014;133:1458–1462. doi: 10.1016/j.jaci.2013.10.045. e1-7. [DOI] [PubMed] [Google Scholar]
- 137.Punwani D., Wang H., Chan A.Y. Combined immunodeficiency due to MALT1 mutations, treated by hematopoietic celltransplantation. J Clin Immunol. 2015;35:135–146. doi: 10.1007/s10875-014-0125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Charbit-Henrion F., Jeverica A.K., Begue B., Markelj G., Parlato M., Avcin S.L. Deficiency in mucosa-associated lymphoid tissue lymphoma translocation 1: a novel cause of IPEX-like syndrome. J Pediatr Gastroenterol Nutr. 2017;64:378–384. doi: 10.1097/MPG.0000000000001262. [DOI] [PubMed] [Google Scholar]
- 139.Hammarström L., Vorechovsky I., Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID) Clin Exp Immunol. 2000;120:225–231. doi: 10.1046/j.1365-2249.2000.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Abolhassani H., Aghamohammadi A., Hammarström L. Monogenic mutations associated with IgA deficiency. Expet Rev Clin Immunol. 2016 Dec;12(12):1321–1335. doi: 10.1080/1744666X.2016.1198696. [DOI] [PubMed] [Google Scholar]
- 141.Wang Y., Ma C.S., Ling Y. Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med. 2016;213(11):2413–2435. doi: 10.1084/jem.20160576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Del Bel K.L., Ragotte R.J., Saferali A. JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immunol. 2017;139(6):2016–2020. doi: 10.1016/j.jaci.2016.12.957. e5. [DOI] [PubMed] [Google Scholar]
- 143.Giardino G., Gallo V., Somma D. Targeted next-generation sequencing revealed MYD88 deficiency in a child with chronic yersiniosis and granulomatous lymphadenitis. J Allergy Clin Immunol. 2016 May;137(5):1591–1595. doi: 10.1016/j.jaci.2015.09.050. e4. [DOI] [PubMed] [Google Scholar]
- 144.Chiriaco M., Di Matteo G., Conti F. First case of patient with two homozygous mutations in MYD88and CARD9 genes presenting with pyogenic bacterial infections, elevated IgE, and persistent EBV. Viremia. Front Immunol. 2019 Feb 14;10:130. doi: 10.3389/fimmu.2019.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang Q., Lenardo M.J., Baltimore D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017 Jan 12;168(1-2):37–57. doi: 10.1016/j.cell.2016.12.012. Epub 2017 Jan 12. PMID: 28086098; PMCID: PMC5268070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Orange J.S., Geha R.S. Finding NEMO: genetic disorders of NF-[kappa]B activation. J Clin Invest. 2003 Oct;112(7):983–985. doi: 10.1172/JCI19960. PMID: 14523034; PMCID: PMC200971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bryant V.L., Tangye S.G. The expanding spectrum of NFkB 1 deficiency. J Clin Immunol. 2016 Aug;36(6):531–532. doi: 10.1007/s10875-016-0310-5. Epub 2016 Jun 23. PMID: 27338826. [DOI] [PubMed] [Google Scholar]
- 148.Döffinger R., Smahi A., Bessia C. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001 Mar;27(3):277–285. doi: 10.1038/85837. PMID: 11242109. [DOI] [PubMed] [Google Scholar]
- 149.Fliegauf M., Bryant V.L., Frede N. Haploinsufficiency of the NF-κB1 subunit p50 in common variable immunodeficiency. Am J Hum Genet. 2015 Sep 3;97(3):389–403. doi: 10.1016/j.ajhg.2015.07.008. Epub 2015 Aug 13. PMID: 26279205; PMCID: PMC4564940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Boztug H., Hirschmugl T., Holter W. NF-κB1 haploinsufficiency causing immunodeficiency and EBV-driven lymphoproliferation. J Clin Immunol. 2016 Aug;36(6):533–540. doi: 10.1007/s10875-016-0306-1. Epub 2016 Jun 23. PMID: 27338827; PMCID: PMC4940442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Klemann C., Camacho-Ordonez N., Yang L. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol. 2019 Mar 19;10:297. doi: 10.3389/fimmu.2019.00297. PMID: 30941118; PMCID: PMC6435015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ma C.A., Xi L., Cauff B. Somatic STAT5b gain-of-function mutations in early onset nonclonal eosinophilia, urticaria, dermatitis, and diarrhea. Blood. 2017;129(5):650–653. doi: 10.1182/blood-2016-09-737817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Castagnoli R., Notarangelo L.D. Updates on new monogenic inborn errors of immunity. Pediatr Allergy Immunol. 2020 Nov;31;(26):57–59. doi: 10.1111/pai.13365. PMID: 33236415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the cited sources are available and reported in the reference list.