

Abstract
Purpose
There are limited genes known to cause primary ciliary dyskinesia (PCD)–associated asthenozoospermia. In the present study, we aimed to expand the spectrum of mutations in PCD and to provide new information for genetic counseling diagnoses and the treatment of male infertility in PCD.
Methods
One sterile patient with typical situs inversus was recruited to our center, and semen sample was collected. We performed whole-exome sequencing (WES) on the patient to identify the pathogenic mutations associated with PCD and used transmission electron microscopy to investigate spermatozoal ultrastructure. In addition, western blotting and immunofluorescence staining were used to confirm the untoward impact of the variant on the expression of LRRC6, as well as on the dynein arm proteins in the patient’s spermatozoa.
Results
We identified a homozygous nonsense variant c.749G>A (p.W250*) of LRRC6 in the PCD patient. This variant severely impaired LRRC6 expression and further led to negative effects on dynein arm protein expression in the spermatozoa of the affected individual, which eventually caused defects in sperm ultrastructure and motility. Moreover, we are the first to report a positive prognosis using intracytoplasmic sperm injection (ICSI) for LRRC6-associated male infertility.
Conclusions
Our findings strongly implicated the homozygous mutation of c.749G>A (p.W250*) in LRRC6 as a new genetic cause of PCD, uncovering its involvement in defective sperm flagella and poor sperm motility. Furthermore, we posit that patients with LRRC6 mutations may have good outcomes with ICSI treatment. These findings add to the literature on the genetic diagnoses and treatment of male infertility associated with PCD.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10815-020-02036-6.
Keywords: Asthenozoospermia, LRRC6, PCD, ICSI, Mutation
Introduction
There is growing international concern with respect to male sterility, which now affects over 20 million people worldwide [1]. Asthenozoospermia is certainly considered to be an important cause of male infertility and is usually observed in approximately 25% of sterile men [2, 3]. A notable type of asthenozoospermia that influences male infertility is associated with primary ciliary dyskinesia (PCD) [4]. Genetically, PCD covers a rare heterogeneous group of autosomal recessive or X-linked disorders that contribute to dysfunctional immotile or dyskinetic cilia and flagella, affecting an estimated 1 in 15,000–30,000 individuals of the general population [5]. PCD is typically characterized by recurrent respiratory infections, chronic cough, bronchiectasis, and sinusitis. Approximately 50% of PCD patients suffer from male infertility that results from aberrant ultrastructural changes in sperm flagella. Additionally, situs inversus is found in approximately 45–55% of subjects with PCD and is defined as Kartagener syndrome [6, 7]. The phenotypes of PCD are generally non-specific, and the available diagnostic approaches are very limited, which further results in delayed or missed diagnoses [8]. Currently, optimal diagnostic testing is based upon transmission electron microscopy (TEM) of ciliary ultrastructure, with the addition of genetic diagnosis. However, the genes known to cause PCD account for fewer than 70% of human PCD cases [9, 10]. The leucine-rich repeat (LRR)–containing 6 (LRRC6) (NM_012472.5) is a major PCD-causative gene, which is distinctly expressed in testis and respiratory cells. Since LRRC6 was first described as a PCD-causing gene in 2012, only 15 pathogenic mutations have been identified [11–14] (Table 1). Therefore, it is of paramount importance to elucidate the unknown genetic factors involved so as to better understand the pathogenic mechanisms underlying PCD and to provide the appropriate therapeutic schedules for it.
Table 1.
Overview of the LRRC6 pathogenic mutations
| Genotype | cDNA changes | Protein changes | Fertility | References |
|---|---|---|---|---|
| Compound heterozygous |
c.574C>T c.576dupA |
p.Gln192* p.Glu193Argfs*4 |
Asthenospermia | Kott et al. [11] |
|
c.598_599delAA c.436G>C |
p.Lys200Glufs*3 p.Lys200Glufs*3 |
Asthenospermia | Kott et al. [11] | |
|
c.259 T>C c.436G>C |
p.Cys87Arg p.Asp146His |
NM | Zariwala et al. [13] | |
|
c.183T>G c.179-1G>A |
p.N61K | Infertility | Liu and Luo [14] | |
| Homozygous | c.220G>C | p.Ala74Pro | Asthenospermia | Kott et al. [11] |
| c.169_173delinsTCCCAAT | p.Gly57Serfs*3 | NM | Zariwala et al. [13] | |
| c.562C>T | p.Gln188* | NM | Zariwala et al. [13] | |
| c.630delG | p.Trp210Cysfs*12 | NM | Zariwala et al. [13] | |
| c.598_599delAA | p.Lys200Glufs*3 | Asthenospermia/hypofertility/NR | Kott et al. [11], Zariwala et al. [13] | |
| c.653+1G>A | splice | NM | Zariwala et al. [13] | |
| c.710_711delCA | p.Thr237Lysfs*7 | NM | Zariwala et al. [13] | |
| c.891delA | p.Ala298Profs*2 | NM | Zariwala et al. [13] | |
| c.169_174delins8 | p.Gly57Serfs*2 | NM | ||
| c.436G>C | p.Asp146His | NM | Horani et al. [12] | |
| c.64dupT | p.S22fs | NM | Yue et al. [15] |
NR not relevant, NM not mentioned
In the present study, we performed whole-exome sequencing (WES) on a PCD-affected family and uncovered a novel homozygous nonsense mutation of c.749G>A in LRRC6 gene in the infertile proband. The expression of LRRC6 was completely absent in the flagella of the spermatozoa, and ultrastructural analysis of the flagella also showed distinct aberrations. Thus, genetic diagnosis is an acceptable way of assisting in the diagnosis of PCD, and the effective treatment can then be provided in a timely manner.
Materials and methods
Subjects
We recruited an infertile male patient from southwest of China, performed routine semen analysis according to WHO guidelines (WHO, 2010), and collected a peripheral whole blood sample for genetic analyses. This study was approved by the Ethical Review Board of West China Second University Hospital, Sichuan University, and written informed consent was obtained from each subject.
Whole-exome sequencing and Sanger sequencing
We isolated genomic DNA from the infertile patient’s peripheral blood using a whole-blood DNA purification kit (Axygen) and performed WES per our previous study [15]. Variants meeting the following criteria were collected for further analysis: (1) any nonsynonymous, frameshift, splicing, start-lost, or stop-gain variations and (2) variations with minor allelic frequencies ≤ 0.01 in any of the public databases—including the 1000 Genome Project, gnomAD, and ExAC Browser. Sanger sequencing was used to validate the mutation detected by WES in the proband. The PCR primers were F, 5'ACCACCTACAGGCACCAGAC-3' and R, 5'GGCTACTGCTTCCCTCTCCTT-3'.
Electron microscopy and concentration Papanicolaou staining
The appearance of patient spermatozoa was assessed with concentration Papanicolaou staining, and we viewed flagellar ultrastructure in cross-sections with transmission electron microscopy (TEM) according to our previously published methods [15].
Immunofluorescence staining
We collected sperm and testicular samples from the patient, a normal control, and from 12-week-old C57BL mice and performed immunofluorescence staining as in our previous study [15]. The primary antibodies used in the present study were anti-LRRC6 (1:50, TA805963S, OriGene), anti-α-tubulin (1:1000, ab24610, Abcam), anti-DNAH1 (1:50, HPA036806, Sigma-Aldrich), anti-DNAH8 (1:50, HPA028447, Sigma-Aldrich), anti-DNAH9 (1:50, PA5-45744, Invitrogen), anti-DNAH10 (1:50, orb3004, Biorbyt), and anti-DNAH17 (1:50, 24488-1-AP, Proteintech).
Western blotting
We executed western immunoblotting on the sperm samples from our patient and the normal control as in our previous study [15], and the primary antibodies were anti-LRRC6 (1:1000, TA805963S, OriGene) and anti-GAPDH (1:5000, 200306-7E4, Zen-Bio).
Results
Presentation in a PCD patient with infertility phenotype
The 32-year-old man recruited to our study had been infertile for 3 years and complained of having symptoms of chronic coughing and recurrent respiratory infections. Further examinations revealed typical manifestations of PCD-situs inversus (Fig. 1a). The proband came from a consanguineous family, and his semen analysis parameters repeatedly exhibited severe asthenozoospermia and even akinetospermia, with almost all spermatozoa observed as immotile (Table 2). The patient otherwise showed normal results for the full set of infertility-related examinations. Although we observed a normal morphology of sperm flagella in the patient using concentration Papanicolaou staining (Supplementary Fig. 1A), defective ultrastructure was shown in the sperm flagellar cross-sections by TEM analysis. Regarding the flagellar mid-piece, the normal “9 + 2” structure of the axoneme was missing in 94% of the sections, with no outer doublet microtubules (ODMTs) and central pair of microtubule doublets (CPs); the remaining ultrastructure was arranged irregularly and surrounded by an incomplete mitochondrial sheath (MS). In the principal piece, CPs were completely absent (with a “9 + 0” structure), and the fibrous sheath (FS) was disorganized in 91% of the cross-sections. Moreover, the end piece showed marked displacement of the peripheral doublets in 92% of the sections (Fig. 1b, c). In summary, these data suggested that the asthenozoospermia of this PCD-affected man was due to the ultrastructural aberrations in his spermatozoa.
Fig. 1.

Phenotype of patient. a CT scan of patient. i, The left lingual lobe and lower bronchi of the right lung show cystic dilatation (red arrow), and the wall manifests uneven thickening with signet-ring sign. ii, The aortic arch is on the right, with the main body of the heart, and the apex of the heart is pointed to the right (red arrow). iii, The liver, stomach, and spleen were all reversed (red arrows). b Results of ultrastructural abnormalities of flagellar cross-sections as observed by TEM in the infertile patient and a normal control (scale bars, 50 nm). c Percent of defective axonemes in a normal control and in our patient
Table 2.
Semen analysis of the PCD patient
| Semen parameters | Patient | Normal control | Normospermic parameters |
|---|---|---|---|
| Sperm volume (mL) | 2.6 | 2.7 | ≥ 1.5 |
| Sperm concentration (million/mL) | 16 | 58 | ≥ 15 |
| Motile sperm (%) | 0 | 50 | ≥ 40 |
| Vitality (%) | 75 | 73 | ≥ 58 |
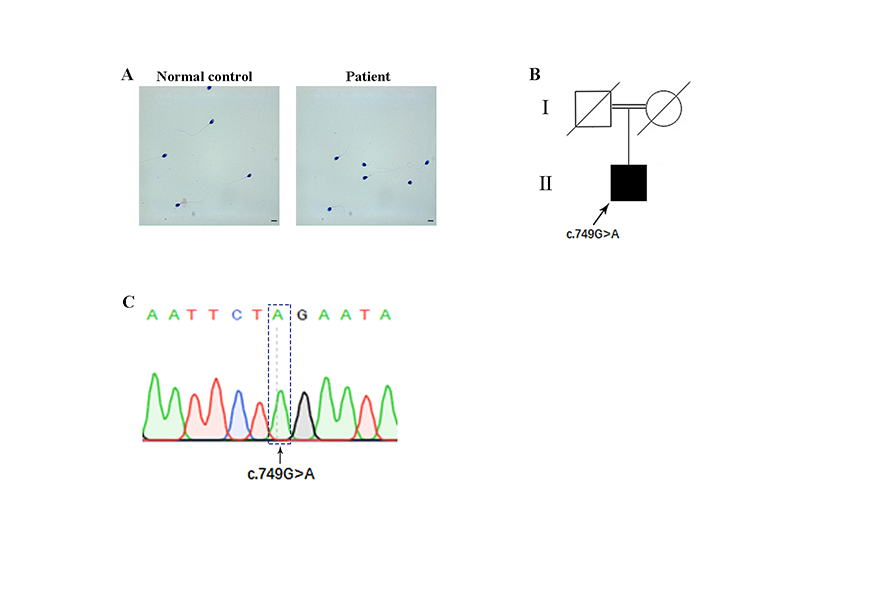
A novel loss-of-function variant of LRRC6 in the PCD-affected individual
Pedigree analysis revealed autosomal recessive inheritance associated with PCD (Supplementary Fig. 1B). To better understand the genetic cause of PCD in this family, we conducted WES on the affected individual. Unexpectedly, we detected a homozygous nonsense variant in exon6 (c.749G>A, p.W250*) of the LRRC6 gene (NM_012472.5), which was not recorded in 1000 Genomes, dbSNP— and with an extremely low frequency in gnomAD (frequency of 0.0000544) and ExAC databases (frequency of 0.00000831)—and predicted to be potentially deleterious. We therefore hypothesized that the novel homozygous mutation of the LRRC6 gene was responsible for the PCD phenotype in this infertile man. Subsequently, Sanger sequencing confirmed the presence of this nonsense mutation in the affected individual (the parents of this patient had already died) (Supplementary Fig. 1C). The nonsense mutation was localized to exon 6, and the corresponding amino acid is highly conserved among different species (Fig. 2a, b). Owing to prematurely terminated translation, the truncated protein with the omission of the important domains of LRRC6, including the poly-Lys and CS domains, might supposedly be easily degraded (Fig. 2a).
Fig. 2.

Negative influence of LRRC6 mutation on its expression. a Exonic organization of the human LRRC6 cDNA, in which the 12 exons are shown as boxes, and the mutation of the corresponding protein is located at the 6th exon (bottom). The domain organization model of LRRC6 exhibits LRR domains, a modified LRR domain, a coiled-coil domain, a polylysine motif, and a CS p23-like domain. b A part of the protein alignment of LRRC6 shows the evolutionary conservation in p.W250* among different species. The black arrow denotes the position of the variant (p. W250*). c Results of immunofluorescence staining obtained from the patient’s sperm showed little expression of LRRC6 compared to the normal control (scale bars, 5 μm; red, LRRC6; green, α-tubulin; blue, DAPI). d We detected no expression of LRRC6 in the patient’s sperm by western blotting analysis. e No immunofluorescence staining was observed for LRRC6 in the testes from the patient compared to the normal control
To determine the impact of this nonsense mutation on LRRC6 expression, we conducted immunofluorescence staining on the spermatozoa collected from the patient as well as from the normal control. As expected, LRRC6 was barely detectable along the flagellum of the patient’s spermatozoa compared to the normal control (Fig. 2c), and western blot analysis of the sperm from the patient consistently showed no LRRC6 expression (Fig. 2d). Furthermore, testis sections were also stained, but the LRRC6 expression in the patient was extremely diminished in comparison with the normal control (Fig. 2e). Therefore, we concluded that this nonsense mutation harmed the expression of LRRC6 and may influence the movement of sperm flagella.
LRRC6 is involved in sperm motility by regulating the expression of dynein arm proteins
As observed in previous study, defective LRRC6 expression is implicated in the transcriptional regulation of some dynein proteins, and it is also presumed to be involved in the preassembly of the dynein arms in the cytoplasm, thereby regulating the movement of sperm flagella indirectly [13, 16, 17]. To further clarify the effects on the expression of dynein arm proteins caused by this mutation, we next examined specific, well-established markers of the inner dynein arm (IDA) polypeptide DNAH1 and outer dynein arm (ODA) polypeptides DNAH8, DNAH9, DNAH10, and DNAH17 by immunofluorescence staining. As we suspected, immunostaining for DNAH1, DNAH8, DNAH9, DNAH10, and DNAH17 in the spermatozoa of the individual carrying the LRRC6 mutation was virtually absent compared to the control subject (Fig. 3). These findings indicated that loss-of-function mutations in LRRC6 could damage sperm motility and cause male infertility by the negative expression of key proteins associated with ODAs and IDAs.
Fig. 3.

Expression of related dyneins in the flagellum of the patient’s sperm. a–e Immunofluorescence staining for DNAH1 (a), DNAH8 (b), DNAH9 (c), DNAH10 (d), and DNAH17 (e) in the sperm tail from the patient as well as for the normal control (scale bars, 5 μm; red, DNAH family; green, α-tubulin; blue, DAPI)
Expression of LRRC6 in various germ cells
With the purpose of analyzing the role of LRRC6 in the formation of sperm flagella, we performed immunofluorescence staining of testis sections from adult mice and found that LRRC6 was visibly detectable in the round and elongating spermatids during the process of spermiogenesis (Fig. 4a). Furthermore, to seek the exact orientation of LRRC6 in diverse stages of sperm transformation, we executed flow cytometry coupled with a cell sorting assay to separate the different stages of sperm cell formation. We first determined that LRRC6 was principally localized in the cytoplasm of early spermatogonia and spermatids of different spermatogenic stages, as well as in the flagella of mature epididymal spermatozoa (Fig. 4b). Overall, these data regarding LRRC6 expression suggested that LRRC6 acts as a crucial component in the formation and motility of spermatozoal flagella.
Fig. 4.

Expression of LRRC6 in mouse tissues. a Representative images of testicular tubules showed that LRRC6 was primarily localized in the cytoplasm of round sperm cells and flagella of different stages of spermatids (scale bars, 50 μm; green, LRRC6; blue, DAPI). b LRRC6 was detected in the cytoplasm of spermatogonia (a) and round spermatids (b) and in the flagella of step 9–16 spermatids (c–f)—as well as in epididymal spermatozoa (g) (scale bars, 50 μm; red, LRRC6; blue, DAPI)
Favorable prognosis after performing intracytoplasmic sperm injection from a man harboring the LRRC6 homozygous mutation
The patient and his wife received assisted reproductive therapy with ICSI at our center. The wife exhibited a regular menstrual cycle and a normal basal endocrine assessment on the third day of her menstrual period and underwent a long gonadotropin-releasing hormone (GnRH) agonist protocol. Our center obtained the patient’s spermatozoa from the biopsy of testicular tissue, which was treated by mechanical method and prepared by suspension, and then isolated normal morphologic spermatozoa by density gradient centrifugation. As shown in Table 3, the couple was counseled following one ICSI cycle, and nine oocytes were collected after GnRH injection. Six mature oocytes (at metaphase II, MII) were successfully microinjected, and five oocytes were subsequently fertilized normally (number of 2PN/injected oocytes = 83.3%). Following extended culture, we obtained one day-3, good-quality embryo (8 cells, Grade II) and three day-5 blastocysts (rated 4BB, 4BC, and 4BC). Ultimately, the 4BB blastocyst was transferred into the woman’s uterus with no obvious complications, and the remaining two blastocysts were cryopreserved. A normal singleton clinical pregnancy was achieved, which is currently ongoing and uneventful. Our findings demonstrated that the male infertility associated with this LRRC6 mutation could be circumvented by ICSI and that loss-of-function in the LRRC6 gene did not hamper embryonic development.
Table 3.
Clinical features of the couple undergoing ICSI treatment
| Female age (y) | 31 | |
|---|---|---|
| Length of primary infertility history (y) | 3 | |
| BMI | 20.4 | |
| Basal hormones | FSH (IU/L) | 2.8 |
| LH (IU/L) | 0.5 | |
| E2 (pg/mL) | 17.8 | |
| Prog (ng/mL) | 0.28 | |
| Cycle 1 | Protocol | Long |
| E2 level on the trigger day (pg/mL) | 2184 | |
| No. of follicles ≥ 14 mm on the trigger day | 8 | |
| No. of follicles ≥ 18 mm on the trigger day | 4 | |
| No. of oocytes retrieved | 9 | |
| ICSI progress | M2 | 6 |
| 2PN | 5 | |
| Oocytes injected | 6 | |
| 4BB fertilization rate (%) | 25% (1/4), 83% (5/6) | |
| Cleavage rate (%) | 100% (5/5) | |
| 4BC | 50% (2/4) | |
| 8 cell formation rate (%) | 20% (1/5) | |
Discussion
In the present study, we attempted to find a novel homozygous mutation in the known PCD candidate gene LRRC6 in an infertile patient. A nonsense mutation was located in the highly conserved domain that caused a marked absence in LRRC6 protein, resulting in defective flagellar ultrastructure and eventually contributing to the sterility phenotype. We noted that LRRC6 was primarily confined to the cytoplasm of germ cells and the flagella of late spermatid stages during the spermiogenesis. According to the evidence, generated from our study, LRRC6 is necessary for the motility of the spermatozoal flagellum, and that this homozygous nonsense variant of c.749G>A in LRRC6 is associated with PCD and sterile phenotype.
The LRRC6 protein principally contains five recognizable domains—including LRR domains, a LRRCT domain, a coiled-coil domain, a polylysine motif, and a CS p23-like domain [18]. The CS p23-like domain was recently reported to be essential for stabilizing LRRC6 via interaction with ZMYND10. Mutations in this domain could then disturb the interaction between ZMYND10 and LRRC6 and result in degradation of LRRC6 [16]. We herein showed that the novel nonsense mutation of c.749G>A caused the lacking of the CS p23-like domain, which led to the degradation of LRRC6 without colocalizing with ZMYND10. LRRC6 is shown to be involved in the transcriptional regulation of IDAs and ODAs such as DNAI1 and DNAH7 [12]. Therefore, we detected the expression of DNAH1, DNAH8, DNAH9, DNAH10, and DNAH17 in spermatozoal flagella and found that the signal of these key proteins associated with IDAs and ODAs was significantly decreased in spermatozoal flagella, strongly suggesting that this loss-of-function mutation in LRRC6 gene plays an important role in the motility of the spermatozoa.
The clinical prognosis associated with ICSI in infertile patients related to PCD is usually favorable. Investigators who studied mutations in PCD-related SPAG6 and RSPH3 that were characterized by multiple flagellar malformations also established a positive pregnancy through ICSI treatment [19]. Patients carrying other PCD candidate gene variants in ZMYND10, DNAH5, or DNAAF6 also consistently achieved normal pregnancies and live births [20, 21]. With regard to mutations in CFAP74 associated with both PCD and MMAF, ICSI treatment was capable of producing a healthy birth despite the use of morphologically abnormal sperm [22]. However, clinical outcomes with ICSI for PCD-affected men with LRRC6 mutations have not been assessed. In this study, we first presented a successful pregnancy associated with ICSI in a sterile man with a nonsense mutation in LRRC6. In the future, access to additional genetic counseling diagnoses will definitely improve our knowledge of genotype-phenotype correlations, and ICSI should then become an optimal management modality resulting in a positive prognosis for infertile men with PCD.
In summary, our results provided explicit evidence that the homozygous nonsense mutation of c.749G>A in LRRC6 is a new genetic cause of PCD. This mutation appears to be responsible for the male infertility that manifests itself in defective axonemal dynein arms and in a motor disturbance of the sperm flagellum. Our study expands the spectrum of LRRC6 mutations in PCD and confirms a favorable clinical prognosis for male infertility caused by LRRC6 mutations using ICSI, thus contributing to genetic counseling diagnoses and reinforcing the importance of better characterizing PCD-affected individuals regarding their fertility status.
Supplementary Information
{kind=link}
(PNG 1537 kb)
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yaqian Li and Chuan Jiang contributed equally to this work.
Contributor Information
Yihong Yang, Email: yyhpumc@foxmail.com.
Ying Shen, Email: yingcaishen01@163.com.
References
- 1.Boivin J, Bunting L, Collins JA, Nygren KG. International estimates of infertility prevalence and treatment-seeking: potential need and demand for infertility medical care. Hum Reprod. 2007;22(6):1506–1512. doi: 10.1093/humrep/dem046. [DOI] [PubMed] [Google Scholar]
- 2.Krausz C, Riera-Escamilla A. Genetics of male infertility. Nat Rev Urol. 2018;15(6):369–384. doi: 10.1038/s41585-018-0003-3. [DOI] [PubMed] [Google Scholar]
- 3.Agarwal A, Mulgund A, Hamada A, Chyatte MR. A unique view on male infertility around the globe. Reprod Biol Endocrinol. 2015;13:37. doi: 10.1186/s12958-015-0032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coutton C, Escoffier J, Martinez G, Arnoult C, Ray PF. Teratozoospermia: spotlight on the main genetic actors in the human. Hum Reprod Update. 2015;21(4):455–485. doi: 10.1093/humupd/dmv020. [DOI] [PubMed] [Google Scholar]
- 5.Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193(4250):317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 6.Raidt J, Wallmeier J, Hjeij R, Onnebrink JG, Pennekamp P, Loges NT, Olbrich H, Häffner K, Dougherty GW, Omran H, Werner C. Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. Eur Respir J. 2014;44(6):1579–1588. doi: 10.1183/09031936.00052014. [DOI] [PubMed] [Google Scholar]
- 7.Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, Zariwala MA, Knowles MR. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med. 2004;169(4):459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 8.Lucas JS, Paff T, Goggin P, Haarman E. Diagnostic methods in primary ciliary dyskinesia. Paediatr Respir Rev. 2016;18:8–17. doi: 10.1016/j.prrv.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 9.Shapiro AJ, Davis SD, Polineni D, Manion M, Rosenfeld M, Dell SD, Chilvers MA, Ferkol TW, Zariwala MA, Sagel SD, Josephson M, Morgan L, Yilmaz O, Olivier KN, Milla C, Pittman JE, Daniels MLA, Jones MH, Janahi IA, Ware SM, Daniel SJ, Cooper ML, Nogee LM, Anton B, Eastvold T, Ehrne L, Guadagno E, Knowles MR, Leigh MW, Lavergne V. Diagnosis of primary ciliary dyskinesia. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2018;197(12):e24–e39. doi: 10.1164/rccm.201805-0819ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J. 2017;49(1). 10.1183/13993003.01090-2016. [DOI] [PMC free article] [PubMed]
- 11.Kott E, Duquesnoy P, Copin B, Legendre M, Dastot-Le Moal F, Montantin G, et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am J Hum Genet. 2012;91(5):958–964. doi: 10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horani A, Ferkol TW, Shoseyov D, Wasserman MG, Oren YS, Kerem B, Amirav I, Cohen-Cymberknoh M, Dutcher SK, Brody SL, Elpeleg O, Kerem E. LRRC6 mutation causes primary ciliary dyskinesia with dynein arm defects. PLoS One. 2013;8(3):e59436. doi: 10.1371/journal.pone.0059436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zariwala MA, Gee HY, Kurkowiak M, Al-Mutairi DA, Leigh MW, Hurd TW, et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am J Hum Genet. 2013;93(2):336–345. doi: 10.1016/j.ajhg.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu L, Luo H. Whole-exome sequencing identified a novel compound heterozygous mutation of LRRC6 in a Chinese primary ciliary dyskinesia patient. Biomed Res Int. 2018;2018:1854269–1854265. doi: 10.1155/2018/1854269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen Y, Zhang F, Li F, Jiang X, Yang Y, Li X, Li W, Wang X, Cheng J, Liu M, Zhang X, Yuan G, Pei X, Cai K, Hu F, Sun J, Yan L, Tang L, Jiang C, Tu W, Xu J, Wu H, Kong W, Li S, Wang K, Sheng K, Zhao X, Yue H, Yang X, Xu W. Loss-of-function mutations in QRICH2 cause male infertility with multiple morphological abnormalities of the sperm flagella. Nat Commun. 2019;10(1):433. doi: 10.1038/s41467-018-08182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho KJ, Noh SH, Han SM, Choi WI, Kim HY, Yu S, Lee JS, Rim JH, Lee MG, Hildebrandt F, Gee HY. ZMYND10 stabilizes intermediate chain proteins in the cytoplasmic pre-assembly of dynein arms. PLoS Genet. 2018;14(3):e1007316. doi: 10.1371/journal.pgen.1007316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serluca FC, Xu B, Okabe N, Baker K, Lin SY, Sullivan-Brown J, Konieczkowski DJ, Jaffe KM, Bradner JM, Fishman MC, Burdine RD. Mutations in zebrafish leucine-rich repeat-containing six-like affect cilia motility and result in pronephric cysts, but have variable effects on left-right patterning. Development. 2009;136(10):1621–1631. doi: 10.1242/dev.020735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohyanagi T, Matsushima N, editors. Classification of tandem leucine-rich repeats within a great variety of proteins. Faseb J. 1997: FEDERATION AMER SOC EXP BIOL 9650 ROCKVILLE PIKE, BETHESDA, MD 20814–3998 USA.
- 19.Wu H, Wang J, Cheng H, Gao Y, Liu W, Zhang Z, Jiang H, Li W, Zhu F, Lv M, Liu C, Tan Q, Zhang X, Wang C, Ni X, Chen Y, Song B, Zhou P, Wei Z, Zhang F, He X, Cao Y. Patients with severe asthenoteratospermia carrying SPAG6 or RSPH3 mutations have a positive pregnancy outcome following intracytoplasmic sperm injection. J Assist Reprod Genet. 2020;37(4):829–840. doi: 10.1007/s10815-020-01721-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Tu C, Nie H, Meng L, Li D, Wang W, Zhang H, Lu G, Lin G, Tan YQ, du J. Novel DNAAF6 variants identified by whole-exome sequencing cause male infertility and primary ciliary dyskinesia. J Assist Reprod Genet. 2020;37(4):811–820. doi: 10.1007/s10815-020-01735-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ozkavukcu S, Celik-Ozenci C, Konuk E, Atabekoglu C. Live birth after laser assisted viability assessment (LAVA) to detect pentoxifylline resistant ejaculated immotile spermatozoa during ICSI in a couple with male Kartagener’s syndrome. Reprod Biol Endocrinol. 2018;16(1):10. doi: 10.1186/s12958-018-0321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sha Y, Wei X, Ding L, Ji Z, Mei L, Huang X, Su Z, Wang W, Zhang X, Lin S. Biallelic mutations of CFAP74 may cause human primary ciliary dyskinesia and MMAF phenotype. J Hum Genet. 2020;65(11):961–969. doi: 10.1038/s10038-020-0790-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PNG 1537 kb)