
Fig. 1.
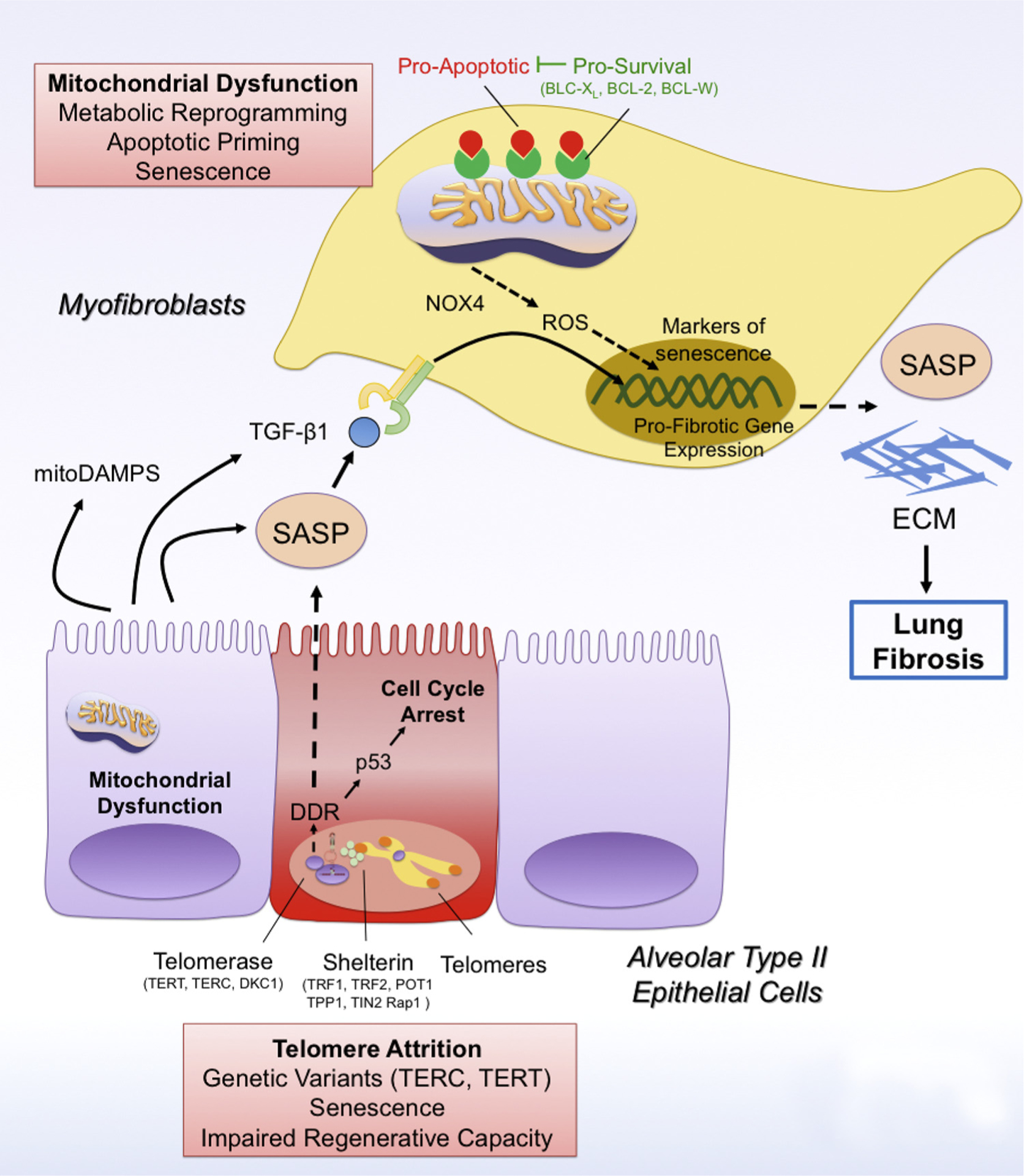
Telomere attrition and mitochondrial dysfunction drive cellular senescence in lung fibrosis.
Telomere attrition due to mutations in telomere genes (TERC, TERT) leads to the activation of the DNA-Damage Response (DDR) pathway, which drives p53-mediated cell cycle arrest and cellular senescence of type II alveolar epithelial cells (AEC2). In addition, PINK1 deficiency in AEC2 cells leads to mitochondrial dysfunction and in these cells. Senescent AEC2 cells are characterized by a senescence-associated secretory phenotype (SASP), which can spread cellular senescence to other AEC2 cells and myofibroblasts. In addition, the SASP has pro-fibrotic factors such as TGFβ1 and mitoDAMPs (such as IL6, mtDNA), which promote the activation of scar-forming myofibroblasts and the development of lung fibrosis. In addition, mitochondrial dysfunction can also drive cellular senescence in myofibroblasts in a NAPDH oxidase 4 (NOX4)/reactive oxygen species (ROS)- and p16-dependent manner by preventing mitophagy and promoting metabolic reprogramming in these cells. Damaged mitochondria in senescent myofibroblasts are characterized by increased levels of pro-apoptotic factors, thus increasing their apoptotic priming. Accordingly, senescent myofibroblasts upregulate expression of pro-survival mitochondrial proteins such as BCL-2, BCL-XL and BCL-W, which block pro-apoptotic signaling and ensure survival of senescent myofibroblasts.