
Fig. 4 |. Molecular control of myofibroblast activation and survival.
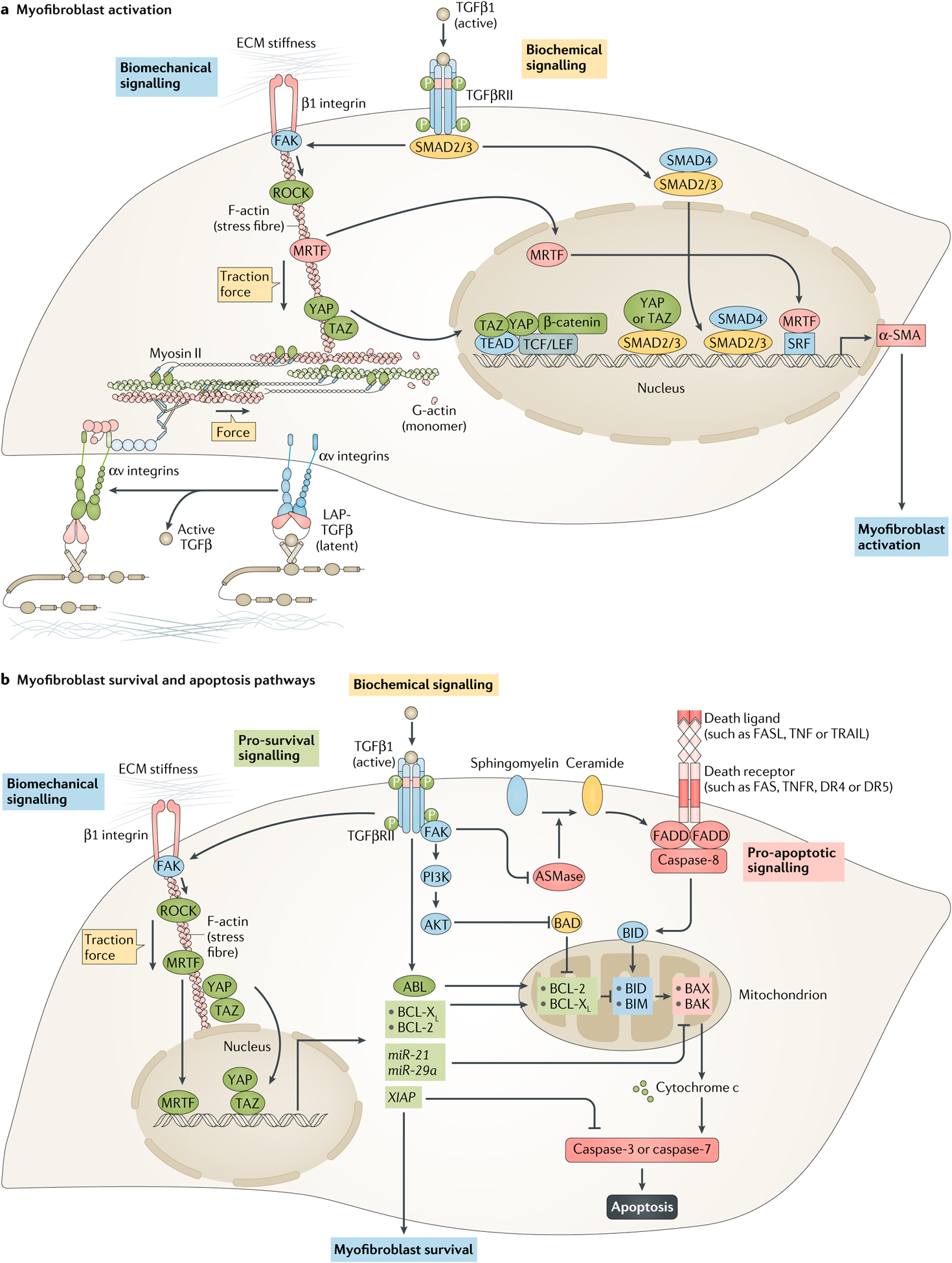
a | Myofibroblast activation is jointly promoted by biomechanical and biochemical cues. Biomechanical signalling induced by extracellular matrix (ECM) stiffness activates mechanotransduction pathways that directly control α-smooth muscle actin (α-SMA) transcription. These pathways involve β1 integrin, focal adhesion kinase (FAK) and RHO-associated protein kinase (ROCK), together with increasing traction forces in fibroblasts. Increased actomyosin activity causes the nuclear translocation of myocardin-related transcription factor (MRTF), as well as the transcriptional co-activators yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ), which regulate α-SMA expression by binding and activating other transcription factors, such as serum response factor (SRF), TEA domain family member (TEAD), T cell factor/lymphoid enhancer-binding factor (TCF/LEF) and β-catenin. ECM stiffness and mechanical forces also regulate force-dependent activation of latent transforming growth factor-β1 (TGFβ1) by increasing resistance to traction forces generated by fibroblasts. In this mechanism, extracellular latent TGFβ1 (TGFβ1 with its latency-associated peptide (L AP)) is released from latent TGFβ1 binding protein stores when αv integrins respond to mechanical pulling forces. Once activated, TGFβ1 binds to TGFβ receptors and promotes canonical mothers against decapentaplegic homolog 3 (SMAD3) activation. Activated SMAD3 binds to SMAD4 and translocates to the nucleus, where it binds to SMAD-binding elements in the promoters of fibrogenic genes, such as ACTA2 (encoding α-SMA). Together, myofibroblast activation is controlled by both the TGFβ–SMAD pathway, as well as biomechanical pathways such as integrin–FAK–ROCK–MRTF–YAP–TAZ signalling. b | Mitochondria in activated myofibroblasts contain large amounts of pro-apoptotic factors, which force these cells to activate pro-survival mechanisms to ensure survival. The intrinsic pathway of apoptosis is directly inhibited by TGFβ1 via activation of ABL signalling, which increases the amount of the pro-survival proteins BCL-2 and BCL-XL. TGFβ1 also blocks the intrinsic pathway by inhibiting the pro-apoptotic protein BCL-2-associated death promoter (BAD) via the FAK–PI3K–AKT signalling pathway, and TGFβ1-mediated FAK signalling blocks the extrinsic pathway of apoptosis by inhibiting sphingomyelinase (ASMase), an enzyme that controls FAS-mediated apoptosis by generating ceramide from sphingomyelin. Biomechanical signalling similarly inhibits the intrinsic pathway via the biomechanically regulated TGFβ–FAK–YAP1–TAZ–BCL-XL and TGFβ–ROCK–MRTF–BCL-2 pathways. Moreover, ECM stiffness induces expression of the microRNAs miR-21 and miR-29a, which promote the survival of myofibroblasts by increasing the expression of pro-survival BCL-2 proteins. Both ECM stiffness and TGFβ1 can also block apoptosis by increasing amounts of X-linked inhibitor of apoptosis protein (XIAP), a direct caspase inhibitor. AKT, pro-survival protein kinase B; BAK, BCL-2 homologous antagonist/killer; BAX, BCL-associated X protein; BID, BH3-interacting domain death agonist; BIM, BCL-2-interacting mediator of cell death (also known as BCL2L11); PI3K, phosphoinositide 3-kinase; TGFβRII, transforming growth factor-β receptor II.