

Abstract
The spread of novel coronavirus SARS-CoV-2 has directed to a state of an unprecedented global pandemic. Many synthetic compounds and FDA-approved drugs have been significantly inhibitory against the virus, but no SARS-CoV-2 solution has been identified. However, small molecule fragment–based derivatives of potent phytocompounds may serve as promising inhibitors against SARS-CoV-2. In the pursuit of exploring novel SARS-CoV-2 inhibitors, we generated small molecule fragment derivatives from potent phytocompounds using neural networking and machine learning–based tools, which can cover unexplored regions of the chemical space that still retain lead-like properties. Out of 300 derivative molecules from withaferin-A, hesperidin, and baicalin, 30 were screened out with synthetic accessibility scores > 4 having the best ADME properties. The withaferin-A derivative molecules 61 and 64 exhibited a significant binding affinity of − 7.84 kcal/mol and − 7.94 kcal/mol. The docking study reveals that withaferin-A mol 61 forms 5 polar H-bonds with the Mpro where amino acids involved are GLU166, THR190, CYS145, MET165, and GLN152 and upon QSAR analysis showed a minimal predicted IC50 value of 7762.47 nM. Furthermore, the in silico cytotoxicity predictions, pharmacophore modeling, and molecular dynamics simulation studies have resulted in predicting the highly potent small molecule derivative from withaferin-A (phytocompound from Withania somnifera) to be the potential inhibitor of SARS-CoV 2 protease (Mpro) and a promising future lead candidate against COVID-19. The rationale of choosing withaferin-A from Withania somnifera (Ashwagandha) was propelled by the innumerous applications of Ashwagandha for the treatment of various antiviral diseases, common cold, and fever since time immemorial.
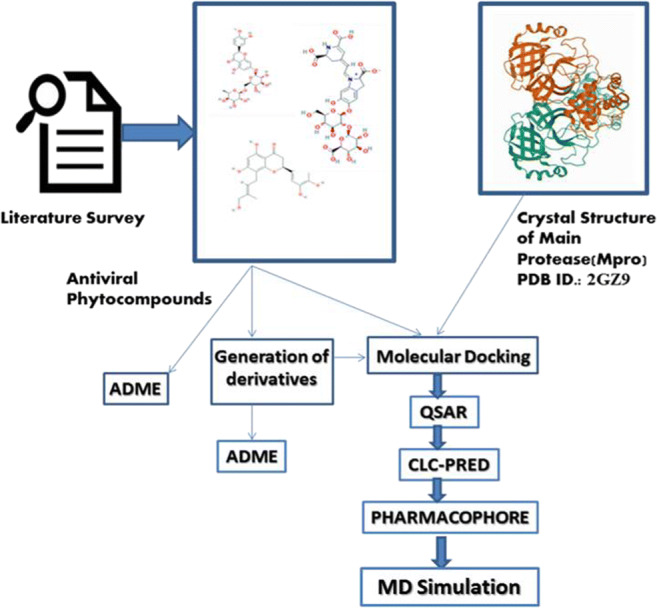
Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s00894-021-04703-6.
Keywords: COVID-19, SARS-CoV-2, Mpro inhibitor, Withania somnifera, Small molecule derivative, QSAR, Machine learning, DNN
Introduction
The novel coronavirus disease 2019 (COVID-19), caused by SARS-CoV-2 which vented in December 2019 in Wuhan, China, is creating disaster by causing significant morbidity and mortality. Cases of COVID-19 are being reported in nearly every country around the globe. As of September 9, 2020, the number of confirmed cases reached 27,486,960 globally, with 894,983 deaths (https://covid19.who.int/). In India, the number of confirmed cases reached 4,462,965, and the death toll reached 75,091 (https://www.worldometers.info/coronavirus/country/india/). Coronaviruses have been implicated in other epidemics in recent decades, such as acute respiratory disease (SARS) and Middle East respiratory disease (MERS). But compared to these, the transmission rate of COVID-19 is much higher, with substantial spreading of the viral infection from one infected individual to two to three healthy individuals on average.
Coronaviruses (CoVs), members of the Coronaviridae family, are among the largest known single-stranded RNA viruses [1]. CoVs contain the biggest genomes among all known RNA viruses, up to 26 to 32 kb in length [2]. The coronavirus genome is composed of four major structural proteins: the spike (S) protein, the nucleocapsid (N) protein, the protein membrane (M), and the protein envelope (E) that are indispensable for the development of a complete viral particle [3, 4]. A significant chunk of the genome of coronavirus is transcribed into polypeptides necessary for viral replication and gene expression. An approximately 306-amino acid polypeptide called main protease (Mpro) has a highly conserved sequence and is a crucial enzyme necessary for coronavirus replication [5]. Due to the known protein structure, main proteases are the primary targets for designing antiviral drugs to combat coronavirus infections [6, 7]. Towards this effort, numerous inhibitors have been designed to block different stages of viral entry, attachment, and replication in host cells. These compounds are then tested in cell-based systems [8, 9]. Currently, the CoV-associated pathologies are not approved for any specific antiviral treatment. The majority of therapies rely mostly on the control of symptoms and support treatments [10]. Few therapeutic agents that are under development are ribavirin, interferon (IFN)-α, and mycophenolic acid. Reports cited the effectiveness of anti-HIV drugs such as ritonavir, lopinavir, either alone or in combination with oseltamivir, remdesivir, and chloroquine [11]. Among these, ritonavir, remdesivir, and chloroquine showed efficacy at the cellular level. However, further experimental support and validation are needed to verify safety and efficacy. Common phytocompound and plant medicines were also used for decades in the fight against normal flulike conditions and fever. Ashwagandha (Withania somnifera) is an Indian Ayurvedic plant, used for herbal therapies in traditional medicine. Ashwagandha is considered to improve the immune system and to have a range of prophylactic and medicinal actions [12]. Withaferin-A is a bioactive withanolide from Ashwagandha that reportedly possesses inhibitory activity for HPV and influenza viruses [12, 13]. Based on these observations, the ability of the SARS-CoV-2 main protease (Mpro) inhibitors withaferin-A and its derivatives has been explored.
In a combination of the docking results reported earlier [14–23], naturally abundant phytocompounds like hesperidin, baicalin, myricitrin, calceolarioside B, methyl rosmarinate, rutin, diosmin, apiin, diacetylcurcumin, withaferin-A, zingiberene, and limonene might be worthy of clinical trials [24]. However, there is no report indicating the potential of important fragments and small molecule derivatives of these phytocompounds as potential agents against SARS-CoV-2 main protease (Mpro).
Several commercially available FDA-approved antiviral drugs such as lopinavir, ritonavir, remdesivir, and several other antiviral drugs are previously predicted to bind to the main protease of SARS-CoV. SARS-CoV-2 3CLpro or Mpro also shows a projected affinity and a strong efficacy value of Kd > 100 nM with these drugs [25, 26]. Prediction suggests that viral proteinase-targeting drugs could effectively influence the viral replication process. This prediction was backed by studies on molecular docking of HIV proteinase inhibitors of CoV protease [27]. This study showed that lopinavir, atazanavir, and ritonavir may inhibit the CoV proteinase. In case studies with inhibitor medicines such as hydroxychloroquin and remdesivir, atazanavir and ritonavir were tested similar to lopinavir [28]. But there is no significant evidence hitherto, whether these drugs can act efficiently as predicted against COVID-19 or not. Here we have reported the molecular interaction studies for both FDA-approved synthetic inhibitors and phytocompounds with the main protease Mpro. In doing so, we aimed to screen out the best phytocompound in comparison to synthetic drugs. In addition, the ADMET profiles of all the compounds were taken into account in order to apply a chemoinformatics approach to find out the small molecule fragments and derivatives of the best docked phytocompounds. Furthermore, quantitative structure-activity relationship (QSAR) analysis was performed to predict the IC50 values of novel derivatives. The CLC-Pred and pharmacophore models, along with molecular dynamics simulations, further supplemented these results in order to ascertain the best derivative compound from the initial set of phytocompounds.
Materials and methods
Retrieval of ligands
The rationale for the selection of 12 phytocompounds and 12 FDA-approved synthetic drugs was based on the reports depicted in Table S-1A (supplementary material). The 12 FDA-approved drugs are reported to have a significant inhibitory effect upon CoV main proteases and are thus considered as reference drugs in the study (Table S-1B, supplementary material). All of the selected molecules were retrieved from the PubChem domain (URL https://pubchem.ncbi.nlm.nih.gov/) [29]. The respective PubChem IDs are also listed in Table S-1A. A flowchart representing the pipeline adopted in the study is depicted in Fig. 1.
Fig. 1.

Flowchart of pipeline adopted in the study to identify small molecule inhibitors of Mpro
ADMET screening of the phytocompounds
A significant step in the production of drugs is the estimation of essential pharmacological properties of the possible small molecules in silico and in vivo. In silico approaches (referring to virtual screening) are preferable over in vivo predictions that are costly and time-consuming. A graph modeling–based tool, pkCSM (predicting small molecule pharmacokinetic properties using graph-based signatures), was used for studying ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties. In this analysis, the respective canonical SMILEs of the compounds were used to calculate the ADMET properties.
Molecular interaction studies of Mpro with naturally derived phytocompounds and FDA-approved drugs
Molecular interaction studies using docking have predicted potential interactions between drug targets through energy minimization and binding energy calculations. The interaction between small molecules (ligands) and the respective protein receptor (which may be an enzyme) is a possible site of inhibition [30]. The molecular docking studies were carried out in AutoDock 4.2.1 [31]. Ligands retrieved from PubChem database for the docking analysis in 3D SDF format were translated and stored in Mol2 format using Open Babel 2.2.3 [32].
The atomic resolution structures resolved in X-ray and NMR 3D coordinates of the target protein molecule CoV main protease (Mpro) (PDB id 2gz9) were downloaded from the protein data bank (RCSB-PDB) and processed using the AutoDock tool. Ligand preparation and molecular docking were done according to the methods of Ghosh and co-workers. [33]. The rationale of this study was based on the ability of phytocompounds and FDA-approved synthetic drugs to bind with the target protein. It was then necessary to evaluate the binding free energy (∆G) in order to establish a comparison that might screen for the best phytocompound against CoV main protease (Mpro). Generation of small molecule derivatives of the best docked phytocompounds.
Generation of small molecule derivatives of the best docked phytocompounds
The 3 best docked phytocompounds (withaferin-A, hesperidin, and baicalin) in the sdf format were uploaded individually to the deep neural networking–based LigDream tool (https://www.playmolecule.org/LigDream/) to generate derivative molecules along with their canonical smiles from each phytocompound. DNNs are used to design effective routes of chemical synthesis where reversed reactions formally decompose the molecule (retrosynthesis) [34]. This process results in a series of reactions which can then be performed in the forward direction in the laboratory to synthesize the target fragments.
ADME analysis of the derived small molecules
The SMILE strings of 100 derivatives from each phytocompound has analyzed in the SwissADME web tool (http://www.swissadme.ch/) to quantify the physicochemical descriptors and estimate ADME parameters, drug-like nature, pharmacokinetic properties, and medicinal chemistry friendliness of multiple small molecules that are the prerequisites for a successful drug discovery. In this study, the ADMET profiles were analyzed for 300 derivatives to further screen out small molecule derivatives on the basis of their synthetic accessibility score and the drug likeliness parameters (Lipinski/Ghose/Veber/Egan/Muegge). Only those molecules that showed a synthetic accessibility score in a range of 1–4 and having no violation of drug likeliness parameters were considered for further studies. A final set of 30 molecules was obtained from 300 derivatives generated from the three preferred phytocompounds.
Molecular docking of Mpro with phytocompound derivatives
The second set of docking was performed to evaluate the binding free energy (∆G) for the selected phytocompound derivatives. A similar docking strategy was adopted as described in the previous section.
Quantitative structure-activity relationship (QSAR) analysis of the screened derivative molecules
Structure-activity relationships (SAR) based upon machine learning and statistical methods are extensively used in many areas of drug development, ranging from primary screening to lead optimization [35]. In this study, the phytocompounds were subjected to QSAR analysis applying a multiple linear regression model using EasyQSAR 1.0. A QSAR equation can be given as:
where the parameters P1 through Pn are computed for each molecule in the series and the coefficients C1 through Cn are calculated by fitting variations in the parameters and the biological activity. These P1, P2, P3, and so on are the descriptor variables of the QSAR equation [35]. For this purpose, molecular descriptors were obtained using the information’s from the tool Marvin 20.15, 2020 (http://www.chemaxon.com), and respective experimental IC50 values of the training dataset were collected from ChEMBL database (https://www.ebi.ac.uk/chembl/). The training dataset included 18 reported inhibitor molecules and 6 ligand descriptors, viz., molecular weight, LOGP, refractivity, polar surface area-2D, polarizability, and molecular surface area-3D which were used to predict the QSAR model. Thereafter, IC50 values of 10 test datasets including 7 best docked derivative molecules and their 3 parent phytocompounds were derived based on the predicted QSAR model. The model was developed from a specific chemical class of SARS-CoV inhibitors and not upon the values of the molecular descriptors. So the influence of the number of training set compounds on the applicability domain of the QSAR model is considered trivial. However, owing to the small training set of compounds, the domain of applicability will naturally be localized.
In silico prediction of cytotoxicity for tumor and non-tumor cell lines
In silico cytotoxicity prediction was carried out for tumor and non-tumor cell lines using CLC-Pred (Cell Line Cytotoxicity Predictor). CLC-Pred tools predict cytotoxicity of tumor cell lines depending on the cytotoxicity relationships between the structure and the cell lines built by Prediction of Activity Spectra for Substances (PASS) special training sets equipped with the leave-one-out cross-validation procedure. The in silico prediction results reportedly accord with the results of in vivo experiments by about 96% [36]. The web service, http://www.way2drug.com/Cell-line/, was used to predict the cytotoxic effects of chemical compounds in non-transformed and cancer cell lines based on the structural formula in silico. CLC-Pred provides a likelihood of the cytotoxicity of a chemical compound to evaluate whether the substance will be used in the experimental screening. The interpretation was done using the default parameters as described in CLC-Pred’s protocol. Using the parameters of this protocol, “Pa” indicates activity, and “Pi” indicates inactivity. Thus, our screening with Pa > Pi indicated that the probability of action is considerably higher than the probability of inactivity.
Pharmacophore modeling
“Pharmacophores” are mostly considered to be molecular fragments or functional groups of a chemical compound. A pharmacophore may be generated in a structural or ligand-based method. [37]. Here we have used pharmacophore modeling based upon the ligand-based approach to establish the pharmacophore models of the phytocompound derivatives. The PDB format file of the target receptor molecule main protease (Mpro) and the two best ligands or the pharmacophore feature molecules in mol2 format were analyzed in the ZINCPharmer Pharmacophore tool (zincpharmer.csb.pitt.edu). The ZINCPharmer utilizes the Pharmer open-source pharmacophore search to screen a large database of fixed conformers such as hydrophobic interactions, hydrogen bond donor/acceptor, positive/negative ions, and other pharmacophore features for pharmacophore matches [38].
MD simulation study
At the atomistic level, the MD simulation results allowed for the investigation of the structural dynamics of the receptor CoV-2 protease (Mpro) upon binding with small ligand (proposed drug) molecules. Molecular dynamics (MD) simulation studies were carried out in order to determine the backbone configuration of SARS-Cov-2 main protease bound to withaferin-A derivative molecule 61. To set up the simulation initially, the systems were built for Mpro with and without the withaferin-A derivative molecule 61 ligand in the system builder. MD simulation study was carried out in Desmond vs 2020–1. To set up the initial parameters of an orthorhombic box of 10x10x10 Å, Desmond system builder was used. The target protein Mpro and target-ligand complex were neutralized with NaCl by adding 0.15 M Na + ions. The prepared systems were relaxed using the Desmond default protocol of relaxation [39]. An MDS run of 50 ns was set up at constant temperature and constant pressure (NPT) for the final production run. The NPT ensemble was set up using the Nosé-Hoover chain coupling scheme at a temperature of 300 K for final production and throughout the dynamics with relaxation time 1 ps. An RESPA integrator was used to calculate the bonding interactions for a time step of 2 fs. All other parameters were associated in the settings followed as described by Shaw and co-workers [39]. After the final production run, the simulation trajectories of SARS-CoV-2 main protease complexed with withaferin-A derivative molecule 61 were analyzed for the final outcome of RMSD, RMSF, and ligand RMSF, derived from the simulation studies.
Results and discussions
The safety and efficacy of drug candidates are the necessary prerequisites for regulatory approval, and an initial ADMET profiling of the chosen reference drugs and phytocompounds provides the ADME properties which confirm the drug likeliness of the selected phytocompounds. The comparable statement from ADMET profiles of the phytocompounds with respect to the FDA-approved synthetic drugs is displayed in Table 1. The water solubility of a compound (logS) reveals the solubility of the molecule in water at 25 °C. The phytocompounds diacetylcurcumin (−6.225), withaferin-A (−5.063), and zingiberene (−5.967) displayed highest solubilities (log mol/L) over the reference set of drugs considered in this study. The intestinal absorptions of the phytocompounds (Table 1) are significantly lower than the synthetic drugs. The phytocompounds diacetylcurcumin, withaferin-A, zingiberene, and limonene shows the highest intestinal absorption among the tested phytocompounds and are comparable to the reference set of drugs. The steady-state volume of distribution (VDss) is the theoretical volume that the total drug would need to be uniformly distributed to give the same concentration as in blood plasma. VDss is considered low if below − 0.15 and high if above 0.45 in a logarithmic scale of L/Kg. The tested phytocompounds (Table 1) depict comparable VDss to the synthetic drugs. Also, all the phytocompounds gave negative results to AMES toxicity which indicated that the compounds are safe to be carried forward in further analysis.
Table 1.
ADMET profiles of synthetic drugs and phytocompounds selected for the study
| Water solubility (log mol/L) | Intestinal absorption (% absorbed) | VDss (human) (log L/kg) | Total clearance (log ml/min/kg) | AMES toxicity (yes/no) | Maximum dosage (human) (log mg/kg/day) | |
|---|---|---|---|---|---|---|
| Synthetic drugs | ||||||
| Bictegravir | −3.495 | 82.275 | −0.241 | 0.543 | NO | −0.016 |
| Tegobuvir | −3.041 | 81.252 | −0.152 | 0.18 | NO | 0.2 |
| Baricitinib | −3.129 | 79.532 | 0.059 | 0.854 | NO | 0.854 |
| Remdesivir | −3.07 | 71.109 | 0.307 | 0.198 | NO | 0.15 |
| Nelfinavir | −3.894 | 70.888 | 0.563 | 0.399 | NO | −0.576 |
| Hydroxychloroquin | −3.627 | 90.217 | 1.076 | 1.152 | YES | −0.091 |
| Prulifloxacin | −3.161 | 60.602 | −0.081 | 0.119 | NO | 0.119 |
| Mefloquine | −4.874 | 85.961 | 0.83 | 0.43 | NO | −0.283 |
| Favipiravir | −2.121 | 91.69 | −0.218 | 0.518 | NO | 1.291 |
| Dexamethasone | −4.147 | 81.31 | −0.078 | 0.658 | NO | 0.097 |
| Chloroquine | −4.249 | 89.95 | 1.332 | 1.092 | YES | −0.167 |
| Methylprednisolone | −3.787 | 73.366 | −0.144 | 0.712 | No | −0.183 |
| Phytocompounds | ||||||
| Hesperidin | −3.014 | 31.481 | 0.996 | 0.211 | NO | 0.525 |
| Baicalin | −2.764 | 26.224 | 0.267 | 0.04 | NO | 0.652 |
| Myricitrin | −2.892 | 43.334 | 1.552 | 0.303 | NO | 0.454 |
| Calceolarioside B | −2.96 | 38.482 | 1.559 | −0.186 | NO | 0.201 |
| Methyl rosmarinate | −3.17 | 64.776 | 0.75 | 0.187 | NO | −0.027 |
| Rutin | −2.892 | 23.446 | 1.663 | −0.369 | NO | 0.452 |
| Diosmin | −2.929 | 29.319 | 1.428 | −0.113 | NO | 0.565 |
| Apiin | −2.851 | 17.411 | 1.004 | −0.054 | NO | 0.446 |
| Diacetylcurcumin | −6.225 | 94.745 | −0.808 | 0.382 | NO | 0.633 |
| Withaferin-A | −5.063 | 85.345 | −0.131 | 0.435 | NO | −0.695 |
| Zingiberene | −5.967 | 95.561 | 0.629 | 1.441 | NO | 0.414 |
| Limonene | −3.568 | 95.898 | 0.396 | 0.213 | NO | 0.777 |
Molecular docking of phytocompounds and reference drugs
Molecular docking is employed to find out the interaction of our chosen reference drugs (bictegravir, tegobuvir, baricitinib, remdesivir, nelfinavir, hydroxychloroquin, prulifloxacin, mefloquine, favipiravir, dexamethasone, chloroquine, methylprednisolone) and selected phytocompounds (hesperidin, baicalin, myricitrin, calceolarioside B, methyl rosmarinate, rutin, diosmin, apiin, diacetylcurcumin, withaferin-A, zingiberene, limonene). Our investigations and results from molecular docking of the synthetic drugs and phytocompounds with the SARS-CoV2 target protein main protease (Mpro) revealed that the docking scores of most of the phytocompounds are comparable to that of the synthetic drugs (Table 2).
Table 2.
Comparative chart depicting the binding energies and inhibitory concentrations of synthetic drugs and phytocompounds
| Synthetic drugs | Phytochemical compounds | ||||
|---|---|---|---|---|---|
| Binding energy (kcal/mol) | Inhibitory concentration (uM) | Binding energy (kcal/mol) | Inhibitory concentration (uM) | ||
| 1. Methylprednisolone | −8.02 | 1.32 | 1. Withaferin-A | −9.22 | 0.175 |
| 2. Dexamethasone | −7.4 | 3.79 | 2. Hesperidin | −6.87 | 9.23 |
| 3. Tegobuvir | −7.33 | 4.24 | 3. Baicalin | −6.68 | 12.72 |
| 4. Bictegravir | −7.26 | 4.79 | 4. Diacetylcurcumin | −6.5 | 17.26 |
| 5. Nelfinavir | −7.01 | 7.32 | 5. Methyl rosmarinate | −6.37 | 21.32 |
| 6. Prulifloxacin | −6.91 | 8.57 | 8. Apiin | −5.88 | 49.19 |
| 7. Baricitinib | −6.82 | 10.0 | 11.Zingiberene | −5.87 | 50.21 |
| 8. Hydroxychloroquin | −5.88 | 49.19 | 3. Myricitrin | −5.41 | 108.38 |
| 9. Mefloquine | −5.83 | 53.08 | 6. Rutin | −5.04 | 201.68 |
| 10. Chloroquine | −5.80 | 56.34 | 4. Calceolarioside B | −4.98 | 223.93 |
| 11. Remdesivir | −5.08 | 188.87 | 12.Limonene | −4.67 | 375.22 |
| 9. Favipiravir | −4.65 | 391.39 | 7. Diosmin | −4.27 | 741.0 |
However, withaferin-A showed best docking score among all the phytocompounds as well as synthetic drugs that were screened (Table 2). The compounds with low binding energy and inhibitory concentration are better suited to act as drug compounds owing to their better binding conformations. Thus, withaferin-A, hesperidin, and baicalin having binding energies − 9.22 kcal/mol, − 6.87 kcal/mol, and − 6.68 kcal/mol, respectively, were selected for further analysis. The synthetic drug methylprednisolone having binding energy − 8.02 kcal/mol was selected to be taken as a reference. The docked ligand molecules with protease were shown in Fig. 2 (a), (c), (e), and (g). While Figs. 2 (b), (d), (f), and (h) highlighted all the amino acids involved in the ligand-receptor binding. Sharma and Deep previously reported withaferin as a potential phytocompound against Mpro, and their experiments generated a dock score of − 8.9 kcal/mol [40]. Our result however is substantiated and better than the previous findings as reported elsewhere [40].
Fig. 2.


Molecular dock pose of a withaferin-A and Mpro complex, b withaferin-A-Mpro binding in 2D, c hesperidin and Mpro complex, d hesperidin-Mpro binding in 2D, e baicalin and Mpro complex, f baicalin-Mpro binding in 2D, g methylprednisolone and Mpro complex, h methylprednisolone-Mpro binding in 2D
The phytocompound withaferin-A binds to the SARS-CoV2 Mpro strongly, and the polar and nonpolar amino acid residues involved are GLN192, THR190, PRO168, LEU167, ALA191, GLN189, ARG188, GLU166, HIS164, MET49, MET165, HIS41, GLY143, CYS145, LEU27, THR25, and THR26, while CYS145, HIS163, ASN142, GLU166, HIS41, THR45, LEU141, PHE140, HIS172, SER144, HIS164, THR25, VAL42, CYS44, GLU47, ALA46, MET49, ASP187, ARG188, GLN189, and MET165 amino acids are involved in hesperidin::Mpro binding. The baicalin and SARS-CoV2 Mpro binding involves ARG88, GLN83, GLU178, TYR37, LYS102, TYR101, ASP33, THR98, PRO99, LYS100, and PHE103 amino acids, while the reference drug methylprednisolone involves THR190, GLN192, GLU166, LEU141, HIS163, ASN142, GLY143, GLN189, ARG188, MET49, MET165, HIS41, HIS164, HIS172, CYS145, SER144, and PHE140 amino acids to bind to the catalytic core of SARS-CoV2 Mpro. Sharma and Deep however previously reported that the withaferin-A shows interactions with residues Thr 24, Thr 25, Cys 44, Ser 46, Met 49, His 41Leu 141, His 164, Phe 140, Asn 142, and Glu 166 of the SARS-CoV2 Mpro.
Table 3 depicts withaferin-A, hesperidin, and baicalin having the best dock scores having comparable amino acids involved in the catalytic core as is present in the reference drug methylprednisolone, and thus these 3 phytocompounds are considered to generate small molecule fragment derivatives.
Table 3.
Binding energy and active site residues present in the catalytic core of Mpro(2gz9)-ligand complex
| Ligand | Predicted free energy of binding (ΔG) kcal/mol | Polar residue involved in hydrogen bonding | Amino acid residues involved in other bonds |
|---|---|---|---|
| Withaferin-A | −9.22 | GLN192, THR190 | PRO168, LEU167, ALA191, GLN189, ARG188, GLU166, HIS164, MET49, MET165, HIS41, GLY143, CYS145, LEU27, THR25, THR26 |
| Hesperidin | −6.87 | CYS145, HIS163, ASN142, GLU166, HIS41, THR45 | LEU141, PHE140, HIS172, SER144, HIS164, THR25, VAL42, CYS44, GLU47, ALA46, MET49, ASP187, ARG188, GLN189, MET165 |
| Baicalin | −6.68 | ARG88, GLN83, GLU178 | TYR37, LYS102, TYR101, ASP33, THR98, PRO99, LYS100, PHE103 |
| Methylprednisolone | −8.02 | THR190, GLN192, GLU166, LEU141, HIS163, ASN142, GLY143 | GLN189, ARG188, MET49, MET165, HIS41, HIS164, HIS172, CYS145, SER144, PHE140 |
The molecular docking and QSAR analysis was performed to screen out the best potential hits against SARS-CoV2 main protease (Mpro) from the phytocompounds by taking reference FDA-approved synthetic drugs. Prashanth and co-workers, 2020 [41] recently reported tenufolin as a highly effective phytocompound against SARS-CoV2 showing a dock score of − 8.8 kcal/mol. Our results for initial molecular docking studies successfully identified withaferin-A to be the best inhibitor of Mpro whose dock score(− 9.22 kcal/mol) surpassed every other synthetic drugs taken as reference. Reports from Kumar and co-workers, 2020 [22, 42], revealed that withaferin-A and withanone could also bind and stably interact to the catalytic site of TMPRSS2 which further validates the strong potential of withaferin compounds against SARS-CoV2. Also, we considered phytocompounds hesperidin (− 6.87 kcal/mol) and baicalin (− 6.68 kcal/mol) for further analysis owing to their significantly better binding energies over other chosen compounds on an average.
Generation of small molecule derivatives
The deep learning and neural networking–based LigDream tool (https://www.playmolecule.org/LigDream/) within the PlayMolecules software generated 100 small molecule derivatives for each of the three phytocompounds. All the derivatives were tested for the adsorption, distribution, metabolism, and excretion analysis using SwissADME software. Only those compounds were chosen which had no violations of Lipinski’s rule of 5 and had synthetic accessibility score between 1 and 4. This step of screening resulted in 8 derivative compounds from withaferin-A, 1 from hesperidin, and 21 derivatives of baicalin. A final set of 30 molecules was obtained from 300 derivatives generated from 3 phytocompounds. All 30 derivatives thus obtained were again docked with the SARS-CoV2 main protease target. Tables S-2(A), S-2(B), and S-2(C) (supplementary material) show the best derivatives screened on the basis of ADME profiling from the 100 derivatives generated from each phytocompounds along with their binding energies and inhibitory concentrations obtained by molecular docking with Mpro.
The best small molecule fragment derivatives from the 3 phytocompounds obtained were re-docked with the main protease of SARS-CoV 2 to identify a set of finest small molecule inhibitors of Mpro based upon the dock scores generated. Two derivates of withaferin-A, 1 derivative of hesperidin, and 4 derivatives from baicalin were considered as the best hits for QSAR analysis.
QSAR analysis
Dataset selection and descriptor calculation
In present work, the dataset selection and descriptor calculation for the QSAR analysis involved the collection of a set of 18 molecules (Table S-3, supplementary material) as inhibitors against the main protease of SARS-CoV 2 virus from the CHEMBL database. The dataset used comprises of varied classes of compounds where the experimental activity of each compound is expressed in IC50 (nM) values. For model development, we have converted the IC50 values to pIC50 (pIC50 = -logIC50) values. All the compounds were drawn using MarvinSketch, followed by cleaning of molecules. The descriptors were computed using MarvinSketch.
Analysis and prediction of IC50 of phytocompounds
The QSAR analysis (Fig. 3) with descriptors molecular weight (MW), LOGP, refractivity, polar surface area (PSA), polarizability, and molar surface area (MSA) reveals an Rsq = 67.25%, adjusted Rsq = 49.39%, F statistics = 3.76, and critical F = 2.70. Among all the descriptors, polarizability demonstrated a negative correlation of − 0.02 with the activity. It is also important to mention that LOGP contributed in activity to a greater extent with a percentage contribution of 44%. The predicted IC50 values of 10 test data including 7 best docked derivative molecules and their 3 parent phytocompounds are depicted in Table 4.
Fig. 3.

QSAR activity plot and governing equation for the derivative molecules and phytocompounds
Table 4.
Predicted IC50 and corresponding values of descriptors obtained through QSAR analysis
| Derivative compounds | MW | LOGP | Refractivity | PSA | Polarizability | MSA | Predicted activity (ic50nm) |
|---|---|---|---|---|---|---|---|
| Withaferin mol 61 | 373.51 | 0.67 | 96.33 | 80.75 | 38.63 | 626.49 | 7762.471166 |
| Withaferin mol 64 | 373.51 | 0.71 | 96.23 | 80.75 | 38.63 | 625.49 | 7943.282347 |
| Hesperidin mol 28 | 461.42 | 2.64 | 114.58 | 116.3 | 41.98 | 613.08 | 45,708.81896 |
| Baicalin molecule 65 | 407.42 | 2.52 | 109.29 | 101 | 42.91 | 557.57 | 30,902.95433 |
| Baicalin molecule 78 | 406.39 | 2.56 | 104.29 | 98.19 | 42.87 | 531.21 | 38,018.93963 |
| Baicalin molecule 79 | 449.41 | 2 | 114.68 | 127.3 | 44.6 | 575.75 | 28,840.31503 |
| Baicalin molecule 99 | 424.4 | 2.11 | 110.93 | 125.4 | 41.62 | 550.22 | 30,902.95433 |
| Phytocompounds | MW | LOGP | Refractivity | PSA | Polarizability | MSA | Predicted activity (ic50nm) |
| Withaferin-A | 470.6 | 3.58 | 127.19 | 96.36 | 50.24 | 705.71 | 38,018.93963 |
| Hesperidin | 610.6 | 0.65 | 140.77 | 234.3 | 56.68 | 804.64 | 14,791.08388 |
| Baicalin | 446.4 | 0.76 | 104.93 | 183.2 | 40.82 | 527.68 | 23,442.28815 |
Molecular docking analysis of withaferin-A derivatives with M PRO
The molecular docking studies of SARS-CoV2 main protease (Mpro) with withaferin-A derivative molecules 61 and 64 (Fig. 4a–d) having the lowest IC50 values as obtained by the QSAR study generated significant binding free energies of − 7.84 kcal/mol and − 7.94 kcal/mol, respectively (Table 5). The docking study reveals that the withaferin-A mol 61 forms 5 polar H-bonds with the Mpro where amino acids involved are GLU166, THR190, CYS145, MET165, and GLN152 (Table 5), and the withaferin-A mol 64 forms 4 polar H-bonds with Mpro involving amino acids THR190, GLN192, CYS145, and GLU166 (Table 5), while Table 3 shows that the parent molecule withaferin-A forms only 2 polar H-bonds with Mpro with amino acids GLN192 and THR190 involved. Thus, the withaferin-A derivatives are predicted to exhibit better binding with the target protease of SARS-CoV2 over the parent withaferin-A molecule.
Fig. 4.

Molecular dock pose of a withaferin-A mol 61 and Mpro complex, b withaferin-A mol 61::Mpro binding in 2D, c withaferin-A mol 64 and Mpro complex, d withaferin-A mol 64::Mpro binding in 2D, e pharmacophore model of withaferin-A derivative molecule 61 showing pharmacophore interaction at the binding site of main protease (Mpro), f pharmacophore model of withaferin-A derivative molecule 64 showing pharmacophore interaction at the binding site of the main protease (Mpro)
Table 5.
Binding energy and active site residues present in the catalytic core of Mpro(2gz9)-withaferin-A derivative molecules 61 and 64
| Ligand | Predicted free energy of binding (ΔG) kcal/mol | Polar residue involved in hydrogen bonding | Amino acid residues involved in other bonds |
|---|---|---|---|
| Withaferin-A mol 61 | −7.84 | GLU166, THR190, CYS145, MET165, GLN152 | LEU141, SER144, PRO168, |
| Withaferin-A mol 64 | −7.94 | THR190, GLN192, CYS145, GLU166 | HIS41, MET49, GLN189 |
Our study identified the 2 withaferin derivatives to be the top hits among all other derivatives owing to their relatively low predicted IC50 values. Further upon comparative analyzing the ligand-receptor interactions of the derivatives and parent withaferin-A molecule with Mpro reveals 4 to 5 polar hydrogen bonds of derivatives in the catalytic region of Mpro while only 2 polar H-bonds were involved in the interaction of the parent withaferin-A molecule with Mpro.
In silico cytotoxicity prediction and analyses
Withaferin-A fragment derivative molecules 61 and 64 that showed the lowest predicted IC50 values were selected to predict the biological spectrum by PASS. It finds the probability of activity and inactivity against tumor and non-tumor cells out of a maximum probability score of 1. In the PASS filter, the significant anticarcinogenic activity was displayed with osteosarcoma for both the withaferin-A derivative molecules 61 and 64 having an active coefficient 0.414 and 0.412, respectively (Table 6). Nonetheless, these derivatives have a significant role in inhibiting the growth of major carcinoma cell lines such as colon adenocarcinoma, gastric carcinoma, stomach carcinoma, prostate carcinoma, ovarian adenocarcinoma, thyroid gland undifferentiated (anaplastic) carcinoma, T-lymphoid carcinoma, and osteosarcoma. These results indicate a strong potential for anticarcinogenic activity. It is also shown in Table 7 that the activity of the fragment derivatives maintained the growth of embryonic lung fibroblasts (0.254, 0.226), embryonic kidney fibroblasts (0.208), umbilical vein endothelial cells (0.163, 0.413), lymphocytes (0.092, 0.085), and fibroblasts (0.135). This bioactivity analysis confirms the role of withaferin derivative molecules 61 and 64 in maintaining human health against tumor generation and inflammation.
Table 6.
PASS prediction coefficient with tumor cell lines based on the best phytocompound derivatives with lowest IC50 obtained after QSAR analysis
| Compounds | Pi* | Pa* | Cell line | Cell line name full | Tissue | Tumor type |
|---|---|---|---|---|---|---|
| Withaferin-A mol 61 | 0.128 | 0.089 | DLD-1 | Colon adenocarcinoma | Colon | Adenocarcinoma |
| 0.155 | 0.130 | NCI-N87 | gastric carcinoma | Stomach | Carcinoma | |
| 0.167 | 0.141 | St-4 | Stomach carcinoma | Stomach | Carcinoma | |
| 0.197 | 0.107 | COLO 205 | Colon adenocarcinoma | Colon | Adenocarcinoma | |
| 0.225 | 0.120 | DU-145 | Prostate carcinoma | Prostate | Carcinoma | |
| 0.312 | 0.091 | OVCAR-5 | Ovarian adenocarcinoma | Ovarium | Adenocarcinoma | |
| 0.317 | 0.061 | 8505C | Thyroid gland undifferentiated (anaplastic) carcinoma | Thyroid | Carcinoma | |
| 0.344 | 0.035 | H9 | T-lymphoid | Hematopoietic and lymphoid tissue | Leukemia | |
| 0.414 | 0.008 | SJSA-1 | Osteosarcoma | Bone | Sarcoma | |
| Withaferin-A mol 64 | 0.412 | 0.009 | SJSA-1 | Osteosarcoma | Bone | Sarcoma |
| 0.353 | 0.031 | H9 | T-lymphoid | Hematopoietic and lymphoid tissue | Leukemia | |
| 0.348 | 0.067 | OVCAR-5 | Ovarian adenocarcinoma | Ovarium | Adenocarcinoma | |
| 0.327 | 0.048 | 8505C | Thyroid gland undifferentiated (anaplastic) carcinoma | Thyroid | Carcinoma | |
| 0.262 | 0.096 | DU-145 | Prostate carcinoma | Prostate | Carcinoma | |
| 0.135 | 0.082 | DLD-1 | Colon adenocarcinoma | Colon | Adenocarcinoma | |
| 0.169 | 0.137 | St-4 | Stomach carcinoma | Stomach | Carcinoma | |
| 0.156 | 0.128 | NCI-N87 | gastric carcinoma | Stomach | Carcinoma | |
| 0.199 | 0.173 | PC-3 | Prostate carcinoma | Prostate | Carcinoma | |
| 0.211 | 0.200 | MKN-7 | Gastric carcinoma | Stomach | Carcinoma |
Pa*, probability “to be active”; Pi**, probability “to be inactive”
Table 7.
PASS prediction coefficient with non-tumor cell lines based on the best phytocompound derivatives with lowest IC50 obtained after QSAR analysis
| Compounds | Pa* | Pi** | Cell line | Cell line name | Tissue/organ |
|---|---|---|---|---|---|
| Withaferin-A mol 61 | 0.254 | 0.071 | WI-38 VA13 | Embryonic lung fibroblast | Lung |
| 0.163 | 0.041 | HUVEC | Umbilical vein endothelial cell | Endothelium | |
| 0.092 | 0.032 | MT2 | Lymphocyte(HTLV-producing cell line) | Blood | |
| Withaferin-A mol 64 | 0.226 | 0.088 | WI-38 VA13 | Embryonic lung fibroblast | Lung |
| 0.208 | 0.097 | HEK293 | Embryonic kidney fibroblast | Kidney | |
| 0.143 | 0.050 | HUVEC | Umbilical vein endothelial cell | Endothelium | |
| 0.085 | 0.043 | MT2 | Lymphocyte (HTLV-1 producing cell line) | Blood | |
| 0.135 | 0.117 | NHDF | Fibroblast | Skin |
Generation of pharmacophore models
Pharmacophore modeling based on the principle of Lipinski’s rule of five displayed a more accurate picture of the withaferin-A derivative ligand interaction with the binding site of the SARS-CoV2 main protease more accurately to predict the drug likeliness. The pharmacophore model of derivative molecule 61 displayed that it has four hydrogen bond acceptors and three hydrophobic interactions which is a major parameter of drug likeliness (Fig. 4e). Moreover, the Pharmer webserver identified 6 new hits for derivative molecule 61 by identifying its hydrophobic, hydrogen bond donor/acceptor, positive/negative ions, and other pharmacophore features and are enlisted in Table S-4 (supplementary material). The pharmacophore model of derivative molecule 64 displayed that it has 4 hydrogen acceptors and also 4 hydrophobic interactions (Fig. 4f). This model did not generate any further hit compound. Withaferin derivative molecule 61 is considered to be more potent than derivative mol 64 and thus carried forward for further investigations.
ADME toxicity prediction of withaferin-A fragment derivatives
In order to be an effective drug, a potent molecule must meet its target in the body in adequate concentration and remain there in a bioactive form sufficiently long to cause the predicted biological events. Early in the drug discovery process, assessment of absorption, distribution, metabolism, and excretion (ADME) occurs at a stage when chosen compounds are abundant, but access to the physical samples is restricted. Withaferin-A derivative molecule 61, using the SwissADME tool showed a significant lipophilicity of 1.47 (Fig. S-5A, supplementary material). Typically, lipophilicity falls in a range between (XLOGP3) − 0.7 and + 5.0. The molecular weight of the molecule was shown to be 373.51 g/mol, where the acceptable range is between 150 and 500 g/mol). The molecule’s polarity, measured as topological polar surface area (TPSA), was found to be 89.13 Å2, where the acceptable range is typically between 20 and 130 Å2. The solubility was derived at log S -2.95, a value not recommended higher than 6.0. Taken together, these data indicate that the molecule is soluble in water and moderately polar with some bond flexibility (measured at 8.0 where a value greater than 9.0 indicates rotatable bonds). Given these analyses, the compound is predicted to be an orally bioavailable or administered drug.
Other pharmacokinetic parameters were found. The drug had a high absorption in the GI tract (white oval shape, Fig. S-5B, supplementary material) and had permeability across the blood-brain barrier. These parameters are typically failed by most drug candidates but were apparent with withaferin-A derivative molecule 61. In contrast, the withaferin-A derivative molecule showed high blood-brain barrier (BBB) permeability (yellow oval shape, Fig. S-5B, supplementary material).
One of the major physiological phenomena in human and higher eukaryotes is to efflux the drug molecule from the central nervous system for protection. This poses a major concern in drug delivery systems. In this case, the derivative molecules are retained within the neurological system where the CNS did not show any signs of efflux (no blue spot within the yellow oval, Fig. S-5B, supplementary material). On the other hand, the molecule also displayed a reasonable skin permeation of log Kp − 7.53 cm/s. Hence, the drug can be considered as a safe and effective potential fragment derivative of withaferin-A.
Molecular dynamics and simulation study
For MD simulation, systems were developed both for SARS-CoV2 main protease (Mpro) and withaferin-A derivative mol 61 bound complex that were analyzed for 50 ns. RMSD plots thus generated from the MDS production displayed the conformational change of the target protein, i.e., SARS-CoV2 Mpro, as displayed in Fig. 5a, whereas withaferin-A derivative molecule 61 bound Mpro displayed more stability as compared to the SARS-CoV2 Mpro alone (Fig. 5b). Initial changes were observed in RMSD from 1.2 to 3.4 Å for the first 18 20 ns, and later large conformational changes observed at 65–78 ns before final stabilized conformation were achieved (Fig. 5a). By contrast, the Mpro-withaferin-A derivative molecule 61 bound complex showed much better stability in the Cα-backbone conformation. The ligand conformation over the aligned position of Cα-backbone of the main protease displayed the conformational difference of 1.6 Å until 60 ns due to the diffused pattern. Later, the stabilized RMSD was observed from 60 to 100 ns due to ligand accommodation at the binding cavity of Mpro (Fig. 5b). A final conformational variation was observed for the bound complex from the beginning of the simulation till the end at 50 ns was 0.8 Å (Fig. 5b). This suggested that the ligand-bound state of the SARS-CoV2 main protease gained much more stability during the simulation as compared to the unbound state. Moreover, RMSF plots exhibited the evidence derived from the RMSD plot where clearly visible that the positional fluctuations of amino acids were more in as compared to the withaferin-A derivative bound state (Fig. 5c and d). The diffusion of ligand at the initial stage of the simulation indicated the entry movement through the SARS-CoV2 main protease receptor complex and later stabilized due to deep entry inside the binding cavity and significant binding. In addition, ligand RMSF plot (Fig. 5e) exhibited the interaction position of withaferin-A derivative molecule 61 within SARS-CoV2 during the simulation. The protein-ligand complex is first aligned on the protein backbone, and then the ligand RMSF is measured on the ligand heavy atoms. The ligand root-mean-square fluctuation reveals that the atomic residues from 1 to 12 illustrate elevated fluctuations. But the fluctuation curve from 12th atom to 13th exhibits a sharp ramp which means that at this point, the ligand is entropically changed and thereby fits the catalytic core of SARS-CoV-2 Mpro (Fig. 5e).
Fig. 5.

a SARS-CoV2 Mpro displaying the Cα backbone of structural conformation showing large a change from beginning to the end of the simulation, b aligned Mpro::withaferin-A mol 61 bound complex displaying the Cα backbone and withaferin-A mol 61 conformation allowing less changes over time during simulation. RMSF plot of c Mpro displaying large fluctuations with respect to particular amino acids on the Cα backbone and d displaying more stable complex in withaferin-A derivative mol 61 bound state, and e ligand RMSD plot displaying the atomic positions throughout the simulation
Our docking results, QSAR studies, and MD simulations are in coherence with the previously published literatures. Our study shows that the binding energy for the ligand withaferin-A and its 2 best derivatives mol 61 and 64 are considerably high (refer to Table 5) and their respective inhibitory constant (ki) values are low (refer to Table S- 2A, supplementary material). Upon QSAR study, the predicted activity (IC50) comes the lowest for withaferin derivative mol 61 and 64. Hence, we conclude that the predicted molecules are having high affinity towards the target and the ligands will show activity even at lower concentration. Results on data mining–based predictions of some hit fragments from natural compounds are reported by Ghosh et al. (2020) [43], but our study is the first to report the effectiveness of small molecule fragment derivative of withaferin-A against SARS-CoV2 Mpro by integrating DNN and machine learning–based tool in screening out derivatives against SARS-CoV2 which still retain lead-like properties. The predictions in this report may open new possibilities for the use of small molecule inhibitor drugs to successfully combat COVID-19.
Conclusion
The COVID-19 outbreak originated by the highly pathogenic SARS-CoV-2 coronavirus has posed a major threat to public health and needs urgent intervention. With the current global crisis, all successful diagnostics and novel treatments need to be produced at a reasonable price with limited to no side effects. Over the last 30 years, structural bioinformatics and cheminformatics have emerged as an effective drug discovery technique. In this regard, 3D target protein frameworks have played important roles in designing as well as the development of novel or alternative drugs. Medicinal plants are considered a significant source for the treatment of various diseases. In the current study, the antiviral potential of some phytoconstituents and their small molecule derivatives was studied. The results of molecular docking, QSAR analysis, and MD simulations suggested that withaferin-A and associated fragment derivatives may act as an inhibitor for the Mpro protease of SARS-CoV-2. Withaferin-A, a bioactive withanolide from Ashwagandha, was shown to possess inhibitory activity for HPV and a wide range of influenza viruses. Based on previous reports as well as the results presented here, we propose withaferin-A derivatives as efficient lead compounds of potential drugs for combatting COVID-19. The six hit compounds generated by the pharmacophore model of withaferin-A derivative molecule 61 from the ZINC database might be used to screen for anti-CoV activities. Further, experimental work for all of the compounds predicted in this study needs to be carried out in order to verify specific drug likeliness in greater depth. The in silico strategy of integrating DNN and machine learning–based tool adopted here might be utilized to explore the potential applications of several other medicinal phytocompounds and also the available drugs against COVID-19. Finally, a line of caution: prior to using any outcome of an in silico study, a rigorous in vivo and in vitro research is obligatory.
Supplementary information
(DOC 1282 kb)
Acknowledgments
The authors would like to acknowledge the Central Instrumentation Facility, Gauhati University. Moreover, we are thankful to ProteinInsights (www.proteininsights.com) for providing computational resources for MDS study.
Authors’ contributions
Concept development, experiment, writing, editing, and revision by AG; experiment by MC; and MD simulation studies by AC and MPA.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Data availability
All the additional data are available as supplementary content.
Code availability
Not applicable.
Declarations
Ethical approval
This study does not contain any studies with animals performed by any of the authors.
Consent to participate
Not applicable.
Consent for publication
All the authors have the consent for publication in the present form.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Arabinda Ghosh and Monoswi Chakraborty, both the authors, contributed equally.
References
- 1.Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. J Virol. 2019;16(1):1–22. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brierley I. Ribosomal frameshifting on viral RNAs. J Gen Virol. 1995;76(8):1885–1892. doi: 10.1099/0022-1317-76-8-1885. [DOI] [PubMed] [Google Scholar]
- 4.Brown TDK, Brierley I (1995) The coronavirus nonstructural proteins. Coronaviridae 191–217. 10.1007/978-1-4899-1531-3_10
- 5.Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the nidovirales. J Gen Virol. 2000;81(4):853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 6.Fields BN, Knipe DM, Howley PM, Griffin DE. Coronaviruses. Fields virology. 4 th. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 1163–1185. [Google Scholar]
- 7.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82(5):2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 9.Kilianski A, Baker SC. Cell-based antiviral screening against coronaviruses: developing virus-specific and broad-spectrum inhibitors. Antivir Res. 2014;101:105–112. doi: 10.1016/j.antiviral.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pillaiyar T, Meenakshisundaram S, Manickam M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov Today. 2020;25(4):668–688. doi: 10.1016/j.drudis.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin MH, Chuang SJ, Chen CC, Cheng SC, Cheng KW, Lin CH, et al. Structural and functional characterization of MERS coronavirus papain-like protease. J Biomed Sci. 2014;21(1):54. doi: 10.1186/1423-0127-21-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prajapat M, Sarma P, Shekhar N, Avti P, Sinha S, Kaur H, et al. Drug targets for corona virus: a systematic review. Indian J Pharm. 2020;52(1):56. doi: 10.4103/ijp.IJP_115_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munagala R, Kausar H, Munjal C, Gupta RC. Withaferin a induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells. Carcinogenesis. 2011;32(11):1697–1705. doi: 10.1093/carcin/bgr192. [DOI] [PubMed] [Google Scholar]
- 14.Lin L, Cai Q, Zhang X, Zhang H, Zhong Y, Xu C, Li Y. Two less common human microRNAs miR-875 and miR-3144 target a conserved site of E6 oncogene in most high-risk human papillomavirus subtypes. Protein Cell. 2015;6(8):575–588. doi: 10.1007/s13238-015-0142-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joshi RS, Jagdale SS, Bansode SB, Shankar SS, Tellis MB, Pandya VK et al (2020) Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J Biomol Struct Dyn 1–16. 10.1080/07391102.2020.1760137 [DOI] [PMC free article] [PubMed]
- 16.Khan SU, Htar T (2020) Deciphering the binding mechanism of dexamethasone against SARS-CoV-2 main protease: computational molecular modelling approach. 10.26434/chemrxiv.12517535.v1
- 17.Devaux CA, Rolain JM, Colson P, Raoult D. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for COVID-19? Int J Antimicrob Agents. 2020;55(5):105938. doi: 10.1016/j.ijantimicag.2020.105938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stancioiu F, Papadakis GZ, Kteniadakis S, Izotov BN, Coleman MD, Spandidos DA, Tsatsakis A. A dissection of SARS-CoV2 with clinical implications. Int J Mol Med. 2020;46(2):489–508. doi: 10.3892/ijmm.2020.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farshi P, Kaya EC, Hashempour-Baltork F, Khosravi-Darani K (2020) A comprehensive review on the effect of plant metabolites on coronaviruses: focusing on their molecular docking score and IC50 values, Preprints, 2020050295. 10.20944/preprints202005.0295.v1 [DOI] [PubMed]
- 20.Das P, Majumder R, Mandal M, Basak P (2020) In-silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid based phytochemical constituents of calendula officinalis. J Biomol Struct Dyn 1–16. 10.1055/a-1170-4624 [DOI] [PMC free article] [PubMed]
- 21.Coppola M, Mondola R. Potential unconventional medicines for the treatment of SARS-CoV-2. Drug Res. 2020;70(6):286. doi: 10.1055/a-1170-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar V, Dhanjal JK, Bhargava P, Kaul A, Wang J, Zhang H et al (2020) Withanone and withaferin-A are predicted to interact with transmembrane protease serine 2 (TMPRSS2) and block entry of SARS-CoV-2 into cells. J Biomol Struct Dyn 1–27. 10.1080/07391102.2020.1775704 [DOI] [PMC free article] [PubMed]
- 23.Ahkam AH, Hermanto FE, Alamsyah A, Aliyyah IH, Fatchiyah F. Virtual prediction of antiviral potential of ginger (zingiber officinale) bioactive compounds against spike and MPro of SARS-CoV2 protein. Berkala Penelitian Hayati J Biol Res. 2020;25(2):52–57. doi: 10.23869/bphjbr.25.2.20207. [DOI] [Google Scholar]
- 24.Renjith MRD, Sankar M. Scope of phytochemicals in the management of covid-19. Pharm Res. 2020;3(1):26–29. [Google Scholar]
- 25.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hendaus MA (2020) Remdesivir in the treatment of Coronavirus Disease 2019 (COVID-19): a simplified summary. J Biomol Struct Dyn 1–10. 10.1080/07391102.2020.1767691 [DOI] [PMC free article] [PubMed]
- 27.Khan RJ, Jha RK, Amera GM, Jain M, Singh E, Pathak A et al (2020) Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J Biomol Struct Dyn 1–14. 10.1080/07391102.2020.1753577 [DOI] [PMC free article] [PubMed]
- 28.Dayer MR, Taleb-Gassabi S, Dayer MS. Lopinavir; a potent drug against coronavirus infection: insight from molecular docking study. Arch Clin Infect Dis. 2017;12(4):e13823. doi: 10.5812/archcid.13823. [DOI] [Google Scholar]
- 29.Kaiser J. Chemists want NIH to curtail database. Science. 2005;308(5723):774. doi: 10.1126/science.308.5723.774a. [DOI] [PubMed] [Google Scholar]
- 30.Chowdhury P (2020) In silico investigation of phytoconstituents from Indian medicinal herb ‘tinospora cordifolia (giloy)‘against SARS-CoV-2 (COVID-19) by molecular dynamics approach. J Biomol Struct Dyn 1–18. 10.1080/07391102.2020.1803968 [DOI] [PMC free article] [PubMed]
- 31.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;16:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Cheminformatics. 2011;3(1):33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18(6):463–477. doi: 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh A, Sutradhar S, Baishya D. Delineating thermophilic xylanase from bacillus licheniformis DM5 towards its potential application in xylooligosaccharides production. World J Microbiol Biotechnol. 2019;35(2):34. doi: 10.1007/s11274-019-2605-1. [DOI] [PubMed] [Google Scholar]
- 35.Guha R. On exploring structure–activity relationships. In: Kortagere S, editor. Silico models for drug discovery. Methods in molecular biology (methods and protocols) Totowa: Humana press; 2013. [Google Scholar]
- 36.Chinnasamy P, Arumugam R. In silico prediction of anticarcinogenic bioactivities of traditional anti-inflammatory plants used by tribal healers in Sathyamangalam wildlife sanctuary, India. Egypt J Basic Appl Sci. 2018;5(4):265–279. doi: 10.1016/j.ejbas.2018.10.002. [DOI] [Google Scholar]
- 37.Wolber G, Seidel T, Bendix F, Langer T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov Today. 2008;13(1–2):23–29. doi: 10.1016/j.drudis.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Koes DR, Camacho CJ. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res. 2012;40(web server issue):W409–W414. doi: 10.1093/nar/gks378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaw DE, Dror RO, Salmon JK, Grossman JP, Mackenzie KM, Bank JA, Chow E (2009) Millisecond-scale molecular dynamics simulations on Anton. In: Proceedings of the conference on high performance computing networking, storage and analysis, 65, 1–11. 10.1145/1654059.1654126
- 40.Sharma S, Deep S (2020) In-silico drug repurposing for targeting SARS-CoV-2 Mpro. ChemRxiv. 10.26434/chemrxiv.12210845.v1
- 41.Prasanth DSNBK, Murahari M, Chandramohan V, Panda SP, Atmakuri LR, Guntupalli C (2020) In silico identification of potential inhibitors from cinnamon against main protease and spike glycoprotein of SARS CoV-2. J Biomol Struct Dyn 1–1. 10.1080/07391102.2020.1779129 [DOI] [PMC free article] [PubMed]
- 42.Amin SA, Ghosh K, Gayen S, Jha T (2020) Chemical-informatics approach to COVID-19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J Biomol Struct Dyn 1–10. 10.1080/07391102.2020.1780946 [DOI] [PMC free article] [PubMed]
- 43.Ghosh K, Amin SA, Gayen S, Jha T (2020) Chemical-informatics approach to COVID-19 drug discovery: exploration of important fragments and data mining based prediction of some hits from natural origins as main protease (Mpro) inhibitors. J Mol Struct 129026. 10.1016/j.molstruc.2020.129026 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 1282 kb)
Data Availability Statement
All the additional data are available as supplementary content.
Not applicable.