

Abstract
Epigenetics is mainly comprised of features that regulate genomic interactions thereby playing a crucial role in a vast array of biological processes. Epigenetic mechanisms such as DNA methylation and histone modifications influence gene expression by modulating the packaging of DNA in the nucleus. A plethora of studies have emphasized the importance of analyzing epigenetics data through genome-wide studies and high-throughput approaches, thereby providing key insights towards epigenetics-based diseases such as cancer. Recent advancements have been made towards translating epigenetics research into a high throughput approach such as genome-scale profiling. Amongst all, bioinformatics plays a pivotal role in achieving epigenetics-related computational studies. Despite significant advancements towards epigenomic profiling, it is challenging to understand how various epigenetic modifications such as chromatin modifications and DNA methylation regulate gene expression. Next-generation sequencing (NGS) provides accurate and parallel sequencing thereby allowing researchers to comprehend epigenomic profiling. In this review, we summarize different computational methods such as machine learning and other bioinformatics tools, publicly available databases and resources to identify key modifications associated with epigenetic machinery. Additionally, the review also focuses on understanding recent methodologies related to epigenome profiling using NGS methods ranging from library preparation, different sequencing platforms and analytical techniques to evaluate various epigenetic modifications such as DNA methylation and histone modifications. We also provide detailed information on bioinformatics tools and computational strategies responsible for analyzing large scale data in epigenetics.
Keywords: Epigenetics, epigenome, DNA methylation, histone modifications, computational epigenetics, machine learning, transcriptional regulation, Next-generation sequencing
1. Introduction
Amongst all the communicable and non-communicable diseases, cancer remains a primary contributing factor to high mortality rates. According to the American Cancer Society, in 2019, approximately 1,762,450 cancer cases and 606,880 mortalities occurred within the United States (1). The investigation of the cancer genome has gained a tremendous amount of interest towards identifying and understanding novel mutations that are related to different types of cancer such as colorectal, breast and ovarian cancer (2). Despite the increased mortality rates, cancer survival rates have improved over the past years. According to the American Cancer Society, from 2007–2016, the cancer death rate declined by 1.4 – 1.8 % annually (1). Various advancements towards early diagnosis and treatment alternatives against cancer are the major contributors to improved cancer survival rates (3).
The global changes in epigenetic machinery are major hallmarks of cancers. Epigenetics is defined as a study of heritable gene expression changes that alter features of DNA without changing the DNA sequence. Besides a genetic anomaly, epigenetic dysregulation is primarily associated with cancer. Epigenetics exploits DNA and histones modifications which are known to be the building blocks of the nucleosomes (4). Nucleosomes are the key unit of chromatin which are comprised of four core histones; H3, H4, H2A and H2B which are closely associated with residual DNA (5). Each of these plays a pivotal role in various cellular processes such as DNA repair and gene expression (6). The accrual of epigenetics and genomic changes can potentially initiate important phenomena associated with various cellular processes that are related to multiple diseases such as cancer (7) and neurological disorders (8).
Despite numerous studies demonstrating the underlying mechanisms associated with DNA and histone modifications, complex epigenetic machinery and its reversible nature leading to disease progression is poorly understood (9). Epigenomics, which integrates multiple conventional genomics with computer science, mathematics, chemistry, biochemistry and proteomics for a broad analysis of genetic alterations in physiology, gene function or gene expression that are not based on gene sequence, opens up new opportunities advancing our knowledge of transcriptional regulation, nuclear structure, growth and disease (10). Various sophisticated computational and bioinformatics technologies such as next generation sequencing (NGS) greatly facilitate handling the genomic data and further understanding of the complex nature of epigenetic modifications at the genomic level. NGS technologies are key players in high-resolution epigenomic profiles across genomes (11). Also, gene expression microarray profiling is another method which aims to identify novel gene targets that can potentially serve as a clinical target for therapeutic intervention (12). These technologies provide great advantages such as parallel computation power thereby minimizing data analysis time and further providing genome-wide epigenetic profiling. These approaches enhance addressing important questions by demonstrating the association of epigenetic change with different chromatin modifications that regulate empirical processes such as transcription.
Computational epigenetics has continued to evolve as an emerging field that focuses on addressing and understanding various bioinformatic challenges which arise during the analysis of epigenetic data. Recent advancements in technology and enhanced developments of high-throughput sequencing methods have provided cost-effective methods for analyzing a vast amount of epigenetics data. In this review, we will cover existing information on the epigenetic patterns and gene regulation mechanisms associated with healthy tissues and their specific relation to disease progression. We will discuss various methodologies responsible for epigenomic profiling in terms of NGS technologies by discussing library preparation techniques and different sequencing platforms. We will also extrapolate recent advancements in computational methods regarding preprocessing and quality control of epigenetics data. Subsequently, we will discuss various computational tools for DNA methylation and histone modifications and further discuss recent online epigenetics databases that are related to DNA methylation and histone modifications.
2. Epigenetics
One of the aims of the study of epigenetic alterations in carcinogenic cells is to identify novel therapeutic targets thereby creating new avenues towards cancer therapy and treatment by availing epigenetic cancer drugs (13). This has been made possible by the recent developments in the field of nanotechnology (14). Studies have provided strong evidence supporting the establishment of differential gene expression changes as one of the primary contributors in epigenetics (14). DNA methylation, histone modification, different chromatin structure states, affiliated protein compositions and gene expression changes associated with transcriptional activity are some of the recent advancements that have provided an in-depth information contributing to epigenetic machineries(15).
DNA methylation is a useful indicator for the evaluation of the specific epigenetic conditions and is often catalyzed by three different DNA methyltransferases (DNMTs); DNMT1, DNMT3a and DNMT3b. During the process of methylation, methyl groups are added to the 5’ position of cytosine (5C) thereby generating 5-methylcytosine (5mC) (16). Cytosine methylation regulates gene transcription by interfering either directly or indirectly with the transcriptional machinery (17). In a mammalian genome, DNA methylation primarily occurs at cytosine residues followed by guanine residues which is symbolized as CpG dinucleotides wherein “p” stands for a phosphate binding deoxycytidine and deoxyguanosine (18). Each of these methylation patterns is crucial for various biological processes with specific molecular activities. For instance, DNMT1 is essential for genomic imprinting, heterochromatin formation and gene silencing. During X chromosome inactivation, DNA methylation plays a crucial role in long-term silencing, thereby affecting cell memory (19). In addition, DNMT3a and DNMT3b are primarily essential for embryonic development and play a crucial role in de novo methylation in the genome (20). However, studies have connected DNA methylation and transcription factor binding variants with the formation of tumor cells that may grow into cancer, as well as changes in the amount of lipids flowing into the blood resulting in cardiovascular disorders, while diabetes has been documented in some cases (21). This was reported after several assays were performed using chromatin immunoprecipitation and microarray analysis (21). It is therefore critical to understand the mechanistic link between DNA methylation and gene silencing followed by their association with diseases.
Besides DNA methylation, covalent histone modifications are another pivotal epigenetic mechanism which consists of histone proteins with a nucleosome (core), N-terminus domain and C-terminus domain. The N-terminus tails undergo post-translational modifications such as acetylation, methylation, ubiquitylation, and phosphorylation on specific residues (22). These amendments enforce key cellular processes such as transcription, repair and replication (23). Histone variations work by modifying the functionality of chromatin, thereby resulting in either activation or repression depending on different residues that undergo modifications(22). For instance, lysine 4 trimethylation on histone H3 (H3K4me3) influences transcriptionally active gene promoters(24), while, trimethylation of H3K9 (H3K9me3) and H3K27 (H3K27me3) occurs on transcriptionally suppressed gene promoters (23). A wide range of histone modifications has been identified, constituting a complex gene regulatory network critical for cell physiological activity (23, 25). The variations in histones are dynamically regulated by enzymes that add and remove covalent changes to the histone proteins. Histone acetyltransferases (HATs) and histone methyltransferases (HMTs) are associated with the addition of acetyl or methyl groups, respectively (26), while histone deacetylases (HDACs) and histone demethylases (HDMs) are associated with removal of either acetyl and methyl groups (27). Changes in histone patterns can result in transcriptional activation or silencing (28). For instance, H3K4me2, H3K4me3, H3R17me, H3K36me3 and H4R3me feature transcriptional activation while H3K9me3, H3K27me3, H4K20me1 and H4K20me3 are related to transcriptional silencing (29). Cancerous cells often show wide variations in patterns of histone methylation. For instance, changes in methylation patterns of H3K9 and H3K27 are related to anomalous gene silencing in different types of cancer (30). Numerous studies have also demonstrated that various HMTs are potentially responsible for abnormal silencing of tumor suppressor genes (TSGs). For instance, an investigation reported the overexpression of EZH2 (an H3K27 HMT) is related to breast cancer and prostate cancer (30). Additionally, elevated levels of G9a (an H3K9 HMT) were observed in hepatic cancer and are associated with a malignant phenotype by regulating chromatin structure (31). Cancer progression is also associated with site-specific demethylases along with overall methylation patterns. For example, LSD1 (lysine demethylase) can potentially remove histone activating and suppressing markers such as H3K4 and H3K9 methylation thereby serving as a co-repressor or co-activator (32). It is therefore imperative to know the specific context-dependent activities of these epigenetic-modulating enzymes and their target to ultimately apply their clinical impact and appropriate strategy for cancer treatment.
2.1. Complex interactions between DNA methylation and histone modifications
Other than conducting their mutually exclusive purposes, histone modifications and DNA methylation interact at numerous levels to modulate gene expression changes and chromatin organization (33). Most research has reported this form of epigenetic interaction and its deleterious means of causing diseases.
For instance, Wen et al., (2016) (34) reported the effect of the interaction in causing neurodegenerative diseases, while Zawisza and Wisnik (2017) have reported the association of both gene hypermethylation and hypomethylation together with histone modifications as the main causes of transformation of cells leading to prostate cancer (35). Numerous HMTs and HDMs also impact DNA methylation levels by modulating the DNMT proteins stability directly or indirectly by engaging HDACs and methyl-binding proteins to silenced genes with chromatin condensation (36). HMTs and HDMs deregulation have been linked with the aberrant effect of Grave’s disease among patients (37). Studies have reported a close association of various HMTs such as G9a/GLP (mediating H3K9 methylation) (38), SUV39H1 (mediating H3K9 methylation) (39) and PRMT5 (mediating H4R3 methylation) (40) by specifically recruiting DNA methyltransferases (DNMTs) to stably silence genomes and further direct DNA methylation at specific genomic targets (such as pericentric heterochromatin). For instance, an investigation in embryonic stem (ES) cells during early embryogenesis reported that silencing of Oct-3/4 (a POU domain homeobox gene, also known as Pou5f1) occurs by recruitment of a repressor complex that is comprised of G9a and other histone deacetylase enzymes which results in recruitment of promotors for DNMT3A and DNMT3b activity (41). Although genetic studies in embryonic stem cells have also demonstrated that a point mutation in the G9a SET domain inhibits heterochromatinization and also assists in de novo methylation, biochemical and functional studies have reported that G9a can assist in de novo methylation by itself due to its ankyrin domain (42) and by recruiting Dnmt3a and Dnmt3b independent of histone methyltransferase activity (43). Another investigation also reported that despite mutations in G9a/GLP that inactivate it during methyltransferase activity, it can potentially interfere with histone H3 lysine 9 (H3K9) methylation without influencing DNA methylation (38, 44). Research performed by Zarchi et al., (2017) reported that DNA methylation discriminates promoters from enhancers through a H3K4me1-H3K4me3 seesaw mechanism, and suggest its possible function in the inheritance of chromatin marks after cell division, an aberrant effect correlated with cancer and aging (45).
Numerous studies have also suggested that DNA methylation can also guide H3K9 methylation by regulating effector proteins such as Methyl-CpG-binding protein 2 (MeCP2), eventually creating a restrictive chromatin state (46). A study demonstrated a bidirectional relationship between DNA methylation and transcription wherein an NGS approach on the oocyte transcriptome and methylome analyses displayed a consistent pattern for CGIs that are methylated in the oocyte which needs to be transcribed and reduced by the active promoter-associated histone H3 lysine 5 methylation (H3K4me3) (47). These results were coherent with studies in different species such as humans and mice, wherein a large fraction of unmethylated intragenic CGIs also exhibited similar binding properties of H3K4me3 and RNA polymerase II (RNA Pol II) (48, 49). Additionally, elongation of RNA Pol II transcript has also made significant contributions to the intragenic aggregation of Histone H3 lysine 36 methylation (H3K36me3) which enhances DNMT3A activity (50). Therefore, complex histone modifications such as lysine methylation contribute to the alterable and dynamic regulation of gene expression in comparison to DNA methylation. These studies suggest that DNA methylation and histone modifications paradigms can strengthen epigenetic regulation by modulating gene expression activity which can further assist in determining and maintaining cellular identity and functionality.
These studies play a crucial role in bringing to the forefront various fundamental biological insights and have resulted in the generation of a tremendous amount of experimental data. Therefore, it is pivotal to design central databases (DB’s) repositories to better interpret and analyze the data using various computational approaches using machine learning approaches and other robust methods such as data mining, text mining and many others. Table 1 summarizes a comprehensive list of epigenetics databases which aim to provide large scale experimental datasets with genome-wide maps of various histone modifications, chromatin accessibility, DNA methylation and mRNA expression in different cell types and tissues across different species. These DB’s are not only restricted to serve as a central repository with large data aggregation but also help with analyzing the data in different ways.
Table 1.
List of Epigenetic Databases and Repositories
| Databases | Description | URL | References |
|---|---|---|---|
| IHEC data portal: International Human Epigenome Consortium | The IHEC DB provides a cumulative list of reference epigenomes applicable in various health and diseases (Humans: hg19, hg 38; Mouse: mm 10) | https://epigenomesportal.ca/ihec/index.html | (51) |
| The NIH ROADMAP Epigenomics Mapping Consortium | This consortium provides high-quality, genome-wide maps of various histone modifications, chromatin accessibility, DNA methylation and mRNA expression in different human cell types and tissues | http://egg2.wustl.edu/roadmap/web_portal/ | (52) |
| CEEHRC: Canadian Epigenetics, Environment and Health Research Consortium Network | A Canadian reference epigenome project with in-depth information about human cells across different tissues | http://www.epigenomes.ca/data-release/ | |
| BLUEPRINT | A European-based consortium that generates epigenomics maps in 100 different blood types | http://www.blueprint-epigenome.eu/ | (53) |
| IHEC CREST: Core Research for Evolutional Science and Technology | A DB consortium with reference genomes of human epithelial cells, vascular endothelial cells and cells of reproductive organs | http://crest-ihec.ip/english/index.html | |
| DeepBlue | A central DB specifically designed for programmatic operations on epigenetics data such as data overlapping and aggregations | https://deepblue.mpi-inf.mpg.de/ | (54) |
| Epigenome Browser | A DB reference sequence and working draft assemblies for a large collection of genomes. | http://www.epigenomebrowser.org/ | (55) |
| WashU Epigenome browser | A DB consortium which is not only limited to human and mouse but also provides extensive epigenome information for cow, chicken, fruit fly, chimpanzee and many others. | http://epigenomegateway.wustl.edu/browser/ | (56) |
| ENCODE Project Consortium | NIH-funded projects aimed at mapping all of the functional elements of the human genome | http://genome.ucsc.edu/ENCODE/ | (57) |
| GenExp | An interactive web-based, genome browser developed that integrates data available on the Distributed Annotation System (DAS) | http://gralggen.lsi.upc.edu/recerca/genexp/ | |
| AHEAD Task Force: Alliance for Human Epigenomics and Disease | A systematic effort to map a human epigenome with its primary goal towards implementing appropriate bioinformatics network and conducting epigenome mapping in a different normal tissue, which can serve as a reference guide for mapping abnormal cells | (58) | |
| HEP Project Consortium: Human Epigenome Project | A European-based consortium high-resolution epigenome data for analyzing DNA methylation in 43 distinctive individuals | (59, 60) | |
| HEROIC Project Consortium: High-Throughput Epigenetic Regulatory Organization IN Chromatin | A European-based DB consortium with chip-on-chip, chromosome interaction analysis and full-genome nuclear localization assays to understand complex human genome | https://cordis.europa.eu/project/id/18883 | (61) |
| dbEM | A DB specifically developed to determine the role of epigenetics proteins in oncogenesis with various genomic information such as mutations, CNV (Copy Number Variants) and gene expression across different tumor samples | http://crdd.osdd.net/raghava/dbem/ | (62) |
| EpiFactors | A DB for corresponding genes and specific epigenetic factors | https://epifactors.autosome.ru/ | (63) |
| HEDD: The Human Epigenetic Drug Database | A DB consortium with primary focus on storage and integration of epigenetics drug datasets | http://hedds.org/ | (64) |
3. Computational techniques for DNA methylation analysis and histone modifications analysis
3.1. Computational techniques for DNA methylation analysis
Numerous studies have reported various machine learning (ML) methods such as predictive modeling methods for determining DNA methylation patterns. The recent development in technology has become crucial in the medical field using machine learning and the aim of medical practioners is to be able to treat individuals based on their genetic and epigenetic profiles (65). Machine learning has eased the epigenetic studies such as DNA methylation due to its massive database and less power input (65). In general, biological and molecular data are obtained as raw datasets. To make biological interpretations, the raw datasets need to be annotated with class labels. Various ML techniques such as Active learning (ACL), Deep learning (DL) and Imbalanced class learning (ICL) have been employed in various cancer-related studies for genomic mapping to methylation patterns. All of these methods have exhibited various applications in biological datasets (66). For instance, a deep ML framework was designed to extract motifs by visualizing the positive classes learned by the network. The study demonstrated effective use of the Deep Motif (DeMo) framework to classify transcription factor binding sites (TFBS) and for further extracting visual representations of positive binding sites (67). Another study implemented in silico methods by using the Most Informative Positive (MIP) ACL approach to identify positive p53 mutants using 33% fewer studies than traditional active non-MIP research. The study also performed in vivo assays which demonstrated that positive regions had more new strong cancer rescue mutants than control regions based on p-value<0.01 in comparison to negative and non-MIP active learning. Additionally, the MIP ACL approach also released un-rescued p53 cancer mutant P152L (68). Another investigation reported the use of the class Based AL (CBAL) method (an ACL-based approach) which uses a mathematical model for calculating the cost of building a training set with a certain size and class ratio to annotate digital histopathology data (69). As a result, the investigation developed a mathematical model to predict the number of annotations required to achieve balanced training classes (69).
ML techniques have also been used efficiently to identify potent imprinted genes that play a vital role in embryonic development (70). For example, a study identified KCK9 (an oncogene expressed in the brain) and DLGAP2 (bladder cancer tumor suppressor) genes. Another study examined the correlation of various features with CpG island DNA methylation. The study extracted features of 190 CpG islands from human chromosome 21 and tested these CpGs on the remaining CpGs islands in the genome to identify methylated CpG islands (71). DNA methylation was associated with bladder cancer, a form of neoplasia and this was mainly due to the hypermethylation of CpG islands at the promoter regions of the genome (72). Other ML models such as artificial neural networks (ANN), linear discriminant analysis (LDA), Hidden markov model (HMM) and support vector machines (SVMs) have been used in predicting DNA methylation patterns. ANN and LDA have been employed for the classification into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) cell lines. The study reported DNA methylation levels across 20 loci in 41 SCLC and 46 NSCLC wherein 10 ANN models and 10 LDA models were trained using 10 different datasets, thereby proving effective use of ANN and LDA for development of automated methods in lung cancer classification (73).
Due to the tremendous amount of experimental data, the usefulness of ML approaches is not only confined to individual studies but also widely used to develop epigenetics databases using DL and Text mining techniques. DNA methylation DB’s play a vital role in studying co-valent modification of the genetic material of a cell, particularly in the complex genomes of vertebrates. Cancer methylation DB’s such as PubMeth (74), MethyCancer (75) and MethCancerDb (76) are advantageous for investigating irregular patterns of methylation associated with different cancers.
Generally, the development in the field of computational methods and machine learning have had a positive impact on the field, particularly in epigenetic studies. Methods such as Active Learning have effectively addressed the expense of generating epigenetic data whereas Imbalanced Class has addressed the problem of occurrence of low epimutations in the data. While supervised learning is more precise and has both predictive and interpretive data, unsupervised learning does not require class labels on data. Also with the advent of the method of deep learning, the problem associated with manually relevant genomic features has been addressed (66). However, machine learning as a newly advanced method of studying epigenetics is vulnerable to some challenges which makes it disadvantageous. For example, deep learning is more responsive to specific parameters of choice and takes quite a long time to learn. Imbalanced class learning is liable to overfitting of data based on the prejudice against minority class (66).These DNA methylation DB’s contain explicit information on DNA methylation patterns and specific genes across different species, individuals, tissues, cells and phenotypes.
3.2. Computational techniques for histone modifications analysis
The use of computational methods such as comparative genomics and ML to examine, model and forecast histone modifications in DNA sequences is of great interest. Computational methods have also been used to develop a simplified stochastic model to analyze the biostability and heredity conditions of an epigenetic memory-dependent silent mating-type region in the yeast. This model demonstrated robust biostability, which is resistant to high noise related to a random increase or loss of nucleosome modifications and a random partitioning after DNA replication (77). Comparative genomics has utilized technological innovations to identify histone markers and regulatory elements in eukaryotic genomes by performing genome-wide chromatin structure analysis. As a result, the study identified a binary pattern of histone modifications between euchromatic genes wherein active and inactive genes were hyperacetylated at histone H3 and H4 and hypermethylated to lysine 4 and lysine 79 of histone H3, respectively (78). It was also found that histone modification patterns of active genes are confined to the transcribed region and their regulation is closely associated with polymerase activity (79)Another investigation demonstrated extensive use of high-resolution genome-wide mapping technique (GMAT) in detecting histone H3 acetylation in active gene promoter regions. The accessibility and gene expression of a genetic domain to chromatin were found to be closely associated with promoter hyperacetylation (80) and further determine lysine-9/14-diacetylated histone H3 distribution in human peripheral T cells (81). The GMAT technique has also proven to be very effective in identifying nucleosome positioning sequences (NPSs) in Saccharomyces cerevisiae genes. Genes with a comparatively compact NPS framework over the promoter region were found to possess TATA box embedded in the NPS and tend to be regulated by chromatin enhancing and transforming factors (82).
Numerous studies have also reported the implementation of ML algorithms/techniques for determining histone positions as well as the various histone modifications such as acetylation, methylation and phosphorylation in DNA sequences (83). For instance, a study demonstrated efficient employment of HMMs models in determining differential histone modification sites (DHMS) by comparing the whole genome and ChIP-seq libraries. A new approach, ChIPDiff was proposed to unravel differential H3K4me3 and H3K36me3 sites between mouse embryonic stem cell (ESC) and neural progenitor cell (NPC) states (84). As a result, H3K27me3 DHMSs had high sensitivity, high specificity and greater technical reproducibility (84). Another investigation reported use of HMMs in conjunction with wavelet analysis to the non-partisan discovery of “domain-level” behavior in genomic functional data, along with activation and/or repressive histone modifications, RNA output and DNA replication timing. It was hypothesized that high-order patterns trends can be mutually studied in the HeLa cells to distinguish between 53 active and 62 repressed functional domains within the ENCODE regions based on the ENCODE project consortium (85). Besides, HMMs and supervised learning, Multilinear (ML) Regression and Multivariate Adaptive Regression Splines (MARS) have been used to build predictive models of gene expression to determine histone modifications and variants levels in human CD4+ T-cells (86) thereby identifying a close association of histone lysine and arginine methylation (H4R3me2) with PRMT5-catalyzed symmetric dimethylation gene expression repression (87).
Besides proving to be beneficial in analyzing and/or interpreting disease-specific (or research-specific) datasets, ML approaches have also been deployed in developing databases for predicting histone modifications (88). For instance, DeepHistone database was developed using a DL framework combining sequence information and usability evidence for chromatin to reliably assess various alteration sites specific to different histone markers (83). Another database, DeepChrome was developed using deep convolutional neural network framework to classify gene expressions by taking histone modifications data as input, thereby providing a visual representation of pattern maps using learnt deep model (83). Besides DeepHistone and DeepChrome, other databases as listed in Table 2 have also been developed to predict genome-wide histone modifications events, chromatin interactions, conserved sequence motifs, chromatin-associated proteins across different organisms.
Table 2.
DNA methylation and histone modifications databases
| Databases | Description | URL | References |
|---|---|---|---|
| DNA methylation databases (DB’s) | |||
| SRA | A DB consortium with integrated data of DNA methylation, cancer-related gene, mutation and cancer information from public resources, and the CpG Island (CGI) clones derived from our large-scale sequencing | https://www.ncbi.nlm.nih.gov/sra | (89) |
| MethDB | DNA Methylation database (MethDB) is currently the only public database for DNA methylation (http://www.methdb.net). This constantly growing database has become a key resource in the field of DNA methylation research | http://www.methdb.net/ | (90) |
| methBank 3.0 | An integration of different DNA methylomes in different species that can be used to predict age. | https://bigd.big.ac.cn/methbank/ | (91) |
| NGSmethDB | Whole-genome bisulfite sequencing (WGBS) database for across different tissues, pathological conditions, and various species | https://bioinfo2.ugr.es/NGSmethDB/ | (92) |
| MethBase | A DB repository with provides methylation level at individual sites, allele specific methylation regions, hypo- or hyper-methylated regions, partially methylated regions, and detailed meta data along summary statistics | http://smithlabresearch.org/software/methbase/ | |
| EWAS | A knowledgebase of epigenome-wide studies. | https://bigd.big.ac.cn/ewas | (93) |
| MethSMRT | An integrated DB for DNA 6mA and 4mC methylomes, generated from SMRT sequencing. The database provides a platform to host, browse, search and download 6mA and 4mC profiles for 156 species | http://sysbio.gzzoc.com/methsmrt/ | (94) |
| ENA | ENA provides a comprehensive record of the world’s nucleotide sequencing information, covering raw sequencing data, sequence assembly information and functional annotation | https://www.ebi.ac.uk/ena/browser/view | (95) |
| PubMeth | A cancer methylation DB which is based on text-mining approaches from Medline/Pubmed abstracts | http://www.pubmeth.org/ | (74) |
| DBCAT | A DB repository of CpG islands and various analytical tools for identifying different methylation profiles in cancer cells | https://bigd.big.ac.cn/databasecommons/database/id/3633 | (96) |
| MethMotif | An integrated DB with information on transcription factor binding motifs along with DNA methylation profiles | https://bioinfo-csi.nus.edu.sg/methmotif | (97) |
| REBASE | A DB consortium for DNA restrictions and modifications | http://rebase.neb.com/ | (98) |
| Histone modifications databases (db) | |||
| 4DGenome | A DB consortium with chromatin interactions across 5 different species. The DB incorporates 3C, 5C, CHIA-PET, Hi-C, Capture-C and I-PET | https://4dgenome.research.chop.edu/ | (99) |
| 3CDB: Chromatin Confirmation Capture Database | A manually curated DB to analyze contact frequencies between specific genomic sites in a genome population | http://3cdb.big.ac.cn/ | (100) |
| HIstome: The Histone Infobase | A DB with contains information about human histone variants, post-translational modifications sites and different histone modifying enzymes. | http://www.actrec.gov.in/histome/index.php | (101) |
| CisRED | A DB consortium with conserved sequence motifs information using genome scale motif discovery, similarity, clustering, co-occurrence and co-expression calculations | http://www.cisred.org/ | (102) |
| DAnCER | A DB with genome-wide information based on histone modifications | http://wodaklab.org/dancer/ | (103) |
| HIstome | Displays information about human histone variants, sites of their post-translational modifications and about various histone modifying enzymes | http://www.iiserpune.ac.in/~coee/histome/index.php | (104) |
| CPLM | A DB with protein lysine modification sites occurring at active amino groups of lysine residues in proteins. The DB contains 203,972 protein modifications on 189,919 lysine-modified sites in 45,748 proteins across 122 different species. | http://cplm.biocuckoo.org/ | (105) |
| ChromDB: Chromatin Database | A DB with chromatin-associated proteins, including RNAi-associated proteins across different species. | http://www.chromdb.org/ | (106) |
| WERAM | A DB for Writers, Erasers and Readers of histone acetylation and methylation in eukaryotes | http://weram.biocuckoo.org/ | (107) |
| PLMD: Protein Lysine Modification Database | An online data source with protein lysine modifications such as phosphorylation, sumolyation, calpain cleavage, pupylation and many others | http://plmd.biocuckoo.org/ | (108) |
| PTMD | A DB for specifically analyzing association of Posttranslational modifications (PTMs) and human diseases | http://ptmd.biocuckoo.org/ | (109) |
Table 2 summarizes DNA methylation and histone methylation DB’s which contains specific information on DNA methylation patterns and histone modification events which are very helpful in understanding various epigenetic events within the specific types of cells and also in an evolutionary context. These DB’s are very helpful in understanding various epigenetic events within the specific types of cells and also in an evolutionary context. These approaches are extremely beneficial for the interpretation of epigenetic events such as DNA methylation and histone modifications.
4. Next-generation sequencing and epigenomics
4.1. Next-generation sequencing platforms
Next generation sequencing NGS describes a recently revolutionized form of sequencing characterized by speed and accuracy of the process such that the human genome can be fully sequenced in one day in contrast to Sanger sequencing that was relatively very slow (110). NGS technologies have made significant contributions in identifying differentially methylated DNA regions along with the discovery of new gene regulatory elements in epigenetic machinery (111). Besides DNA methylation, histone modifications and transcriptomes are also being consistently investigated using NGS genome-wide methodologies (111). Due to the lack of preservation of the methylation signature in PCR amplification, NGS methods have been extensively used to conserve the epigenetic landmark in the DNA. These approaches have also been employed in other aspects of chromatin organization such as DNA accessibility (112) and high-order chromatin complexes globally (113). Illumina genome analyzer, soLiD and Roche:454 Genome sequencer FLX (GS FLX) are three NGS platforms which have various advantages and disadvantages. NGS involves the implementation of specific protocols pertaining to library preparation, sequencing, read alignment and data analysis techniques using different software for varying sequencing platforms as demonstrated in Figure 1.
Figure 1.
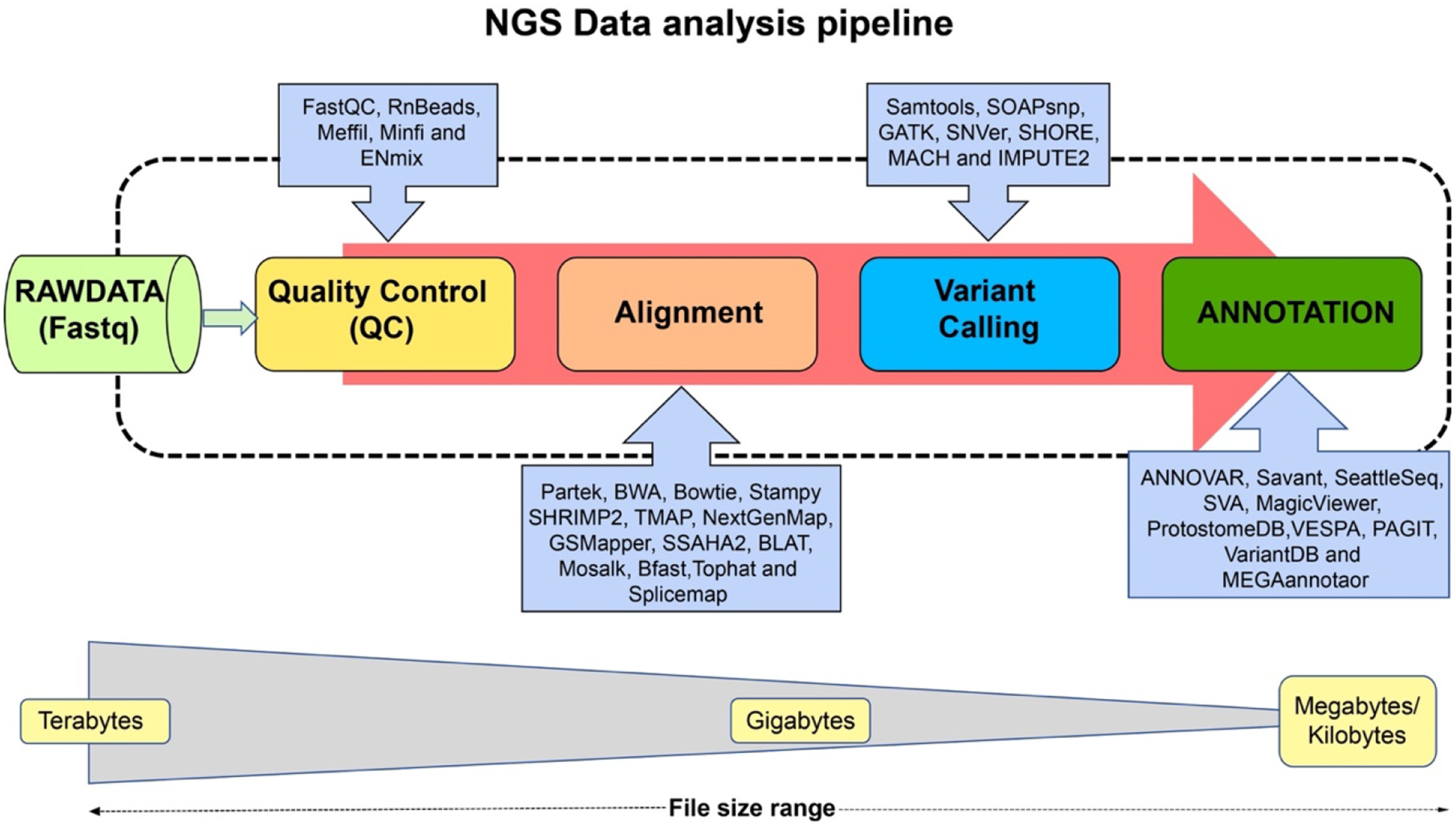
Bioinformatics pipeline NGS analysis. Raw datasets are generated either using single-end or pair-end sequencing which is then tested for quality control. Afterward, the data are aligned to the reference genome. The process of variant calling is diverse and based on the experiment and/or clinical research, numerous softwares or methods are employed. Finally, depending on different requirements, annotation is performed using different softwares as mentioned above.
Consequently, various computational approaches have recently emerged that are developed for the detection of DNA methylation and histone modifications. While these technologies are not yet ubiquitous due to relatively high costs and are still in the development phase, these methods are encouraging towards identifying approximately 10% higher methylation states in comparison to conventional methods. Table 3 summarizes a comprehensive list of different computational tools that can be used for analysis using NGS methodologies.
Table 3.
List of various tools used during NGS pipeline analysis
| Tools | Description | URL | References |
|---|---|---|---|
| Quality control (QC) | |||
| FastQC | A QC tool for high-throughput data | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | |
| RnBeads | A QC tool for DNA methylation data | https://rnbeads.org/ | (114) |
| Meffil | A QC tool specifically designed to handle gigantic DNA methylation data | https://github.com/perishky/meffil/ | (115) |
| Alignment | |||
| BWA | Alignment tool for mapping low-divergent sequences | http://bio-bwa.sourceforge.net/bwa.shtml | (116) |
| Hisat2 | Alignment tool for RNA-seq reads | https://github.com/DaehwanKimLab/hisat2/blob/master/docs/_data/collaborate.yml | (117) |
| Bowtie | Ultra-fast, memory efficient short read aligner | http://bowtie.cbcb.umd.edu/ | (118) |
| DNAscan | fast and efficient bioinformatics pipeline that allows for the analysis of DNA Next Generation sequencing data, requiring very little computational effort and memory usage. | https://github.com/KHP-Informatics/DNAscan | (119) |
| Variant calling | |||
| SAMtools | Variant calling tool which is based on UNIX commands | https://samtools.github.io/bcftools/howtos/variant-calling.html | (120) |
| GATK | A genome analysis toolkit for variant detection | https://gatk.broadinstitute.org/hc/en-us | (121) |
| SNVer | A statistical tool for calling rare variant in analyzing poor or individual NGS data | https://sourceforge.net/projects/snver/ | (122) |
| Annotation | |||
| ANNOVAR | A tool for functionally annotating genetic variants | https://annovar.openbioinformatics.org/en/latest/user-guide/filter/ | (123) |
| SAVANT | Sequence annotation and visualization analysis tool for genomic data | http://compbio.cs.toronto.edu/savant | (124) |
| SVA | NGS tool for annotating and visualizing human genomic data | https://www.ebi.ac.uk/ena/browser/sva | (125) |
| VariantDB | A flexible annotation and filtering tool for NGS data | http://www.biomina.be/app/variantdb/ | (126) |
Despite numerous variations in their technicalities, the three platforms display a basic workflow starting from preparation of sequencing library, which is built from DNA fragments wherein the ends are modified by a ligating platform using specific PCR and sequencing adapters. Conventional methods usually involve splitting of breaking genomic DNA (gDNA) randomly into smaller sizes followed by generation of either fragment templates or mate-pair templates. The primary characteristic of NGS technologies is that the template is linked to a solid surface thereby assisting in immobilization which in turn facilitates numerous sequencing reactions/protocols. Table 4 summarizes primary features of sequencing platforms along with their advantages and disadvantages.
Table 4.
Characteristics of primary NGS platforms and their comparison based on different parameters
| Sequencing Platforms | Read length | Sequencing | Reads, Output data/ run and time/run | Primary characteristics, applications and strength | Weakness |
|---|---|---|---|---|---|
| Solexa | ~ 75–100 bp (pair-end) | Sequencing by synthesis | Reads: 3 G Output data/run: 600 Gb Time/run: 2–10 Days |
Read counting Variant detection by genome re-sequencing |
Short read assembly Relatively high error rate |
| MiSeq | ~ 250–600 bp (single-end and pair-end) | Reads: 2.5 G Output data/run: 600 Gb Time/run: 4 ~ 12 Days |
Fastest Illumina run-time Longest Illumina read lengths |
Fewer reads than HiSeq Higher cost/megabyte run in comparison to HiSeq |
|
| NextSeq500 | ~ 300 bp (pair-end) | Easy to handle Moderate instrumental and run-time cost |
Higher cost/megabyte run Higher instrument cost |
||
| HiSeq 2500 | ~ 50 bp (single-end) ~ 50–100 bp (pair-end) |
Reads: 3 G Output data/run: 600 Gb Time/run: 4 ~ 12 Days |
High- throughput sequencing with parallel processing of samples with higher output (8 lane) and Lower cost/megabyte run Flexibility for flow cells with various read-length configuration |
Higher instrument cost Higher cost/run |
|
| Roche | |||||
| 454 GS FLX | ~700–100 bp | Pyrosequencing | Reads: 1 Million Output data/run: 0.7 Gb Time/run: 24–28 hours |
Generates long reads Short run time De novo genome Transcriptome assembly |
High instrument cost Expensive reagents |
| 454 GS Jr | ~ 700 bp | Reads: 0.1 Million Output data/run: 0.35 Gb Time/run: 3–10 hours |
Lower cost than 454 GS FLX Longest contiguous reads |
Higher cost/megabyte run Fewer reads Shorter reads than 454 GS FLX |
|
| SOLiD 5500/ 5500xl/ 5500W | |||||
| SOLiD 5500/ 5500xl/ 5500W | 75 × 35 bp (pair-end) | Ligation and two-base coding | Reads: 1200~1400 Million Output data/run: 120 Gb Time/run: 7–14 days |
Each flow-chip lane can operate independently; high accuracy; bases output (not color-space) | Higher accuracy in case of coverage > 30x Generates shorter reads Less data coverage Higher capital cost |
| °° | |||||
| Proton | ~ 200 bp | Semiconductor sequencing | Reads: 300–500 Mb Output data/run: 1.2–2 Gb Time/run: 2–8 hours |
Lower instrumental cost Provides greater coverage than MiSeq |
Shorter reads Higher error rates Higher cost/megabyte run |
| PGM | ~ 200–400 bp | Reads: 0–6 - 3 M Output data/run: 10 Gb Time/run: 2–4 hours |
|||
4.2. DNA methylation profiling and Next-generation sequencing
Despite recent advancements, 5mC analysis possess various limitations such as robustness and lack of the consistency as the methyl group prevents the direct labeling for subsequent affinity purification and detection (111). Initially DNA methylation profiles were being studied using methods such as microarrays and methyl-specific polymerase chain reaction which were cost effective and required small DNA input. The method however have limitations in that they have low levels of accuracy and low sample input (127). Next-generation sequencing (NGS) platforms are now advancing that facilitate a significant investigation of the methylation patterns of numerous CpG sites and the creation of genomic maps of DNA methylation at a single base resolution (127). Integration of NGS and methylation studies can therefore be a potent approach for DNA methylation studies involving clinical research due to their versatility in clinical diagnosis and prognosis while in pharmacogenomics, targeting certain CpG islands on promoters of certain genes of tumors may predict the likelihood of disease response to treatment. NGS approaches to the genome-wide profile of DNA methylation can be broadly classified by focusing on affinity enrichment-based methods, restriction enzyme-based methods and direct bisulfite-based conversion methods (128). Studies have suggested that these methods can also be assimilated to enhance single method resolution and performance. For instance, methylated immunoprecipitation of DNA (MeDIP-seq) and MRE-seq methods can be employed in combination for profiling of methylated and unmethylated regions within the genome (49). Therefore, the use of NGS and methylation arrays can be a powerful approach for DNA methylation studies which are primarily involving clinical research in terms of data exploration, data integration and screening methods Figure 2 represents a historical description of NGS-based methods applied to DNA methylation profiling.
Figure 2.

NGS methods in DNA methylation profiling. Affinity-based enrichment methods: MeDIP-Seq: methylated DNA immunoprecipitation sequencing, MBD-Seq: methyl-CpG binding domain sequencing; Busulfite conversion-based methods: RRBS-Seq: reduced representation bisulfite sequencing, WGBS: whole-genome bisulfite sequencing.
4.2.1. Affinity-based enrichment methods
Affinity-based enrichment methods are comprised of MeDIP-Seq and methylated-CpG binding proteins (MBD-Seq) to determine the methylated genomic regions. MeDIP-Seq is an immune-precipitation based technique that uses antibodies against a single-stranded methylcytosine and thereby performing immunoprecipitation in a denatured state (127). MeDIP-seq method can determine approximately 80% of the 28 million CpGs in the human genome at around 100–300 bases (129). MBD-Seq is very similar to MeDIP-Seq wherein the enrichment of genomic fragments occurs depending on its methylation content. Upon the enrichment of genomic fragments, standard library construction methods are employed to generate a library determining the methylation regions within the genome (130). Unlike MeDIP-Seq, in the MBD-Seq technique, the fragments of weakly methylated DNA are elucidated at lower salt concentrations relative to fragments of highly or strongly methylated DNA such as methylated CpG Islands. These methods extensively characterize enriched CpGs which includes their islands and promoter regions (127). Affinity-based enrichment techniques have been applied in assays to determine methyl-CpG binding activity and DNA methylation in early Xenopus embryos (131). In this experiment, methylated DNA affinity precipitation method was implemented to assay binding of proteins to methylated DNA. Endogenous MeCP2 and MBD3 were precipitated from Xenopus oocyte extracts and conditions for methylation-specific binding were optimized. For a reverse experiment, DNA methylation in early Xenopus embryos was assessed by MBD affinity capture (131).
The main limitations of these approaches are the limited quantification across regions and lack of base-specific data analysis, which ultimately diminishes the insight that could be gathered from them. Poor quantification across genomic regions and lack of base-specific data analysis are the major drawbacks of affinity-based enrichment methods. Such methodologies, therefore, require significant experimental work and extensive use of bioinformatics approaches for in-depth analysis (129).
4.2.2. Restriction enzyme (RE)-based methods
(132)RE-based approaches use restriction enzymes that can cleave recognition sequences at the DNA methylation site which can potentially identify 5mC in a particular sequence of interest. This method is either based on single methyl-sensitive RE digestion such as HpaII; HELP-seq, Methyl-seq and MSCC or multiple RE digestion such as AciI, Hinc6I and MRE-seq (132). Restriction- sensitive endonucleases such as Hpall digest the high-quality DNA at an unmethylated region followed by ligation with an adaptor which assists other restriction enzymes such as EcoP15I or MmeI (132). RE-based methods are the most cost-effective and time-consuming sequencing method which involves minute amounts of DNA (133). RE-based methodologies pose unique challenges while sequencing on either Illumina Genome Analyzer or SOLiD platforms. The primary drawback of this method is the inability to adapt coverage regions of interest as this method relies on the restriction sites positions within the genome which makes it incapable of determining the genes with sparse CCGG motifs (127). To overcome this restriction enzyme challenge, LpnPI was used to perform restriction digestion and was blocked by fragment sizes less than 32 bp to prevent complete digestion. The results showed that methylated DNA sequencing (MeD-seq) of LpnPI-digested fragments revealed highly reproducible genome-wide CpG methylation profiles for >50% of all potentially methylated CpGs, at a sequencing depth less than one-tenth required for whole-genome bisulfite sequencing (WGBS) (134).
4.2.3. Bisulfite conversion-based methods
The protocol of bisulfite sequencing (BS-Seq) begins with the treatment of denatured DNA with bisulfite, during which the non-modified cytosine is converted to uracil; although, methylated cytosines remain stable without undergoing any changes leading to the identification of 5mC resolution (135). As a result, the genomic regions treated with bisulfite are amplified by site-specific PCR, cloned, and imperiled to Sanger sequencing. Additionally, sequencing reads are evaluated periodically and visualized as a matrix with each clone’s CpG material depicted as a row. After the treatment with bisulfite, the sequencing library is subjected to PCR amplifications which extend the sequencing of the adapter thereby allowing clonal amplification and sequence. Recent advancements in NGS systems have enhanced the performance and reliability of this method thereby reducing the costs associated with experimental procedures.
Whole-genome bisulfite sequencing (WGBS) is by far the most informative that targets the whole genome, which in turn makes it the most expensive technology for base resolution. The process begins with the creation of genomic DNA libraries that are further subjected to bisulfite conversion, following sequencing and aligning to the reference genome (136). Even though BS-seq is by far the most direct assay by exhibiting the highest methylation detection resolution, this approach is specifically restricted to various studies which aim to answer specific questions related to a comprehensive DNA methylation profiling (127).
In the reduced representation of bisulfite sequencing (RRBS) method, the genome is digested by Msp1 (a methylation-insensitive restriction enzyme) and specific DNA fragments approximately ranging from 100–300 bps are selected to generate a fragment library using NGS platforms (137). These shortlisted DNA fragments are considered as the input for the library construction using adapters (methylated) and are further subjected to bisulfite conversion (138). Although RRBS covers only 12% of CpGs, the CpGs are enormously enriched in CpG islands (139). The method has been applied in an experiment to detect methylome profiling in plants which is termed as plant-reduced representation bisulfite sequencing (plant-RRBS), using optimized double restriction endonuclease digestion, fragment end repair and adapter ligation, followed by bisulfite conversion, PCR amplification and NGS. The results have produced tens of millions of RRPS methylated regions using multiple samples (140). Application of the method is limited to the high cost of performing the assays to large number of patients (141).
4.3. Histone modifications profiling and Next-generation sequencing
The underlying beliefs for mapping these genome-wide post-translation modifications involve an NGS methodology such as chromatin immunoprecipitation (ChIP) (142). The process begins by either integrating histones biologically with DNA via the intervention of a cross-linking reagent (such as formaldehyde) or releasing the histones in their native form by nuclease addition which results in digestion of genomic DNA (gDNA) at unprotected linker sequences (142). The protein / DNA mixture is subjected to immunoprecipitation after gDNA fragmentation using antibodies raised against post-translational modification (143). Subsequently, during the process of immunoprecipitation, DNA fragments associated with histone peptides are subjected to purification and library construction followed by direct sequencing (144). Single-cell ChIP-sequencing in different stem cells and progenitor cells have led to the identification of population sub-groups that are differentiated based on variations in pluripotent chromatin signatures and primer distinction (145).
Numerous studies have suggested that direct sequencing of ChIP fractions have remarkable benefits over alternative techniques such as ChIP-chip wherein fragments obtained from ChIP are potentially determined by hybridization to the microarray. A study on the same experiments reported that ChIP-chip obstructed unique and biologically distinct peaks that were observed in ChIP-Seq. Furthermore, the study also reported that in ChIP-Seq, fragments of the DNA are sequenced explicitly, rather than being hybridized on an array. Unlike hybridization, ChIP-Seq does not have background noise which arises during the hybridization process due to the higher frequency of inconsistently matched sequences. In comparison to ChIP, Nucleic acid hybridization is dynamic process that depends on several variables such as GC-content, concentration secondary structure of the target and test sequences (146). Genome-wide CHIP-Seq has been applied in histone-modification profiles, including mapping of H3K4 acetylation and H3K4 trimethylation, H3K9 acetylation, and H3K27 methylation, which have been used for defining breast cancer subtypes and recognition of special players in tumorigenesis (141). Past research has reported the use of next generation sequencing in breast cancer studies. This was done using cell based models transcribing three oncogenes and reductions in histones H3K9me2 and H3K9me3, and elevations in the demethylases for H3K9me1 and H3K9me2 (KDM3A, or JMJD1A) as important events in breast cancer (141).
Recent advancements in technologies have offered innovative and more efficient NGS platforms such as the Illumina Genome Analyzer and SOLiD offers significant advantages by offering parallel processing of shorter reads and facilitating direct library construction from the immuno-precipitated products unlike early methods of ChIP such as capillary sequencing (147). The process of library construction for Illumina Genome Analyzer and SOLiD is very similar to typical approaches of the whole genome shotgun sequencing. The process begins with the repairing of the disheveled ends (within low nanogram range) of the enriched fragmented DNA and further linking the resulting fragments on either A-tailed end in Illumina Genome Analyzer or blunt end in SOLiD DNA fragments. The adapter-ligated product is later amplified with PCR primers and conjugated to the adapter sequences. Additionally, in SoLiD, the addition of two independent adapter sequences during ligation enables the inclusion of 50% of adapted fragments in PCR amplification. Unlike SoLiD, Illumina genome Analyzer library preparation utilizes adapters which are partially analogous generating a “fork” in the adapter which is ultimately resolved during PCR (148).
Either of these platforms facilitate enhanced spatial resolution which is crucial for the epigenome characterization comprising post-translation modifications of chromatin and nucleosome positioning. For instance, a study exhibited profiling of twenty histone methylation marks, histone variant H2A.Z, RNA Polymerase II and the DNA-binding protein CTCF in human T cells, totaling close to 8 million tags per sample using Solexa (149). Another investigation demonstrated the role of histone modifications during cell differentiation by profiling seven lysine trimethylation marks in mouse ES cells in pluripotent and lineage-committed cells (150). ChIP-seq has also been implemented in conjugation with Roche 454 pyrosequencing for generating histone variant H2A.Z maps in yeast (151) and common fruit fly (152). These studies have significantly highlighted the advantages of NGS methodologies in histone modification profiling.
NGS technologically has fundamentally pioneered DNA methylation and histone modification research leading to the determination of new approaches which can potentially extend understanding and characterization of epigenetic machinery at a global level. However, the biggest limitation for NGS in clinical therapeutics is the tremendous cost associated with infrastructure, requirements of high-performance computing concerning data handling capacity and storage, and requirement of expert bioinformatician support to analyze and interpret the vast amount of data generated using NGS technologies. In addition, the efficacious assessment of datasets generated by various research facilities employing common DNA profiling and histone modification techniques involves the application of experimental and computational methods specifications to facilitate significant relationships between the experiments (141).
5. Concluding remarks
Investigators have made extensive use of computational methods ranging from different databases designed specifically for demonstration of epigenetic machineries such as DNA methylation and histone modifications. Furthermore, in-depth use of NGS technologies has contributed significantly towards DNA methylation profiling and histone modification profiling at a relatively affordable price. This is a paramount in the field of medicine, particularly with respect to oncology studies. Such massive-parallel high-throughput sequencing technologies offer promising results to decipher the existence and trends of epigenetic modifications as well as their effects on the different processes of pathology and physiology. However, some challenges do exist for the next generation sequencing in that it can be technologically demanding. A fundamental principle to this endeavor is the creation of statistically validated quality methods for assessing data quality and further rendering significant efforts towards enrichment-dependent epigenomic profiles which are mainly used in genomic studies. As a result, the increasing amount of epigenomic datasets facilitates bioinformatics-based methodologies being deployed regularly for continuous development of new methods and/or techniques for proper curation and maintenance of existing databases and methodologies.
Highlights.
Implementation of computational approaches towards understanding the complex interactions between DNA methylation and histone modifications.
Different computational techniques such as machine learning for analyzing epigenetic machinery.
Various epigenetics databases and resources for analyzing large scale datasets in epigenetics.
NGS approaches in DNA methylation and histone modifications profiling.
Funding source
This work was supported in part by grants from the National Cancer Institute (R01 CA178441 and R01 CA204346)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement:
Declarations of interest: none. The authors have no conflict of interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: a cancer journal for clinicians. 2019;69(1):7–34. [DOI] [PubMed] [Google Scholar]
- 2.Chakravarthi BV, Nepal S, Varambally S. Genomic and epigenomic alterations in cancer. The American journal of pathology. 2016;186(7):1724–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM, et al. Cancer treatment and survivorship statistics, 2019. CA: a cancer journal for clinicians. 2019;69(5):363–85. [DOI] [PubMed] [Google Scholar]
- 4.Kumar S, Singh AK, Mohapatra T. Epigenetics: history, present status and future perspective. Indian J Genet Plant Breed. 2017;77:445–63. [Google Scholar]
- 5.Zhu P, Li G. Structural insights of nucleosome and the 30-nm chromatin fiber. Current opinion in structural biology. 2016;36:106–15. [DOI] [PubMed] [Google Scholar]
- 6.Lai WK, Pugh BF. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nature reviews Molecular cell biology. 2017;18(9):548–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harbor perspectives in biology. 2016;8(9):a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banik A, Kandilya D, Ramya S, Stünkel W, Chong YS, Dheen ST. Maternal factors that induce epigenetic changes contribute to neurological disorders in offspring. Genes. 2017;8(6):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teschendorff AE. Computational and Statistical Epigenomics: Springer; 2015. [Google Scholar]
- 11.Mensaert K, Denil S, Trooskens G, Van Criekinge W, Thas O, De Meyer T. Next-generation technologies and data analytical approaches for epigenomics. Environmental and molecular mutagenesis. 2014;55(3):155–70. [DOI] [PubMed] [Google Scholar]
- 12.Irigoyen A, Jimenez-Luna C, Benavides M, Caba O, Gallego J, Ortuño FM, et al. Integrative multi-platform meta-analysis of gene expression profiles in pancreatic ductal adenocarcinoma patients for identifying novel diagnostic biomarkers. PloS one. 2018;13(4):e0194844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laird PW. The power and the promise of DNA methylation markers. Nature Reviews Cancer. 2003;3(4):253–66. [DOI] [PubMed] [Google Scholar]
- 14.Roberti A, Valdes AF, Torrecillas R, Fraga MF, Fernandez AF. Epigenetics in cancer therapy and nanomedicine. Clinical Epigenetics. 2019;11(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng A, Mao X, Zhong J, Fan S, Hu Y. Single-Cell Multi-Omics and Its Prospective Application in Cancer Biology. Proteomics. 2020:1900271. [DOI] [PubMed] [Google Scholar]
- 16.Hoang NM, Rui L. DNA methyltransferases in hematological malignancies. Journal of Genetics and Genomics. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewsey MG, Hardcastle TJ, Melnyk CW, Molnar A, Valli A, Urich MA, et al. Mobile small RNAs regulate genome-wide DNA methylation. Proceedings of the National Academy of Sciences. 2016;113(6):E801–E10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321(6067):209–13. [DOI] [PubMed] [Google Scholar]
- 19.Park Y, Kuroda MI. Epigenetic aspects of X-chromosome dosage compensation. Science. 2001;293(5532):1083–5. [DOI] [PubMed] [Google Scholar]
- 20.Veland N, Lu Y, Hardikar S, Gaddis S, Zeng Y, Liu B, et al. DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic acids research. 2019;47(1):152–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brasa S, Mueller A, Jacquemont S, Hahne F, Rozenberg I, Peters T, et al. Reciprocal changes in DNA methylation and hydroxymethylation and a broad repressive epigenetic switch characterize FMR1 transcriptional silencing in fragile X syndrome. Clinical epigenetics. 2016;8(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corujo D, Buschbeck M. Post-translational modifications of H2A histone variants and their role in cancer. Cancers. 2018;10(3):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. [DOI] [PubMed] [Google Scholar]
- 24.Ricketts MD, Han J, Szurgot MR, Marmorstein R. Molecular basis for chromatin assembly and modification by multiprotein complexes. Protein Science. 2019;28(2):329–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128(4):669–81. [DOI] [PubMed] [Google Scholar]
- 26.DesJarlais R, Tummino PJ. Role of Histone-Modifying Enzymes and Their Complexes in Regulation of Chromatin Biology. Biochemistry. 2016;55(11):1584–99. [DOI] [PubMed] [Google Scholar]
- 27.Woo H, Ha SD, Lee SB, Buratowski S, Kim T. Modulation of gene expression dynamics by cotranscriptional histone methylations. Experimental & molecular medicine. 2017;49(4):e326–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lubecka K, Kurzava L, Flower K, Buvala H, Zhang H, Teegarden D, et al. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis. 2016;37(7):656–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Z, Zhou J, Jiang J, Yuan J, Zhang Y, Wei X, et al. Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nature communications. 2019;10(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui H, Hu Y, Guo D, Zhang A, Gu Y, Zhang S, et al. DNA methyltransferase 3A isoform b contributes to repressing E-cadherin through cooperation of DNA methylation and H3K27/H3K9 methylation in EMT-related metastasis of gastric cancer. Oncogene. 2018;37(32):4358–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bárcena-Varela M, Caruso S, Llerena S, Álvarez-Sola G, Uriarte I, Latasa MU, et al. Dual Targeting of Histone Methyltransferase G9a and DNA-Methyltransferase 1 for the Treatment of Experimental Hepatocellular Carcinoma. Hepatology. 2019;69(2):587–603. [DOI] [PubMed] [Google Scholar]
- 32.Pirola L, Ciesielski O, Balcerczyk A. The methylation status of the epigenome: Its emerging role in the regulation of tumor angiogenesis and tumor growth, and potential for drug targeting. Cancers. 2018;10(8):268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature Reviews Genetics. 2009;10(5):295–304. [DOI] [PubMed] [Google Scholar]
- 34.Wen K-x, Miliç J, El-Khodor B, Dhana K, Nano J, Pulido T, et al. The role of DNA methylation and histone modifications in neurodegenerative diseases: a systematic review. PLoS One. 2016;11(12):e0167201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nowacka-Zawisza M, Wiśnik E. DNA methylation and histone modifications as epigenetic regulation in prostate cancer. Oncology reports. 2017;38(5):2587–96. [DOI] [PubMed] [Google Scholar]
- 36.De Smedt E, Lui H, Maes K, De Veirman K, Menu E, Vanderkerken K, et al. The epigenome in multiple myeloma: impact on tumor cell plasticity and drug response. Frontiers in oncology. 2018;8:566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan N, Mu K, An X-f, Li L, Qin Q, Song R-h, et al. Aberrant Histone Methylation in Patients with Graves’ Disease. International journal of endocrinology. 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. The EMBO journal. 2008;27(20):2681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumari D, Sciascia N, Usdin K. Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Alleles in Fragile X Syndrome Patient Derived Cells. Genes. 2020;11(4):356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nature structural & molecular biology. 2009;16(3):304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patra SK. Roles of OCT4 in pathways of embryonic development and cancer progression. Mechanisms of Ageing and Development. 2020;189:111286. [DOI] [PubMed] [Google Scholar]
- 42.Almeida GPd. Modulation of an essential histone methyltransferase in mouse embryonic stem cells: lmu; 2018.
- 43.Eisenberg CA, Eisenberg LM. G9a and G9a-Like Histone Methyltransferases and Their Effect on Cell Phenotype, Embryonic Development, and Human Disease. The DNA, RNA, and Histone Methylomes: Springer; 2019. p. 399–433. [Google Scholar]
- 44.Dong KB, Maksakova IA, Mohn F, Leung D, Appanah R, Lee S, et al. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. The EMBO journal. 2008;27(20):2691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharifi-Zarchi A, Gerovska D, Adachi K, Totonchi M, Pezeshk H, Taft RJ, et al. DNA methylation regulates discrimination of enhancers from promoters through a H3K4me1-H3K4me3 seesaw mechanism. BMC Genomics. 2017;18(1):964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Experimental & Molecular Medicine. 2017;49(4):e324–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart KR, Veselovska L, Kelsey G. Establishment and functions of DNA methylation in the germline. Epigenomics. 2016;8(10):1399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai H, Wang Z. Histone modification patterns and their responses to environment. Current environmental health reports. 2014;1(1):11–21. [Google Scholar]
- 49.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhattacharya S, Zhang N, Li H, Workman J. Regulation of SETD2 stability by its intrinsically disordered regions maintains the fidelity of H3K36me3 deposition. bioRxiv. 2020. [Google Scholar]
- 51.Bujold D, de Lima Morais DA, Gauthier C, Côté C, Caron M, Kwan T, et al. The international human epigenome consortium data portal. Cell systems. 2016;3(5):496–9. e2. [DOI] [PubMed] [Google Scholar]
- 52.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH roadmap epigenomics mapping consortium. Nature biotechnology. 2010;28(10):1045–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A, et al. BLUEPRINT to decode the epigenetic signature written in blood. Nature biotechnology. 2012;30(3):224–6. [DOI] [PubMed] [Google Scholar]
- 54.Albrecht F, List M, Bock C, Lengauer T. DeepBlue epigenomic data server: programmatic data retrieval and analysis of epigenome region sets. Nucleic acids research. 2016;44(W1):W581–W6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou X, Maricque B, Xie M, Li D, Sundaram V, Martin EA, et al. The human epigenome browser at Washington University. Nature methods. 2011;8(12):989–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li D, Hsu S, Purushotham D, Sears RL, Wang T. WashU epigenome browser update 2019. Nucleic acids research. 2019;47(W1):W158–W65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Consortium EP. The ENCODE (ENCyclopedia of DNA elements) project. Science. 2004;306(5696):636–40. [DOI] [PubMed] [Google Scholar]
- 58.Jones PA, Archer TK, Baylin SB, Beck S, Berger S, Bernstein BE, et al. Moving AHEAD with an international human epigenome project. Nature. 2008;454(7205):711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nature genetics. 2006;38(12):1378–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rakyan VK, Hildmann T, Novik KL, Lewin J, Tost J, Cox AV, et al. DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol. 2004;2(12):e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Consortium HP. High-throughput Epigenetic Regulatory Organisation In Chromatin-Project Fact Sheet. 2005.
- 62.Nanda JS, Kumar R, Raghava GP. dbEM: A database of epigenetic modifiers curated from cancerous and normal genomes. Scientific reports. 2016;6:19340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Medvedeva YA, Lennartsson A, Ehsani R, Kulakovskiy IV, Vorontsov IE, Panahandeh P, et al. EpiFactors: a comprehensive database of human epigenetic factors and complexes. Database. 2015;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qi Y, Wang D, Wang D, Jin T, Yang L, Wu H, et al. HEDD: the human epigenetic drug database. Database. 2016;2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rauschert S, Raubenheimer K, Melton PE, Huang RC. Machine learning and clinical epigenetics: a review of challenges for diagnosis and classification. Clinical epigenetics. 2020;12(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holder LB, Haque MM, Skinner MK. Machine learning for epigenetics and future medical applications. Epigenetics. 2017;12(7):505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lanchantin J, Singh R, Lin Z, Qi Y. Deep motif: Visualizing genomic sequence classifications. arXiv preprint arXiv:160501133. 2016. [Google Scholar]
- 68.Danziger SA, Baronio R, Ho L, Hall L, Salmon K, Hatfield GW, et al. Predicting positive p53 cancer rescue regions using Most Informative Positive (MIP) active learning. PLoS Comput Biol. 2009;5(9):e1000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doyle S, Monaco J, Feldman M, Tomaszewski J, Madabhushi A. An active learning based classification strategy for the minority class problem: application to histopathology annotation. BMC bioinformatics. 2011;12(1):424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Argyraki M, Damdimopoulou P, Chatzimeletiou K, Grimbizis GF, Tarlatzis BC, Syrrou M, et al. Inutero stress and mode of conception: impact on regulation of imprinted genes, fetal development and future health. Human reproduction update. 2019;25(6):777–801. [DOI] [PubMed] [Google Scholar]
- 71.Wrzodek C, Büchel F, Hinselmann G, Eichner J, Mittag F, Zell A. Linking the epigenome to the genome: correlation of different features to DNA methylation of CpG islands. PloS one. 2012;7(4):e35327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martinez VG, Munera-Maravilla E, Bernardini A, Rubio C, Suarez-Cabrera C, Segovia C, et al. Epigenetics of Bladder Cancer: Where Biomarkers and Therapeutic Targets Meet. Frontiers in Genetics. 2019;10(1125). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marchevsky AM, Tsou JA, Laird-Offringa IA. Classification of individual lung cancer cell lines based on DNA methylation markers: use of linear discriminant analysis and artificial neural networks. The Journal of Molecular Diagnostics. 2004;6(1):28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ongenaert M, Van Neste L, De Meyer T, Menschaert G, Bekaert S, Van Criekinge W. PubMeth: a cancer methylation database combining text-mining and expert annotation. Nucleic acids research. 2007;36(suppl_1):D842–D6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He X, Chang S, Zhang J, Zhao Q, Xiang H, Kusonmano K, et al. MethyCancer: the database of human DNA methylation and cancer. Nucleic acids research. 2007;36(suppl_1):D836–D41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lauss M, Visne I, Weinhaeusel A, Vierlinger K, Noehammer C, Kriegner A. MethCancerDB–aberrant DNA methylation in human cancer. British journal of cancer. 2008;98(4):816–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dodd IB, Micheelsen MA, Sneppen K, Thon G. Theoretical analysis of epigenetic cell memory by nucleosome modification. Cell. 2007;129(4):813–22. [DOI] [PubMed] [Google Scholar]
- 78.Cui P, Li J, Sun B, Zhang M, Lian B, Li Y, et al. A quantitative analysis of the impact on chromatin accessibility by histone modifications and binding of transcription factors in DNase I hypersensitive sites. BioMed research international. 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Z, Chu T, Choate LA, Danko CG. Identification of regulatory elements from nascent transcription using dREG. Genome research. 2019;29(2):293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ferrari R, de Llobet Cucalon LI, Di Vona C, Le Dilly F, Vidal E, Lioutas A, et al. TFIIIC binding to Alu elements controls gene expression via chromatin looping and histone acetylation. Molecular cell. 2020;77(3):475–87. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roh T-Y, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes & development. 2005;19(5):542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ioshikhes IP, Albert I, Zanton SJ, Pugh BF. Nucleosome positions predicted through comparative genomics. Nature genetics. 2006;38(10):1210–5. [DOI] [PubMed] [Google Scholar]
- 83.Yin Q, Wu M, Liu Q, Lv H, Jiang R. DeepHistone: a deep learning approach to predicting histone modifications. BMC genomics. 2019;20(2):11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Z, Manaf A, Li Y, Perez SP, Suganthan R, Dahl JA, et al. Histone methylations define neural stem/progenitor cell subtypes in the mouse subventricular zone. Molecular neurobiology. 2020;57(2):997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Whalen S, Truty RM, Pollard KS. Enhancer–promoter interactions are encoded by complex genomic signatures on looping chromatin. Nature genetics. 2016;48(5):488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chitsazian F, Sadeghi M, Elahi E. Confident gene activity prediction based on single histone modification H2BK5ac in human cell lines. BMC bioinformatics. 2017;18(1):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gogol-Döring A, Ammar I, Gupta S, Bunse M, Miskey C, Chen W, et al. Genome-wide profiling reveals remarkable parallels between insertion site selection properties of the MLV retrovirus and the piggyBac transposon in primary human CD4+ T cells. Molecular Therapy. 2016;24(3):592–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Singh R, Lanchantin J, Robins G, Qi Y. DeepChrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics. 2016;32(17):i639–i48. [DOI] [PubMed] [Google Scholar]
- 89.Information NCfB. SRA (Sequence Read Archive). 2011.
- 90.Genetics IoH. MethDB - the database for DNA methylation and environmental epigenetic effects. 2001.
- 91.Information NcfB. 2018.
- 92.Lebrón R, Gómez-Martín C, Carpena P, Bernaola-Galván P, Barturen G, Hackenberg M, et al. NGSmethDB 2017: Enhanced methylomes and differential methylation. Nucleic Acids Research. 2016:gkw996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Atlas E. Epigenome wide atlas. 2018.
- 94.Commons D. MENT (Methylation and Expression database of Normal and Tumor tissues). 2013. [DOI] [PubMed]
- 95.Archives EN. ENA (European Nucleotide Archive). 2018. [DOI] [PMC free article] [PubMed]
- 96.Commons D. DBCAT (database of CpG islands and analytical tools). 2011. [DOI] [PubMed]
- 97.Xuan Lin QX, Sian S, An O, Thieffry D, Jha S, Benoukraf T. MethMotif: an integrative cell specific database of transcription factor binding motifs coupled with DNA methylation profiles. Nucleic acids research. 2019;47(D1):D145–D54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic acids research. 2015;43(D1):D298–D9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Teng L, He B, Wang J, Tan K. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics. 2015;31(15):2560–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yun X, Xia L, Tang B, Zhang H, Li F, Zhang Z. 3CDB: a manually curated database of chromosome conformation capture data. Database. 2016;2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Khare SP, Habib F, Sharma R, Gadewal N, Gupta S, Galande S. HIstome—a relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic acids research. 2012;40(D1):D337–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Robertson G, Bilenky M, Lin K, He A, Yuen W, Dagpinar M, et al. cisRED: a database system for genome-scale computational discovery of regulatory elements. Nucleic acids research. 2006;34(suppl_1):D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Institute Wr. Disease Annotated Chromatin Epigenetics Resource. 2010.
- 104.Advanced Center for Treatment RaEiCA, Navi Mumbai and Center of Excellence in Epigenetics (CoEE), Indian Institute of Science Education and Research (IISER), Pune. HIstome: the histone infobase. 2011.
- 105.Liu Z, Wang Y, Gao T, Pan Z, Cheng H, Yang Q, et al. CPLM: a database of protein lysine modifications. Nucleic acids research. 2014;42(D1):D531–D6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gendler K, Paulsen T, Napoli C. ChromDB: the chromatin database. Nucleic acids research. 2008;36(suppl_1):D298–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu Y, Zhang S, Lin S, Guo Y, Deng W, Zhang Y, et al. WERAM: a database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic acids research. 2016:gkw1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu H, Zhou J, Lin S, Deng W, Zhang Y, Xue Y. PLMD: An updated data resource of protein lysine modifications. Journal of Genetics and Genomics. 2017;44(5):243–50. [DOI] [PubMed] [Google Scholar]
- 109.Xu H, Wang Y, Lin S, Deng W, Peng D, Cui Q, et al. PTMD: a database of human disease-associated post-translational modifications. Genomics, proteomics & bioinformatics. 2018;16(4):244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Behjati S, Tarpey PS. What is next generation sequencing? Arch Dis Child Educ Pract Ed. 2013;98(6):236–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hsu F-M, Gohain M, Chang P, Lu J-H, Chen P-Y. Chapter 4 - Bioinformatics of Epigenomic Data Generated From Next-Generation Sequencing. In: Tollefsbol TO, editor. Epigenetics in Human Disease (Second Edition). 6: Academic Press; 2018. p. 65–106. [Google Scholar]
- 112.Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nature methods. 2009;6(4):283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sparks TM, Harabula I, Pombo A. Evolving methodologies and concepts in 4D nucleome research. Current Opinion in Cell Biology. 2020;64:105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Müller F, Scherer M, Assenov Y, Lutsik P, Walter J, Lengauer T, et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome biology. 2019;20(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Min JL, Hemani G, Davey Smith G, Relton C, Suderman M. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics. 2018;34(23):3983–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jo H, Koh G. Faster single-end alignment generation utilizing multi-thread for BWA. Bio-medical materials and engineering. 2015;26(s1):S1791–S6. [DOI] [PubMed] [Google Scholar]
- 117.Biology JHUCfC. Hisat2. 2016.
- 118.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9(4):357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.GitHub. DNAscan. 2014.
- 120.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.De Summa S, Malerba G, Pinto R, Mori A, Mijatovic V, Tommasi S. GATK hard filtering: tunable parameters to improve variant calling for next generation sequencing targeted gene panel data. BMC bioinformatics. 2017;18(5):119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wei Z, Wang W, Hu P, Lyon GJ, Hakonarson H. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic acids research. 2011;39(19):e132–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38(16):e164–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fiume M, Williams V, Brook A, Brudno M. Savant: genome browser for high-throughput sequencing data. Bioinformatics. 2010;26(16):1938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Archive EN. Sequence Versions Archive. 2018.
- 126.Vandeweyer G, Van Laer L, Loeys B, Van den Bulcke T, Kooy RF. VariantDB: a flexible annotation and filtering portal for next generation sequencing data. Genome medicine. 2014;6(10):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Barros-Silva D, Marques CJ, Henrique R, Jerónimo C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes. 2018;9(9):429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bubancova I, Kovarikova H, Laco J, Ruszova E, Dvorak O, Palicka V, et al. Next-Generation Sequencing Approach in Methylation Analysis of HNF1B and GATA4 Genes: Searching for Biomarkers in Ovarian Cancer. Int J Mol Sci. 2017;18(2):474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xing X, Zhang B, Li D, Wang T. Comprehensive whole DNA methylome analysis by integrating MeDIP-seq and MRE-seq. DNA Methylation Protocols: Springer; 2018. p. 209–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic acids research. 2010;38(2):391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bogdanović O, Veenstra G. Affinity-based enrichment strategies to assay methyl-CpG binding activity and DNA methylation in early Xenopus embryos. BMC research notes. 2011;4:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kurdyukov S, Bullock M. DNA Methylation Analysis: Choosing the Right Method. Biology (Basel). 2016;5(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang L, Sun J, Wu H, Liu S, Wang J, Wu B, et al. Systematic assessment of reduced representation bisulfite sequencing to human blood samples: A promising method for large-sample-scale epigenomic studies. Journal of biotechnology. 2012;157(1):1–6. [DOI] [PubMed] [Google Scholar]
- 134.Boers R, Boers J, De Hoon B, Kockx C, Ozgur Z, Molijn A, et al. Genome-wide DNA methylation profiling using the methylation-dependent restriction enzyme LpnPI. Genome research. 2018;28(1):88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Olova N, Krueger F, Andrews S, Oxley D, Berrens RV, Branco MR, et al. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biology. 2018;19(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Suzuki M, Liao W, Wos F, Johnston AD, DeGrazia J, Ishii J, et al. Whole-genome bisulfite sequencing with improved accuracy and cost. Genome research. 2018;28(9):1364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chappell L, Russell AJ, Voet T. Single-cell (multi) omics technologies. Annual Review of Genomics and Human Genetics. 2018;19:15–41. [DOI] [PubMed] [Google Scholar]
- 138.Sun K, Jiang P, Cheng SH, Cheng TH, Wong J, Wong VW, et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome research. 2019;29(3):418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Harris R, Wang T, Coarfa C, Zhou X, Xi Y, Nagarajan R, et al. Sequence-based profiling of DNA methylation: comparisons of methods and catalogue of allelic epigenetic modifications. Nature Biotechnology. 2010;10:1097–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schmidt M, Van Bel M, Woloszynska M, Slabbinck B, Martens C, De Block M, et al. Plant-RRBS, a bisulfite and next-generation sequencing-based methylome profiling method enriching for coverage of cytosine positions. BMC Plant Biology. 2017;17(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Davalos V, Martinez-Cardus A, Esteller M. The Epigenomic Revolution in Breast Cancer: From Single-Gene to Genome-Wide Next-Generation Approaches. The American Journal of Pathology. 2017;187(10):2163–74. [DOI] [PubMed] [Google Scholar]
- 142.Solomon MJ, Larsen PL, Varshavsky A. Mapping proteinDNA interactions in vivo with formaldehyde: Evidence that histone H4 is retained on a highly transcribed gene. Cell. 1988;53(6):937–47. [DOI] [PubMed] [Google Scholar]
- 143.O’Neill L, Turner BM. Histone H4 acetylation distinguishes coding regions of the human genome from heterochromatin in a differentiation-dependent but transcription-independent manner. The EMBO journal. 1995;14(16):3946–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ramani V, Qiu R, Shendure J. High sensitivity profiling of chromatin structure by MNase-SSP. Cell reports. 2019;26(9):2465–76. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature biotechnology. 2015;33(11):1165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee O-K, Kharchenko P, et al. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008;134(4):599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ghosh M, Sharma N, Singh AK, Gera M, Pulicherla KK, Jeong DK. Transformation of animal genomics by next-generation sequencing technologies: a decade of challenges and their impact on genetic architecture. Critical Reviews in Biotechnology. 2018;38(8):1157–75. [DOI] [PubMed] [Google Scholar]
- 148.Leamon J, Andersen M, Thornton M. Methods and compositions for multiplex PCR. Google Patents; 2018. [Google Scholar]
- 149.Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–37. [DOI] [PubMed] [Google Scholar]
- 150.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Courtney AJ, Kamei M, Ferraro AR, Gai K, He Q, Honda S, et al. Normal Patterns of Histone H3K27 Methylation Require the Histone Variant H2A. Z in Neurospora crassa. Genetics. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453(7193):358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]