
Figure 1.
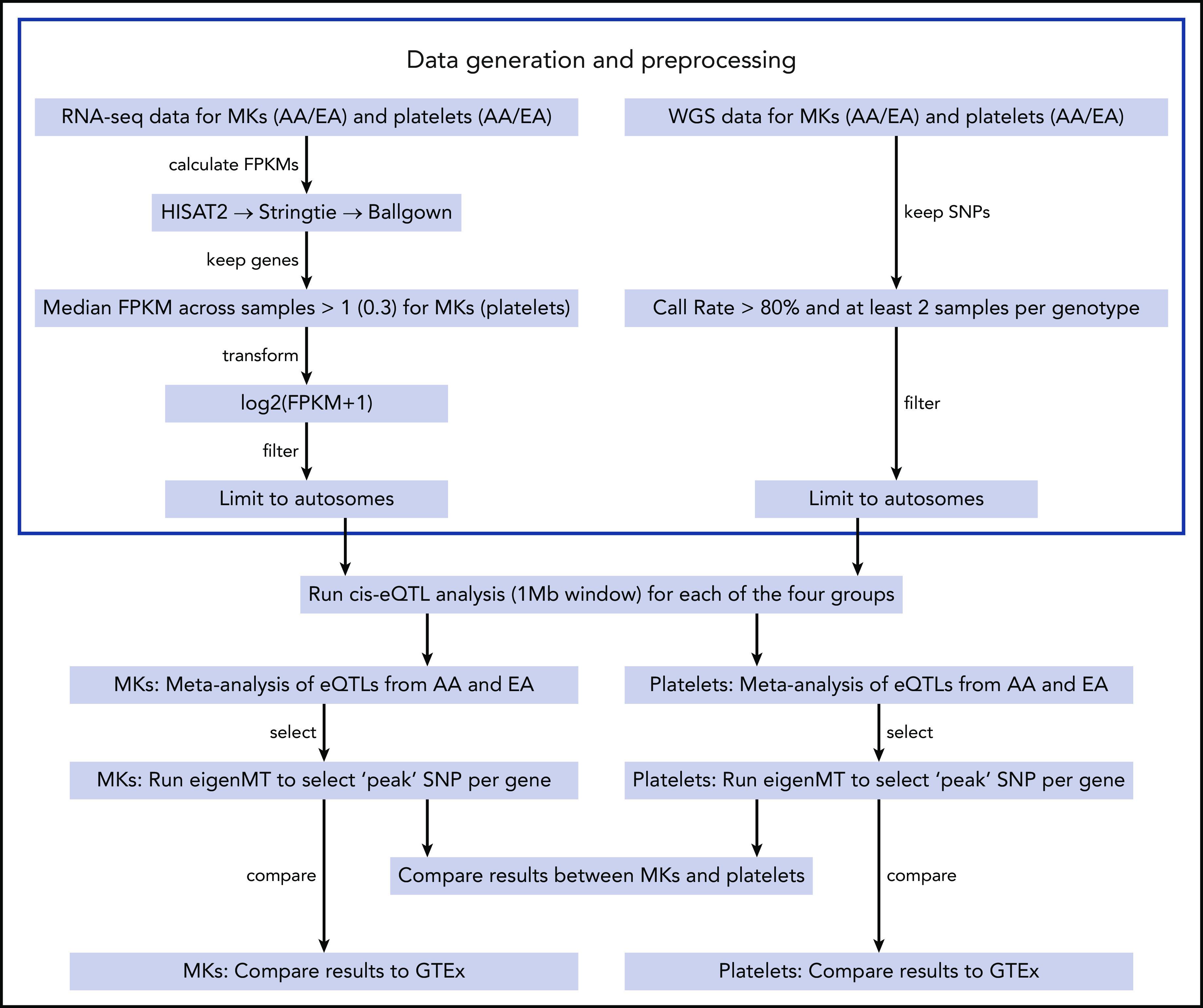
eQTL analysis pipeline. Samples were processed and analyzed separately by ethnicity (African American [AA] and European American [EA]) and tissue type (megakaryocytes [MKs] and platelets). Reads of each ethnicity/sample were aligned and assembled using the standard steps of the Tuxedo suite and the results were loaded into R using the Ballgown package. Only autosomal genes with median FPKM across samples larger than 1 (0.3 for platelets) were kept and logarithmically transformed. SNPs with less than 2 samples per genotype were excluded as were nonautosomal SNPs. Differences in gene expression between genotypes were assessed with linear models adjusting for clinical covariates, batch information, and principal components derived from gene-expression and genotype data. Meta-analysis was performed to combine results from AAs and EAs for MKs and platelets, respectively. Finally, significance of “peak” SNPs per gene were calculated by eigenMT software. Shared eQTLs between MKs and platelets were detected using these results, and the comparison with eQTLs reported at the GTEx portal was performed as well.