

Abstract
Pan-Gyn cancers entail 1 in 5 cancer cases worldwide, breast cancer being the most commonly diagnosed and responsible for most cancer deaths in women. The high incidence and mortality of these malignancies, together with the handicaps of taxanes—first-line treatments—turn the development of alternative therapeutics into an urgency. Taxanes exhibit low water solubility that require formulations that involve side effects. These drugs are often associated with dose-limiting toxicities and with the appearance of multi-drug resistance (MDR). Here, we propose targeting tubulin with compounds directed to the colchicine site, as their smaller size offer pharmacokinetic advantages and make them less prone to MDR efflux. We have prepared 52 new Microtubule Destabilizing Sulfonamides (MDS) that mostly avoid MDR-mediated resistance and with improved aqueous solubility. The most potent compounds, N-methyl-N-(3,4,5-trimethoxyphenyl-4-methylaminobenzenesulfonamide 38, N-methyl-N-(3,4,5-trimethoxyphenyl-4-methoxy-3-aminobenzenesulfonamide 42, and N-benzyl-N-(3,4,5-trimethoxyphenyl-4-methoxy-3-aminobenzenesulfonamide 45 show nanomolar antiproliferative potencies against ovarian, breast, and cervix carcinoma cells, similar or even better than paclitaxel. Compounds behave as tubulin-binding agents, causing an evident disruption of the microtubule network, in vitro Tubulin Polymerization Inhibition (TPI), and mitotic catastrophe followed by apoptosis. Our results suggest that these novel MDS may be promising alternatives to taxane-based chemotherapy in chemoresistant Pan-Gyn cancers.
Keywords: tubulin, sulfonamide, antitumor, taxane, combretastatin A-4, breast cancer, gynecologic cancer
1. Introduction
Gynecologic (i.e., ovarian, uterine, vulva, and vagina) and women breast cancer share relevant similarities at the molecular level that cluster them together (Pan-Gyn) and distinguish them from other tumor types [1,2]. According to GLOBOCAN estimates, Pan-Gyn cancers accounted for 19% of new diagnoses worldwide in 2018 and entailed 40% of new cancer cases among females [3]. 39% of women with Pan-Gyn cancers died from them, representing 30% of all female cancer deaths and 13% of total cancer deaths globally. The high incidence and mortality of female breast [4,5], ovarian [6,7], and uterine [8,9] cancers and the acquired resistance to first-line treatments highlight the need for new therapeutic alternatives.
Since the FDA approved the use of Taxol® for advanced ovarian carcinoma in 1992 and metastatic breast cancer in 1994 [10], taxane-based chemotherapy has become the basis for treating breast and ovarian cancers [11]. First-line chemotherapy involves a platinum-taxane combination for ovarian cancer [12,13,14], preferably carboplatin/paclitaxel [15], and taxanes sequenced or combined with anthracyclines for advanced and metastatic breast cancer [16,17,18,19]. Furthermore, the combination of cisplatin and paclitaxel is considered a standard regimen in the palliative setting for patients with metastatic, recurrent, or persistent uterine cancer [9,20,21,22].
However, the clinical efficacy of taxanes is limited by an intrinsic low aqueous solubility (<2 µg/mL [23,24,25]), dose-limiting toxicities, and the development of drug resistance [26]. The low solubility (remaining challenging) requires complex solvent-system formulations that can alter the pharmacokinetic profiles [27,28] and have been associated with toxicity [29,30]. The development of resistance to these drugs is usually ascribed to Pgp-mediated efflux and alterations in tubulin (mutations in the tubulin genes, altered tubulin isotype expression, or aberrant levels of tubulin) [31,32,33]. Specific resistance to taxane-based chemotherapy has been described for female breast cancer, with an approximately 30% of recurrence [34,35], ovarian cancer, in which 50% of patients relapse with the chemoresistant disease within the first two years post-diagnosis [36,37], and uterine cancer [38].
Taxanes bind to microtubules, stabilizing lateral contacts between protofilaments, thus promoting assembly and inhibiting depolymerization at high concentrations. At lower concentrations, they alter microtubule dynamics and preclude cells from properly completing mitosis [39,40], which leads to cell cycle arrest at G2/M and, subsequently, to cell death mainly triggered by apoptosis [41,42]. There are other drug binding sites in tubulin that affect microtubule dynamics similarly to taxanes, but whose engagement results in microtubule depolymerization at high concentrations, such as the colchicine, the vinca alkaloids, the maytansine, the pironetin, and the hemiasterlin sites [43]. The only approved drugs belong to the vinca alkaloids site binding ligands, which are used against hematological malignancies and solid tumors, including breast cancer. However, they share with taxanes the problems of toxic side effects, difficult pharmacokinetics, and complex hydrophobic structures that make them substrates of drug efflux pumps [44].
The colchicine site offers advantages for drug design since its ligands exhibit simple chemical structures and are synthetically accessible. Colchicine-site ligands display antiproliferative effects (characterized by cell cycle arrest at G2/M phase followed by the induction of apoptosis [45]) and act as vascular disrupting agents, affording additional odds against solid tumors [46]. Representatives such as the stilbene combretastatin A-4 (CA-4) and the sulfonamide ABT-751 have reached clinical trials [47]. However, the Z olefin of combretastatins is chemically unstable and readily isomerizes to the more stable but inactive E isomer. The low solubility of CA-4 requires the use of phosphate prodrugs on the phenolic hydroxyl group, which is, in turn, the target for metabolic inactivation by glucuronidation in resistant cells, such as the colon adenocarcinoma HT-29 cell line [48]. ABT-751 is an orally administered sulfonamide with modest potency against human cancer cell lines and xenograft models. Despite its favorable pharmacokinetics, ABT-751 has not found clinical application due to insufficient potency [49].
In this work, we have designed and synthesized a new family of Microtubule Destabilizing Sulfonamides (MDS) hybrids of CA-4 and ABT-751. The effects of replacing the chemically unstable CA-4 olefin with a sulfonamide bridge, the removal or substitution of the phenolic hydroxyl group, and the introduction of several modifications on the aromatic rings and the sulfonamide bridge have been explored while maintaining the 3,4,5-trimethoxyphenyl ring that has been long considered essential for high potency [50,51] (Figure 1). The resulting compounds have been evaluated for tubulin inhibition in vitro and antiproliferative activity against several human tumor cell lines. We have also studied whether MDR1 pumps could compromise their effectiveness by the pharmacological inhibition of Pgp using verapamil. After a comprehensive preliminary evaluation, three promising MDS have been further screened against several representative cancer cell lines representative of the tumor types that are associated with the highest mortalities: breast, ovarian, and uterine, accounting for 51%, 15%, and 32% of cancer deaths in women, respectively. The effect of the compounds on cancer cell proliferation has been studied and compared with paclitaxel, CA-4, and ABT-751. The mechanism of action of these novel MDS has been studied by ascertaining their effect on the microtubule network in vitro. These MDS induce mitotic arrest, followed by apoptotic cell death with differences arising from different genetic backgrounds of the studied cell lines. The favorable pharmacodynamic and pharmacokinetic profiles compared to reference drugs, including solubility, absence of Pgp-mediated resistance, and improved potency indicate that MDS are promising candidates for the treatment of this kind of malignancies.
Figure 1.

Representative ligands binding at the colchicine site used as a starting point for the rational design of new Microtubule Destabilizing Sulfonamides (MDS). General structure and structure variations of new MDS.
2. Results
2.1. Synthesis of MDS
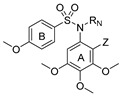
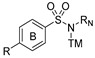
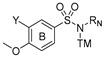
52 new MDS (Figure 1) were prepared following the synthetic approach shown in Figure 2 (detailed synthetic procedures and NMR spectra can be found in Supplemental Figure S1 and Methods SP1,2). The synthesized compounds were divided into three series according to the substituents on the aromatic B ring (ArB): series 1 (compounds 1a-24), series 2 (25–38), and series 3 (39–48b) (Table 1). Sulfonamides were built up by the reaction between 4-methoxy- (series 1), 4-nitro- (series 2), or 4-methoxy-3-nitro- (series 3) benzenesulfonyl chlorides and 3,4,5-trimethoxyaniline, providing crystalline products in excellent yields (90–96%). Nitro groups were reduced to amines by palladium-catalyzed hydrogenation (82–98% yields). The subsequent amino derivatization by alkylation, acylation, and/or formylation-reduction sequences allowed the introduction of varied Z, R, and Y substituents (Figure 1). Substitutions at the sulfonamide nitrogen were conducted by alkylation reactions with alkyl halides in KOH/CH3CN (methylations with methyl iodide in 63–98% yields) or K2CO3/DMF (ethyl, acetyl, acetonitrile, benzyl, or ethyl acetate substituents in 40–99% yields).
Figure 2.

General synthetic approach. Reagents, conditions, and yields: (a) Pyridine, CH2Cl2, rt, 4–8 h, 90–96% (b) RN = CH3, CH3I, KOH, CH3CN, rt, 24 h, 63–98%; RN = Ac, acetic anhydride, pyridine, CH2Cl2, reflux, 8–12 h, 61–83%; RN ≠ CH3 and Ac, RN-halogen, K2CO3, dry DMF, rt, 24–48 h, 40–99% (c) tert-Butyl nitrite, CH3CN, 45 °C, 24 h, 60% (d) H2, Pd/C, EtOAc, rt, 48–72 h, 82–98% (e) Formic acid, CH2Cl2, rt, 24–48 h, 62–82% (f) Trichloroacetic acid, NaBH4, dry THF, 0°C, 24 h, 97% (g) Paraformaldehyde, NaBH3CN, AcOH, MeOH, rt, 72–96 h, 95% (h) Ethyl 2-bromoacetate, NaI, acetone/THF 1:1, reflux, 48 h, 19% (i) NBS, CH2Cl2, rt, 6 h, 43%.
Table 1.
Chemical structure, antiproliferative activity against human tumor cell lines, and Tubulin Polymerization Inhibition (TPI) of novel Microtubule Destabilizing Sulfonamides (MDS). Compounds have been divided into three different series according to the substituents on the aromatic B ring (ArB). TM: 3,4,5-trimethoxyphenyl.
Series 1:
|
Antiproliferative Activity
IC50 (nM) |
TPI | ||||
| TPI % | ||||||
| Z | RN | Compound | HeLa | MCF7 | 10 µM | IC50 (µM) |
| H | H | 1a | 240 | 375 | 0 | >20 |
| H | SO2–4-OMePh | 1b | >1000 | >1000 | 0 | >20 |
| H | Me | 2a | 71 | 127 | 35 | >20 |
| H | CH2-Dim | 2b | >1000 | >1000 | 0 | >20 |
| H | Et | 3 | 99 | 87 | 25 | >20 |
| H | EtBr | 4 | 94 | 340 | 30 | >20 |
| H | Ac | 5 | 287 | 270 | 0 | >20 |
| H | CH2CN | 6 | 143 | 275 | 0 | >20 |
| H | CH2COOEt | 7 | 217 | 335 | 21 | >20 |
| H | CH2COOH | 8 | >1000 | >1000 | 0 | >20 |
| H | Benzyl | 9 | 750 | 830 | 21 | >20 |
| H | COOBenzyl | 10 | >1000 | >1000 | 11 | >20 |
| NO2 | H | 11 | >1000 | >1000 | 10 | >20 |
| NH2 | H | 12 | >1000 | >1000 | 0 | >20 |
| NH2 | Me | 13 | >1000 | >1000 | 0 | >20 |
| NH2 | CH2-Dim | 14 | >1000 | >1000 | 0 | >20 |
| NHAc | H | 15 | >1000 | >1000 | 0 | >20 |
| NHAc | Me | 16 | >1000 | >1000 | 0 | >20 |
| NHCHO | H | 17 | >1000 | >1000 | 4 | >20 |
| N(CH3)2 | H | 18 | >1000 | >1000 | 0 | >20 |
| N(CH3)2 | Me | 19 | >1000 | >1000 | 0 | >20 |
| Br | H | 20 | >1000 | >1000 | 0 | >20 |
| Gly-tBOC | H | 21 | >1000 | >1000 | 0 | >20 |
| Succinic | H | 22 | >1000 | >1000 | 0 | >20 |
| -N=N- | 23 | >1000 | >1000 | 0 | >20 | |
| -N=CH- | 24 | >1000 | >1000 | 0 | >20 | |
Series 2:
|
Antiproliferative Activity
IC50 (nM) |
TPI | ||||
| TPI % | ||||||
| R | RN | Compound | HeLa | MCF7 | 10 µM | IC50 (µM) |
| NO2 | H | 25 | >1000 | >1000 | 0 | >20 |
| NH2 | H | 26 | >1000 | >1000 | 0 | >20 |
| NH2 | Me | 27 | >1000 | >1000 | 0 | >20 |
| N(CH3)2 | H | 28 | 230 | 173 | 0 | >20 |
| N(CH3)2 | Me | 29a | 63 | 30 | 40 | >20 |
| N(CH3)2 | CH2-Dim | 29b | >1000 | >1000 | 0 | >20 |
| N(CH3)2 | Et | 30 | 66 | 81 | 45 | >20 |
| N(CH3)2 | Ac | 31 | 183 | 135 | 38 | 14 |
| N(CH3)2 | CH2CN | 32 | 55 | 120 | 17 | >20 |
| N(CH3)2 | CH2COOEt | 33 | 99 | 91 | 14 | >20 |
| N(CH3)2 | CH2COOH | 34 | >1000 | >1000 | 6 | >20 |
| N(CH3)2 | Benzyl | 35a | 330 | 100 | 72 | 5.3 |
| N(CH3)2 | COOBenzyl | 35b | >1000 | >1000 | 0 | >20 |
| NHCHO | H | 36 | >1000 | >1000 | 0 | >20 |
| NHCH3 | H | 37 | 607 | >1000 | 7 | >20 |
| NHCH3 | Me | 38 | 44 | 61 | 47 | 10 |
Series 3:
|
Antiproliferative Activity
IC50 (nM) |
TPI | ||||
| TPI % | ||||||
| Y | RN | Compound | HeLa | MCF7 | 10 µM | IC50 (µM) |
| NO2 | H | 40 | >1000 | >1000 | 0 | >20 |
| NH2 | H | 41 | 260 | 96 | 15 | >20 |
| NH2 | Me | 42 | 23 | 26 | 41 | 12 |
| NH2 | Et | 43 | 38 | 14 | 37 | >20 |
| NH2 | CH2CN | 44 | 60 | 8 | 5 | >20 |
| NH2 | Benzyl | 45 | 25 | 48 | 75 | 3.7 |
| NHCH2COOEt | Me | 46 | 210 | 48 | 13 | >20 |
| NHCH2COOEt | CH2COOEt | 47 | >1000 | >1000 | 0 | >20 |
| NHCH3 | Me | 48a | 440 | 1190 | 2 | >20 |
| N(CH3)2 | Me | 48b | >1000 | >1000 | 0 | >20 |
| Paclitaxel [52] | 2.6 | 2.5 | n.d. 1 | n.d. | ||
| Combretastatin A-4 | 2 | 1 | 100 | 3 | ||
| ABT-751 | 388 | 180 | 69 | 4.4 | ||
1 n.d.: not determined.
2.2. Replacement of CA-4 Olefin by a Sulfonamide Highly Increased Aqueous Solubility
Aqueous solubility of representative compounds was spectrophotometrically measured in pH 7.0 phosphate buffer (Table 2). UV absorbance at three maximum absorbance wavelengths of selected compounds was measured after 48 h shaking and subsequent microfiltration. Replacement of the CA-4 olefinic bridge by a sulfonamide group highly increased aqueous solubility when compared to paclitaxel and to CA-4 (4- up to 1700-fold increase). Similar or even better results were obtained when compared with the orally administered sulfonamide ABT-751 (up to 42-fold increase). The best results were achieved by the introduction of amino acid derivatives at the Z position (compounds 21 and 22).
Table 2.
Aqueous solubility in µg/mL of representative compounds.
2.3. MDS Inhibit Cell Proliferation in Breast, Cervix, and Ovarian Tumor Cells
The in vitro antiproliferative activity of MDS at 1 µM after 72 h treatments was evaluated by a colorimetric method against the human tumor cell lines HeLa (cervix epithelioid carcinoma) and MCF7 (breast adenocarcinoma). Compounds showing more than 40% inhibition at 1 µM in at least three independent assays were evaluated in a range of concentrations between 0.1 nM and 10 µM for the determination of half-maximal inhibitory concentrations (IC50) (Table 1). Paclitaxel [52], CA-4, and ABT-751 were used as references.
HeLa and MCF7 cells showed similar sensitivities to MDS, both cell lines displayed less than two-fold changes in IC50 values between them in most cases. The most potent compounds (e.g., 29a, 30, 38, 42, 43, 44, and 45) showed IC50 values in the double-digit nanomolar range, less potent than paclitaxel and CA-4 but more than ABT-751. Substitutions on the trimethoxyphenyl ring resulted in inactive compounds (11–24). Alkylations of the sulfonamide improved the potency, with methyl groups yielding the lowest IC50 values in most cases (e.g., 29a, 30, and 42). Larger substituents gave worse results except for some benzyl (e.g., 45) and cyanomethyl (e.g., 44) derivatives. Replacement of the 4-methoxy group on the B ring in series 1 by an amino substituent (series 2) and introduction of an amino group on position 3 as a primary amine (series 3) gave favorable results. Substituents with a hydrogen bond donor, such as methylamines (38) in series 2, or primary amines in series 3 (42–45), resulted in the most potent MDS. Based on antiproliferative results, we selected 3 out of the 7 most potent compounds (38, 42, and 45) as representative leaders for further investigation.
The in vitro antiproliferative activity of compounds 38, 42, and 45 against a panel of five human ovarian carcinoma cell lines (SKOV3, IGROV-1, A2780, OVCAR-8, and OVCAR-3) was evaluated by a colorimetric method (Table 3) and compared with paclitaxel [14]. Paclitaxel and MDS showed a similar comparative behavior against the highly paclitaxel-sensitive A2780 and OVCAR-8 and the less sensitive IGROV-1 cell line, with paclitaxel being one order of magnitude more potent than MDS. Concerning OVCAR-3, paclitaxel is less potent, but MDS maintain their potency range, thus resulting in similar effects of both types of drugs. For SKOV3, the potency reduction observed for paclitaxel together with a sustained or improved (42 with a 7 nM IC50 value) potency for MDS results in a more favorable profile of the MDS compared to paclitaxel.
Table 3.
Antiproliferative activity of the lead compounds and paclitaxel against several human ovarian tumor cell lines.
| Antiproliferative Activity IC50 (nM) | |||||
|---|---|---|---|---|---|
| Compound | SKOV3 | IGROV-1 | A2780 | OVCAR-8 | OVCAR-3 |
| 38 | 46 | 248 | 68 | 74 | 31 |
| 42 | 7 | 400 | 42 | 37 | 72 |
| 45 | 48 | 492 | 104 | 48 | 58 |
| Paclitaxel 1 | 81 | 39 | 3 | 6 | 17 |
1 IC50 values against A2780, OVCAR-8, and OVCAR-3 cell lines from Bicaku, E. et al. evaluation [14].
2.4. Lead Compounds Overcome MDR-Mediated Resistance
The antiproliferative activity of MDS against the CA-4-resistant cell line HT-29 (colon adenocarcinoma) in the absence or the presence of 10 µM verapamil, a non-selective Pgp/MDR1 inhibitor, was measured 72 h post-treatment by a colorimetric method (Supplementary Table S1). Verapamil itself did not affect cell proliferation at that concentration. Detecting changes in drug potency when inhibiting MDR-mediated efflux reflect that these transporters reduce the intracellular drug concentration. HT-29 has been reported to express moderate levels of MDR1, accounting for a resistant phenotype [53]. A comparison of the antiproliferative IC50 values of each MDS in the presence or absence of verapamil provides an estimate of the susceptibility of each compound to efflux pumps. Most of the compounds, including leading derivatives 38, 42, and 45 (Table 4 and Supplementary Table S1), showed no significant differences (up to two-fold change) between pairs of IC50 values, thus suggesting that they are not substrates of MDR pumps. Compounds 3, 29a, and 30 showed higher potencies against HT-29 cells when combined with verapamil (comparables to that of obtained against sensitive HeLa an MCF7 cells), thus validating the assay as positive controls.
Table 4.
Antiproliferative activity against the CA-4 resistant cancer cell line HT-29 and sensitivity of the lead MDS to MDR pumps.
| Antiproliferative Activity IC50 (nM) | ||
|---|---|---|
| Compound | HT-29 | HT-29 Verapamil 10 µM |
| 38 | 59 | 50 |
| 42 | 81 | 79 |
| 45 | 300 | 276 |
| CA-4 | 305 | 327 |
2.5. Lead Compounds Induce G2/M Arrest in Breast, Ovarian and Cervix Tumor Cells
The effect of the lead MDS on the cell cycle of MCF7, SKOV3, and HeLa cells was analyzed in time-course experiments at 24, 48, and 72 h post-treatment by DNA staining and flow cytometry quantification. The working concentration for each compound (600 nM for 38, 50 nM for 42, and 400 nM for 45) was established at the complete inhibition of cell proliferation in view of individually assays run in parallel for each cell line.
For breast adenocarcinoma MCF7 cells (Figure 3a) a pronounced and sustained G2/M arrest was observed for all the treatments from 24 h (70–80%) to 72 h (60–76%), compared to untreated MCF7 cells, which maintained a 32–40% population at G2/M throughout the time course. A moderate increase of the percentage of cells at the SubG0/G1 region was observed, which slightly increased over time (from 4–6% at 24 h to 6–12% at 72 h) accompanied by the decrease in the G2/M population.
Figure 3.

Cell cycle analysis. (a) MCF7 cell cycle distribution. Cells were arrested in G2/M phase. (b) SKOV3 cell cycle distribution. Cells arrested in G2/M phase experienced time-dependent cell death (SubG0/G1 region). (c) HeLa cell cycle distribution. Cells were arrested in the G2/M phase followed by cell death (SubG0/G1 region). Cells were incubated with the lead compounds at 600 nM (38), 50 nM (42), or 400 nM (45) for 24, 48, and 72 h, stained with propidium iodide (PI) and their DNA content was analyzed by flow cytometry. Untreated samples were analyzed in parallel. The different cell cycle populations were quantified, expressed in percentages, and represented in bar charts. Data shown are representative of three independent experiments.
Compounds 38, 42, and 45 (Figure 3b) arrested ovarian carcinoma SKOV3 cells at the G2/M phase 24 h post-treatment (44%) compared to untreated cells (30%). The percentage of SubG0/G1 treated SKOV3 cells was already elevated at 24 h (20% to 22%) compared to untreated controls (always <14%). Treatments with compounds 38 and 42 increased the percentage of SubG0/G1 modestly at 48 h (35% and 34%, respectively), and notably at 72 h (43% and 53%, respectively). This increase presumably occurred at the expense of cells arrested at the G2/M phase. The effect of compound 45 was notably higher, with 46% of cells at SubG0/G1 region 48 h post-treatment and 63% at 72 h.
Treatment of HeLa cells with 38 and 45 (Figure 3c) caused a significant arrest at the G2/M phase after 24 h (80%) and a more modest one for compound 42 (44%) accompanied by a limited increase in the percentage of cells of the SubG0/G1 population (9–16% vs. <4% in untreated controls). At subsequent time points, around 50% of the cells for all three treatments were allocated to the SubG0/G1 region of the cell cycle histogram at the expense of the G2/M phase.
2.6. Lead Compounds Trigger Apoptotic Cell Death
To determine whether the cytotoxic/antiproliferative effect of the MDS was due to apoptosis induction, cells were treated with variable concentrations of the drug for 72 h or 6 days, stained with Annexin V-FITC (AnV) and propidium iodide (PI), and analyzed by flow cytometry. Double negative cells (AnV−/PI−) were classified as live cells (L), cells positive for AnV and negative for PI (AnV+/PI−) were considered in Early Apoptosis (EA), cells negative for AnV and positive for PI (AnV−/PI+) were considered necrotic (N), and double-positive cells (AnV+/PI+) as Late Apoptosis (LA). In all cases, the compounds were used at the same concentrations previously used in cell cycle studies (Table 5).
Table 5.
Cell death quantification in Annexin V-FITC/PI double staining experiments. MCF7, SKOV3, and HeLa cells were incubated with the lead compounds for 72 h and/or 6 days, stained with Annexin V-FITC (AnV) and PI, and analyzed by flow cytometry. The results are expressed in percentage as the average of three independent experiments. Untreated samples were analyzed in parallel. Cells were classified into double-negatives (live cells), AnV-positive cells (early apoptosis), double-positives (late apoptosis), and PI-positive cells (necrosis).
| Compound | Annexin V-FITC/PI 72 h (%) | Annexin V-FITC/PI 6 Days (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Live | EA 1 | LA 2 | Necrosis | Live | EA | LA | Necrosis | ||
| MCF7 | 38 (600 nM) | 60.5 | 29.0 | 5.4 | 5.1 | 52.0 | 45.0 | 2.7 | 0.3 |
| 42 (50 nM) | 87.7 | 8.5 | 2.3 | 1.4 | 79.1 | 18.9 | 1.7 | 0.3 | |
| 45 (400 nM) | 70.5 | 20.1 | 7.5 | 1.9 | 56.7 | 39.3 | 3.8 | 0.3 | |
| CA-4 (50 nM) | 58.3 | 31.7 | 3.3 | 6.7 | 44.5 | 52.4 | 2.7 | 0.4 | |
| Control | 93.6 | 1.8 | 2.9 | 1.7 | 97.4 | 0.2 | 0.8 | 1.6 | |
| SKOV3 | 38 (600 nM) | 62.3 | 30.5 | 5.4 | 1.8 | ||||
| 42 (50 nM) | 85.1 | 12.2 | 2.0 | 0.7 | |||||
| 45 (400 nM) | 72.7 | 23.4 | 2.6 | 1.3 | |||||
| Paclitaxel (10 nM) | 90.7 | 3.8 | 3.1 | 2.4 | |||||
| Control | 99.2 | 0.5 | 0.3 | 0.1 | |||||
| HeLa | 38 (600 nM) | 0.6 | 1.1 | 87.6 | 10.6 | ||||
| 42 (50 nM) | 7.3 | 1.4 | 82.0 | 9.4 | |||||
| 45 (400 nM) | 0.8 | 1.5 | 84.9 | 12.7 | |||||
| Control | 90.5 | 1.7 | 5.5 | 2.4 | |||||
1 Early Apoptosis (EA). 2 Late Apoptosis (LA).
Breast cancer MCF7 cells treated with compound 42 for 72 h were mostly classified as live cells (88%), whereas lower percentages of live cells were found when treated with compounds 38 (60%), 45 (71%) and the reference compound CA-4 (58%). This lower percentage of live cells was accompanied by a higher proportion of EA cells for 38 (29%), 45 (20%), and CA-4 (32%) compared to the treatment with 42 (9%). Longer treatment exposures of 6 days confirmed this trend, with 38, 45, and CA-4 showing significantly lower percentages of live cells (45–57%) and higher EA populations (39–52%) than 42, which showed live-cell percentages of 79% and EA of 19%.
Similar results were found 72 h post-treatment for SKOV3 ovarian tumor cells for MCF7 cells. SKOV3 cells treated with compounds 38 and 45 showed modest percentages of EA cells (31% and 23%, respectively) whereas a lower apoptotic population was found for compound 55 (12%). These percentages of apoptotic cells for compounds 38 and 55 were accompanied by low percentages of live cells (62% and 73%, respectively), with 10 nM paclitaxel showing high percentages of live cells (91%).
Treatment of the cervical cancer HeLa cells with compounds 38, 42, and 45 resulted in a severe reduction of live cells down to less than 7% and an accumulation of LA (82–88%) and necrotic (9–13%) cells after 72 h.
2.7. Lead Compounds Disrupt the Microtubule Network and Inhibit Tubulin Polymerization
The effect of MDS 38 and 45 on the tubulin cytoskeleton was analyzed in MCF7, SKOV3, and HeLa cells after 72-h treatments. The microtubule network was visualized by confocal microscopy staining α-tubulin in green, and cell nuclei were stained in blue with DAPI (4’,6-diamidino-2-phenylindole) (Figure 4). Compounds 38 and 45 disrupted the microtubule system of MCF7, SKOV3, and HeLa cells (Figure 4a), which loses the hairy appearance of untreated cells and shows up as a diffuse gauze. Treated cells showed multilobulated nuclei resembling bunches of grapes (Figure 4b).
Figure 4.

Confocal microscopy experiments in MCF7, SKOV3, and HeLa cells. Cells were incubated in the absence (Control) or the presence of 38 (600 nM) and 45 (400 nM) for 72 h. (a) Visualization of the microtubule network. α-Tubulin was stained in green, and nuclei were stained with DAPI (blue fluorescence). (b) Multilobulated nuclei (blue) resembling bunches of grapes in MCF7 cells. Photomicrographs are representative of three independent experiments. Scale bar: 25 µm.
The effect of all the synthesized compounds on the in vitro tubulin polymerization was evaluated at 10 µM. All the compounds that inhibited polymerization more than 30% were also assayed at 20 µM. IC50 values were calculated for those compounds inhibiting tubulin polymerization by at least 50% at 20 µM (Table 1 and Supplemental Figure S2). Most antiproliferative N-alkylated sulfonamides interfere to a greater or lesser extent with tubulin polymerization, with the highest effect shown by compound 45 (IC50 = 3.7 µM), with a benzyl group on the sulfonamide nitrogen, close to CA-4 (IC50 = 3 µM) and better than ABT-751 (IC50 = 4.4 µM). Compounds 38 and 42 showed moderate to good IC50 values of 10 and 12 µM, respectively.
The binding mode of the active compounds at the colchicine site of tubulin was studied by flexible docking studies. The protein conformational variability was represented by using 55 protein structures with different binding sites, as previously described [54,55]. Two frequently used docking programs with different scoring functions were used for the flexible docking of the ligands in the binding sites. Several thousand poses were generated for each ligand which were automatically classified according to their occupation of the subsites of the colchicine domain, and the poses with the best consensus scores were selected (Figure 5). The selected ligands bind in similar dispositions to combretastatin A-4, with a good overlap of the trimethoxyphenyl ring, which is inserted edgewise towards the surface of sheets S8 and S9 between the sidechains of Ala316β, Val318β, Ala354β, and covered by helices H7 and H8 and the H7-H8 loop and interacting with Cys241β, Leu242β, Leu248β, Ala250β, and Leu255β. The sulfonamide bridges are placed close to the olefin binding zone of CA-4, with the other aromatic ring behind helix H8 and above the sidechains of Ala316β and the methylenes of Lys352β, with the substituents on the sulfonamide nitrogen projecting towards a hydrophobic region of the interdimer interface, thus explaining the preference for small hydrophobic substituents such as methyl or cyanomethyl groups.
Figure 5.

Docking poses for compounds 38, 42, and 45 at the colchicine site of tubulin. Combretastatin A-4 (CA-4) is shown in diffuse green as a reference.
3. Discussion
Taxanes—usually paclitaxel alone or in combination regimens—entail the first-line chemotherapy for breast and gynecological cancers [9,13,18,20]. However, limitations related to toxicity, pharmacokinetic problems, and the appearance of resistance [31] prompt the search for new treatments. This work has yielded a new family of tubulin inhibitors with a sulfonamide scaffold as hybrids of combretastatin A-4 and ABT-751, two colchicine site ligands in clinical trials that present shortcomings of opposite nature whose hybridization might alleviate (Figure 1).
Therefore, a focused library of sulfonamides was designed, and 52 representatives were synthesized following a synthetic methodology that involves first the assembly of the sulfonamide followed by functional group manipulations at different points of the main scaffold. A breadth of substituents was thus introduced to explore the chemical space for optimal pharmacokinetic and pharmacodynamic properties (Figure 1). One of the main drawbacks of taxanes and CA-4 is their low water solubility, making necessary drug administration as formulations with several associated adverse effects [27,28,47]. New sulfonamide derivatives show moderate to good solubilities (Table 2). Improvements up to 1700 times in solubility were found when compared with reference drugs paclitaxel and CA-4 and even surpass the solubility of the orally administered ABT-751 up to 42 times.
The antiproliferative activity of the synthesized compounds was evaluated against two Pan-Gyn representative human tumor cell lines (HeLa and MCF7) (Table 1). Many compounds showed growth inhibitory activities at submicromolar concentrations, thus validating the adequacy of the selected diarylsulfonamide scaffold. Most potent derivatives inhibited cell proliferation at very low drug concentrations, with IC50 values in the low to medium nanomolar range. Compounds 2a, 29a, 30, 32, 38, and 42–45 display antiproliferative IC50 values comparable to those of paclitaxel and CA-4 and better than ABT-751, a tubulin colchicine-site inhibiting sulfonamide in clinical trials used as a reference [49]. Alkylation of the sulfonamide nitrogen significantly increases potency compared to the unsubstituted sulfonamides, probably due to a combination of a favoring of the cisoid disposition of both aromatic rings, an essential requirement for colchicine-site-binding drugs, with a more favorable interaction with the target [56]. Small alkyl groups, such as methyl or ethyl (e.g., 2a, 29a, 38, or 42), are usually preferred over longer chains such as carboxylic acid derivatives. All the modifications attempted on the trimethoxyphenyl ring abolished the activity, whereas the introduction of hydrogen bond donor amines on the B ring translated into more potent analogs, such as the secondary amines in the para position of the sulfonamide group (38) and the primary amines ortho to the methoxy group of B ring (42–45). Both modifications improve the polarity and aqueous solubility. The potency improvement suggests favorable hydrogen bonding with the target. Thus, B-ring amines improve both the pharmacokinetics and the pharmacodynamics.
The antiproliferative IC50 values of each compound against HeLa and MFC7 cell lines were quite similar, whereas the CA-4-resistant HT-29 cell line was less or at best equally sensitive to MDS (Supplementary Table S1). When differences in antiproliferative potencies occur, they are not due to multidrug-resistance by the described moderate expression of MDR1 or MDR3 proteins in HT-29 colon carcinoma cell line [48,53,57], as only a handful of compounds experienced a potency increase in the presence of the Ppg/MDR1 inhibitor, verapamil [58]. Therefore, none of our lead compounds (38, 42, and 45) seemed to be MDR substrates and even compounds 38 and 42 overcame the resistance shown by CA-4 in the HT-29 cell line (IC50 = 59, 81, and 305 nM, respectively) (Table 4).
The microtubule disruption caused by the lead compounds in MCF7 cells winded up in the accumulation of cells in the G2/M region (Figure 3a), as previously reported for CA-4 and ABT-751, used as references [59,60]. This sustained mitotic arrest lasted at least 72 h with percentages of cells showing DNA fragmentation below 12%. The mild response regarding this apoptotic marker is in line with the results observed in AnV/PI staining experiments (Table 5). Compounds 38 and 45 triggered phosphatidylserine exposure in 28–34% of MCF7 cells; 11% in the case of 42. The lack of PI staining within the apoptotic population reveals the integrity of the plasma membrane in most Annexin V-positive cells. This slight apoptotic response might be related to the expression levels of caspase 3, the hub effector of the apoptotic machinery. MCF7 cells have been reported to be deficient in caspase 3 [61,62]. Despite MCF7 cells suffer a weak apoptotic response upon treatment with the lead compounds for 72 h, the percentage of early apoptotic cells increases after 6-day treatments, still preserving the integrity of the plasma membrane. This slow build-up of the apoptotic response in MCF7 cells may putatively rely on the other two effector caspases 6 and 7, as described for DNA damaging agents [11,63,64].
Lead compounds 38, 42, and 45 were then assayed against a panel of human ovarian tumor cell lines. Ovarian serous adenocarcinoma cell lines (SKOV3, OVCAR-8, and OVCAR-3), the most common type of ovarian cancer, were more sensitive than endometrioid cell lines (IGROV-1 and A2780). The metastatic cell line SKOV3 was found quite resistant to paclitaxel and CA-4 (IC50 of 81 and 380 nM [65], respectively) but was sensitive to MDS (Table 3). Time-course analysis of the cell cycle distribution (Figure 3b) showed a strong mitotic arrest for 42, 38, and 45 24 h after treatment. The mitotic arrest readily triggered apoptosis 48 h after treatment. Compound 45 elicits a higher SubG0/G1 population than 38 and 42 at 72 h. Dual staining experiments confirm the apoptotic response with Annexin V-positive cells at 72 h ranging from 14–36%, notably higher when compared with paclitaxel (7%).
Studies on the mechanism of action of MDS on the cervical cancer cell line HeLa were also performed (Figure 3c). After the common mitotic arrest at G2/M phase 24 h after treatment (more pronounced for 38 and 45 than for 42) HeLa cells exhibited strikingly high percentages of SubG0/G1 cells (around 50%) from 48 h after drug exposure. AnV/PI staining experiments revealed a strong and advanced apoptotic response with rates of double-positives cells above 82%, indicating the permeabilization of the plasma membrane.
At this point, we wished to verify if the observed effect was due to the disruption of microtubule assembly. Tubulin immunofluorescence analysis of cells treated with compounds 38 and 45 revealed a clear disruption in the microtubule network (Figure 4a). Furthermore, as interference with microtubule dynamics is expected to affect chromatin organization, cell nuclei were stained with DAPI. Accumulation of multiple micronuclei characteristic of mitotic catastrophe was observed, widely linked to tubulin-binding drugs [66,67]. In vitro tubulin polymerization inhibition experiments confirmed the effect of the compounds on tubulin, and molecular docking experiments suggest a similar binding mode to that of CA-4 to the colchicine site of tubulin (Figure 5). However, no correlation was observed between TPI and antiproliferative IC50 values, as has previously been found for many tubulin polymerization inhibitors [68]. This discrepancy can be explained by an antiproliferative effect dependent on the alteration of microtubule dynamics and not on polymer mass, as measured in TPI assays.
In conclusion, a new family of MDS has been designed and synthesized. MDS antiproliferative effects against several representative cell lines of Pan-Gyn cancers have been studied. The effects of the compounds vary depending on the structural modifications, the most potent derivatives being those with a small alkyl substituent on the sulfonamide nitrogen. The molecular mechanism of action studies revealed a pronounced G2/M arrest mainly followed by a SubG0/G1 increase over time. The different behavior between MCF7, SKOV3 and HeLa cells seems to be related to the rate at which each cell line builds up its apoptotic response to the initial unsolved mitotic arrest. Inhibition of tubulin polymerization in vitro was observed for most potent MDS, as well as disruption of microtubule network by immunofluorescence, probably by binding similarly to CA-4 to the colchicine site of tubulin. The improved aqueous solubility, lack of Pgp-mediated resistance, and antiproliferative potencies comparable to paclitaxel—first-line treatment of Pan-Gyn cancers—make MDS interesting for future application as antitumor agents.
4. Materials and Methods
4.1. Chemistry
4.1.1. Chemical Synthesis
Detailed synthetic procedures and characterization of compounds can be found in Supplemental methods SP1 and SP2.
4.1.2. Chemical Characterization of Lead MDS 38, 42, and 45
MDS 38. M.p.: 147–150 °C (CH2Cl2/Hexane). IR (KBr): 3390, 1599, 1502, 835 cm−1. 1H NMR (400 MHz, CDCl3): δ 2.83 (3H, s), 3.07 (3H, s), 3.71 (6H, s), 3.80 (3H, s), 6.28 (2H, s), 6.51 (2H, d, J = 8.8), 7.35 (2H, d, J = 8.8). 13C NMR (100 MHz, CDCl3): δ 30.0 (CH3), 38.4 (CH3), 56.1 (2CH3), 60.8 (CH3), 104.6 (2CH), 110.8 (2CH), 122.6 (C), 130.0 (2CH), 137.2 (C), 137.8 (C), 152.7 (C), 152.8 (2C). HRMS (C17H22N2O5S + H+): calcd 367.1322 (M + H+), found 367.1325.
MDS 42. M.p.: 124–126 °C (MeOH). 1H NMR (400 MHz, CDCl3): δ 3.11 (3H, s), 3.73 (6H, s), 3.83 (3H, s), 3.90 (3H, s), 3.94 (2H, s), 6.30 (2H, s), 6.78 (1H, d, J = 8.8), 6.90 (1H, d, J = 2.4), 6.99 (1H, dd, J = 8.8 and 2.4). 13C NMR (50 MHz, CDCl3): δ 38.5 (CH3), 55.7 (CH3), 56.0 (2CH3), 60.8 (CH3), 104.6 (2CH), 109.1 (CH), 113.2 (CH), 118.8 (CH), 128.0 (C), 136.5 (C), 137.3 (C), 137.7 (C), 150.2 (C), 152.8 (2C). HRMS (C17H22N2O6S + H+): calcd 383.1271 (M + H+), found 383.1263.
MDS 45.1H NMR (200 MHz, CDCl3): δ 3.63 (6H, s), 3.78 (3H, s), 3.93 (3H, s), 4.63 (2H, s), 6.14 (2H, s), 6.83 (1H, d, J = 8.4), 7.02 (1H, d, J = 2), 7.11 (1H, dd, J = 8.4 and 2), 7.23 (5H, bs). 13C NMR (100 MHz, CDCl3): δ 55.3 (CH2), 55.8 (CH3), 55.9 (2CH3), 60.8 (CH3), 106.7 (2CH), 109.3 (CH), 113.1 (CH), 118.8 (CH), 127.6 (CH), 128.3 (2CH), 128.7 (2CH), 130.5 (C), 134.9 (C), 136.2 (C), 136.6 (C), 137.6 (C), 150.3 (C), 152.7 (2C). HRMS (C23H26N2O6S + H+): calcd 459.1584 (M + H+), found 459.1589.
4.1.3. Aqueous Solubility
The aqueous solubility of the sulfonamides was determined using an approach based on the saturation shake-flask method. Tested compounds (1–2 mg) were stirred in pH 7.0 phosphate buffer (300 µL) for 48 h at room temperature. The resulting suspension was filtered over a 45 µm filter to discard insoluble residues, and the concentration in the supernatant was measured by UV absorbance. To determine the concentration, a scan between 270 and 400 nm was performed in a Helios-α UV-320 Spectrophotometer (Thermo-Spectronic, Thermo Fischer Scientific, Waltham, MA, USA) for each tested compound. Then, three maximum wavelengths of absorbance per compound were selected from the previous scan and calibration curves were performed at these three wavelengths. Solubility results are given as the average of the three measurements.
4.2. Biological Evaluation
4.2.1. Cell Lines and Cell Culture Conditions
MCF7 (human breast carcinoma), SKOV-3 (human ovarian carcinoma), OVCAR-3 (human ovarian carcinoma), OVCAR-8 (human ovarian carcinoma), and HeLa (human cervical carcinoma) cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Thermo Fischer Scientific) containing 10% (v/v) heat-inactivated fetal bovine serum (HIFBS) (Lonza-Cambrex, Karlskoga, Sweden), 2 mM L-glutamine (Lonza-Cambrex, Karlskoga, Sweden), 100 μg/mL streptomycin, and 100 IU/mL penicillin (Lonza-Cambrex) at 37 °C in humidified 95% air and 5% CO2. HT-29 (human colon carcinoma), IGROV-1 (human ovarian carcinoma), and A2780 (human ovarian carcinoma) cell lines were cultured in RPMI 1640 medium (Gibco, Thermo Fischer Scientific) supplemented with 10% HIFBS, 100 μg/mL streptomycin, and 100 IU/mL penicillin at 37 °C in humidified 95% air and 5% CO2 atmosphere. The presence of mycoplasma was routinely checked with MycoAlert kit (Lonza-Cambrex) and only mycoplasma-free cells were used in the experiments. Ovarian tumor cell lines were originally acquired from Dr Atanasio Pandiella from Centro de Investigación del Cáncer (CIC), Salamanca, Spain. HeLa, HT-29 and MCF7 tumor cell lines were provided by Dr Faustino Mollinedo from Centro de Investigaciones Biológicas Margarita Salas, Madrid, Spain.
4.2.2. Cell Proliferation Assay
MCF7, HeLa, and HT-29 cell proliferation, when treated with the corresponding compounds, were determined using the XTT (sodium 3’-[1(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro)-benzenesulfonic acid hydrate) cell proliferation kit (Roche Molecular Biochemicals, Mannheim, Germany). A freshly prepared mixture of XTT labeling reagent with 0.02% (v/v) PMS (N-methyl-dibenzopyrazine methyl sulfate) electron coupling reagent was added to cells (50 µL/well in 96-well plates, total volume of 160 µL/well). Cells were incubated under standard culture conditions for 4 h (MCF7 and HeLa) or 6 h (HT-29). The absorbance of the formazan product generated was measured at 450 nm using a multi-well plate reader. SKOV-3, OVCAR-3, OVCAR-8, IGROV-1, A-2780 cell proliferation, when treated with the corresponding compound, were determined using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) (Sigma-Aldrich, St. Louis, MO, USA). MTT in Phosphate Buffer Saline (PBS) (5 mg/ml) was added to cells (110 µL/well in 24-well plates, total volume of 1110 µL/well). After 1 hour of incubation, the medium was aspirated, and formazan violet crystals were dissolved in dimethyl sulfoxide (DMSO) (500 µL/well). Absorbance was measured at 570 nm in a plate reader (Ultra Evolution, Tecan, Männedorf, Suiza). To determine cell viability, cells in exponential growth phase were seeded (100 µL/well in 96-well plates and 1000 µL/well in 24-well plates) with appropriate cell line concentration (1.5·104 cells/mL for MCF7 and HeLa, 3·104 cells/mL for HT-29, and 1·104 cells/mL for SKOV-3, OVCAR-3, OVCAR-8, IGROV-1, and A-2780) in complete RPMI 1640 or DMEM medium at 37°C and 5% CO2 atmosphere. After 24 h incubation, to allow cells to attach to the plates, all compounds were added at 1 µM concentration and the effect on the proliferation was evaluated 72 h post-treatment. Compounds showing antiproliferative effects at tested concentration were selected for IC50 calculation (50% inhibitory concentration with respect to the untreated controls) from 10−5 to 10−10 M concentration. Non-linear curves fitting the experimental data were carried out for each compound. Compounds were dissolved in DMSO and the final solvent concentrations never exceeded 0.5% (v/v). The control wells included treated cells with 0.5% (v/v) DMSO and the positive control. 10 µM verapamil was included as a control for the HT-29 cell line. Measurements were performed in triplicate, and each experiment was repeated three times.
4.2.3. Cell Cycle Analysis
Cell cycle analysis was performed in MCF7, SKOV-3, and HeLa cells by quantifying the DNA content by flow cytometry. Cells in the exponential growth phase at 2·104 cells/mL were seeded in 24-well plates (1 mL/well). After 24 h incubation, cells were treated with different concentrations of the selected compounds and the effect of treated and untreated cells was measured 24, 48, and 72 h post-treatment. Live and dead cells were collected and fixed in ice-cold ethanol/PBS (7:3) and stored at 4 °C for later use. Cells were rehydrated with PBS, treated with 100 µg/mL RNase A (Sigma-Aldrich Co., St. Louis, MO, USA) and stained overnight in darkness at room temperature with 50 µg/mL PI (Sigma-Aldrich Co.). Cell cycle profiles were then analyzed by flow cytometry using BD Accuri™ C6 Plus Flow Cytometer (BD Biosciences, San José, CA, USA). Data were analyzed with BD Accuri™ C6 Software (version 1.0.264.21, BD Biosciences, San José, CA, USA) and compared to control cells. Compounds were dissolved in DMSO and the final solvent concentration never exceeded 0.5% (v/v). Control wells included cells with 0.5% (v/v) DMSO.
4.2.4. Apoptotic Cell Death Quantification
MCF7, SKOV-3, and HeLa apoptotic cells were quantified using an Annexin V-FITC/PI apoptosis detection kit (Immunostep, Salamanca, Spain) according to the manufacturer’s guidelines. 1.5 mL/well of cells in the exponential growth phase at 2·104 cells/mL were seeded onto 12-well plates and left to attach overnight. After 24 h incubation, cells were treated with selected compounds. After 72 h (MCF7, SKOV-3, and HeLa) and 6 days (MCF7) post-treatment, attached and floating cells of treated and untreated wells were collected, centrifuged, resuspended in Annexin V binding buffer, and stained with Annexin V-FITC/PI. Cells were then incubated in darkness at room temperature for 15 min and a total of 30,000 cells were acquired and analyzed using the BD Accuri™ C6 Plus Flow Cytometer and Software (version 1.0.264.21, BD Biosciences, San José, CA, USA), respectively. Compounds were dissolved in DMSO and the final solvent concentration never exceeded 0.5% (v/v). Control wells included cells with 0.5% (v/v) DMSO.
4.2.5. Immunofluorescence
MCF7, SKOV-3, and HeLa cells were grown on glass coverslips coated with poly-L-lysine (12 mm diameter). To reach appropriate cell confluence, coverslips were manipulated in 6-well plates (3 coverslips/well) seeding 3 mL/well of cells in the exponential growth phase at 2 × 104 cells/mL. After 24 h incubation, cells were treated with selected compounds and incubated for 72 h. Cells were washed with PBS, fixed with 4% formaldehyde in PBS for 10 min, permeabilized with 0.5% Triton X-100 (Boehringer Mannheim, Ingelheim am Rhein, Germany) in PBS for 10 min and blocked with 10% BSA in PBS for 30 min. Microtubule detection was performed by incubation with anti-α-tubulin mouse monoclonal antibody (1:200 in 3% BSA/PBS) (Sigma-Aldrich) for 1.5 h. After PBS washing, coverslips were incubated with fluorescent secondary antibody Alexa Fluor 488 goat anti-mouse IgG (1:400 in 1% BSA/PBS) (Molecular Probes, Invitrogen, Thermo Fischer Scientific) for 1 h in darkness. After being washed with PBS, cell nuclei were stained with DAPI (dihydrochloride of 4′,6-diamidino-2-phenylindole) (diluted 1:10,000 in mq H2O) (Roche, Basel, Switzerland) for 5 min in darkness. DAPI excess was removed by washing with PBS. Mowiol reagent (Calbiochem, Sigma-Aldrich) was used to mount preparations on slides. Cells were analyzed by confocal microscopy using a LEICA SP5 microscope DMI-6000V model coupled to a LEICA LAS AF software computer.
4.2.6. Tubulin Isolation
Microtubular protein was isolated from calf brain according to the modified Shelanski method [69,70] by two cycles of temperature-dependent assembly/disassembly and stored at −80 °C. Before each use, protein concentration was determined by the Bradford method [71] taking BSA as standard.
4.2.7. Tubulin Polymerization Inhibition (TPI) Assay
Tubulin polymerization was monitored using a Helios α spectrophotometer by measuring the increase in turbidity at 450 nm, caused by a shift from 4 °C to 37 °C, which allows the in vitro microtubular protein to depolymerize and polymerize, respectively. The assay was carried out in quartz cuvettes containing 1.5 mg/mL microtubular protein and the ligand (except for control cuvette with only DMSO at the same concentration) in a mixture of 0.1 M MES buffer, 1 mM EGTA, 1 mM MgCl2, 1 mM β-ME, and 1.5 mM GTP at pH 6.7 (final volume 500 µL). Cuvettes were preincubated at 20 °C for 30 min, to allow ligand binding to tubulin, and subsequently cooled on ice for 10 min. Then, the experiment starts at 4 °C to establish the initial baseline. The assembly process was initiated by a temperature shift to 37 °C and the turbidity produced by tubulin polymerization can be measured by an absorbance increase. After reaching a stable plateau, the temperature was switched back to 4 °C to return to the initial absorption values (to confirm the reversible nature of the monitored process and to determine whether or not the microtubular tubular protein has stabilized). The difference in amplitude between the stable plateau and the initial baseline of the curves was taken as the degree of tubulin polymerization for each experiment. Comparison with control curves under identical conditions but without ligands yielded TPI as a percentage value. All compounds were tested at 10 µM concentration. Compounds that inhibited tubulin polymerization by at least 30% at 10 µM were tested at 20 µM. IC50 values were calculated for compounds that inhibit tubulin polymerization by more than 50% at 20 µM. Compounds were dissolved in DMSO, and the final solvent concentration never exceeded 4% (v/v), which has been reported not to interfere with the assembly process. All the measurements were carried out in at least two independent experiments using microtubular protein from different preparations.
4.3. Computational Studies
Docking studies were carried out as previously described [45]. Briefly, dockings were performed in parallel with PLANTS [72] with default settings and 10 runs per ligand and with AutoDock 4.2 [73] runs applying the Lamarckian genetic algorithm (LGA) 100−300 times for a maximum of 2.5 × 106 energy evaluations, 150 individuals, and 27,000 generations maximum. The retrieved poses were automatically assigned to the colchicine subzones using in-house KNIME pipelines [74]. The programs’ docking scores were converted into Z-scores and the poses with best consensus scores were selected as the docking results. Docked poses were analyzed with Chimera [75], Marvin [76], OpenEye [77], and JADOPPT [78].
Acknowledgments
We thank the people at Frigoríficos Salamanca S.A slaughterhouse for providing us with the calf brains, “Servicio General de NMR” and “Servicio General de Espectrofotometría de Masas” of the University of Salamanca for equipment. M.G. acknowledges a predoctoral fellowship from the Junta de Castilla y León (ORDEN EDU/529/2017 de 26 de junio). M.O.-S. acknowledges a predoctoral fellowship from the IBSAL (IBpredoc17/00010). A.V.-B. acknowledges a predoctoral fellowship from the Spanish Ministerio de Educación, Cultura y Deporte (FPU15/02457).
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/22/4/1907/s1, Figure S1: Synthetic procedures of new Microtubule Destabilizing Sulfonamides, Table S1: Antiproliferative activity against HT-29 and sensitivity to MDR pumps, Figure S2: Tubulin Polymerization Assay data. IC50 calculation, Methods SP1: Chemistry. General chemical techniques and chemical synthesis, Methods SP2: 1H NMR and 13C NMR spectra
Author Contributions
Conceptualization, M.G. and R.P.; Data curation, M.G. and R.P.; Formal analysis, M.G., M.O.-S., A.V.-B. and A.B.H.; Funding acquisition, R.G.-S. and R.P. Investigation, R.Á., M.M., R.G.-S. and R.P.; Methodology, M.G. and M.O.-S.; Project administration, R.G.-S. and R.P.; Resources, R.G.-S. and R.P.; Software, R.P.; Supervision, R.G.-S. and R.P.; Validation, M.M., R.G.-S. and R.P.; Visualization, M.G. and M.O.-S.; Writing—original draft, M.G.; Writing—review and editing, A.V.-B., A.B.H., M.M., R.G.-S. and R.P.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Consejería de Educación de la Junta de Castilla y León (SA030U16, SA262P18 and SA116P20), co-funded by the EU’s European Regional Development Fund-FEDER, the Spanish Ministry of Science, Innovation and Universities (RTI2018-099474-BI00) and the health research program of the Instituto de Salud Carlos III (Spanish Ministry of Economy and Competitiveness, PI16/01920 and PI20/01569) co-funded with FEDER founds.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Berger A.C., Korkut A., Kanchi R.S., Hegde A.M., Lenoir W., Liu W., Liu Y., Fan H., Shen H., Ravikumar V., et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell. 2018;33:690–705.e9. doi: 10.1016/j.ccell.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoadley K.A., Yau C., Hinoue T., Wolf D.M., Lazar A.J., Drill E., Shen R., Taylor A.M., Cherniack A.D., Thorsson V., et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell. 2018;173:291–304.e6. doi: 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 4.Rivera E., Gomez H. Chemotherapy resistance in metastatic breast cancer: The evolving role of ixabepilone. Breast Cancer Res. 2010;12:S2. doi: 10.1186/bcr2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Angulo A.M., Morales-Vasquez F., Hortobagyi G.N. Breast Cancer Chemosensitivity. Springer; Berlin/Heidelberg, Germany: 2013. Overview of Resistance to Systemic Therapy in Patients with Breast Cancer. Madame Curie Biosci. Database. [DOI] [PubMed] [Google Scholar]
- 6.Agarwal R., Kaye S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer. 2003;3:502–516. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 7.Pokhriyal R., Hariprasad R., Kumar L., Hariprasad G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer. 2019;11:1179299X1986081. doi: 10.1177/1179299X19860815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaudhry P., Asselin E. Resistance to chemotherapy and hormone therapy in endometrial cancer. Endocr. Relat. Cancer. 2009;16:363–380. doi: 10.1677/ERC-08-0266. [DOI] [PubMed] [Google Scholar]
- 9.Moxley K.M., McMeekin D.S. Endometrial Carcinoma: A Review of Chemotherapy, Drug Resistance, and the Search for New Agents. Oncologist. 2010;15:1026–1033. doi: 10.1634/theoncologist.2010-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.U.S. Food and Drug Administration. [(accessed on 27 May 2020)]; Available online: https://www.fda.gov/
- 11.Ofir R., Seidman R., Rabinski T., Krup M., Yavelsky V., Weinstein Y., Wolfson M. Taxol-induced apoptosis in human SKOV3 ovarian and MCF7 breast carcinoma cells is caspase-3 and caspase-9 independent. Cell Death Differ. 2002;9:636–642. doi: 10.1038/sj.cdd.4401012. [DOI] [PubMed] [Google Scholar]
- 12.Jayson G.C., Kohn E.C., Kitchener H.C., Ledermann J.A. Ovarian cancer. Lancet. 2014;384:1376–1388. doi: 10.1016/S0140-6736(13)62146-7. [DOI] [PubMed] [Google Scholar]
- 13.McGuire W.P., Markman M. Primary ovarian cancer chemotherapy: Current standards of care. Br. J. Cancer. 2003;89:S3–S8. doi: 10.1038/sj.bjc.6601494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bicaku E., Xiong Y., Marchion D.C., Chon H.S., Stickles X.B., Chen N., Judson P.L., Hakam A., Gonzalez-Bosquet J., Wenham R.M., et al. In vitro analysis of ovarian cancer response to cisplatin, carboplatin, and paclitaxel identifies common pathways that are also associated with overall patient survival. Br. J. Cancer. 2012;106:1967–1975. doi: 10.1038/bjc.2012.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ozols R.F., Bundy B.N., Greer B.E., Fowler J.M., Clarke-Pearson D., Burger R.A., Mannel R.S., DeGeest K., Hartenbach E.M., Baergen R., et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2003;21:3194–3200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- 16.Ghersi D., Wilcken N., Simes R.J. A systematic review of taxane-containing regimens for metastatic breast cancer. Br. J. Cancer. 2005;93:293–301. doi: 10.1038/sj.bjc.6602680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedrichs K., Hölzel F., Jänicke F. Combination of taxanes and anthracyclines in first-line chemotherapy of metastatic breast cancer: An interim report. Eur. J. Cancer. 2002;38:1730–1738. doi: 10.1016/S0959-8049(02)00144-2. [DOI] [PubMed] [Google Scholar]
- 18.Telli M.L., Carlson R.W. First-line chemotherapy for metastatic breast cancer. Clin. Breast Cancer. 2009;9:S66–S72. doi: 10.3816/CBC.2009.s.007. [DOI] [PubMed] [Google Scholar]
- 19.Piccart-Gebhart M.J., Burzykowski T., Buyse M., Sledge G., Carmichael J., Lück H.J., Mackey J.R., Nabholtz J.M., Paridaens R., Biganzoli L., et al. Taxanes alone or in combination with anthracyclines as first-line therapy of patients with metastatic breast cancer. J. Clin. Oncol. 2008;26:1980–1986. doi: 10.1200/JCO.2007.10.8399. [DOI] [PubMed] [Google Scholar]
- 20.Pectasides D., Pectasides E., Economopoulos T. Systemic therapy in metastatic or recurrent endometrial cancer. Cancer Treat. Rev. 2007;33:177–190. doi: 10.1016/j.ctrv.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Serkies K., Jassem J. Systemic therapy for cervical carcinoma—Current status. Chin. J. Cancer Res. 2018;30:209–221. doi: 10.21147/j.issn.1000-9604.2018.02.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boussios S., Seraj E., Zarkavelis G., Petrakis D., Kollas A., Kafantari A., Assi A., Tatsi K., Pavlidis N., Pentheroudakis G. Management of patients with recurrent/advanced cervical cancer beyond first line platinum regimens: Where do we stand? A literature review. Crit. Rev. Oncol. Hematol. 2016;108:164–174. doi: 10.1016/j.critrevonc.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Konno T., Watanabe J., Ishihara K. Enhanced solubility of paclitaxel using water-soluble and biocompatible 2-methacryloyloxyethyl phosphorylcholine polymers. J. Biomed. Mater. Res. Part A. 2003;65:209–214. doi: 10.1002/jbm.a.10481. [DOI] [PubMed] [Google Scholar]
- 24.Hamada H., Ishihara K., Masuoka N., Mikuni K., Nakajima N. Enhancement of water-solubility and bioactivity of paclitaxel using modified cyclodextrins. J. Biosci. Bioeng. 2006;102:369–371. doi: 10.1263/jbb.102.369. [DOI] [PubMed] [Google Scholar]
- 25.Yang T., Cui F.-D., Choi M.-K., Lin H., Chung S.-J., Shim C.-K., Kim D.-D. Liposome Formulation of Paclitaxel with Enhanced Solubility and Stability. Drug Deliv. 2007;14:301–308. doi: 10.1080/10717540601098799. [DOI] [PubMed] [Google Scholar]
- 26.Eric K., Rowinsky M.D., Ross C., Donehower M.D. Paclitaxel (Taxol®) N. Engl. J. Med. 1995;332:1004–1014. doi: 10.1056/nejm199504133321507. [DOI] [PubMed] [Google Scholar]
- 27.Hennenfent K.L., Govindan R. Review Novel formulations of taxanes: A review. Old wine in a new bottle? Ann. Oncol. 2006;17:735–749. doi: 10.1093/annonc/mdj100. [DOI] [PubMed] [Google Scholar]
- 28.Ten Tije A.J., Verweij J., Loos W.J., Sparreboom A. Pharmacological effects of formulation vehicles: Implications for cancer chemotherapy. Clin. Pharmacokinet. 2003;42:665–685. doi: 10.2165/00003088-200342070-00005. [DOI] [PubMed] [Google Scholar]
- 29.Weiss R.B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990;8:1263–1268. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- 30.Gligorov J., Lotz J.P. Preclinical Pharmacology of the Taxanes: Implications of the Differences. Oncologist. 2004;9:3–8. doi: 10.1634/theoncologist.9-suppl_2-3. [DOI] [PubMed] [Google Scholar]
- 31.Orr G.A., Verdier-Pinard P., McDaid H., Horwitz S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene. 2003;22:7280–7295. doi: 10.1038/sj.onc.1206934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer. 2010;10:194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 33.Yusuf R., Duan Z., Lamendola D., Penson R., Seiden M. Paclitaxel Resistance: Molecular Mechanisms and Pharmacologic Manipulation. Curr. Cancer Drug Targets. 2003;3:1–19. doi: 10.2174/1568009033333754. [DOI] [PubMed] [Google Scholar]
- 34.Murray S., Briasoulis E., Linardou H., Bafaloukos D., Papadimitriou C. Taxane resistance in breast cancer: Mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat. Rev. 2012;38:890–903. doi: 10.1016/j.ctrv.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 35.Tommasi S., Mangia A., Lacalamita R., Bellizzi A., Fedele V., Chiriatti A., Thomssen C., Kendzierski N., Latorre A., Lorusso V., et al. Cytoskeleton and paclitaxel sensitivity in breast cancer: The role of β-tubulins. Int. J. Cancer. 2007;120:2078–2085. doi: 10.1002/ijc.22557. [DOI] [PubMed] [Google Scholar]
- 36.Chong T., Sarac A., Yao C.Q., Liao L., Lyttle N., Boutros P.C., Bartlett J.M.S., Spears M. Deregulation of the spindle assembly checkpoint is associated with paclitaxel resistance in ovarian cancer. J. Ovarian Res. 2018;11:27. doi: 10.1186/s13048-018-0399-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kavallaris M., Kuo D.Y.S., Burkhart C.A., Regl D.L., Norris M.D., Haber M., Horwitz S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific β-tubulin isotypes. J. Clin. Investig. 1997;100:1282–1293. doi: 10.1172/JCI119642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng X., Gong F., Chen Y., Jiang Y., Liu J., Yu M., Zhang S., Wang M., Xiao G., Liao H. Autophagy promotes paclitaxel resistance of cervical cancer cells: Involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis. 2014;5:1–11. doi: 10.1038/cddis.2014.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horwitz S.B., Lothstein L., Manfredi J.J., Mellado W., Parness J., Roy S.N., Schiff P.B., Sorbara L., Zeheb R. Taxol: Mechanisms of Action and Resistance. Ann. New York Acad. Sci. 1986;466:733–744. doi: 10.1111/j.1749-6632.1986.tb38455.x. [DOI] [PubMed] [Google Scholar]
- 40.Schiff P.B., Fant J., Horwitz S.B. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 41.Dumontet C., Jordan M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mollinedo F., Gajate C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis. 2003;8:413–450. doi: 10.1023/A:1025513106330. [DOI] [PubMed] [Google Scholar]
- 43.Vicente-Blázquez A., González M., Álvarez R., del Mazo S., Medarde M., Peláez R. Antitubulin sulfonamides: The successful combination of an established drug class and a multifaceted target. Med. Res. Rev. 2019;39:775–830. doi: 10.1002/med.21541. [DOI] [PubMed] [Google Scholar]
- 44.Lee C.-T., Huang Y.-W., Yang C.-H., Huang K.-S. Drug Delivery Systems and Combination Therapy by Using Vinca Alkaloids. Curr. Top. Med. Chem. 2015;15:1491–1500. doi: 10.2174/1568026615666150414120547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Álvarez R., Aramburu L., Gajate C., Vicente-Blázquez A., Mollinedo F., Medarde M., Peláez R. Potent colchicine-site ligands with improved intrinsic solubility by replacement of the 3,4,5-trimethoxyphenyl ring with a 2-methylsulfanyl-6-methoxypyridine ring. Bioorg. Chem. 2020;98:103755. doi: 10.1016/j.bioorg.2020.103755. [DOI] [PubMed] [Google Scholar]
- 46.Lee R.M., Gewirtz D.A. Colchicine site inhibitors of microtubule integrity as vascular disrupting agents. Drug Dev. Res. 2008;69:352–358. doi: 10.1002/ddr.20267. [DOI] [Google Scholar]
- 47.Tron G.C., Pirali T., Sorba G., Pagliai F., Busacca S., Genazzani A.A. Medicinal chemistry of combretastatin A4: Present and future directions. J. Med. Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 48.Cummings J., Zelcer N., Allen J.D., Yao D., Boyd G., Maliepaard M., Friedberg T.H., Smyth J.F., Jodrell D.I. Glucuronidation as a mechanism of intrinsic drug resistance in colon cancer cells: Contribution of drug transport proteins. Biochem. Pharmacol. 2004;67:31–39. doi: 10.1016/j.bcp.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 49.ABT-751 ClinicalTrials.gov. [(accessed on 21 May 2020)]; Available online: https://clinicaltrials.gov/
- 50.Seddigi Z.S., Malik M.S., Saraswati A.P., Ahmed S.A., Babalghith A.O., Lamfon H.A., Kamal A. Recent advances in combretastatin based derivatives and prodrugs as antimitotic agents. Medchemcomm. 2017;8:1592–1603. doi: 10.1039/C7MD00227K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaudhary A., Pandeya S., Kumar P., Sharma P., Gupta S., Soni N., Verma K., Bhardwaj G. Combretastatin A-4 Analogs as Anticancer Agents. Mini Rev. Med. Chem. 2007;7:1186–1205. doi: 10.2174/138955707782795647. [DOI] [PubMed] [Google Scholar]
- 52.Liebmann J.E., Cook J.A., Lipschultz C., Teague D., Fisher J., Mitchell J.B. Cytotoxic studies of paclitaxel (Taxol®) in human tumour cell lines. Br. J. Cancer. 1993;68:1104–1109. doi: 10.1038/bjc.1993.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naret T., Khelifi I., Provot O., Bignon J., Levaique H., Dubois J., Souce M., Kasselouri A., Deroussent A., Paci A., et al. 1,1-Diheterocyclic Ethylenes Derived from Quinaldine and Carbazole as New Tubulin-Polymerization Inhibitors: Synthesis, Metabolism, and Biological Evaluation. J. Med. Chem. 2019;62:1902–1916. doi: 10.1021/acs.jmedchem.8b01386. [DOI] [PubMed] [Google Scholar]
- 54.Alvarez R., Medarde M., Pelaez R. New Ligands of the Tubulin Colchicine Site Based on X-Ray Structures. Curr. Top. Med. Chem. 2014;14:2231–2252. doi: 10.2174/1568026614666141130092637. [DOI] [PubMed] [Google Scholar]
- 55.Goodsell D.S., Zardecki C., Di Costanzo L., Duarte J.M., Hudson B.P., Persikova I., Segura J., Shao C., Voigt M., Westbrook J.D., et al. RCSB Protein Data Bank: Enabling biomedical research and drug discovery. Protein Sci. 2020;29:52–65. doi: 10.1002/pro.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen T.L., McGrath C., Hermone A.R., Burnett J.C., Zaharevitz D.W., Day B.W., Wipf P., Hamel E., Gussio R. A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach. J. Med. Chem. 2005;48:6107–6116. doi: 10.1021/jm050502t. [DOI] [PubMed] [Google Scholar]
- 57.Schobert R., Effenberger-Neidnicht K., Biersack B. Stable combretastatin A-4 analogues with sub-nanomolar efficacy against chemoresistant HT-29 cells. Int. J. Clin. Pharmacol. Ther. 2011;49:71–72. doi: 10.5414/CPP49071. [DOI] [PubMed] [Google Scholar]
- 58.Sharom F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011;50:161–178. doi: 10.1042/bse0500161. [DOI] [PubMed] [Google Scholar]
- 59.Wei R.J., Lin S.S., Wu W.R., Chen L.R., Li C.F., De Chen H., Chou C.T., Chen Y.C., Liang S.S., Chien S.T., et al. A microtubule inhibitor, ABT-751, induces autophagy and delays apoptosis in Huh-7 cells. Toxicol. Appl. Pharmacol. 2016;311:88–98. doi: 10.1016/j.taap.2016.09.021. [DOI] [PubMed] [Google Scholar]
- 60.Kanthou C., Greco O., Stratford A., Cook I., Knight R., Benzakour O., Tozer G. The tubulin-binding agent combretastatin A-4-phosphate arrests endothelial cells in mitosis and induces mitotic cell death. Am. J. Pathol. 2004;165:1401–1411. doi: 10.1016/S0002-9440(10)63398-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jänicke R.U., Sprengart M.L., Wati M.R., Porter A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 62.Yang X.H., Sladek T.L., Liu X., Butler B.R., Froelich C.J., Thor A.D. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Res. 2001;61:348–354. [PubMed] [Google Scholar]
- 63.Walker P.R., Leblanc J., Carson C., Ribecco M., Sikorska M. Neither Caspase-3 nor DNA Fragmentation Factor Is Required for High Molecular Weight DNA Degradation in Apoptosis. Ann. N. Y. Acad. Sci. 1999;887:48–59. doi: 10.1111/j.1749-6632.1999.tb07921.x. [DOI] [PubMed] [Google Scholar]
- 64.Kagawa S., Gu J., Honda T., McDonnell T.J., Swisher S.G., Roth J.A., Fang B. Deficiency of caspase-3 in MCF7 cells blocks Bax-mediated nuclear fragmentation but not cell death. Clin. Cancer Res. 2001;7:1474–1480. [PubMed] [Google Scholar]
- 65.Stefański T., Mikstacka R., Kurczab R., Dutkiewicz Z., Kucińska M., Murias M., Zielińska-Przyjemska M., Cichocki M., Teubert A., Kaczmarek M., et al. Design, synthesis, and biological evaluation of novel combretastatin A-4 thio derivatives as microtubule targeting agents. Eur. J. Med. Chem. 2018;144:797–816. doi: 10.1016/j.ejmech.2017.11.050. [DOI] [PubMed] [Google Scholar]
- 66.Vakifahmetoglu H., Olsson M., Zhivotovsky B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008;15:1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 67.Castedo M., Perfettini J.L., Roumier T., Andreau K., Medema R., Kroemer G. Cell death by mitotic catastrophe: A molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 68.González M., Ellahioui Y., Álvarez R., Gallego-Yerga L., Caballero E., Vicente-Blázquez A., Ramudo L., Marín M., Sanz C., Medarde M., et al. The Masked Polar Group Incorporation (MPGI) Strategy in Drug Design: Effects of Nitrogen Substitutions on Combretastatin and Isocombretastatin Tubulin Inhibitors. Molecules. 2019;24:4319. doi: 10.3390/molecules24234319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shelanski M.L., Gaskin F., Cantor C.R. Microtubule assembly in the absence of added nucleotides. Proc. Natl. Acad. Sci. USA. 1973;70:765–768. doi: 10.1073/pnas.70.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dumortier C., Gorbunoff M.J., Andreu J.M., Engelborghs Y. Different Kinetic Pathways of the Binding of Two Biphenyl Analogues of Colchicine to Tubulin. Biochemistry. 1996;35:4387–4395. doi: 10.1021/bi952451f. [DOI] [PubMed] [Google Scholar]
- 71.Bradford M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 72.Korb O., Stützle T., Exner T.E. Empirical scoring functions for advanced Protein-Ligand docking with PLANTS. J. Chem. Inf. Model. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 73.Forli S., Huey R., Pique M.E., Sanner M.F., Goodsell D.S., Olson A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berthold M.R., Cebron N., Dill F., Gabriel T.R., Kötter T., Meinl T., Ohl P., Sieb C., Thiel K., Wiswedel B. Studies in Classification, Data Analysis, and Knowledge Organization. Springer; Berlin/Heidelberg, Germany: 2007. KNIME: The Konstanz Information Miner; pp. 319–326. [Google Scholar]
- 75.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 76.Marvin 17.8 ChemAxon. [(accessed on 2 May 2020)]; Available online: https://www.chemaxon.com.
- 77.OpenEye Scientific Software, Inc. Santa Fe. [(accessed on 2 May 2020)]; Available online: https://www.eyesopen.com/
- 78.Garcia-Perez C., Pelaez R., Theron R., Luis Lopez-Perez J. JADOPPT: Java based AutoDock preparing and processing tool. Bioinformatics. 2017;33:583–585. doi: 10.1093/bioinformatics/btw677. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.