

Abstract
Background:
The clinical and biological significance of the newly described small cell lung cancer (SCLC) subtypes, SCLC-A, SCLC-N, SCLC-Y and SCLC-P, defined respectively by the dominant expression of transcription factors ASCL1, NeuroD1, YAP1 or POU2F3, remain to be established.
Methods:
We generated new RNA-Seq expression data from a discovery set of 59 archival tumor samples of neuroendocrine tumors and new protein expression data by immunohistochemistry in 99 SCLC cases. We validated the findings from this discovery set in two independent validation sets consisting of RNA-Seq data generated from 51 SCLC cell lines and 81 primary human SCLC samples.
Results:
We successfully classified 71.8% of SCLC and 18.5% of carcinoid cases in our discovery set into one of the four SCLC subtypes. Gene Set Enrichment Analysis (GSEA) for differentially expressed genes between SCLC survival outliers (top and bottom decile) matched for clinically relevant prognostic factors, showed significant upregulation of IFN-γ response genes in long-term survivors. SCLC-Y subtype was associated with high expression of IFN-γ response genes, highest weighted score on a validated 18-gene T-cell inflamed gene expression profile score as well as high expression of HLA and T-cell receptor genes. YAP1 protein expression was more prevalent and more intensely expressed in limited stage versus extensive stage SCLC (30.6% vs. 8.5%; p=0.0058) indicating good prognosis for the SCLC-Y subtype. We replicated the inflamed phenotype of SCLC-Y in the two independent validation datasets from SCLC cell lines and tumor samples.
Conclusion:
SCLC subtyping using transcriptional signaling hold clinical relevance with the inflamed phenotype associated with SCLC-Y subset.
Keywords: SCLC, subtype, inflamed phenotype, biomarker
Introduction
Outcomes for small cell lung cancer (SCLC) remains poor, in part because of limited understanding of both the biology of the disease and the key determinants of response to treatment. Recent advances in the treatment of this disease have come from the use of immune checkpoint blockade (ICB) agents in the relapsed and frontline settings. However, the benefit of this new treatment paradigm is limited to a small subset of patients.1–4 Chemotherapy remains the key backbone of treating this disease with more than 60% of extensive stage SCLC patients responding to frontline cytotoxic chemotherapy, yet 5-year survival remains under 10%. Moreover, approximately 30% of patients treated with cytotoxic chemotherapy still progress and die within a few months of treatment. The biological underpinning of the widely different outcomes for early and late survivors despite their treatment with the same platinum doublet chemotherapy has not been well explored.
Several recent studies of the genomics of SCLC have elucidated a consistent pattern of widespread loss of tumor suppressor genes and transcriptional factor deregulation but very rare presence of mutations in oncogenic drivers of tumor growth.5,6 While these findings have not yielded targets for therapy, they have informed an emerging consensus in subtyping SCLC.7 Thus, four distinct subtypes of SCLC defined by the predominant transcriptional regulatory mechanism operating in the cancer cells have now been described based on the expression of achaete-scute homologue 1 (ASCL1), neurogenic differentiation factor 1 (NeuroD1), yes-associated protein 1 (YAP1) and POU class 2 homeobox 3 (POU2F3).7 The data that informed this subtyping came from studies using predominantly immortalized human and murine SCLC cell lines and limited number of primary tumor samples.5–12 The small number of human tissue samples did not allow for detailed correlation with patient characteristics and outcome.13,14 These limitations represent major knowledge gaps that can be bridged through clinical translational research studies to properly validate the proposed SCLC subtypes and make this concept applicable for clinical use.
We, therefore, designed this translational study to systematically elucidate transcriptomic differences between the four SCLC subtypes using archival tumor samples of carcinoid and SCLC. We also sought to establish if there is any association between SCLC subtypes and short and long survival outliers matched for prognostic factors and treatment received as well as identify transcriptomic differences between these survival outliers. We planned to use an initial set of tumor samples as our discovery cohort followed by confirmation of promising findings in independent validation sets.
Methods
Patient election and archival tumor sample collection
This study was conducted under a human subject study protocol approved by Emory University IRB under protocol number IRB00076758. All cases were reviewed by a single pathologist to confirm the original diagnosis of SCLC or carcinoid and sufficient tumor sample of at least 20% enrichment in the resection or core biopsy samples. Patient demographic and clinical information including date of diagnosis, treatment received, vital status and date of death were retrieved from the electronic medical records. When not recorded in the medical records, vital status was established using the social security death index.
RNA-sequencing
RNA was extracted with the Omega Mag-Bind FFPE RNA extraction kit (Omega Bio-Tek, Catalog M2551), according to the manufacturer’s protocol. RNAseq libraries were prepared by Omega Bioservices.
RNA library preparation and sequencing:
RNA sequencing libraries were prepared using Illumina TruSeq RNA Exome kit (formerly TruSeq RNA Access library kit) (Illumina, Inc., San Diego, CA, USA) according to the manufacturer`s protocol. The RNA concentration was measured with Nanodrop 2000c spectrophotometer (Thermo Scientific Inc., Waltham, MA, USA). Integrity was assessed using Agilent 2200 Tapestation instrument (Agilent Technologies, Santa Clara, CA, USA) and the percentages of fragments larger than 200 nucleotides (DV200) were calculated. 20~100ng RNA samples were used as input based on the DV200 value. First-strand cDNA syntheses were performed at 25°C for 10 minutes, 42°C for 15 minutes and 70°C for 15 minutes, using random hexamers and ProtoScript II Reverse Transcriptase (New England BioLabs Inc.). In a second strand cDNA synthesis the RNA templates were removed and a second replacement strand was generated by incorporation dUTP (in place of dTTP, to keep strand information) to generate ds cDNA. The blunt-ended cDNA was cleaned up from the second strand reaction mix with beads. The 3’ends of the cDNA were then adenylated and followed by the ligation of indexing adaptors. PCR (15 cycles of 98°C for 10 seconds, 60°C for 30 seconds and 72°C for 30 seconds) was used to selectively enrich those DNA fragments that have adapter molecules on both ends and to amplify the amount of DNA in the library.
The library was qualified using an Agilent 2200 Tapestation instrument and quantified using the QuantiFluor dsDNA System (Promega). A 4-plex pool of libraries was made by combining 200 ng of each DNA library. The pooled DNA libraries were then mixed with capture probes to target regions of interest. Hybridization was performed by 18 cycles of 1-minute incubation, starting at 94°C, and then decreasing 2°C per cycle. Streptavidin-coated magnetic beads were then used to capture probes hybridized to the target regions. The enriched libraries were then eluted from the beads and prepared for a second round of hybridization and capture to ensure the high specificity of capture regions. The enriched libraries were amplified by a second 10 cycles of PCR amplification(98°C for 10 seconds, 60°C for 30 seconds and 72°C for 30 seconds) followed by clean-up of beads. The final libraries were validated using Agilent High Sensitivity D1000 ScreenTape on an Agilent 2200 Tapestation instrument. The size distribution of the library ranges from approximately 200 bp–1 kb. Libraries were normalized, pooled and subjected to cluster and pair read sequencing was performed for 150 cycles on a HiSeqX10 instrument (Illumina, Inc. San Diego, CA, USA), according to the manufacturer’s instructions.
Immunohistochemistry
We assessed YAP1 protein expression by immunohistochemistry as previously described in a blinded fashion using archival tumor samples (resection and core biopsy samples) of 96 neuroendocrine tumors including cases of 13 cases of carcinoid, 36 cases of LS-SCLC and 47 cases of ES-SCLC. We employed a mouse anti-YAP1 monoclonal antibody (targeting amino acids 53–162 of Human YAP1; ABCAM cat# 56701; Abcam Inc. Cambridge, MA 02139-1517 USA) at a 1:320 dilution as primary antibody. Staining, antigen retrieval and detection were performed on a DAKO autostainer using the EnVision FLEX, High pH (Link) kit for mouse (Agilent DAKO part number K80021) per manufacturers direction. Further details regarding the immunohistochemistry procedure, assessment and quantitation of protein expression have been previously described.15
Bioinformatics data analysis and workflow
Data processing, normalization & transformation:
FASTQ files were trimmed for adaptor sequences and low quality using Trim Galore and FASTQC. Data were mapped to the GRCh38 human reference genome using STAR aligner (v. 2.5.3a) and annotated with Gencode v29 transcript database. Raw gene counts were estimated using the summarizeOverlaps function of the GenomicAlignments R package (v. 1.14.2). Genes with no expression and/or low expression were filtered out. Genes were filtered with (CPM<0.1) in at least 25 samples prior to normalization. Expression data was normalized based on the negative binomial distribution with the DESeq2 package. Normalized data was log2 plus one transformed and used as input in all downstream analyses. Differential expression analysis was done with DESeq2 while clustering analysis was performed using ConsensusClusterPlus and heatmap.3 statistical computing packages. Heatmaps were generated using row-scaled data with Pearson correlation distance and ward.D clustering method.
Gene Set Enrichment Analysis (GSEA) was employed to identify enriched pathways between the late vs. early survival outliers. GSEAPreranked was performed using modified ranking statistics. Ranked list of genes was generated from log10(p-value)*direction of fold change of all the tested and differentially expressed genes between the late vs. early survival outliers. The C2.all.v7.0 curated gene sets (from MSigDB; 5501 gene sets) in GSEA version 3.0 with classic enrichment statistic was used for this analysis. For duplicate row identifiers in the input ranked list, one id was arbitrarily chosen. The top 20 enriched up and down-regulated pathways were visualized as Dotplot to see the list of top 20 pathways by their significance/statistics and by UpsetPlot to see the overlap among the top 20 pathway gene members.
Unsupervised analysis of expression data was performed using the top 1000 most variable genes selected using median absolute deviation approach among the 33 SCLC samples (15 from matched survival outliers and 18 additional samples from other patients outside of the matched outliers), carcinoid samples and the whole sample together as appropriate to the research question. Unsupervised PAM-based consensus cluster analysis (1000 reps, 90% sampling, and maximum k=10) was performed with Pearson correlation and average linkage method. The optimal number of clusters were detected and assessed based on different measures. Supervised analysis of the RNA-expression was conducted between defined subgroups including SCLC subtypes and patients in the outlier group i.e. in the top and bottom decile of the survival curve matched for age, gender and treatment received).
SCLC Subtyping:
We used the shinySISPA technique (http://bbisr.shinyapps.winship.emory.edu/shinySISPA/) to classify the 59 cases of carcinoid and SCLC based on the expression profile for ASCL1, NeuroD1, YAP1 and POU2F3 genes.
T-cell Inflamed GEP:
Expression score for each sample (SCLC and carcinoid) was calculated as weighted sum of the normalized expression values (log2(count+1)) of the 18 genes (PSMB10, HLA-DQA1, HLA-DRB1, CMKLR1, HLA-E, NKG7, CD8A, CCL5, CXCL9, CD27, CXCR6, IDO1, STAT1, TIGIT, LAG3, CD274, PDCD1LG2, CD276) as originally described and validated as a predictor of clinical efficacy in patients treated with anti PD-1 immunotherapy drug, pembrolizumab.16,17 The weightings for each gene in the signature was obtained from (claim #21c) published in the patent filed under “WO201609437” https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016094377&tab=PCTCLAIMS.
Cancer-testis antigen (CTA):
We interrogated all genes designated as CTAs in the CTdatabase (http://www.cta.lncc.br). Out of 276 genes listed on the database, 70 genes were excluded from the analysis, because they were not mappable (by Gene symbol) to the Ensemble annotation, duplicate entries or were expressed at very low levels or not all. We therefore analyzed the expression profile for 206 CTAs across 59 samples.
Bioinformatic analysis of publicly available expression data as validation dataset
We validated our findings in cell line and primary tumor RNA-Seq expression datasets. The human SCLC cell lines (n=51) count data (version dated September 29 2019) was from CCLE (https://portals.broadinstitute.org/ccle) normalized by TMM and log2 transformed, and the human primary SCLC (n=81) FPKM data was from George et al., 2015.6 The data was used for SCLC subtyping, GEP scores and for clustering by subtypes, IFN-γ, and CTA genes using workflow as described above.
Biostatistics
Statistical analysis was conducted using SAS Version 9.4. Descriptive statistics for each variable were reported. Chi-square test and t-test were performed for univariate analysis. Kaplan-Meier Survival curve and Log-rank test were used to explore association of each covariate with OS or PFS. A p-value <0.05 was considered significant without correction for multiple comparisons given the preliminary discovery stage of this work. Cox Proportional hazards model was performed to detect the association among covariates with OS using a backward variable selection method with an alpha of 0.20 as removal criterion. In order to select matching extreme outliers, we employed three prognostic variables (age, gender, and treatment with first line chemotherapy) to match patients from an institutional database of 579 patients limiting to 271 patients with non-missing values on the three variables. Age was matched on an interval (two-year difference). First 70 and last 140 subjects were treated as top OS and bottom OS, respectively in other to find 54 pairs of patients matched on the 3 variables. An additional 46 pairs of patients were matched from the remainder of the non-outlier population.
Results
Patient and sample selection:
We interrogated our institutional database of SCLC patients under an IRB-approved protocol to identify SCLC patients who were survival outliers, defined as patients in the top and bottom deciles of overall survival. Outliers in the top and bottom decile were matched for known clinical prognostic factors, including, age, gender, stage and treatment received in the frontline setting. Subsequently, archival tumor samples from the matched outlier patients were retrieved for gene expression profiling using an RNA-Seq platform. We included additional non-matched patients with intermediate survival between the outlier groups for internal validation of any observed differences between the outlier groups. As an additional control, we also included archival tumor samples from 27 consecutive patients with confirmed diagnosis of pulmonary carcinoid who underwent surgical resection of their tumor at our center. Available tumor samples from a total of 59 patients with pulmonary neuroendocrine tumors (27 carcinoid and 34 SCLC) were employed for this study. The median age was 64.82 years with gender breakdown of 54.2%/45.8% for females and males. Additional breakdown by subtypes of carcinoid versus SCLC is detailed in Supplementary Table S1.
SCLC subtypes identified using expression phenotype for key transcription regulators
Supervised analysis of 59 samples of neuroendocrine tumors based on the expression of transcription factors ASCL1, NeuroD1, YAP1 and POU2F3 showed that the majority of SCLC cases clustered together under one of the four subtypes ASCL1 (SCLC-A), NeuroD1 (SCLC-N), YAP1 (SCLC-Y) and POU2F3 (SCLC-P) whereas most of the cases of carcinoid clustered together but separate from the SCLC clusters (Figure 1a). Of the 59 cases, ten were classified as SCLC-A (16.9%), three cases as SCLC-N (5.1%), six cases as SCLC-Y (10.2%), four cases classified as SCLC-P (6.8%) and 14 cases of SCLC failed to cluster with any of the four subtypes. The vast majority of carcinoid cases clustered together but not into any of the transcriptionally defined SCLC subtypes (Figure 1a). Only five of the 27 cases of carcinoid clustered under a SCLC subtype, four as SCLC-Y (14.8%) and a single case clustered with the SCLC-N subtype (3.7%). These cases were further reviewed, and the original diagnosis of typical carcinoid was confirmed with no necrosis and <2 mitosis/2mm2. The five cases of atypical carcinoid clustered along with the bulk of cases of typical carcinoid in the unassigned group that did not fall into any of the four transcriptionally-defined SCLC subtypes. Unsupervised analysis of the transcriptomic expression data from the SCLC cases showed three main clusters (Figure 1b), which did not align with the SCLC subtypes (Figure 1c). We then focused on the SCLC extreme outliers (samples from patients in the top and bottom deciles of survival) to identify unique expression profiles that could explain the clinical outcomes in these patients who were otherwise matched for prognostic factors and treatment received. Unsupervised analysis showed no discernible clustering by survival groups but supervised analysis based on outlier subgroups (early death vs. late survivor) showed generally higher gene expression in early death patients (Figure 1d).
Figure 1:
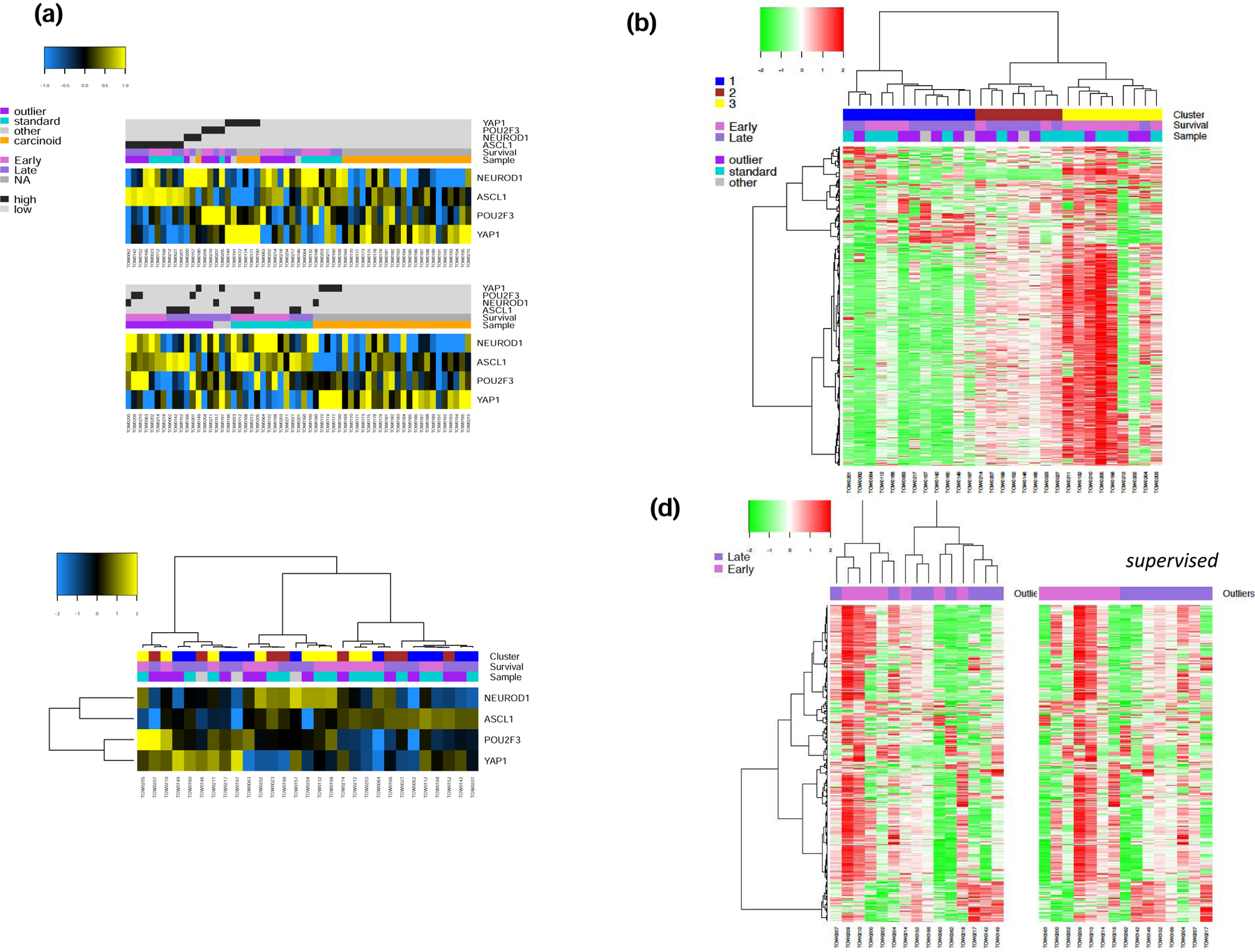
Gene expression of 59 neuroendocrine pulmonary tumors. a). Supervised and unsupervised analyses of 33 SCLC and 26 carcinoid cases displayed as a heatmap in which blue and yellow represent low and high expression, respectively of the four transcription factors, ASCL1, NeuroD1, YAP1 and POU2F3 that define SCLC subtypes. b). Unsupervised analysis limited to SCLC with cases arranged in columns revealed three main clusters based on their expression profiles in which green and red represent low and high expression, respectively; samples were annotated for survival (matched survival outliers selected from the bottom decile, top decile and the intermediate groups on the survival curve). c). Unsupervised analysis limited to SCLC with cases arranged in columns based on their expression profiles for the four transcription factors in which blue and yellow represent low and high expression, respectively; samples were annotated for the clusters defined in B; there was no correlation between the clusters defined in B and SCLC subtypes. d). Unsupervised and supervised analysis of expression data in SCLC limited to matched survival outliers showed generally increased gene expression in patients with early death.
Interferon gamma pathways significantly upregulated in long-term SCLC survivors
Heatmap showing differentially expressed genes between the short and long survival outliers (Figure 2a). Gene Set Enrichment Analysis looking for differentially activated pathways in tumor tissue samples between Early and Late SCLC survivors showed that one of the significantly upregulated pathways was interferon pathways (Figure 2b). Specifically, IFN-γ response genes (DER_IFN_gamma_UP: ADAR, ATP6V0B, B2M, BAG1, BAK1, BBC3, BST2, BTG1, C1S, CASP8, CEBPD, COL16A1, CSRP3, CYCS, EIF2B1, ELK4, EPS15, FAS, FOSL1, GBP1, HADH, HADHB, HLA-A, HLA-C, HLA-E, ICAM1, IFI16) were upregulated (Figure 2b). Contrarily, the top 20 downregulated pathways showed the most downregulated pathway to be the WEBER_METHYLATED_HCP_IN_FIBROBLAST_UP. This pattern of inactive unmethylated CpG island promoters is associated with elevated levels of dimethylation of Lys4 of histone H3, suggesting some protection of the DNA from methylation (Figure 2c).
Figure 2:
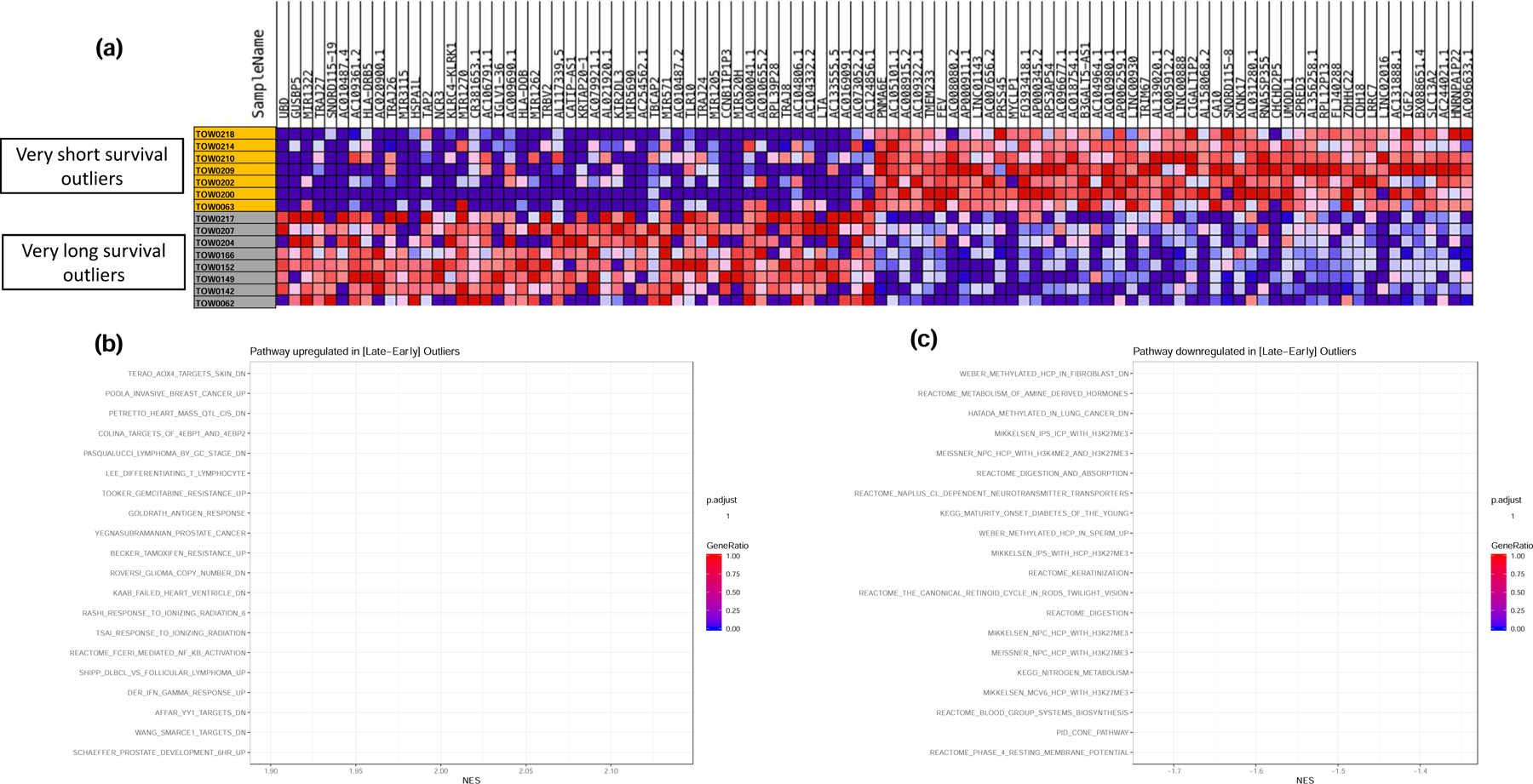
GSEA for differentially regulated pathways between matched survival outliers (i.e. patients within the top and bottom deciles of the OS curves matched for key prognostic factors of gender, age and treatment). a). heatmap of differentially expressed genes in tumor samples from the matched outliers. b). Top 20 up-regulated pathways in very long survival outliers over very short survival outliers. Note that IFN-γ response genes were significantly upregulated in late survivors. c). Top 20 down-regulated pathways in very long survival outliers over very short survival outliers.
T-cell inflamed gene expression profile is enriched in late survivors and SCLC-Y subtype of SCLC
The results of the GSEA showing upregulation of interferon response pathway genes in late survivors led us to assess whether a previously reported and validated 18-gene T-cell inflamed gene expression profile (GEP) signature,17 which was associated with benefit of ICB in SCLC and other tumor types,16 is differentially expressed between SCLC outliers i.e. early and late survivors, between SCLC subtypes and between SCLC and carcinoid tumors. In unsupervised analysis, there was higher expression of this validated 18-gene panel in carcinoid versus SCLC cases and an enrichment for late survivors (Figure 3a). Supervised analysis based on SCLC subtypes also showed the highest expression in the SCLC-Y subtype (Figure 3b). In a final step, we employed the weighted sum of the normalized expression values of the 18 genes for each sample using the weightings described in the original derivation of the signature for pembrolizumab studies.16 Consistent with the results obtained with supervised and unsupervised analysis using the unweighted expression score, the 18-gene GEP signature was highest in the extreme outliers who were late SCLC survivors compared to outliers who suffered early death (Figure 3c). Similar comparison within the intermediate SCLC subgroup showed association of longer survival and higher GEP score (Figure 3c). Comparison by SCLC subtypes showed the highest GEP score recorded in the SCLC-Y subtype followed by the POU2F3 subtype whereas the SCLC-N and SCLC-A subtypes had the lowest GEP score (Figure 3d). The indeterminate category, which comprised 81% of carcinoid cases and 32% of SCLC, also showed an intermediate GEP score between the low (N and A) and high (P and Y) GEP scoring SCLC subtypes (Figure 3d).
Figure 3:
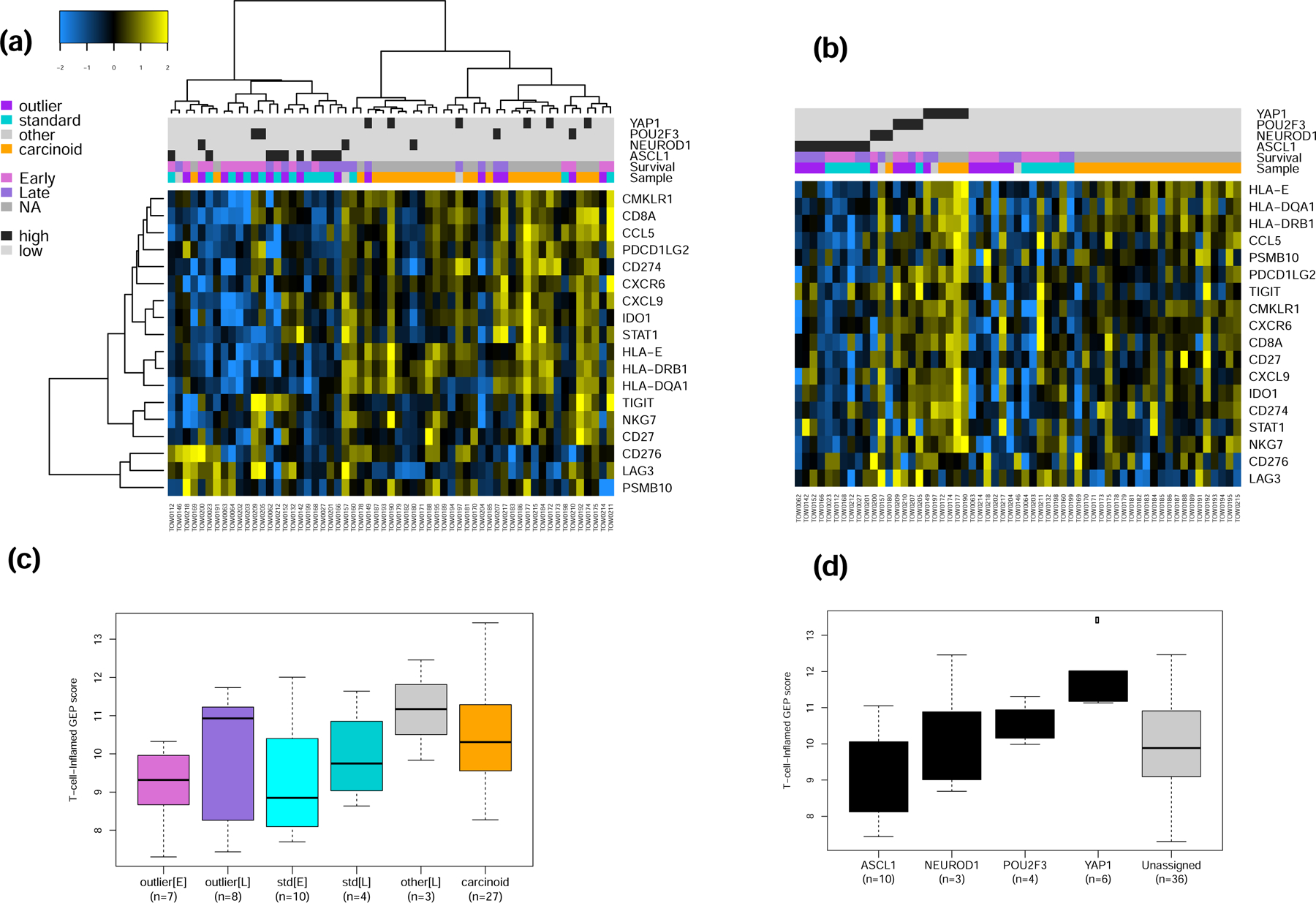
a&b) Unsupervised and supervised analysis of the expression of 18 IFN-γ related genes that were previously validated as a pan-tumor T cell-inflamed gene-expression profile (GEP) to predict clinical efficacy of ICB. The normalized expression of this 18-gene panel was relatively higher in carcinoid tumors compared to SCLC but the highest level of expression overall was noted in the SCLC-Y subtype. c&d). Weighted score of the T-cell-inflamed GEP in SCLC showed a higher score in samples from late survivors compared to patient who suffered early deaths while SCLC-Y had the highest GEP score of all the SCLC subtypes.
SCLC-Y subtype and long-term outliers show high expression of HLA gene family and low expression of cancer testis antigens
Based on the finding that the SCLC-Y subtype enriches for T-cell inflamed GEP and long-term survival in our dataset, we sought to assess whether this subtype is also characterized by other phenotypes that would suggest potent antitumor immunity. We therefore analyzed the expression profile for 28 HLA family genes. In an initial unsupervised analysis of HLA gene expression, the 59 cases of SCLC and carcinoid clustered into 4 major subgroups (Figure 4a). The cluster with the highest expression of HLA genes was enriched for long-term SCLC survivors; (6 of 18 (33%) cases that defined the cluster. Moreover, all the SCLC cases (100%) included in this cluster were late survivors. There was also an enrichment for YAP1 expressing tumors (4 carcinoid and 2 SCLC cases) in the high HLA gene expression cluster. YAP1 expression was previously reported to correlate with the morphology and differentiation of SCLC whereby YAP1 expressing cell lines were more adherent and well differentiated. Knockdown of YAP1 in these cell lines led to dedifferentiation and morphological transformation into floating cells.18 Cancer testis antigens (CTA) are protein groups that are expressed in normal embryonic cells but repressed in normal adult cells. These molecules are implicated in the regulation of diverse cellular processes during cell development and differentiation and carcinogenesis, though the biological roles and cell functions of CTA families remain largely unclear. Aberrant CTA expression patterns may be associated with cancer transformation and failure of the developmental program of cell lineage specification and germ line restriction.19 The expression of specific CTAs in resected non-small cell lung cancer was associated with adverse prognostic factors such as higher stage of disease and lymph node involvement as well as poor overall survival.20 We therefore hypothesized that the SCLC-Y subtype will be less likely to express CTA. Unsupervised analysis of a family of 206 CTA genes showed three main clusters with high, intermediate and low expression but without any discernible correlation with SCLC subtypes, although the carcinoid tumors generally had the lowest expression levels (Figure 4c). In supervised analysis of SCLC cases, we observed that SCLC-Y had the lowest expression of this family of genes while SCLC-P had the highest expression and SCLC-A and SCLC-N had intermediate level of expression (Figure 4d). Analysis of T cell receptor genes (Tcra, Tcrb, Tcrg, and Tcrd) expression showed lowest expression in carcinoid and the highest expression in SCLC-Y subtype suggesting increased immune cell infiltrate in this subset of SCLC (Figures 4e and 4f).
Figure 4:
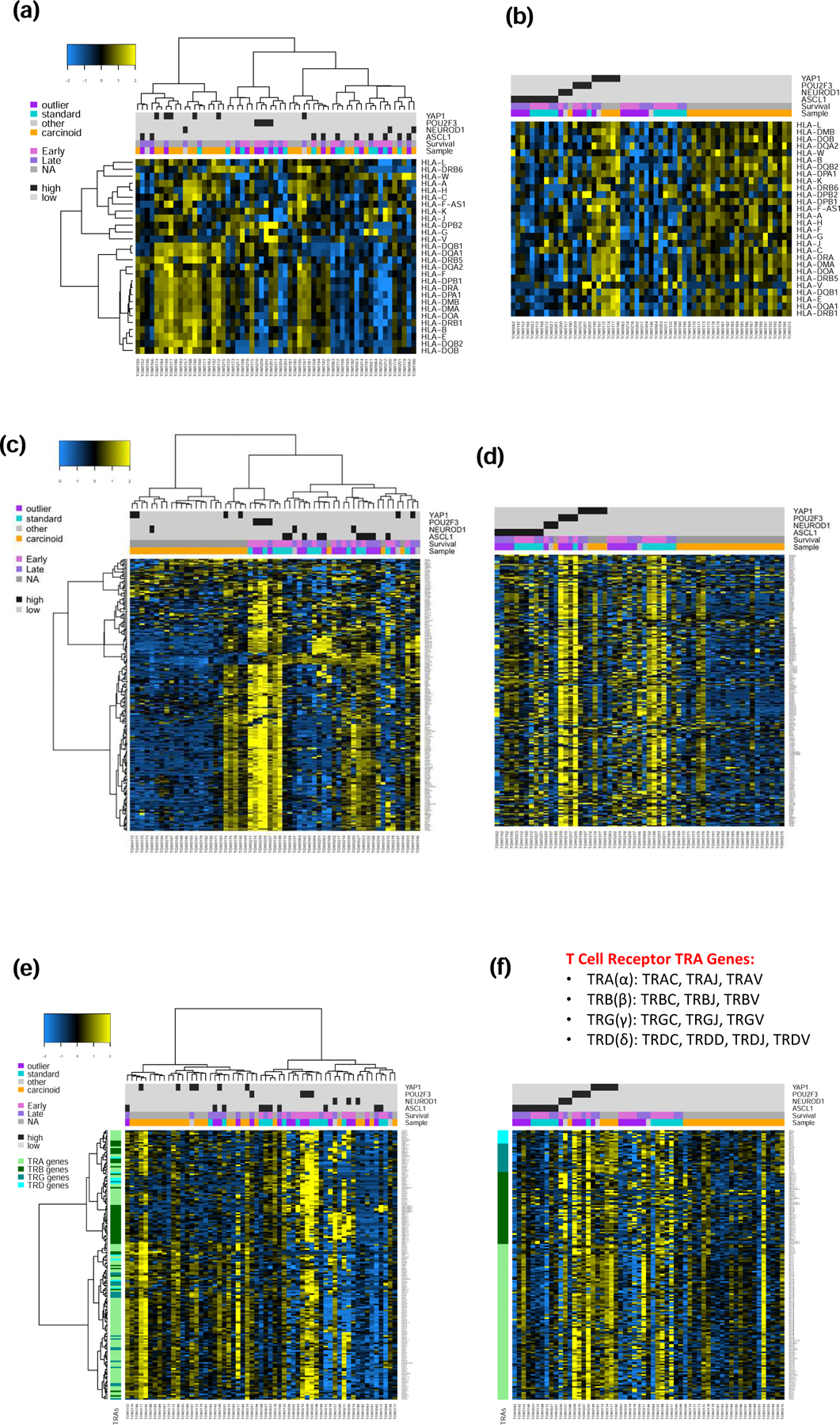
Unsupervised and supervised analyses of expression profile in SCLC and carcinoid samples displayed as a heatmap in which blue and yellow represent low and high expression, respectively. Samples were annotated for histology, survival (outliers including early survival i.e. bottom decile of OS and late survival i.e. top decile of OS and the intermediate group), and SCLC subtypes. a&b): Expression of HLA genes showed no clear correlation with survival but the highest expression was observed in SCLC-Y and carcinoid tumors. c&d). Contrarily, the expression of a panel of 206 genes that encode for cancer testis antigens, a type of oncofetal protein, showed low expression in SCLC-Y and carcinoid but high expression in SCLC-P and a subset of indeterminate SCLC patients who suffered early death. e&f). Unsupervised analysis showed intense expression of Tcrb gene in a subset made up predominantly of SCLC-N and SCLC-P subtypes; on supervised analyses, the highest expression for the four T cell receptor genes (Tcra, Tcrb, Tcrg, and Tcrd) was observed in the SCLC-Y and SCLC-P subtypes.
SCLC-Y subtype and YAP1 protein expression associated with better prognosis in SCLC
We employed the clinical outcome dataset retrieved from the patients’ electronic medical records to interrogate the prognostic significance of the transcriptionally defined subtypes of SCLC. There was no statistically significant difference in PFS or OS between the four subtypes in this small cohort of patients. However, a consistent trend was noted towards a better outcome for SCLC-Y subtype both for OS and PFS: Median (95%CI) OS of 14 (4.3, 28.8), 16.7 (0.9, NA), 8.1 (2, 9.7) and 20.1 (0.6, 39.5) months and median (95%CI) PFS of 7.9 (5.9, 33.8), 7.1 (1.2, 12.7), 7.8 (4.2, NA), and 15.1 (NA, NA) months for SCLC-A, N, P and Y subtypes respectively (Supplementary Figure S1). We employed 96 samples of neuroendocrine tumors (SCLC and carcinoid) to assess YAP1 protein expression by immunohistochemistry. There was no YAP1 protein expression in carcinoid cases similar to the gene expression data (Figure 1). However, the intensity and frequency of YAP1 expression was higher in limited stage SCLC (LS-SCLC) compared to extensive stage (ES-SCLC) with a 30.6% versus 8.5% positivity rate; p=0.0058 and mean immunoscore of 15.5 versus 0.6; p=0.03 (Figure S1).
External validation in SCLC cell lines
Given the limited sample size in our discovery group, we sought to validate our findings in additional sample using publicly available expression datasets derived from SCLC cell lines. We analyzed RNA-Seq expression datasets from 51 human SCLC cell lines available as part of the Broad Institute Cancer Cell Line Encyclopedia (CCLE; version dated September 29 2019) available at https://portals.broadinstitute.org/ccle. We successfully classified these cell lines into the four transcriptionally defined subsets (Figure 5a and Supplementary Table S2). Supervised analysis showed that cell lines that fall into the SCLC-Y subtype had the highest expression of IFN-γ gene signature (Figure 5b). The weighted normalized expression values for these genes was also highest in SCLC-Y compared to the other subtypes (Figure 5c) consistent with the observation in the discovery set. Similarly, SCLC cell lines that fall into the SCLC-Y subtype had the highest expression of HLA genes (Figure 5d). There was a very low expression of the 206 CTA genes (Figure 5e) and negligible expression of the four T cell receptor genes across the entire dataset of 51 SCLC cell lines (Figure 5f). Additional results from the unsupervised analyses of the expression data are provided as supplementary data (Supplementary Figure S2).
Figure 5:
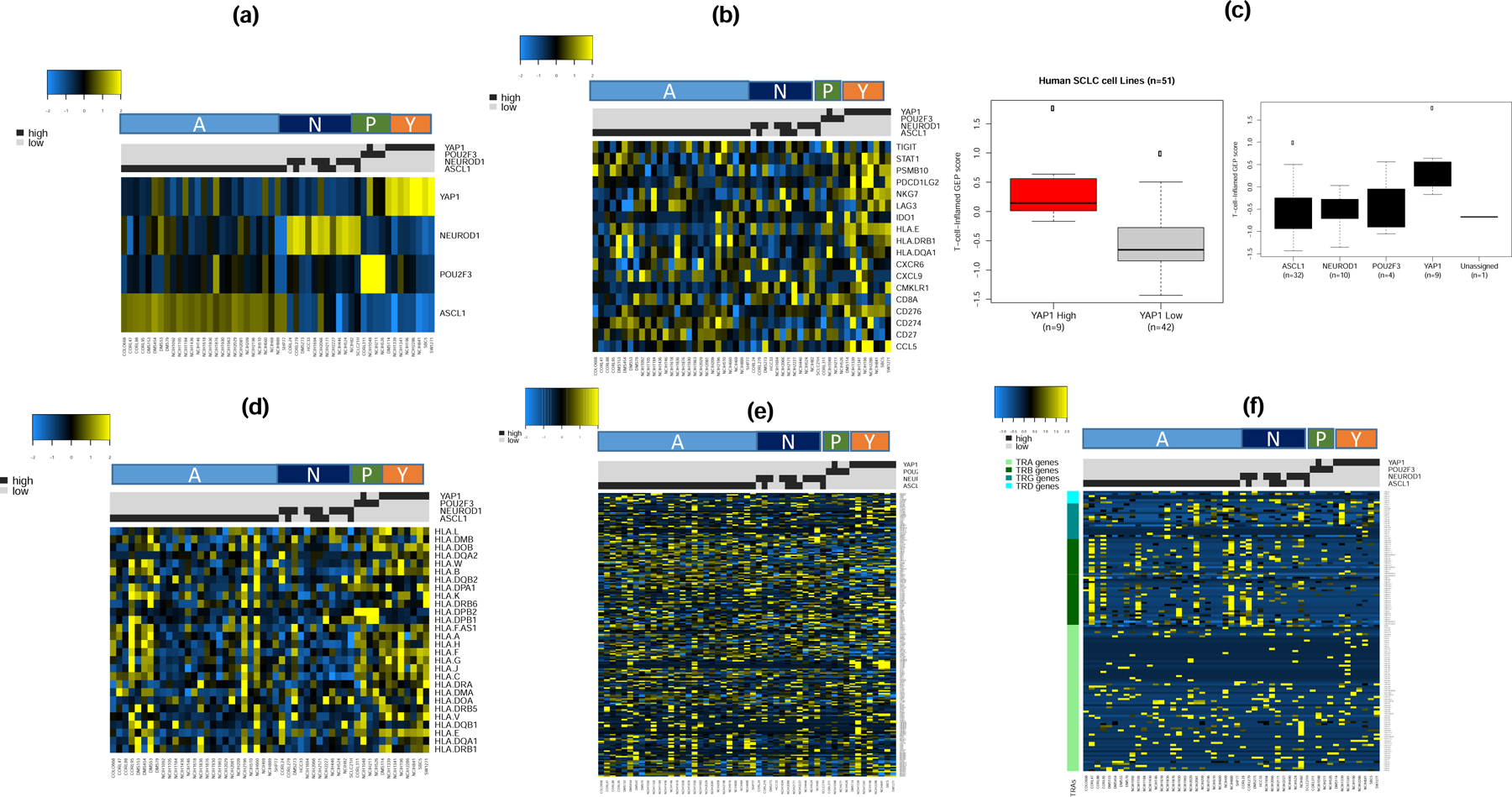
Supervised analysis of expression dataset from 51 SCLC cell lines from CCLE by SCLC subtypes (A, N, P and Y) as a validation step. a). The cell lines were classified into one of four subtypes with the vast majority being of the SCLC-A subtype; note that few cases showed concurrent expression of ASCL1 and NeuroD1; b). The highest expression of 18 IFN-γ related genes cells was observed in the SCLC-Y subtype; c). T-cell-inflamed GEP score was higher in YAP-1 expressing cell lines (left panel) and overall trend of expression consistent with the discovery dataset with highest score in SCLC-Y followed by SCLC-P while SCLC-A and SCLC-N had lowest GEP score (right panel); d). HLA gene expression analysis showed highest expression in SCLC-Y and SCLC-P subtypes, lowest expression in SCLC-N and a variable expression in the SCLC-A subtype; e&f). Supervised analysis by SCLC subtypes showed no differential expression patterns for CTA and T-cell receptor genes.
External validation in primary human tumor samples
Additional validation of the results from the discovery dataset was obtained by analyzing publicly available normalized expression data from 81 human primary SCLC cases.6 The cases were successfully classified into the four subtypes and consistent with the results from the discovery datasets, majority of cases were of the SCLC-A and SCLC-N subtypes while a smaller subset were classified as SCLC-Y and SCLC-P (Figure 6a). We replicated the observation from the discovery dataset of highest expression of IFN-γ 18-gene signature in the SCLC-Y subtype (Figure 6b) as well as the highest weighted expression score in SCLC-Y compared to the other subtypes (Figure 6c and 6d). Consistent with the results from the discovery cohort, analysis of HLA genes expression also showed highest levels in the SCLC-Y subtype (Figure 6e) but no discernible differential expression pattern was noted for the 206 CTA genes (Figure 6f). We were unable to validate the differential T-cell receptor expression demonstrated in the discovery dataset because the publicly available dataset did not contain expression data for T cell receptor genes. Additional results from the unsupervised analyses of the expression data are provided as supplementary data (Supplementary Figure S3).
Figure 6:
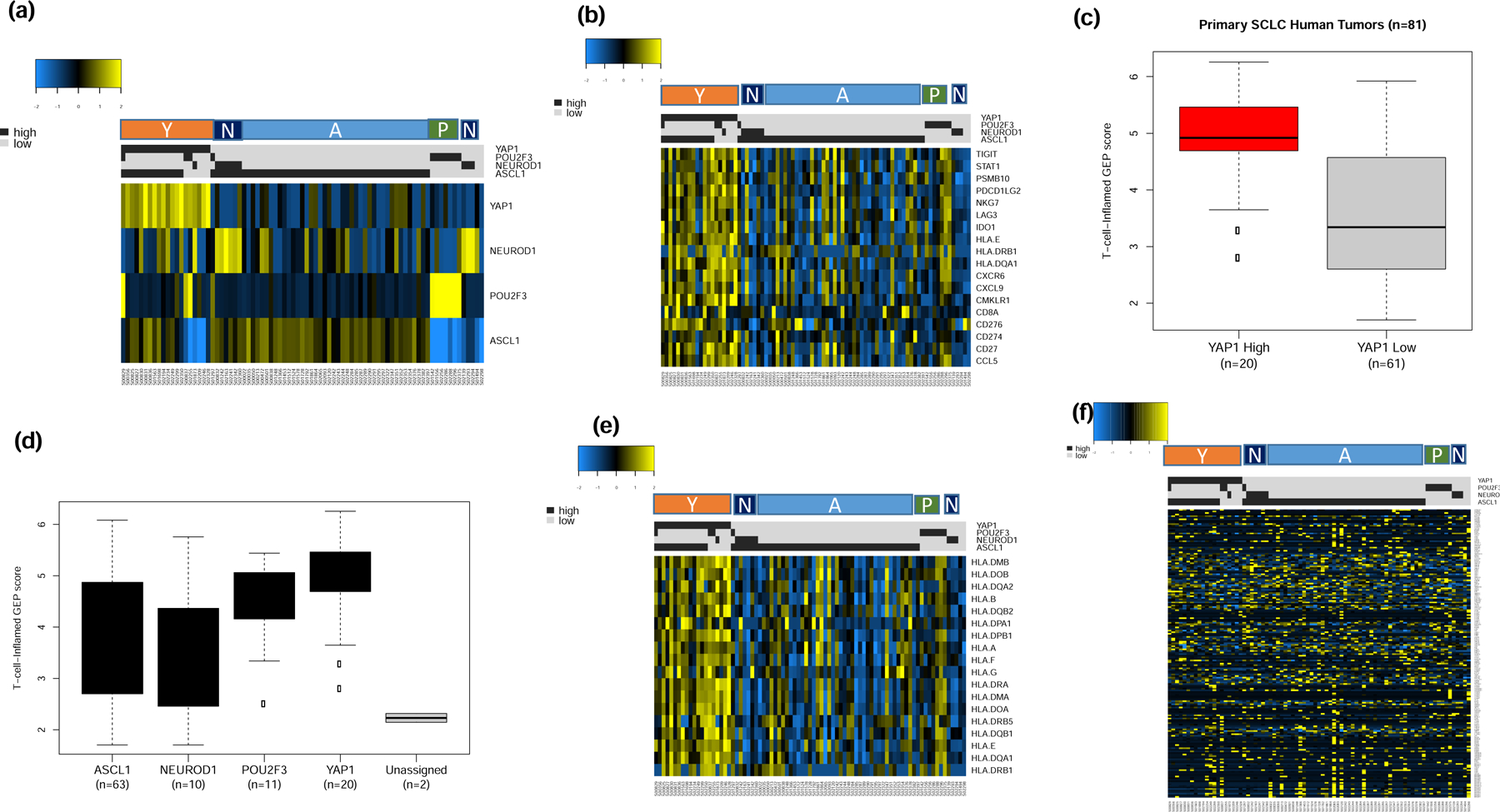
Supervised analysis of publicly available expression dataset from 81 primary tumor samples by SCLC subtypes (A, N, P and Y) as a validation step. a). The tumor samples were classified into one of four subtypes with the vast majority being of the SCLC-A subtype; cases with concomitant expression of two lineage markers were classified based on the marker with dominant expression; b). The highest expression of 18 IFN-γ related genes cells was observed in the SCLC-Y subtype; c). T-cell-inflamed GEP score was higher in YAP-1 expressing tumors; note that cases with concurrent expression of YAP-1 and ASCL-1 were classified as YAP1 positive due to the extreme dominance of YAP1 expression in these tumors; d). overall trend of T-cell-inflamed GEP score was consistent with the findings in the discovery dataset and cell line datasets whereby SCLC-Y had the highest score followed by SCLC-P while SCLC-A and SCLC-N had the lowest GEP score (right panel); e). HLA gene expression analysis with the highest expression in SCLC-Y followed by SCLC-P subtypes and negligible expression in SCLC-N and SCLC-A subtypes similar to the result from the initial analysis in the discovery dataset; f). Supervised analysis showed no differential expression patterns for CTA genes between the four SCLC subtypes.
Discussion
New treatment options for SCLC are now emerging after a long period of stagnation.1–4 Additionally, elucidation of SCLC biology using newer genomic testing platforms has informed the proposed subtyping of this disease into four subtypes.7 Our work represents an initial attempt to establish clinical relevance for this new classification of SCLC. We successfully classified the majority of SCLC cases in our study into one of the four proposed subtypes of SCLC-N, SCLC-A, SCLC-Y and SCLC-P. However, 32% of the SCLC cases did not fall into any of these four subtypes. Importantly, 81% of typical carcinoid tumors interrogated as part of this study also failed to match any of the SCLC phenotypes. This provides further validation that the described subtypes represent unique biology of SCLC that is probably not relevant for carcinoid, a well-differentiated variant of neuroendocrine tumors.
Outlier analysis based on clinical and genomic data as employed in this study is an unbiased approach that can be used to elucidate unique biologic drivers of outcome and enable the discovery of tumor vulnerabilities to guide the development of novel targeted therapies.21 We systematically identified exceptional responders and non-responders (outliers) from discovery samples collected from our institutional patient clinical database. Without any a priori knowledge of likely genomic differences between these subgroups, we successfully uncovered differential upregulation of IFN-γ response genes in exceptional responders who survived long term. This finding is biologically plausible and consistent with observations in other tumors where active adaptive antitumor immunity was associated with better clinical outcome.22–24 Further support for this finding came from our analysis of the 18-gene T cell-inflamed GEP signature, which also showed higher expression in long-term survivors. The T cell-inflamed GEP includes IFN-γ-responsive genes that mediate antigen presentation, chemokine expression, cytotoxic activity, and adaptive immune resistance.17 This signature was previously further validated in a tumor agnostic manner using 9000 tumor samples from the TCGA database and high expression was noted in tumor types known to be vulnerable to ICB.25 It has also been validated as a predictor of clinical efficacy of ICB with pembrolizumab in SCLC and other tumor types.16 Since none of the patients included in this analysis received ICB therapy, the higher levels of the T-cell inflamed GEP score in long term survivors suggest a prognostic value in SCLC. This suggestion is further supported by the trend towards a better survival outcome for the YAP1 subtypes which showed the highest T cell-inflamed GEP score of all subtypes. Furthermore, we noted YAP1 protein expression to be highest in patients with limited stage-SCLC, which is the better prognostic subset of SCLC and the patient subset more likely to manifest paraneoplastic syndromes, which correlates with strong antitumor immunity.
The clinical implication of the various SCLC subtypes remains to be elucidated. This work is an initial attempt to link tumor subtypes to outcome. We noted that the SCLC-Y subtype is characterized by enrichment in long term survivors, high T-cell inflamed GEP score and HLA gene expression as well as low levels of CTA expression. This overall phenotype is consistent with a better differentiated tumor histology, an inflamed tumor microenvironment and likely vulnerability to ICB. We successfully validated most of these findings in two independent datasets. In particular the correlation between SCLC-Y subtype and the inflamed gene signature was reproduced across the three independent samples. This key finding strongly suggest that the SCLC-Y subtype could be for the key subset of SCLC patients who benefit from ICB. However, this hypothesis requires further validation in patients treated with ICB. The SCLC-P subtype also showed moderately high T-cell inflamed GEP score, however, it also showed the highest expression of CTA suggesting a poorly differentiated tumor that is unlikely to respond to ICB alone. Perhaps the combination of cytotoxic chemotherapy and ICB will be more effective in the SCLC-P subtype. The SCLC-N and SCLC-A subtypes constitute the largest proportion of cases in our study similar to the distribution previously reported for the different subtypes. These subtypes appear to be immunologically cold, with low T-cell inflamed GEP score and low HLA gene expression. Interestingly, a recent analysis of large cell neuroendocrine carcinomas (LCNEC) identified two subtypes, I and II. The type I LCNEC harbored bi-allelic alterations in TP53 and STK11/KEAP1 along with high ASCL1 and DLL3 gene expression typical of classic SCLC (subtypes A and N) while the type II LCNEC showed bi-allelic inactivation of TP53 and RB1 genes and reduced expression of neuroendocrine markers similar to the variant non-neuroendocrine SCLC subtypes (SCLC-P and SCLC-Y).26 Similar to our findings, there was an upregulation of immune-related pathway genes and YAP1 in the type II LCNEC.
Our study validated the proposed subtyping of SCLC using human tissue samples along with clinical outcome data. Nonetheless, there are some important limitations of our work, which should be acknowledged. The retrospective nature of the study raises the risk for selection and recall bias in patient and sample selection. Also, the limited number of cases included in our initial discovery cohort and the fact that none of these patients received treatment with ICB are weaknesses to be addressed in follow-up studies that we have planned. However, some of these weaknesses were mitigated by the internal validation steps such as outlier analysis to identify non-overlapping patient subgroups along with comparison with cases of carcinoid. We also successfully validated the key results using two independent external datasets.
In conclusion, this study used human tissue samples and outcome data to provide independent verification for the proposed classification of SCLC using the expression pattern of key transcriptional regulators. Our discovery and validation cohorts demonstrated clinical and biological differences between the four subtypes of SCLC in particular, prognostic differences with respect to patient survival and evidence of adaptive antitumor immunity.
Supplementary Material
Acknowledgement:
We thank Anthea Hammond, PhD for proofreading the manuscript.
Funding for this study was provided by Novartis Oncology under a grant award to TKO (CLDK378AUS39T). Additional support was provided by the DoD grant award to TKO (LC150291); the NIH SPORE grant award to SSR (P50CA217691) and the NIH NCI CTD2 network to HF (U01CA217875). Research reported in this publication was supported in part by the Biostatistics and Bioinformatics Shared Resource and the Tissue Procurement and Pathology Shared Resource of Winship Cancer Institute of Emory University, Atlanta, GA 303022 and NIH/NCI under award number P30CA138292. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures: Paid advisory board participation by BMS, AstraZeneca, Roche, Merck and Novartis to TKO. Research grant funding from Merck, AstraZeneca, BMS to institution (TKO and SSR).
This work was presented as an abstract as part of the poster discussion session at the 2020 Virtual Annual Meeting of the American Society of Clinical Oncology.
References:
- 1.Horn L et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. The New England journal of medicine 379, 2220–2229, doi: 10.1056/NEJMoa1809064 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Paz-Ares L et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet, doi: 10.1016/S0140-6736(19)32222-6 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Ready NE et al. Nivolumab Monotherapy and Nivolumab Plus Ipilimumab in Recurrent Small Cell Lung Cancer: Results From the CheckMate 032 Randomized Cohort. J Thorac Oncol, doi: 10.1016/j.jtho.2019.10.004 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Chung HC et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol 37, 1470–1478, doi: 10.1200/JCO.18.01265 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Rudin CM et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nature genetics 44, 1111–1116, doi: 10.1038/ng.2405 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George J et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53, doi: 10.1038/nature14664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudin CM et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nature reviews. Cancer 19, 289–297, doi: 10.1038/s41568-019-0133-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peifer M et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature genetics 44, 1104–1110, doi: 10.1038/ng.2396 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poirier JT et al. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene, doi: 10.1038/onc.2015.38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borromeo MD et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep 16, 1259–1272, doi: 10.1016/j.celrep.2016.06.081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mollaoglu G et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer cell 31, 270–285, doi: 10.1016/j.ccell.2016.12.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang YH et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes & development 32, 915–928, doi: 10.1101/gad.314815.118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wooten DJ et al. Systems-level network modeling of Small Cell Lung Cancer subtypes identifies master regulators and destabilizers. PLoS Comput Biol 15, e1007343, doi: 10.1371/journal.pcbi.1007343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McColl K et al. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 8, 73745–73756, doi: 10.18632/oncotarget.20572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owonikoko TK et al. A Translational, Pharmacodynamic, and Pharmacokinetic Phase IB Clinical Study of Everolimus in Resectable Non-Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 21, 1859–1868, doi: 10.1158/1078-0432.CCR-14-1998 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ott PA et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J Clin Oncol 37, 318–327, doi: 10.1200/JCO.2018.78.2276 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Ayers M et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127, 2930–2940, doi: 10.1172/JCI91190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horie M, Saito A, Ohshima M, Suzuki HI & Nagase T YAP and TAZ modulate cell phenotype in a subset of small cell lung cancer. Cancer science 107, 1755–1766, doi: 10.1111/cas.13078 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordeeva O Cancer-testis antigens: Unique cancer stem cell biomarkers and targets for cancer therapy. Semin Cancer Biol 53, 75–89, doi: 10.1016/j.semcancer.2018.08.006 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Jin S et al. Cancer/testis antigens (CTAs) expression in resected lung cancer. Onco Targets Ther 11, 4491–4499, doi: 10.2147/OTT.S159491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Z, Ihle NT, Rejto PA & Zarrinkar PP Outlier analysis of functional genomic profiles enriches for oncology targets and enables precision medicine. BMC genomics 17, 455, doi: 10.1186/s12864-016-2807-y (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dieu-Nosjean MC et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol 26, 4410–4417, doi: 10.1200/JCO.2007.15.0284 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Fridman WH, Pages F, Sautes-Fridman C & Galon J The immune contexture in human tumours: impact on clinical outcome. Nature reviews. Cancer 12, 298–306, doi: 10.1038/nrc3245 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Giraldo NA et al. The immune contexture of primary and metastatic human tumours. Curr Opin Immunol 27, 8–15, doi: 10.1016/j.coi.2014.01.001 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Danaher P et al. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). Journal for immunotherapy of cancer 6, 63, doi: 10.1186/s40425-018-0367-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.George J et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nature communications 9, 1048, doi: 10.1038/s41467-018-03099-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.