
Fig. 1. Activation of CB2Rs after neurological disorders.
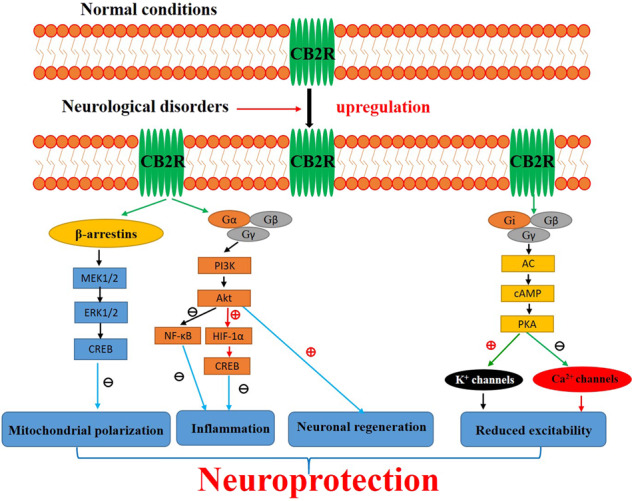
Although brain CB2R levels are low in healthy individuals, they are significantly upregulated in glial elements in response to various neurological insults. Existing evidence demonstrates that signaling through CB2Rs can be mediated via G protein and β-arrestin, each with their own downstream effectors [147–149]. As shown in Fig. 1, CB2Rs are coupled to Gαi/o to inhibit adenylyl cyclase (AC) activity, leading to a decrease in cAMP levels. On the other hand, the Gβγ subunits, upon dissociation from Gαi/o, are known to activate G protein-gated inwardly rectifying K+ channels and PI3K and inhibit voltage-gated Ca2+ channels. Alternatively, β-arrestin 2 recruitment to CB2Rs results in activation of the ERK pathway. Overall, activation of CB2Rs plays neuroprotective roles, including attenuation of neuroinflammation, amelioration of mitochondrial depolarization and stimulation of neurogenesis, which could eventually reduce deficits in cognition, memory and motor inhibition. In addition, the activation of CB2Rs also modulates ion channel functions, thereby altering neuronal excitability, and leads to neuroprotection.