

Abstract
Pancreatic cancer has poor prognosis and treatment outcomes due to its highly metastatic nature and resistance to current treatments. The RNA binding protein (RBP) Hu-antigen R (HuR) is a central player in posttranscriptional regulation of cancer related gene expression, and contributes to tumorigenesis, tumor growth, metastasis and drug resistance. HuR has been suggested to regulate pancreatic cancer epithelial to mesenchymal transition (EMT) but the mechanism was not well understood. Here we further elucidated the role HuR plays in pancreatic cancer cell EMT, and developed a novel inhibitor specifically interrupting HuR-RNA binding. The data showed that HuR binds to the 3’-UTR of the mRNA of the transcription factor Snail, resulting in stabilization of Snail mRNA and enhanced Snail protein expression, thus promoted EMT, metastasis and formation of stem-like cancer cells (CSCs) in pancreatic cancer cells. siRNA silencing or CRISPR/Cas9 gene deletion of HuR inhibited pancreatic cancer cell EMT, migration, invasion, and inhibited CSCs. HuR knockout cells had dampened tumorigenicity in immunocompromised mice. A novel compound KH-3 interrupted HuR-RNA binding, and KH-3 inhibited pancreatic cancer cell viability, EMT, migration/invasion in vitro. KH-3 showed HuR-dependent activity and inhibited HuR-positive tumor growth and metastasis in vivo.
Keywords: RNA binding protein (RBP), HuR, pancreatic cancer, HuR inhibitor
Introduction
Pancreatic cancer has the highest fatality rate among all cancers, with a 5-year overall survival rate less than 8%1. Whereas advancements in targeted therapies and immunotherapies have greatly improved outcomes in patients with many types of cancers, the benefits have not been gained for pancreatic cancer patients. First-line chemotherapies such as gemcitabine plus nab-paclitaxel2, or FOLFIRINOX3, only achieve a median overall survival of 9–13 months in patients with advanced disease, and have multiple significant toxicities.
An enrichment of stem-like cancer cells (CSCs) in pancreatic cancer has been proposed to root the poor prognosis and treatment outcomes of this disease4, 5. The presence of CSCs is highly associated with cancer cell epithelial-mesenchymal transition (EMT) that contributes to chemoresistant tumors prone to metastasis and recurrence. EMT is typified by loss of cell-cell junctions and apico-basolateral polarity, resulting in the formation of migratory mesenchymal cells with invasive properties6. EMT is a major process underlying the heterogeneity of cancer cells, and is an important initial step for cancer cell dissemination and metastasis. Through EMT, cancer cells de-differentiate, gain motility and invasiveness, evade senescence, apoptosis, and immune surveillance, and become resistant to conventional and targeted therapies7–10. Induction of EMT in neoplastic cells also resulted in the enrichment of CSCs11. Given the highly aggressive, drug resistant and metastatic nature of pancreatic cancer, targeting EMT would cast hope for the treatment for this devastating disease.
EMT initiation and progress are regulated by complex signaling networks. HuR (the RBP Hu antigen R), an RNA binding protein, is increasingly recognized as a pivotal factor in cancer-related gene expression, and is proposed to play a role in EMT regulation. HuR is a member of the embryonic lethal abnormal vision (ELAV) family, and is highly expressed in virtually all malignancies tested, including pancreatic cancer12, 13. HuR contains three RNA recognition motifs (RRM), of which RRM1 and RRM2 are involved in RNA binding, and RRM3 is needed for cooperative assembly of HuR oligomers on RNA14. HuR target mRNAs bear adenine- and uridine-rich elements (AREs) in their 3′- or 5′-UTRs15. Cytoplasmic binding of HuR to these mRNAs generally confers the ARE-mediated mRNA decay and leads to mRNA stabilization and increased translation16. HuR promotes tumorigenesis by interacting with a subset of mRNAs which encode proteins implement in cell proliferation, cell survival, angiogenesis, invasion, metastasis, and treatment resistance17. It has been reported that HuR stabilizes mRNAs of MMPs, uPA, and probably Snail18–20. Snail is a central EMT-promoting transcription factor. This suggested a regulatory role of HuR in cancer cell EMT. However, so far there is only one published study demonstrating stabilization of Snail by HuR in a breast cancer cell line20. The mechanisms that HuR could be involved in cancer cell EMT have not been well understood.
HuR is highly expressed in pancreatic cancer as well as many other types of cancers, compared to normal tissues21. In normal cell, HuR is mostly located in the nucleus, while in cancer cells the cytoplasmic level is elevated. The translocation of HuR from nucleus to cytoplasm is mediated by a nuclear-cytoplasmic shuttling sequence (NCS) between RRM2 and RRM322. The elevated HuR expression levels and its cytoplasmic translocation enhances pro-inflammatory and oncogenic protein expressions23, 24, and is correlated with advanced clinical pathology, and patient prognosis and survival rate in various types of cancers25. Therefore, HuR is considered a putative target for cancer treatment. However, despite many efforts, there has been limited success in small molecules that directly disrupt the HuR interaction with AREs of its target mRNAs26–30. In this study, we aim to fill the gap of understanding of the role HuR plays in pancreatic cancer cell EMT and metastasis, and explore a novel approach for pharmacological inhibition of HuR.
Materials and Methods
Cell culture, detection of cell viability, migration/invasion, and tumor spheres formation
Pancreatic cancer cell lines were from the American Type Culture Collection (Manassas, VA). hTERT-HPNE cells (immortalized human pancreatic ductal epithelial cells) were donated by Dr. Anant at the University of Kansas Medical Center. MTT assay was used for cell viability detection, with starting cell number in 96-well plate of 3000/well (for 72 h treatment) or 5000/well (for 48 h treatment).
Wound healing assay was performed by scratching confluent monolayer with a 100 μL pipette tip. Wound recovery was calculated by 100% − (Remaining Area ÷ Original Area) × 100% at each time point.
Matrigel invasion assay was performed using Boyden chambers (BD Biosciences, San Jose, CA) either pre-coated or uncoated with 0.1 mg/ml Matrigel, with 0.5% FBS inside and 10% FBS outside. Starting cell density was 1 × 104 /well.
For tumor spheres formation, single cell suspension was plated into 96-well ultra-low attachment plates (Corning Inc., Corning, NY) at 100 cells/well in stem cell media, supplemented with B27 Supplement, 20 ng/ml human basic fibroblast growth factor, 20 ng/ml epidermal growth factor, 100 units/ml penicillin/streptomycin (Invitrogen, Grand Island, NY), and 4 μg/ml heparin calcium salt (Fisher Scientific, Pittsburg, PA). Tumor spheres were counted after 14 days, and size was measured using Image J software.
RNA isolation, cDNA synthesis, and Real-Time PCR
Total RNA was extracted using TRIZOL reagent (Invitrogen, Grand Island, NY). cDNA synthesis was performed with 1 μg RNA using Omniscript RT kit (Qiagen, Valencia, CA), and diluted 1:5 for further use. Real-time PCR was performed using Bio-Rad iQ iCycler detection system with iQ SYBR green supermix (Bio-Rad Laboratories Ltd, Hercules, CA). Data was normalized to 18S rRNA.
To detect the decay of mRNAs, cells were treated with 5 μg/mL actinomycin D to block transcription (at - 0.5 h). Total RNA was extracted at 0, 0.5, 1, 2, and 3 h. KH-3 (2 μM) was added 30 m after actinomycin D (at 0 h).
HuR knockdown/overexpression
Recombinant pcDNA3.1 HuR-flag Plasmid (pHuR) was provided by Dr. Dixon at the University of Kansas Cancer Center. The vector pcDNA3.1+ (pVec) was purchased from Addgene (Cambridge, MA), and HuR siRNA from Qiagen (Valencia, CA). Plasmids were transfected by lipofectamine™ 3000 reagent for 48 h, and siRNA by lipofectamine™ RNAiMAX reagent for 24 h (Invitrogen, Grand Island, NY). HuR levels were verified by western blot.
CRISPR/Cas9 deletion of HuR gene was performed using the lentiCRISPRV2 vector (AddGene). The control single guide RNAs (sgRNAs) and HuR sgRNAs were cloned into the vector following reported procedures31. The HuR lentiviral sgRNA or control sgRNA were co-transfected into HEK293FT cells with the packaging plasmids pMD2.G and psPAX2 (AddGene). MIA PaCa-2 cells were infected with virus-containing medium and then selected with 1.0 μg/mL puromycin. Single clones were generated by limited dilution.
RNP-IP Assay
Total cell lysate was used for immunoprecipitation with anti-HuR or normal rabbit IgG (Cell Signaling Technology, Beverly, MA), using the Immunoprecipitation Kit (Protein G) (Roche, Basel, Switzerland), supplemented with RNaseOUT™ Recombinant Ribonuclease Inhibitor (Invitrogen, Grand Island, NY) in all steps (100 U/mL). In the KH-3 treatment groups, KH-3 (2 μM) was supplemented in all steps. Total RNA was then extracted from the immunoprecipitation products by TRIZOL reagent and subjected to qRT-PCR analysis.
Dual-Glo luciferase reporter assay
The full-length Snail mRNA 3’-UTR was synthesized by Genewiz (South Plainfield, NJ). The two truncated Snail mRNA 3’- UTRs (ΔAREs, and AREs) were cloned from total RNA of MIA PaCa-2 cells and amplified by PCR, and then constructed into the pmirGLO dual luciferase reporter plasmid (Fig. 2C). MIA PaCA-2 HuR KO cells were co-transfected with pmirGLO dual luciferase reporter with or without the constructions (full length, ΔAREs, AREs, or empty reporter) (Promega, Madison, WI) and pCDNA-3.1+-HuR (or empty vector) using Lipofectamine 3000 reagent (Invitrogen, Grand Island, NY). KH-3 was added at 24 h, and the dual-glo luciferase reporter assay was performed at 48 h using Dual-Glo® Luciferase Assay System (Promega, Madison, WI).
Fig. 2. HuR regulates the expression of Snail.
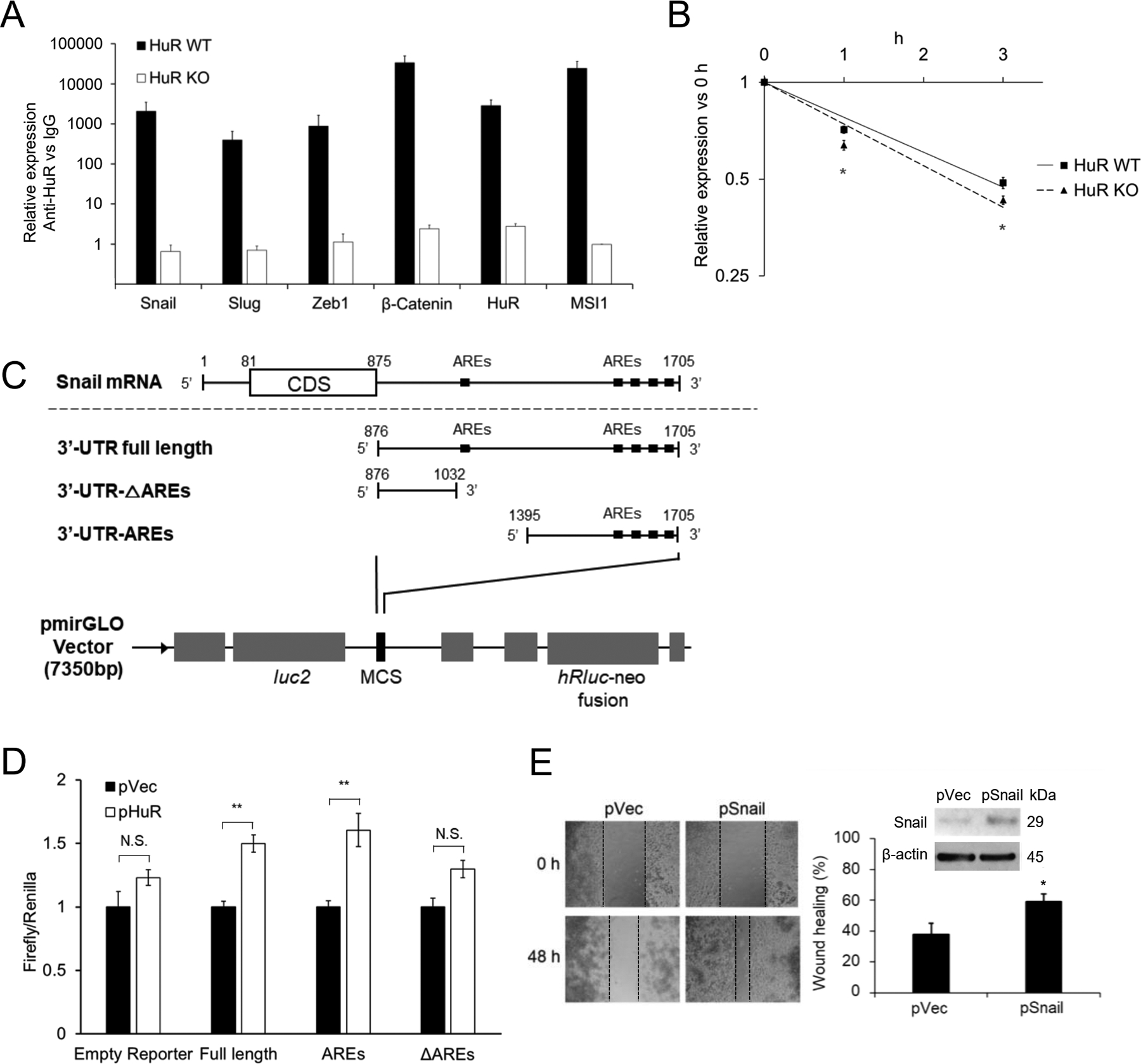
A. RNP-IP detection of HuR binding RNAs of EMT related genes. Data for each individual mRNA was normalized to the IgG pull-down product of that mRNA. Bar graphs show Mean ± SEM of 9 repeats. B. Stability of Snail mRNA in MIA PaCa-2 HuR WT or HuR KO cells. Transcription was blocked by actinomycin D (5 μg/ml) treatment 30 min before the first sample was collected (0 h). Data shows Mean ± SEM of 9 repeats. C. Schematic diagram of the constructions of the full length and 2 truncations of 3’-UTR of Snail mRNA into the dual-luciferase reporter. D. Luciferase reporter assay. MIA PaCa-2 HuR KO cells were co-transfected with HuR (or vector) and the dual-luciferase reporter with Snail 3’-UTR constructions (either the Full length, AREs or ΔAREs, or empty reporter). E. Scratch assays in MIA PaCa-2 HuR-KO cells with Snail overexpression. Cells were transfected with empty vector (pVec) or Snail gene (pSnail) or 48 h before seeded at 3×105 cell/ml in 24 well plate to form monolayer. *, p<0.05; **, p<0.01 with one-way ANOVA-Tukey’s test or Student’s t-test.
Western blot, Immunofluorescence and immunohistochemistry
Cells were lysed with RIPA buffer (Sigma Al), and total protein was subjected to western blotting. BCA method was used for protein quantification (Pierce BCA protein assay kit, Waltham, MA). Blots were established using either Pierce ECL substrate or Pierce ELC+ substrate (Thermo Scientific, Rockford, IL).
Immunofluorescence detection of protein expression was performed with cells grown on 6-well chamber slides as routine. Blocking was performed using 5% Goat serum+0.3%Triton X-100. Nucleus were stained with ProLong® Gold Antifade Reagent containing DAPI (Cell Signaling Technology, Beverly, MA).
Immunohistochemistry was performed with paraffin-embedded tissue sections (5 μM thick), as routine. DAB were used to develop the sections (HRP/DAB (ABC) detection IHC kit, Abcam, Cambridge, UK). All the sections were then counterstained with hematoxylin.
KH-3 synthesis and structure validation
The compound KH-3 was synthesized in-house by a collaborator. The synthesis and structure validation were reported in another publication32.
Mouse tumor models and KH-3 treatment
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center under the protocol #2015–2247. A pilot MTD (maximum tolerant dose) experiment was carried out to determine dose regimen to be used in treatment. Mice (n=3) were started at 50 mg/kg body weight of KH-3 daily, intraperitoneal injection (IP) for 3 consecutive days. No clinical signs of toxicities were observed. Dose was then escalated to 75 mg/kg for 3 days and when no signs of toxicities observed, dose was increased to 100 mg/kg for 5 consecutive days. On day 5 of 100 mg/kg, reduction in activities were observed in mice. Dose was reduced to 3x weekly at 100 mg/kg. No signs of toxicities were observed. The 100 mg/kg 3x weekly IP was determined as the treatment dose. All treatment concerning KH-3 used this dose regimen.
A subcutaneous tumor model was used to determine tumor formation rate. MIA PaCa-2 HuR WT cells or MIA-PaCa-2 HuR KO cells were inoculated into the flank of female Ncr nu/nu mice at the number of 2×106 cell in PBS. Tumor formation was monitored daily, tumor size was measured 3 times/week by using a digital caliper.
An orthotopic pancreatic tumor model was used to determine treatment effects of KH-3. Luciferase-expressing PANC-1 cells (PANC-1–Luc, multi-clones) were established by the Preclinical Proof of Concept Core Laboratory (University of Kansas Medical Center, Kansas City, KS). A small subcostal laparotomy was performed in female Ncr nu/nu mice to expose the pancreas, and 2 × 105 PANC-1-Luc cells in 50 μL PBS were injected into the tail of pancreas. After 11 days, the localized tumors inside the pancreas of these donor mice were removed and minced into small pieces of 1 mm3 cube. One tumor cube was implanted into the pancreas of one recipient nude mouse by laparotomy. After 11 days, the recipient mice were scanned for xenograft formation using an IVIS imaging system (Waltham, MA) upon IP injection of 150 mg/kg D-luciferin. Mice were grouped based on tumor burden and treatment commenced as described, with weekly follow-up imaging. Body weight was measured 3 times weekly, and mice were monitored for clinical signs of toxicity during treatment, including guarding, abnormal appearance (hunched), restlessness, and reluctance to move. Treatment lasted for 5 weeks, and gross necropsy was performed at the end of treatment.
Data analysis
Statistical analysis was performed using SPSS software for student’s T-test, Log Rank test, one-way Anova with Turkey’s Method, or Mann-Whitney’s U test as each condition applies. A difference was considered significant at the p < 0.05 level. Correlation was analyzed by Pearson Test.
Results
HuR enhances pancreatic cancer cell EMT, migration, and CSCs
To study the role of HuR in pancreatic cancer cell EMT, HuR expression was first silenced by transfecting siRNAs targeting HuR mRNA (siHuR), and down-regulation of HuR protein was validated by western blots. In two human pancreatic cancer cell lines PANC-1 and MIA PaCa-2 transfected with siHuR, the cellular morphology changed to a more epithelium-like state compared to each of their parent cells, characterized by less spindle-like cells, shortened cell length and/or enlarged cell diameter (Suppl Fig 1). Consistent with this phenotypical change, the expressions of signature EMT genes in both cells were altered (Fig 1A): the epithelial marker Claudin1 was significantly upregulated, the mesenchymal marker Vimentin was downregulated, and the EMT enhancing transcription factor Snail was significantly decreased. We then performed permanent deletion of HuR gene in MIA Paca-2 cells by a CRISPR/Cas-9 method. As expected, the deletion caused a depletion in HuR protein in the cells (Fig. 1A). The morphology of HuR-deleted cells (HuR KO) showed a more epithelium-like state compared to the control cells (Suppl Fig. 1), again with increase in Claudin1, decrease in Vimentin and Snail, consistent with the results in siHuR transfection (Fig. 1A). Claudin1 expression was further confirmed by immunofluorescence staining in the siHuR transfected MIA PaCa2 cells. Results clearly showed increase of Claudin1 expression (Fig. 1B).
Fig. 1. HuR enhances pancreatic cancer cell EMT, migration, and CSCs.
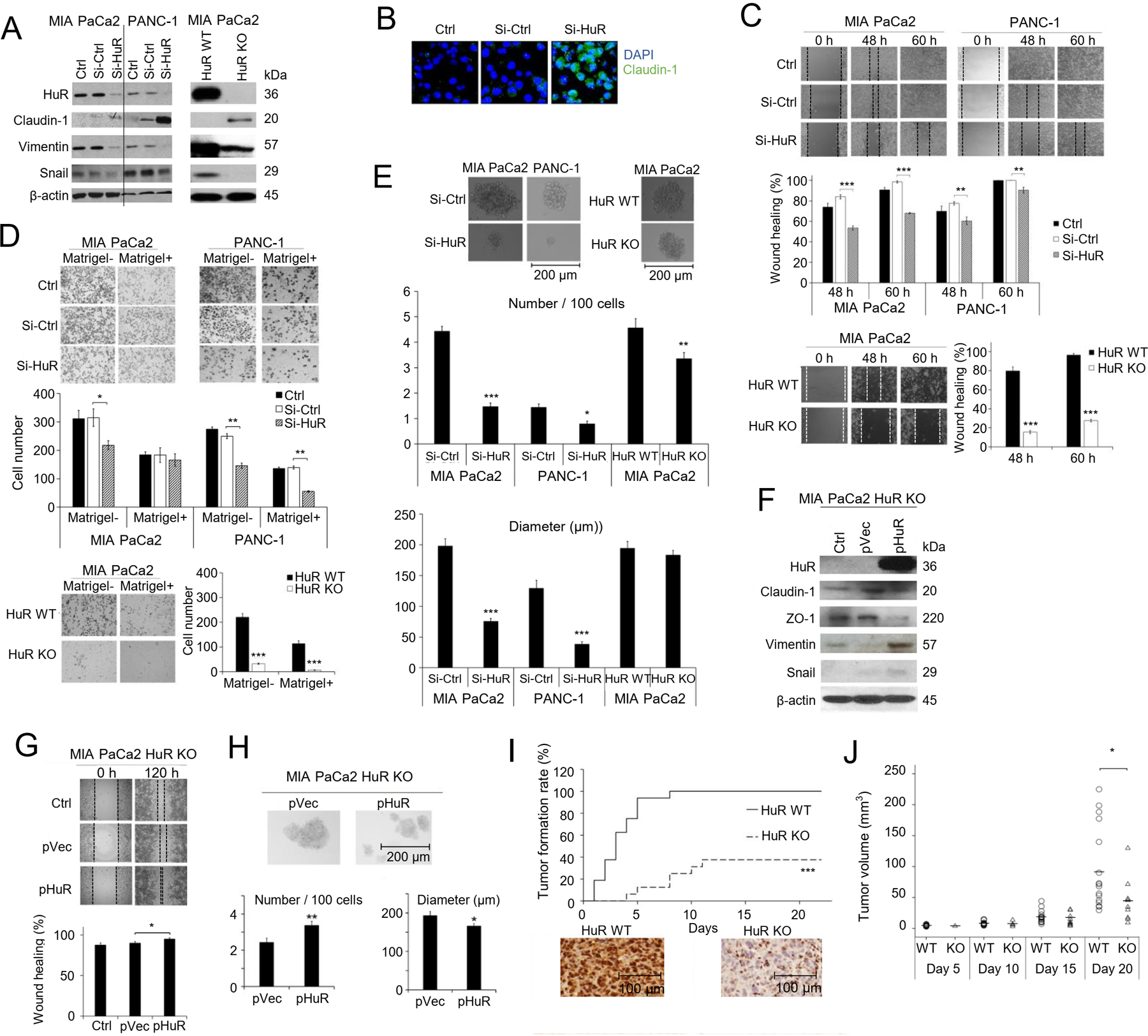
A. Western blot in MIA PaCa-2 and PANC-1 cells showing expression of HuR and markers of EMT. β-actin was a loading control. Left: Cells were transfected with Si-Ctrl or Si-HuR for 24 h, or un-transfected (Ctrl). Right: HuR KO were cells knockout of HuR gene by CRISPR/Cas9 procedure. HuR WT were cells transfected with control sgRNA. B. Immunofluorescence staining for Clauind-1. Cell nuclei were DAPI stained. C. Scratch assays. Bar graphs represent Mean ± SEM of ≥ 3 repeats. D. Matrigel invasion assays. Cell migration (Matrigel-) and invasion (Matrigel+) were detected at 24 h for PANC-1 cells and 48 h for MIA PaCa-2 cells. Bar graphs show the Mean ± SEM of migrated/invaded cells per field of > 3 fields per experiment for 3 experiments. E. Tumor spheres formation assay. Bar graphs show Mean ± SEM of 36 repeats. *, p<0.05; **, p<0.01; ***, p<0.001 with one-way ANOVA-Tukey’s test. F-H. HuR re-expression rescues the effects of HuR knockdown. F. Western blot in MIA PaCa-2 HuR-KO cells after HuR re-expression. Ctrl were HuR KO cells, pVec were HuR KO cells transfected with empty vector, and pHuR were HuR KO cells transfected with HuR gene. β-actin was a loading control. Scratch assays (G) and tumor spheres formation (H) in MIA PaCa-2 HuR KO cells with HuR re-expression. Bar graphs represent Mean ± SEM of 3 – 36 repeats. *, p<0.05; **, p<0.01; ***, p<0.001 with one-way ANOVA-Tukey’s test. I. In vivo tumor formation of MIA PaCa-2 HuR WT cells and HuR KO cells in nude mice (n=16 per group). Cells were subcutaneously inoculated at 2×106 cells per injection. Immunohistochemistry blotted for HuR expression in tumor tissues. ***, p<0.001 with Log-rank test. J. Volume of the tumors formed in I. Each circle or triangle represents a tumor. The short bars show the mean tumor volume of each group. *, p<0.05 with Mann-Whitney U test.
As EMT promotes cancer cells migration and invasion, we expected HuR downregulation would inhibit pancreatic cancer cell migration and invasion, and HuR overexpression would enhance them. Indeed, siHuR significantly decreased the migration of PANC-1 and MIA PaCa-2 cells in a wound healing assay (Fig 1C). Consistently, HuR KO MIA PaCa-2 cells also had decreased ability to migrate (Fig 1C). Migration/invasion were further assessed using matrigel uncoated and coated Boyden chambers. siHuR inhibited migration and/or invasion in both MIA PaCa2 cells and PANC-1 cells, and HuR gene deletion in MIA PaCa2 cells greatly impaired cell migration and invasion (Fig 1 D). Because HuR also regulates cell proliferation, there is a possibility that the innihbiton in migration/invasion deteced here were due to inhibition in proliferation. To address this question, we examined the proliferation of siHuR and HuR KO cells. SiHuR MIA PaCa-2 cells had the same growth rate as the SiCtrl cells and untreated cells (Ctrl) up to 72 hours (Suppl Fig 2A), suggesting the inhibition in migration/invason was independent of cell proliferation. A difference in growth was observed between the HuR KO MIA PaCa-2 cells and the wild type cells at 48 hours and 60 hours. At 60 hours, HuR KO cells had ~40% growth inhibition compared to wild type cells (Suppl Fig 2B), and could contribute to the inhibition of gap-closing detected in the HuR KO cells. Considering HuR KO cells were permanently and completely deleted with HuR, while siHuR was temporary and not a 100% inhibition, it is likely that inhibition of HuR may first influence EMT and migration/invasion in the tested cells, and when the depletion of HuR is more severe, growth/proliferation were affected.
We then examined the cancer stem-like cell population (CSCs) using tumor spheroid formation assay. Data showed that the number and size of tumor spheres were both significantly reduced in PANC-1 and MIA PaCa2 cells with siHuR transfection (Fig. 1E), indicating inhibition in CSCs. The HuR gene deletion also decreased the number of spheres, but did not influence the sizes of the spheres formed (Fig. 1E).
We then re-expressed HuR in the HuR KO MIA PaCa2 cells and examined the EMT markers, migration and CSCs. HuR re-expression decreased the epithelial markers Claudin1 and ZO-1, and increased the mesenchymal marker Vimentin, and Snail (Fig. 1F). The restore of HuR expression also enhanced migration (Fig. 1G), and increased number of tumor spheres (Fig 1H) compared to HuR KO cells while the size of the formed spheres slightly decreased.
As the number of CSCs is responsible for tumorigenicity in vivo, we compared the tumor formation rate of MIA PaCa2 HuR KO cells to that of the CRSPR/Cas9-control cells (HuR WT cells) in nude mice. At the inoculation number of 2 × 106 cells subcutaneously, the HuR WT cells yield 100% (16/16) tumor formation in 8 days after injection (day 8). The HuR KO cells had a tumor formation rate of 25% (4/16) at day 8, and only reached a final tumor formation rate of 37.5% (6/16) at day 21 (Fig. 1I). At day 20, the average tumor volume of HuR KO tumors was significantly smaller than that of the WT tumors (Fig. 1J).
HuR regulates the expression of Snail
HuR typically stabilizes its targeting mRNAs and promotes translation by binding to adenine- and uridine-rich elements (AREs) located in the 3’untranslated region (UTR) of the target mRNA. We examined whether HuR binds to the mRNAs of important regulators of EMT and CSC, using ribonucleoprotein immunoprecipitation (RNP-IP) assay33. Pull-down products from MIA PaCa2 total cell lysate using anti-HuR antibody were quantified for RNA components by qRT-PCR. mRNAs of a panel of EMT/CSC regulators showed strong association with HuR protein, i.e. Snail, Slug, Zeb1, and β-catenin, as well as the mRNAs of the known HuR targets Msi1 and HuR itself34, 35 (Fig. 2A). In HuR KO cells, this panel of mRNAs were not pulled down (Fig. 2A).
We postulated that the binding to HuR stabilized these mRNAs. HuR WT and HuR KO MIA PaCa2 cells were treated with actinomycin D to block transcription, and then the stability of these mRNAs was detected during time. Data showed significantly enhanced degradation of Snail mRNA (Fig. 2B), but the decay of the mRNAs of Slug, Zeb1 and β-catenin did not change by the knockdown of HuR (Sppl Fig. 3A) despite binding of their mRNAs to HuR. Consistent with these results, the protein expression of Snail was decreased with HuR knockdown (Fig. 1A), whereas the protein levels of Slug, Zeb1 and β-catenin were minimally influenced (Sppl Fig. 3B).
The direct interaction of HuR with Snail mRNA 3’-UTR was examined with a luciferase reporter assay. The full length 3’-UTR, and two truncated Snail mRNA 3’- UTRs (ΔAREs, and AREs) were each constructed into the pmirGLO vector, which contains a firefly luciferase gene under the PGK promoter (Fig. 2C). The sequence of ΔAREs did not contain the AU-rich HuR binding elements, and the sequence of AREs contained the major part of the AU-rich elements in the 3’-UTR (Fig. 2C). MIA PaCa2 HuR KO cells were then co-transfected with HuR and the pmirGLO plasmid containing each of the constructed Snail UTRs. Data clearly showed that only with the full length 3’-UTR and the AREs, HuR transfection could enhance luminescence signal, and when there lacked the HuR binding elements (ΔAREs), the luminescence signal did not change with HuR transfection (Fig. 2D).
To further determine the functional importance of Snail in the HuR regulated EMT and migration. We re-expressed Snail in HuR KO MIA PaCa2 cells and detected the migration ability of the cells. The restoration of Snail significantly increased migration of the HuR KO cells (Fig. 2E).
A novel HuR inhibitor KH-3 disrupts HuR-mRNA interaction, and inhibits pancreatic cancer cell viability depending on endogenous HuR levels
Using a reported fluorescent polarization assay30, a novel compound KH-3 (Fig. 3A) was identified that bound to HuR and interrupt HuR-mRNA interaction. The direct interaction of KH-3 to HuR was identified and validated using multiple in-vitro binding assays: Surface Plasmon Resonance (SPR) analysis, Fluorescence Polarization Assay (FP), and Amplified Luminescent Proximity Homogeneous Assay (Alpha assay). The Alpha assay and FP assay showed that KH-3 interfered HuR binding to one of its known target Msi1 mRNA (Ki ~300–700 nM). The SPR assay identified that KH-3 directly bound to the RNA Recognition Motifs of HuR (RRM1/2). These data are reported in another paper32.
Fig. 3. A novel HuR inhibitor KH-3 disrupts HuR-mRNA interaction, and inhibits pancreatic cancer cell viability.
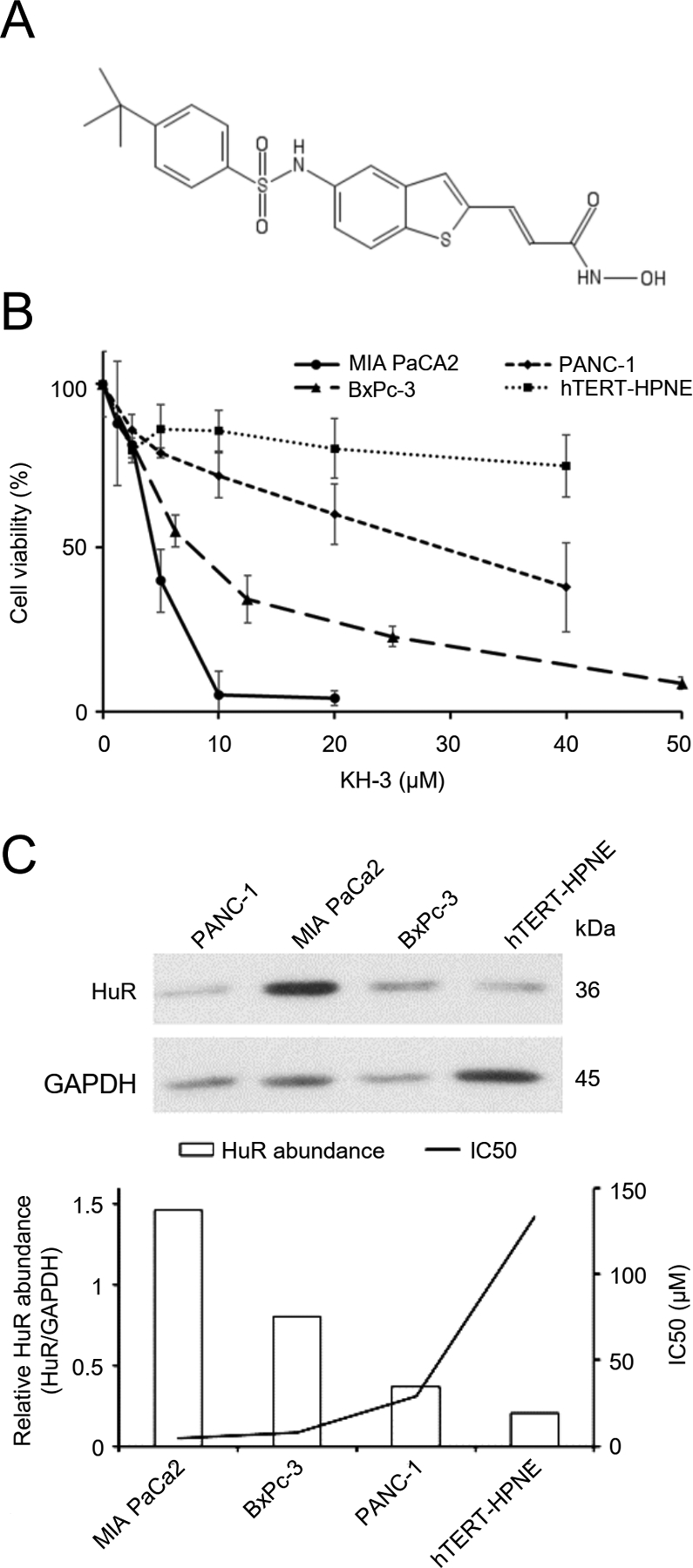
A. Chemical structure of KH-3. B. Inhibition of cell viability by KH-3. C. Upper: Western blot showing endogenous HuR levels of the tested cell lines. Lower: The correlation between HuR levels and the sensitivity of cells to KH-3 treatment. Bars show relative band density of HuR normalized to GAPDH, and the line shows IC50 values of KH-3.
Pancreatic cancer cell lines with different endogenous HuR expression levels were then treated with serial concentrations of KH-3 for 48 hours. KH-3 induced cytotoxicity in pancreatic cancer cells, with the sensitivity correlated to endogenous HuR protein levels (Fig. 3B, C). MIA PaCa2 cells have the highest HuR protein abundance among the tested cell lines and were the most sensitive to HuR treatment (IC50 = 5 μM). PANC-1 cells have the lowest HuR expression level and were the most resistant among the tested cancer cells (IC50 = 25 μM). BxPC-3 cells, another human pancreatic cancer cell line, have HuR expression level in the middle, and the IC50 of KH-3 was in the middle (10 μM). A non-cancerous human pancreatic ductal epithelial cell line (hTERT-HPNE) was tested under the same conditions. hTERT-HPEN cells have the lowest abundance of HuR protein compared to the cancer cells, and the cytotoxicity of KH-3 to these cells were minimal (IC50 >> 40 μM). There is an inverse correlation in the tested cell lines between the HuR expression levels and the sensitivity to KH-3 treatment (Fig. 3C) (R = −0.71 by Pearson Tests).
KH-3 inhibits pancreatic cancer EMT, invasion, and CSCs by inhibiting HuR functions
EMT signature gene expression was altered by KH-3 treatment in both MIA PaCa2 and PANC-1 cells showing Vimentin and Snail decreases, and Claudin1 increase (Fig. 4A, B), mimicking the consequences of HuR knockdown shown above (Fig. 1A). The alternation indicated EMT inhibition. HuR expression was not changed (Fig. 4A), confirming that KH-3 works through interrupting HuR-mRNA binding but does not alter HuR expression.
Fig. 4. KH-3 inhibits pancreatic cancer EMT, invasion, and CSCs by inhibiting HuR functions.
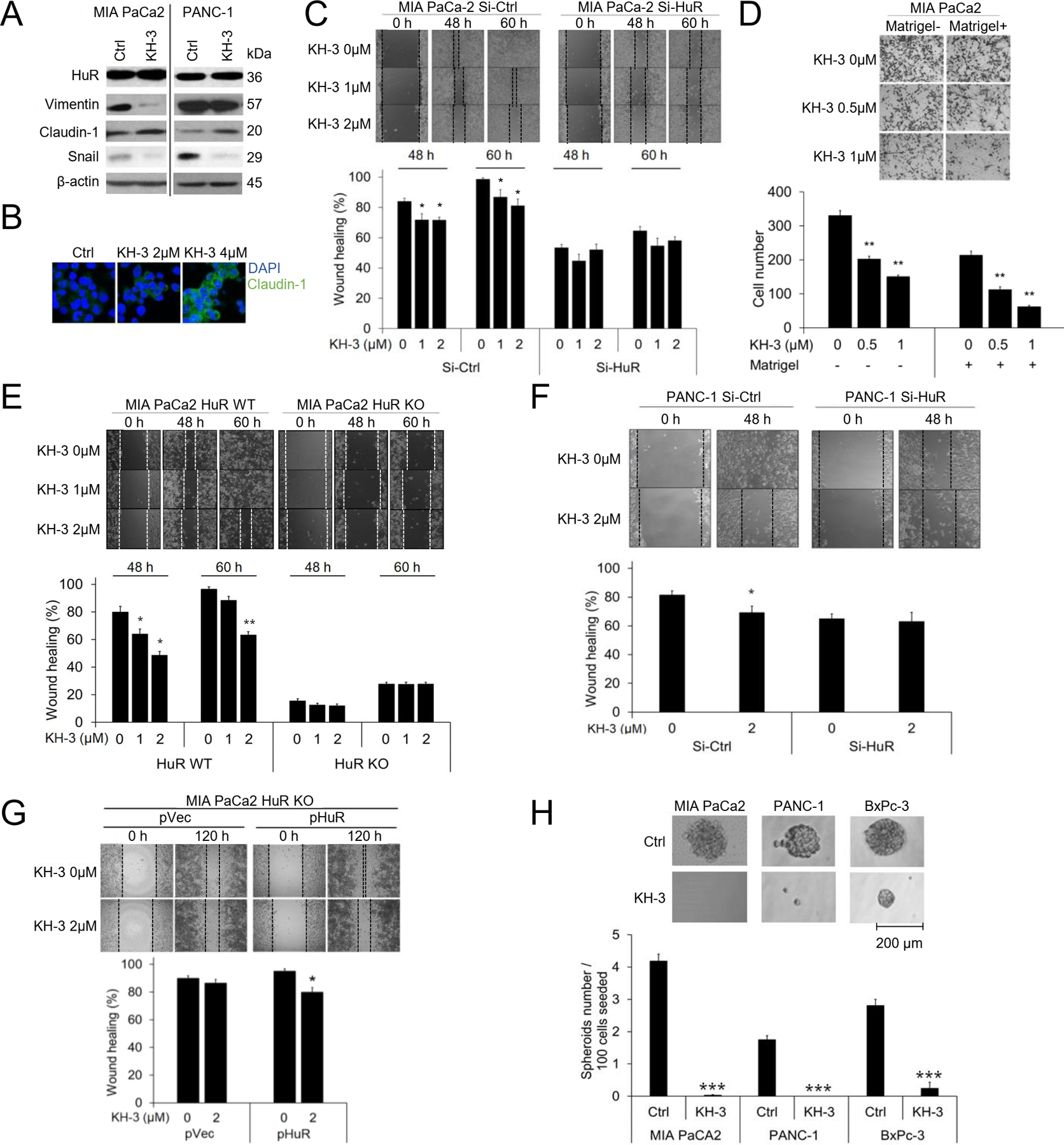
A. Western blot showing EMT markers with and without KH-3 treatment. MIA PaCa-2 cells were treated with 2 μM KH-3 for 24 h, and PANC-1 5 μM, based on their IC50s. β-actin was a loading control. B. Immunofluorescence staining for Clauind-1 in MIA PaCa-2 cells. Cells were treated with 2 μM KH-3 for 24 h, and nuclei were DAPI stained. C. Scratch assays in MIA PaCa-2 cells with KH-3 treatment. Cells were transfected with Si-Ctrl or Si-RNA for 24 h before seeded to form monolayer. Bar graphs show Mean ± SEM of 3 repeats. D. Matrigel invasion assays. Cell migration (Matrigel-) and invasion (Matrigel+) were detected at 48 h post treatment. Bar graphs show the Mean ± SEM of migrated/invaded cells per field of at least 3 fields per experiment for 3 repeated experiments. E, F. Scratch assays in MIA PaCa-2 HuR WT cells and HuR KO cells treated with KH-3 (E), and in PANC-1 cells with HuR knockdown and KH-3 treatment (F). Bar graphs represent Mean ± SEM of 3 repeats. G. Re-expression of HuR in MIA PaCa-2 HuR KO cells. Lower right: Western blots showing expression of HuR and EMT markers without transfection (Ctrl), with empty vector transfection (pVec), or with HuR transfection (pHuR). Upper: Scratch assay in cells re-expressing HuR with KH-3 treatment. Lower right: Bar graphs showing Mean ± SEM of 3 repeats. H. Tumor spheres formation. Cells were seeded at 100 cells/well. MIA PaCa-2 cells were treated with 4 μM of KH-3, PANC-1 cells with 10 μM, and BxPC-3 cells 8 μM. Spheres were imaged and counted 14 days post seeding. Bar graphs show Mean ± SEM of 36 repeats. *, p<0.05; **, p<0.01 with one-way ANOVA-Tukey’s test.
KH-3 inhibited MIA PaCa2 and PANC-1 cells migration and invasion in the wound healing assay as well as in the Boyden chamber trans-well assay (Matrigel assay) (Fig. 4C, D). To examine the target specificity of KH-3, HuR knockdown cells were used. In both the siHuR cells (MIA PaCa2 and PANC-1) and the CRISPER/Cas9 HuR KO cells (MIA PaCa2), the knockdown of HuR itself resulted in dampened migration compared to the wild type cells, as expected. Importantly, in the HuR knocked down cells, KH-3 lost its target, and did not show additional effects to the effect of the knockdown (Fig. 4C, E, F). We then re-expressed HuR in the HuR KO cells by transfecting the cells with an HuR-expressing plasmid. At the restore of HuR, KH-3 showed inhibitory effect again to the migration of the cells (Fig. 4G).
Tumor spheres formation was inhibited by KH-3 treatment. In PANC-1 cells, 10 μM of KH-3 eliminated tumor spheres formation, while in MIA PaCa2 cells 4 μM of KH-3 had the similar effects. As in BxPC-3 cells, 8 μM of KH-3 significantly inhibited both the number and the size of tumor spheres (Fig. 4H).
KH-3 decreases Snail mRNA stability and protein expression
RNP-IP assay was recruited to examine the interruption of binding between HuR and its target mRNAs with KH-3 treatment. It is expected the HuR downstream EMT-related mRNAs will less likely to be co-precipitated with HuR protein upon KH-3 treatment. Indeed, KH-3 treatment at 2 μM for 24 hours significantly decreased the pull-down amounts of mRNAs of Snail, Slug, Zeb1, β-catenin, HuR and Msi1 in MIA PaCa2 cells (Fig. 5A), consistent with but slightly less efficient than the HuR KO. Parallel with HuR KO, the KH-3 treatment (2 μM) enhanced Snail mRNA decay (Fig. 5B), and decreased the protein level of Snail (Fig. 4A).
Fig. 5. KH-3 decreases Snail mRNA stability and protein expression.
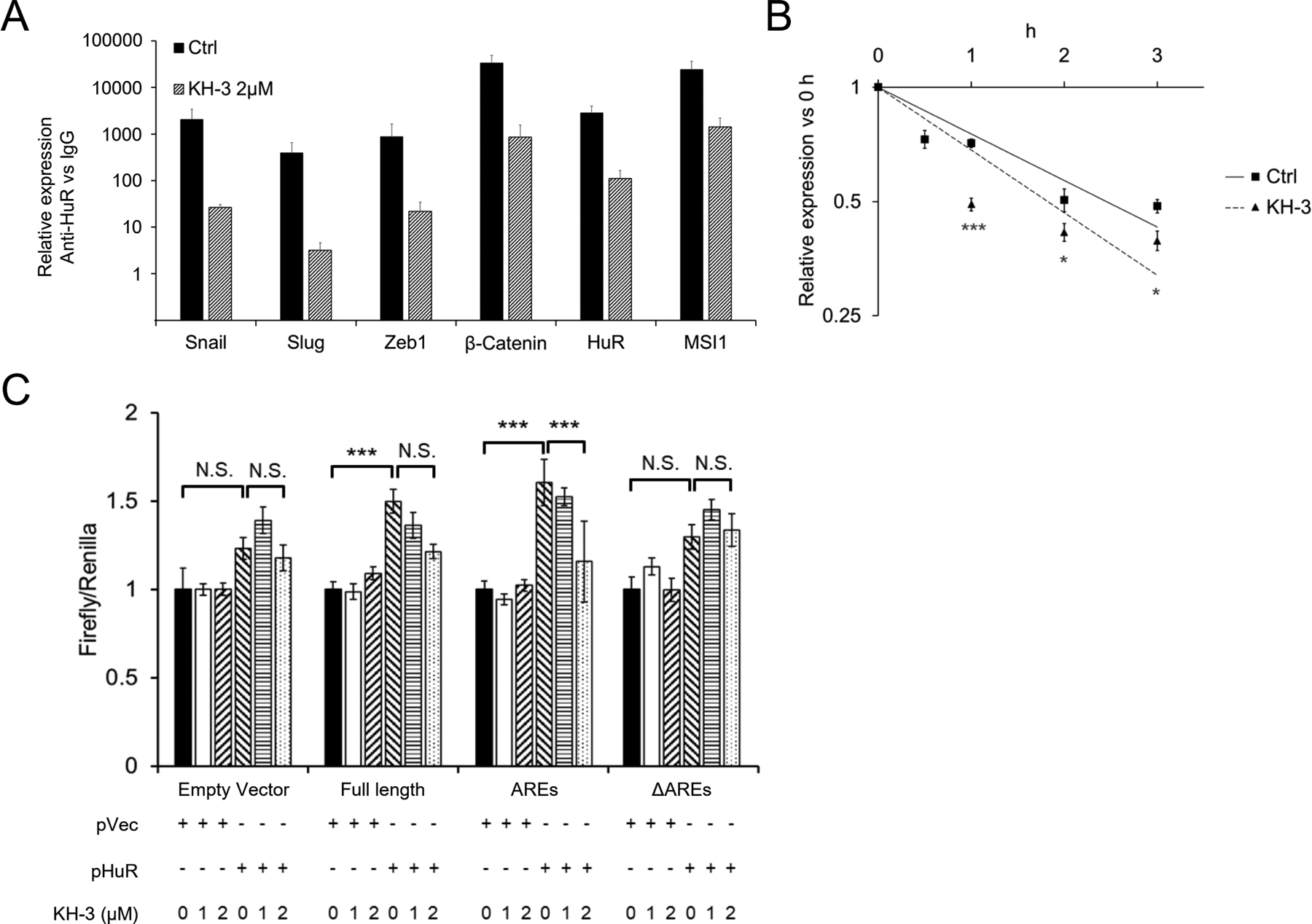
A. RNP-IP assay. MIA PaCA-2 cells were treated with 2 μM of KH-3 for 24 h. Pull-down products of whole cell lysate were subjected qRT-PCR detection. Data for each individual mRNA was normalized to the IgG pull-down product of that mRNA. Bar graphs show Mean ± SEM of 9 repeats. B. Stability of Snail mRNA in MIA PaCa-2 cells treated with KH-3. Transcription was blocked by actinomycin D (5 μg/ml) treatment 30 min before the cells were exposed to KH-3 (2 μM) (0 h). Data represents Mean ± SEM of 9 repeats. C. Luciferase reporter assay. MIA PaCa-2 HuR KO cells were co-transfected with HuR (or vector) and the dual-luciferase reporter with Snail 3’-UTR constructions (either the Full length, AREs or ΔAREs, or empty reporter). At 24 h of the co-transfection, cells were treated with KH-3 at indicated concentrations for an additional 24 h. *, p<0.05 ; ***, p<0.001 with one-way ANOVA-Tukey’s test or Student T-test.
Interruption of KH-3 to the binding of HuR with Snail 3’-UTR was further examined in the luciferase reporter assay. With co-transfection of HuR and full-length Snail 3’-UTR or AREs, KH-3 treatment inhibited the luminescence signal (Fig. 5C), clearly demonstrating interruption of HuR interaction with the 3’-UTR. When there lacked HuR binding elements (with ΔAREs), KH-3 had no influence on the luminescence signal (Fig. 5C).
KH-3 inhibits an HuR positive pancreatic cancer progression and metastasis in vivo.
The in vivo tumor inhibitory effects of KH-3 was tested in a highly metastatic orthotopic model of pancreatic cancer. To avoid peritoneal lesions resulted from leak of injection, 5 mice (donor mice) were injected with luciferase expressing PANC-1 cells (PANC-1-Luc, 2 × 105) and tumors developed in the pancreas of these donor mice were harvested and cut into ~1 mm3 and implanted into the pancreatic parenchyma of recipient mice. After 2 weeks, the recipient mice were imaged for tumor development and grouped to have equip average tumor burden (n = 9 for control group, and n = 10 for KH-3 treated group). Treatment then commenced with KH-3 at 100 mg/kg, IP, 3x weekly, determined based on MTD as described in Materials and Methods. The treatment continued for 5 weeks, and mice were euthanized, and gross necropsy was performed. The data showed that KH-3 treatment significantly inhibited longitudinal tumor growth and reduced tumor burden compared to the vehicle treated group (Control) (Fig. 6A, B). The final tumor weight was significantly reduced (Fig. 6C). In the control group, 5/9 mice developed uncountable lesions of metastasis in the liver (56%), whereas in the KH-3 treated group, only 1/10 mouse developed metastasis (10%) (Fig. 6D). At the end of the study, tumor tissues were examined for EMT alternations by Western Blots. The epithelial markers Claudin1 and ZO1 trended towards increase, and Snail trended toward downregulation with the KH-3 treatment (Fig. 6E). This is consistent with the expected EMT inhibition. Immunohistochemistry confirmed the high expression level of HuR in the tumor tissues compared to the adjacent normal pancreatic tissues. KH-3 treatment did not change the expression level of HuR in the tumor tissues (Fig. 6F).
Fig. 6. KH-3 inhibits an HuR positive pancreatic cancer growth and metastasis in vivo.
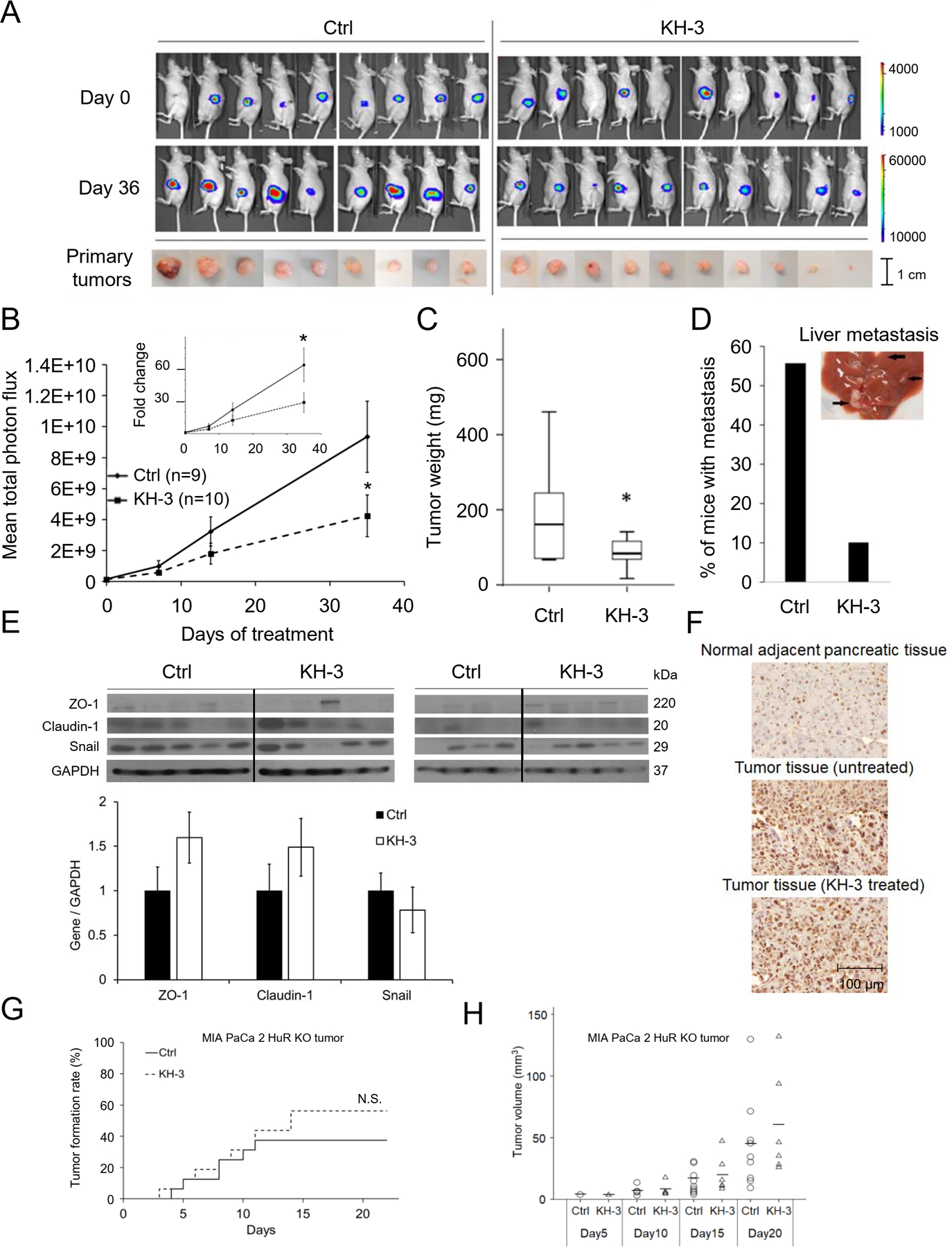
A. Upper: Bioluminescence images of mice bearing PANC-1-Luc orthotopic pancreatic xenografts treated with KH-3 (100mg/kg, 3 × weekly, n=10), or vehicle (Ctrl, n=9). Lower: Tumor in the mice pancreas at the end of the treatment (Day 36). B. Average tumor burden by IVIS imaging, quantified as photons/sec/cm2 (Mean ± SEM). *, p<0.05 with Student’s t-test. C. Average tumor weight at the end of the treatment. D. Percentage of mice having metastatic lesions in the liver at the end of the treatment. E. Western blot in mice tumor tissues showing EMT markers. Bar graphs show average band intensity of each gene relative to GAPDH. F. Immunohistochemistry showing HuR expression in tumor tissues and adjacent normal pancreatic tissues. G. Subcutaneous tumor formation of MIA PaCa-2 HuR KO cells with and without KH-3 treatment (n=16 for each group). Log Rank test resulted in no significant difference. H. Volume of the subcutaneous tumors formed in G. Each circle or triangle represents a tumor. The short lines represent average tumor volume of each group. Mann–Whitney U tests on each day demonstrated no significant differences.
No clinical signs of toxicity were observed during the treatment. There is a difference in body weight between control and treatment group after 21 days of treatment. However, because of tumor growth, it is yet difficult to attribute the changes in body weight to toxicity. Histological examination of the liver found no changes in the treatment group (Suppl Fig 3).
Further, an in vivo treatment was performed using MIA PaCa2 HuR KO tumors, to examine whether the inhibitory effects of KH-3 were dependent on HuR. Because the HuR KO cells did not form tumors orthotopically, cells were subcutaneously inoculated, and tumor formation and growth were monitored with caliper measurement. The KH-3 treatment started on the same day the cells were inoculated. KH-3 (100 mg/kg, IP, 3x weekly) did not influence either tumor formation (Fig. 6G) or tumor growth (Fig 6H) of the HuR KO tumors. This data, together with the data with the orthotopic tumor model, strongly indicated that the KH-3 effects were dependent on HuR.
Discussion
In pancreatic cancer, higher HuR level is associated with higher tumor T stage in patients21 and resistance to gemcitabine treatment36, 37. HuR is not likely to be a tumor initiator in pancreas by itself, but rather facilitates tumor development38. Pancreas specific transgenic HuR mouse did not form spontaneous tumor, instead, the elevated intra-pancreas HuR level promoted a pancreatitis-like inflammatory microenvironment that could facilitate tumor development38. Specific silencing or knockout of HuR inhibited pancreatic cancer cell proliferation, migration, invasion, and disabled in vivo xenograft formation39. We hypothesized that HuR plays an important role in regulating pancreatic cancer cell EMT and stemness, and this regulation underlines the aggressiveness of the tumor in terms of invasion, metastasis, drug resistance and new tumor generation. Data here reveal that HuR enhances pancreatic cancer cell EMT, mainly by stabilizing Snail mRNA and enhancing its protein expression. This enhancement in EMT promotes pancreatic cancer cell migration and invasion. The enhancement of EMT process by HuR also has implications in pancreatic CSC formation and maintenance. Inhibition of HuR dampened the ability of pancreatic cancer cell to migrate and invade, and inhibited CSCs. These data add to our knowledge of the role that HuR is playing in tumor metastasis and cancer stem cells, which was not well understood before.
HuR is more and more recognized as a responder in the cells to various stresses, and is involved in many physiological and pathological processes. Apparently, its function and the downstream genes regulated are different in different tissues and under different conditions. For example, under hypoxia HuR enhances VEGF and HIF-1α expression40, whereas under oxidative stress HuR enhances expression of cyclins and sirtuins41. HuR is also shown to causally linked to the onset of inflammation in kidney disease42, and contributes to liver fibrosis43, as well as many other pathological processes related to inflammation, whereas in adipose tissue HuR protects against diet-induced obesity and insulin resistance40. The oncologic microenvironment and the cancer cell itself might provide a “stress” condition that upregulates and activates HuR, with mechanisms not yet understood. Data here showed that HuR was differentially expressed in cancerous and normal pancreatic cells and mice tissues, consistent with clinical reports of the association between HuR levels and pancreatic cancer. The results here showed that in pancreatic cancer cells, HuR bound to a panel of mRNAs of regulator genes in the process of cancer cell EMT, but apparently the binding did not influence protein expressions of all the bound mRNAs. Here, only Snail mRNA was stabilized and protein expression increased. The mechanism of this selectivity remains to be understood. Another unexplored area in this study is the potential interaction and crosstalk between the tumor microenvironment and HuR expression and function in the cancer cells and in the surrounding cells such as pancreatic stellate cells, immune cells, and fibroblasts.
Given the importance of HuR in EMT and CSC, along with its roles in other signaling pathways involved in cancer progress, it is intriguing to develop pharmacological inhibitors of HuR for the treatment of cancer. However, there has been little success in small molecules that inhibit HuR functions, partially due to the unclear structure of the RNA interacting pocket in the HuR protein26. The only small molecule advanced to early phase clinical trial is MS-444, a myosin light chain kinase inhibitor. MS-444 interferes nuclear-cytoplasmic translocation of HuR26, however it has multiple issues on specificity and toxicities. Our data showed that a novel compound KH-3 works in a different mechanism from MS-444. KH-3 binds to RNA recognition motifs (RRM1/2) of HuR protein, and interrupts HuR-mRNA interaction. Consequently, KH-3 inhibited pancreatic cancer cell EMT, migration, invasion, and CSCs, mimicking the in vitro effects of HuR knockdown. KH-3 also inhibited cell viability dependent on endogenous HuR levels. As an HuR inhibitor, KH-3 may comprehensively impact pancreatic cancer cell proliferation, migration and EMT.
Importantly, KH-3 showed good target specificity to HuR. Our in vitro data showed that when HuR was knocked down in pancreatic cancer cells, KH-3 lost its inhibitory activity against cell migration. In vivo, KH-3 inhibited a HuR positive tumor growth and metastasis, but did not inhibit the formation of HuR knocked-down tumors, nor inhibited the growth of these tumors. Good target specificity is likely to predict less off-target effects that are undesirable in drug development.
As a single-drug treatment, KH-3 inhibited pancreatic cancer growth and reduced metastasis rate in vivo, with good tolerability in mice at the dose of 100 mg/kg 3x IP weekly. Combination treatments with currently available chemo-regimens are worth testing for synergy. Being the first of its class, KH-3 showed the promise of using target-specific small molecule to inhibit HuR function by direct interruption of HuR-RNA binding. Pharmacological inhibition of HuR holds the promise to comprehensively inhibit pancreatic cancer progress, metastasis, drug resistance and tumor recurrence.
Supplementary Material
Acknowledgement
This work was supported in part by National Institutes of Health grant R21 CA198265 (to QC) and R01 CA191785 (to LX, JA). We thank Dr. Shrikant Anant at the University of Kansas Cancer Center for providing cells used in our experiments. We thank Dr. Lisa Zhang at the University of Kansas Medical Center for providing HuR antibody, and help with troubleshooting in immune-blots experiments. We thank our previous postdoctoral fellow Dr. Ying Zhang for exploratory work on this project.
Abbreviations:
- HuR
H-antigen R
- EMT
epithelial to mesenchymal transition
- CSC
stem-like cancer cells
- RNP-IP
ribonucleoprotein immunoprecipitation
- RBP
RNA binding protein
- ARE
adenine- and uridine-rich elements (AREs)
- UTR
untranslated region
- RRM
RNA recognition motifs
- MTT
3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide
- KO
knockout
- WT
wild type
Footnotes
Conflict of Interest: The authors have no conflict of interest in relating to this project.
References
- 1.ACS. American Cancer Society. Cancer Facts and Figures 2018. 2018.
- 2.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim R. FOLFIRINOX: a new standard treatment for advanced pancreatic cancer? Lancet Oncol 2011;12:8–9. [DOI] [PubMed] [Google Scholar]
- 4.Lee CJ, Dosch J, Simeone DM. Pancreatic cancer stem cells. J Clin Oncol 2008;26:2806–12. [DOI] [PubMed] [Google Scholar]
- 5.Li L, Hao X, Qin J, et al. Antibody against CD44s inhibits pancreatic tumor initiation and postradiation recurrence in mice. Gastroenterology 2014;146:1108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010;29:4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009;9:265–73. [DOI] [PubMed] [Google Scholar]
- 8.Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 2004;118:277–9. [DOI] [PubMed] [Google Scholar]
- 9.Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 2009;69:5820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Radisky DC, Levy DD, Littlepage LE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005;436:123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nabors LB, Gillespie GY, Harkins L, et al. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3’ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res 2001;61:2154–61. [PubMed] [Google Scholar]
- 13.Lopez de Silanes I, Fan J, Yang X, et al. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene 2003;22:7146–54. [DOI] [PubMed] [Google Scholar]
- 14.Fialcowitz-White EJ, Brewer BY, Ballin JD, et al. Specific protein domains mediate cooperative assembly of HuR oligomers on AU-rich mRNA-destabilizing sequences. J Biol Chem 2007;282:20948–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doller A, Pfeilschifter J, Eberhardt W. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal 2008;20:2165–73. [DOI] [PubMed] [Google Scholar]
- 16.Barker A, Epis MR, Porter CJ, et al. Sequence requirements for RNA binding by HuR and AUF1. J Biochem 2012;151:423–37. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Guo Y, Chu H, et al. Multiple Functions of the RNA-Binding Protein HuR in Cancer Progression, Treatment Responses and Prognosis. Int J Mol Sci 2013;14:10015–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Annabi B, Currie JC, Moghrabi A, et al. Inhibition of HuR and MMP-9 expression in macrophage-differentiated HL-60 myeloid leukemia cells by green tea polyphenol EGCg. Leuk Res 2007;31:1277–84. [DOI] [PubMed] [Google Scholar]
- 19.Tran H, Maurer F, Nagamine Y. Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase 2. Mol Cell Biol 2003;23:7177–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong R, Lu JG, Wang Q, et al. Stabilization of Snail by HuR in the process of hydrogen peroxide induced cell migration. Biochem Biophys Res Commun 2007;356:318–21. [DOI] [PubMed] [Google Scholar]
- 21.Richards NG, Rittenhouse DW, Freydin B, et al. HuR status is a powerful marker for prognosis and response to gemcitabine-based chemotherapy for resected pancreatic ductal adenocarcinoma patients. Ann Surg 2010;252:499–505; discussion 505–6. [DOI] [PubMed] [Google Scholar]
- 22.Fan XC, Steitz JA. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc Natl Acad Sci U S A 1998;95:15293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez de Silanes I, Lal A, Gorospe M. HuR: post-transcriptional paths to malignancy. RNA Biol 2005;2:11–3. [DOI] [PubMed] [Google Scholar]
- 24.Lal A, Kawai T, Yang X, et al. Antiapoptotic function of RNA-binding protein HuR effected through prothymosin alpha. EMBO J 2005;24:1852–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotta-Loizou I, Giaginis C, Theocharis S. Clinical significance of HuR expression in human malignancy. Med Oncol 2014;31:161. [DOI] [PubMed] [Google Scholar]
- 26.Meisner NC, Hintersteiner M, Mueller K, et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat Chem Biol 2007;3:508–15. [DOI] [PubMed] [Google Scholar]
- 27.Muralidharan R, Mehta M, Ahmed R, et al. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci Rep 2017;7:9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lang M, Berry D, Passecker K, et al. HuR Small-Molecule Inhibitor Elicits Differential Effects in Adenomatosis Polyposis and Colorectal Carcinogenesis. Cancer Res 2017;77:2424–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanco FF, Preet R, Aguado A, et al. Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis. Oncotarget 2016;7:74043–74058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu X, Lan L, Wilson DM, et al. Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS Chem Biol 2015;10:1476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X, Gardashova G, Lan L, et al. Targeting the interaction between RNA-binding protein HuR and FOXQ1 suppresses breast cancer invasion and metastasis. Commun Biol 2020;3:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hassan MQ, Gordon JA, Lian JB, et al. Ribonucleoprotein immunoprecipitation (RNP-IP): a direct in vivo analysis of microRNA-targets. J Cell Biochem 2010;110:817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vo DT, Abdelmohsen K, Martindale JL, et al. The oncogenic RNA-binding protein Musashi1 is regulated by HuR via mRNA translation and stability in glioblastoma cells. Mol Cancer Res 2012;10:143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dai W, Zhang G, Makeyev EV. RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res 2012;40:787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costantino CL, Witkiewicz AK, Kuwano Y, et al. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR Up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res 2009;69:4567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zarei M, Lal S, Parker SJ, et al. Posttranscriptional Upregulation of IDH1 by HuR Establishes a Powerful Survival Phenotype in Pancreatic Cancer Cells. Cancer Res 2017;77:4460–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng W, Furuuchi N, Aslanukova L, et al. Elevated HuR in Pancreas Promotes a Pancreatitis-Like Inflammatory Microenvironment That Facilitates Tumor Development. Mol Cell Biol 2018;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lal S, Cheung EC, Zarei M, et al. CRISPR Knockout of the HuR Gene Causes a Xenograft Lethal Phenotype. Mol Cancer Res 2017;15:696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hung KF, Sun YC, Liou HM, et al. Human antigen R protein modulates vascular endothelial growth factor expression in human corneal epithelial cells under hypoxia. J Formos Med Assoc 2019. [DOI] [PubMed] [Google Scholar]
- 41.Abdelmohsen K, Kuwano Y, Kim HH, et al. Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol Chem 2008;389:243–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shang J, Zhao Z. Emerging role of HuR in inflammatory response in kidney diseases. Acta Biochim Biophys Sin (Shanghai) 2017;49:753–763. [DOI] [PubMed] [Google Scholar]
- 43.Woodhoo A, Iruarrizaga-Lejarreta M, Beraza N, et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology 2012;56:1870–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.