

Abstract
Background
Human breast milk (HBM) contains optimal nutrients for infant growth. Probiotics are used to prevent disease and, when taken by the mother, they may affect infant microbiome as well as HBM. However, few studies have specifically investigated the effect of probiotic intake by the mother on HBM and infant microbiota at genus/species level. Therefore, we present a comprehensive analysis of paired HBM and infant feces (IF) microbiome samples before and after probiotic intake by HBM-producing mothers.
Methods
Lactating mothers were administered with Lactobacillus rhamnosus (n = 9) or Saccharomyces boulardii capsules (n = 9), for 2 months; or no probiotic (n = 7). Paired HBM and IF samples were collected before and after treatment and analyzed by next-generation sequencing.
Results
Forty-three HBM and 49 IF samples were collected and sequenced. Overall, in 43 HBM samples, 1,190 microbial species belonging to 684 genera, 245 families, 117 orders, and 56 classes were detected. In 49 IF samples, 372 microbial species belonging to 195 genera, 79 families, 42 orders, and 18 classes were identified. Eight of 20 most abundant genera in both HBM and IF samples overlapped: Streptococcus (14.42%), Lactobacillus, Staphylococcus, and Veillonella, which were highly abundant in the HBM samples; and Bifidobacterium (27.397%), Bacteroides, and Faecalibacterium, which were highly abundant in the IF samples. Several major bacterial genera and species were detected in the HBM and IF samples after probiotic treatment, illustrating complex changes in the microbiomes upon treatment.
Conclusion
This is the first Korean microbiome study in which the effect of different probiotic intake by the mother on the microbiota in HBM and IF samples was investigated. This study provides a cornerstone to further the understanding of the effect of probiotics on the mother and infant microbiomes.
Keywords: Human Breast Milk, Infant Feces, Breastfeeding, Probiotics, Microbiota, Korean
Graphical Abstract
INTRODUCTION
Human breast milk (HBM) contains optimal nutrients for infant growth. It is nutritionally and immunologically superior to any other artificial nutrition products. It is also fresh and readily fed to the infant. Consequently, the American Academy of Pediatrics and the World Health Organization recommend breastfeeding of all infants for at least 4–6 months.1,2,3
The microbiome is associated with various phenomena and diseases in humans. The understanding of mechanisms underpinning these effects constitutes a hot research topic. In addition to being linked with gut-related diseases, such as intestinal infection, inflammatory bowel disease, and colorectal cancer,4,5,6 the microbiome affects multiple sclerosis, rheumatoid arthritis, systemic lupus erythematosus,7 atopic dermatitis,8,9,10 metabolic diseases such as diabetes, obesity,11,12,13 asthma,14 and breast cancer.15
Probiotics have been shown to be effective in preventing diseases by compensating for pathological problems associated with gut dysbiosis and by balancing microbial composition in the intestinal tract.16 Of note, administration of probiotics during the lactation period changes the composition of HBM microbiome.17,18,19,20,21,22,23,24,25
The composition of a neonatal intestinal microbiome depends on several factors, such as the maternal age, gestational age,26 type of delivery,27,28,29 and exposure to the external environment.30 However, the most important factor is lactation.27,29,31 The HBM microbiome passes with the milk to the infant, and plays an important role in the primary colonization of the newborn intestine. However, little is known about the effect of probiotic intake by the mother on the HBM microbiome and on the intestinal microbiome of a nursed infant. Further, to the best of our knowledge, no Korean microbiome studies to date have investigated the HBM and infant feces (IF) microbiomes together.
Here, we present a comprehensive analysis of paired HBM and IF microbiome samples before and after probiotic intake by HBM-producing mothers in this study.
METHODS
Participants and sample collections
The participants were healthy women (19–45 year old) who were currently breastfeeding their children. They agreed to take part in the study between June 2018 and June 2019 at the HBM Research Institute of Chung-Ang University, Seoul, Korea. Participants who had twin pregnancy, pregnancy diseases (eclampsia and gestational diabetes that required insulin administration), other chronic illnesses, who used probiotics or herbal medicines during the study period, less than 1 week after the start of breastfeeding (colostrum), or who were planning to start solid food within the study period were excluded.
Except for 1 participant who dropped out of the control group, 25 pairs of breastfeeding mothers and their infants were enrolled in the study (Supplementary Table 1). The pairs were randomly assigned to the Lactobacillus casei variety rhamnosus group (administered Ramnos capsule [1 capsule = 2 × 108 as living germs 250 mg, Hanwha Pharma Co., Seoul, Korea]), the Saccharomyces boulardii group (administered Bioflor capsule [1 capsule = 5 × 109 as living germs 282.5 mg, Kuhnil Pharma Co., Seoul, Korea]), and the control group (no treatment). The probiotics used in this study are the two most prescribed probiotics in South Korea and the dosage was based on the product recommendations. The mothers in the probiotic groups were administered probiotics orally, twice a day, for 2 months (120 capsules in total). HBM and IF samples were obtained two times per pair: once before the probiotic treatment (before, 1st) and once after the probiotic treatment (after, 2nd).
Before the HBM sample collection, the nipple and areola were cleaned by wiping with a swab soaked in sterile water. The first few drops of HBM were discarded to prevent contamination. Subsequently, 10 mL of HBM were collected directly into a sterile plastic bag, at an outpatient breast-feeding room, by using a manual breast-pump. The stool samples were collected over 30 g. All samples were immediately frozen at −80°C.
Information on the past medical history, drug administration, fertility history, maternal age, gestational age, type of delivery, infant sex, birth weight, and dietary habits of the participants was also collected.
DNA extraction and next-generation sequencing for microbiome analysis
Microbial DNA was extracted using a PowerMax Soil DNA Isolation kit (MO BIO, Carlsbad, CA, USA), following the manufacturer's instructions. For sequencing, the samples were prepared according to the Illumina 16S Metagenomic Sequencing Library protocols (Illumina, San Diego, CA, USA). Briefly, the V3 and V4 region was amplified using primers 519F and 816R. The DNA quality was determined by PicoGreen and Nanodrop. Input gDNA (10 ng) was amplified by polymerase chain reaction (PCR) with the following barcoded fusion primers: 341F, 5′-CCTACGGGNGGCWGCAG-3′, and 806R, 5′-GACTACHVGGGTATCTAATCC-3′. The final purified product was quantified by quantitative PCR (qPCR), according to the qPCR Quantification Protocol Guide (KAPA Library Quantification kits for Illumina Sequencing platforms) and evaluated by using the LabChip GX HT DNA High Sensitivity Kit (PerkinElmer, Waltham, MA, USA). Next, paired-end (2 × 300 bp) sequencing was performed at Macrogen (Seoul, Korea), by using the MiSeq™ platform (Illumina).
Identification of microbial species
Fastq files were generated from raw MiSeq sequences by Real-Time Analysis and bcl2fastq 2.20.0.422. FLASH 1.2.1132 was used to merge raw sequences from multiple samples. The merged raw sequences were filtered by using CD-HIT-OUT,33 to remove low-quality bases and chimeric sequences, and were then clustered at 97% similarity threshold to generate operational taxonomic units (OTUs). Representative sequences from each OTU were matched to the reference database, NCBI 16S Microbial, using BLASTN 2.4.0.34 Top matched sequences were used for taxonomic assignment. When the query coverage and identification of the best hit were below 85%, it was concluded that the queried sequence was not similar to any other sequence in the database.
Microbiome data analysis
Sample Shannon index was calculated using the phylogenetic distance and the number of observed OTUs and analysis of variance (ANOVA) test was conducted with R package 4.0.2. Rarefaction analyses were conducted based on customized perl scripts which can calculate average, minimum, and maximum values of OTUs in each group (in total, 12 groups) for each 100 reads. ANOVA test was conducted based on 12 groups of rarefaction analysis results.
Comparative microbiome analysis
To analyze the microbiome data, QIIME 1.935 and customized perl scripts were utilized for 1) the analysis of the overall characteristics of microbial communities in HBM and IF samples; 2) comparison of HBM and IF microbiomes in the three treatment groups; 3) comparisons of HBM and IF microbiomes before probiotic administration and in the control group with those after probiotic administration; and 4) comparison of the microbial community structure in the three groups. For detailed analysis, customized perl scripts were developed to combine microbial species into groups, and calculate the abundance and proportion of the bacteria on the species, genus, family, and order levels. Results of each script analyses were exported to Excel format for additional analyses, e.g., identification of the top 20 most prevalent genera in the groups.
Ethics statement
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Institutional Review Board (IRB) of Chung-Ang University Hospital, Seoul, Korea (IRB No. 1820-001-313). Informed consent was submitted by all subjects when they were enrolled.
RESULTS
The overall characteristics of microbial communities in HBM and IF samples
Overall, 25 pairs were included in the study (Supplementary Table 1). They consist of three groups: the L. casei variety rhamnosus group (n = 9), the S. boulardii group (n = 9), and the control group (n = 7). Further, HBM samples of seven participants were not properly collected, or processed during preparation and sequencing and one IF sample collection was missed. Consequently, 43 HBM and 49 IF samples were collected and sequenced (Supplementary Table 2).
In total, 16,623,119 reads were obtained from the 43 HBM and 49 IF samples displaying 180,686 average reads per sample (Supplementary Table 2). In total, 8,563 types of OTUs were clustered from these reads. Based on Shannon indexes calculated for each sample, difference of Shannon indexes between HBM and fecal samples is significant (one-way ANOVA test, P value = 3.16e-06) as expected; while differences among three groups, L. rhamnosus, S. boulardii, and control groups, and between before- and after-treatments displayed not significant (One-way ANOVA test, P value = 0.91 and 0.977, respectively). Three-way ANOVA test with interactions among variables present that differences of HBM-fecal and treatment timepoint and the three groups and treatment are slightly significant (P value = 0.014 and 0.034, respectively). In addition, rarefaction analysis based on the 12 groups defined by sample type, probiotics, and treatment, displays (Fig. 1). Based on these trend of microbiome differences, we investigated the configuration of bacterial species in detail.
Fig. 1. Rarefaction analyses based on the 12 groups defined by sample type, probiotics, and treatment. X-axis is number of sequence reads per sample and Y-axis is observed OTUs (A) displays rarefraction analysis results of the six groups of human BM samples and (B) presents rarefraction analysis results of the six groups of infant fecal samples. X-axis indicates sequences per samples and Y-axis means observed OTUs.

OTU = operational taxonomic unit, BM = breast milk.
Overall, in 43 HBM samples, 1,190 microbial species belonging to 684 genera, 245 families, 117 orders, and 56 classes were detected. The most abundant genera, accounting for a large proportion of the microbial population in these samples, were Streptococcus, Lactobacillus, Staphylococcus, Acinetobacter, and Bacteroides (Fig. 2A and Supplementary Table 3). The most frequent detected genera in these samples were Bacteroides, Streptococcus, Staphylococcus, Lactobacillus, and Corynebacterium (Supplementary Table 4). Lactobacillus, Staphylococcus, and Streptococcus genera were both abundant and commonly detected in the HBM samples.
Fig. 2. Distribution of microbial genera detected in HBM and IF samples. (A) Pie graph displaying the proportion of genera detected in HBM samples. (B) Pie graph displaying the proportion of genera detected in IF samples. (C) X-axis of graph shows genera name identified from both HBM and IF samples. Y-axis indicates relative proportion of each genus. Streptococcus (14.415%) max proportion identified from HBM samples, Bifidobacterium (27.397%) max proportion identified from IF samples, respectively.

HBM = human breast milk, IF = infant fecal.
In 49 IF samples, 372 microbial species belonging to 195 genera, 79 families, 42 orders, and 18 classes were detected. The most abundant genera in these samples were Bifidobacterium, Bacteroides, Escherichia, Enterococcus, and Lactobacillus (Fig. 2B and Supplementary Table 5). The most frequent detected genera in these samples were Streptococcus, Escherichia, Bifidobacterium, Lactobacillus, and Bacteroides (Supplementary Table 6). All of the latter, except for Streptococcus, were among the top 5 most abundant genera.
Eight of the 20 most abundant genera in HBM and IF samples overlapped: Streptococcus (14.415%), Lactobacillus, Staphylococcus, and Veillonella, which were highly abundant in HBM samples; and Bifidobacterium (27.397%), Bacteroides, and Faecalibacterium, which were highly abundant in IF samples (Fig. 2C). The relative abundance of Blautia was similar in both sample types (Fig. 2C).
The effect of two types of probiotics, L. rhamnosus and S. boulardii, on mother and infant microbiota
The microbiomes of the control group, L. rhamnosus group, and S. boulardii group were analyzed. In the control group, the relative abundances of ten genera (Faecalibacterium, Barnesiella, Ruminococcus, Acinetobacter, Gemmiger, Tidjanibacter, Pseudoflavonifractor, Fournierella, Butyricicoccus, and Erysipelatoclostridium) decreased sharply to almost zero in HBM samples after 2 months (Fig. 3A). In contrast, the relative abundances of Parabacteroides, Rothia, Aerosakkonema, Selenomonas, Dialister, Megasphaera, Escherichia, Veillonella, Prevotella, and Streptococcus increased in that time period. Interestingly, the relative abundance of Staphylococcus genus decreased after 2 months but remained relatively high, whereas the genera Bifidobacterium and Lactobacillus exhibited a reverse trend. In the IF samples, the relative abundances of Bifidobacterium, Enterococcus, and Faecalibacterium decreased by more than half, and those of Clostridium, Erysipelatoclostridium, Abyssivirga, Veillonella, and Phascolarctobacterium decreased to zero over 2 months (Fig. 3B). In contrast, Bacteroides, Blautia, Lactobacillus, Lachnoclostridium, and Gemmiger were detectable only after 2 months, and the relative abundances of Escherichia, Streptococcus, Enterobacter, and Alistipes genera increased over the 2-month period.
Fig. 3. Bacterial genera detected in the treatment groups, and their relative proportions in the microbiome. Readings before and after probiotic treatment are shown. (A) Relative genus proportions in HBM samples in the control group. (B) Relative genus proportions in IF samples in the control group. (C) Relative genus proportions in HBM samples in the L. rhamnosus group. (D) Relative genus proportions in IF samples in the L. rhamnosus group. (E) Relative genus proportions in HBM samples in the S. boulardii group. (F) Relative genus proportions in IF samples in the S. boulardii group. Y-axis indicates the proportion of genera of microbial species of which range is from 0 to 1.

HBM = human breast milk, IF = infant fecal.
After 2-month administration of L. rhamnosus, in the HBM samples, the relative abundances of Streptococcus, Staphylococcus, Bacteroides, Prevotella, Muribaculum, and Corynebacterium genera decreased (Fig. 3C). The Plesiomonas, Lachnoclostridium, Dialister, Blautia, Megasphaera, Cetobacterium, Akkermansia, and Bifidobacterium genera became detectable only after 2 months in these HBM samples. In the IF samples, those of Bifidobacterium, Enterococcus, Parabacteroides, Blautia, and Veillonella genera decreased; and the Enterobacter, Faecalibacterium, Lachnoclostridium, and Erysipelatoclostridium genera ceased to be detectable after 2 months (Fig. 3D). Further, the relative abundances of Escherichia, Staphylococcus, Streptococcus, Lactobacillus, Bacteroides, and Intestinibacter genera increased after 2 months.
After 2-month administration of S. boulardii, the relative abundances of Streptococcus, Lactobacillus, Staphylococcus, Prevotella, and Corynebacterium genera decreased in the HBM samples (Fig. 3E). In addition, the relative abundances of Acinetobacter, Bacteroides, Faecalibacterium, Blautia, and Bifidobacterium increased after 2 months in the HBM samples. In the IF samples, the relative abundances of Bacteroides, Escherichia, Enterococcus, and Blautia genera decreased after 2 months (Fig. 3F). In contrast, the relative abundances of Bifidobacterium, Lactobacillus, Streptococcus, Enterobacter, Veillonella, Faecalibacterium, Erysipelatoclostridium, and Phascolarctobacterium genera increased after 2 months IF samples.
Hierarchical clustering of microbiota changes in six groups (two probiotic treatments and one control, two sample types each) revealed that all HBM and IF samples, except for the control samples, clustered together (Fig. 4). Specifically, the relative abundances of Enterococcus, Blautia, and Lachnoclostridium genera decreased after probiotic treatment in IF samples, and those of Streptococcus and Phascolactobacterium genera increased. Further, the relative abundances of Akkermansia, Blautia, and Bifidobacterium genera increased after probiotic treatment in HBM samples, and those of Prevotella, Streptococcus, Anaerococcus, Corynebacterium, Sphingomonas, and Staphylococcus genera decreased in these samples. Of note, the abundance of Streptococcus genus showed opposing trends in HBM and IF samples: it decreased before and after probiotic administration, respectively (Figs. 2E and 3C). The relative abundance of Bifidobacterium increased upon probiotic treatment in both sample types, except in the IF samples in S. boulardii group (Figs. 2E and 3C).
Fig. 4. Hierarchical clustering heat-map of genera that became more or less abundant under different experimental conditions. Microbial genera detected in the current study are shown along the y-axis. The experimental conditions are shown along the x-axis. Hierarchical clustering dendrograms are displayed on the left and top. Blue and red colors indicate a decrease or increase of a population in the microbiome, respectively, upon probiotic treatment.

HBM = human breast milk, IF = infant fecal, Control = the control group, LR = the L. rhamnosus group, SB = the S. boulardii group.
Species-level comparison of microbiomes in the control, L. rhamnosus, and S. boulardii groups
To evaluate the changes in microbiomes in the three groups (the control, L. rhamnosus, and S. boulardii groups) in detail, species compositions under before and after treatment were compared.
In the control group, in HBM samples, 94 microbial species were detected both at time 0 and after 2 months; 77 species were detected only at time 0 and 301 species were detected only after 2 months (Fig. 5A). Similar, 128 species were detected in IF samples at both time points; 47 and 102 microbial species were detected only at time 0 or after 2 months, respectively (Fig. 5A). The changes in bacterial species composition during the 2 months in HBM and IF samples were substantial.
Fig. 5. Comparison of microbial community structure under three experimental conditions. Blue boxes with transparent effect, analysis of HBM samples; green boxes, analysis of IF samples. Venn diagrams in each box show detected bacterial species before and after probiotic treatment. (A) Analysis of the control group samples. (B) Analysis of the L. rhamnosus group samples. (C) Analysis of the S. boulardii group samples.

HBM = human breast milk, IF = infant fecal.
Fifty-one microbial species were specific to HBM samples in the control group (Supplementary Table 7). The most common species among these were Cutibacterium acnes (present in 8 out of 9 samples) and Streptococcus oralis, which were also highly abundant among these species. In addition, 85 bacterial species were specific to IF samples in the control group (Supplementary Table 8), with Streptococcus lactarius detected in 12 out of 13 samples. Further, 43 microbial species were detected in both HBM and IF samples (Supplementary Table 9), and were commonly detected in the control group: Escherichia fergusonii was detected in 21 out of 22 samples, and Bifidobacterium longum and B. breve were most frequently detected in 43 samples (in 17 and 13 samples, respectively).
In the L. rhamnosus group, in HBM samples, 197 microbial species were detected both before and after probiotic treatment; 486 species were detected only before the treatment; and 122 species were detected only after the treatment (Fig. 5B). These numbers were much higher than those in the control group samples, indicating that the species diversity in HBM samples in the L. rhamnosus group was higher than that in the control group. Prevotella copri and Corynebacterium tuberculostearicum were detected in 12 out of 17 HBM samples and ranked third and fourths in terms of frequency (Supplementary Table 10). In the L. rhamnosus group, in IF samples, 141 species were detected both before and after treatment; 48 and 66 species were only detected before and after probiotic treatment, respectively (Fig. 5B). Among the common detected species (141), 59 species were only detected in IF samples (Supplementary Table 11).
The changes in proportions of microbial species under each condition exhibited different trends. Before L. rhamnosus treatment, HBM samples contained less microbial species with a read number below 32 (log2 value is 5) than those with a read number above 32 (Fig. 5B). This was also the case for the common species in HBM samples and microbial species detected in IF samples only after taking L. rhamnosus (Fig. 5B). Further, 82 out of 256 microbial species (32.03%) were detected in both HBM and IF samples before and after L. rhamnosus treatment (Fig. 5B and Supplementary Table 12).
In the S. boulardii group, in HBM samples, 208 microbial species were detected both, at time 0 and after 2 months; 147 species were detected only before treatment; and 280 species were detected only after treatment (Fig. 5C). In IF samples, 143 species were detected both, before and after S. boulardii treatment; 49 and 43 species were detected only before or after treatment, respectively (Fig. 5C). Further, 135 species, among the 208 species commonly detected before and after treatment in HBM samples, were only detected in HBM samples (Supplementary Table 13). C. acnes was commonly detected in 15 out of 17 samples, and ranked 21st with respect to abundance. Further, 70 species, among 143 species commonly detected in IF samples before and after treatment, were only detected in IF samples (Supplementary Table 14). Taken together, these observations indicate that in the S. boulardii group, bacterial species composition changed dynamically in HBM and IF samples.
In addition, 73 out of 278 microbial species (26.26%) were detected in both HBM and IF samples before and after S. boulardii treatment (Fig. 5C and Supplementary Table 15). E. fergusonii and Streptococcus salivarius were abundant, detected in 29 out of 35 samples, with a large proportion in these microbiomes, ranking second and sixth in abundance, respectively (Supplementary Table 15).
Overall, 105 microbial species were commonly detected under the three conditions. Among these, 31 species were detected in all three groups, and were common microbial species in both HBM and IF samples in the 2-month period (Fig. 6). Eight microbial species were detected only in the control group, indicating that they may be affected by probiotic intake (Fig. 6). In addition, 21 and 15 species were only detected in the L. rhamnosus and S. boulardii groups, respectively (Fig. 6), which can be promoted by probiotics in some ways.
Fig. 6. Microbial species that were unique or common to the three experimental groups. Venn diagram in the center displays species detected under the three conditions. Species specific to the three groups and common species are listed.

DISCUSSION
HBM is an important source of colonizing microflora for the infant. We here investigated the effect of probiotic intake by the mother on IF microflora. We show that probiotic intake had profound impact on the composition of HBM and IF microflora, highlighting the importance of maternal nutrition on infant microflora.
Lactobacillus, Staphylococcus, and Streptococcus genera were abundant and commonly detected in 43 HBM samples analyzed in the current study. Similar, in another study, Weissella, Leuconostoc, Staphylococcus, Streptococcus, and Lactococcus genera were predominant in colostrum samples, and Streptococcus and Staphylococcus were abundant.36 Pseudomonas, Staphylococcus, and Streptococcus genera were predominant in ten human milk samples analyzed in another study,37 with two of these genera also commonly detected in the current study. In addition, Staphylococcus is a common constituent of the milk microbiota, as determined by both, culture-dependent and independent investigations.36,38,39,40 This indicates that the overall trend of predominant bacterial genera in different HBM samples was similar. In addition, the dataset in the current study was larger than that of previous studies.36,37
Seven out of twenty most abundant genera in both HBM and IF samples overlapped. These were: Streptococcus (14.415%), Lactobacillus, Staphylococcus, and Veillonella, which were highly abundant in the HBM samples; and Bifidobacterium (27.397%), Bacteroides, and Faecalibacterium, highly abundant in the IF samples. The major bacterial genera detected in both 43 HBM and 49 IF samples were Streptococcus, Lactobacillus, Staphylococcus, and Veillonella, similar to those reported in previous studies.38,39,40 The analysis of a large number of microbiome samples indicated the presence of some common bacterial genera, including Bifidobacterium and Lactobacillus. That is different from the findings of previous studies that emphasize “microbial diversity” instead of “core microbiome.”39,41,42,43 Despite some dynamic differences in bacterial composition in individual samples, the data from the current study provide evidence on the existence of a “core microbiome” in HBM and IF.
Staphylococcus is a common constituent of milk microbiota.38,39,40 In a study of breast-fed Swedish infants, delivered both vaginally and by caesarean section, 100% of IF were colonized by Staphylococcus from day 3 of life. It has been suggested that Staphylococcus epidermis in the breast milk may originate from the maternal skin. Its presence appears to be biologically relevant, as it is the predominant species in both HBM and IF of breast-fed infants, but is less common in the stool of formula-fed infants.38,39,40,41,42,43,44 Microbial genera associated with the oral cavity, such as Streptococcus and Veillonella,36,37,45 were also detected in the current study: unlike the former, the latter showed different patterns of changing abundance in HBM and IF samples under the conditions tested.
The detection of high levels of the typical inhabitants of the skin and oral cavity in the current study may imply that their origin in samples was a secondary contamination. However, anaerobic gut-associated microbiota, such as Bacteroides, Blautia, Faecalibacterium, Ruminococcus, Enterococcus, and Escherichia, were also detected, here and in other studies.36,39 Bifidobacterium was also consistently detected in IF samples, as in previous studies39,45; however, several studies did not report the presence of Bifidobacterium,36,46 which should be investigated further.
In the current study, the IF microbiota were less diverse than the HBM microbiota. This was in agreement with a previous study.37 The Streptococcus, Lactobacillus, Staphylococcus, Bacteroides, Bifidobacterium, Veillonella, and Faecalibacterium genera were abundant in both, HBM and IF samples. In previous studies, the Lactobacillus, Staphylococcus, Enterococcus, and Bifidobacterium genera were frequently detected in both, HBM and IF samples.44,47 One of the reasons why more genera were detected in the current study than in previous studies is the large number of samples analyzed. Other reasons include environmental differences, also genetic background, suggesting a research topic for further evaluation. Additional studies are required to understand the effect of genetic background on the HBM and IF microbiomes.
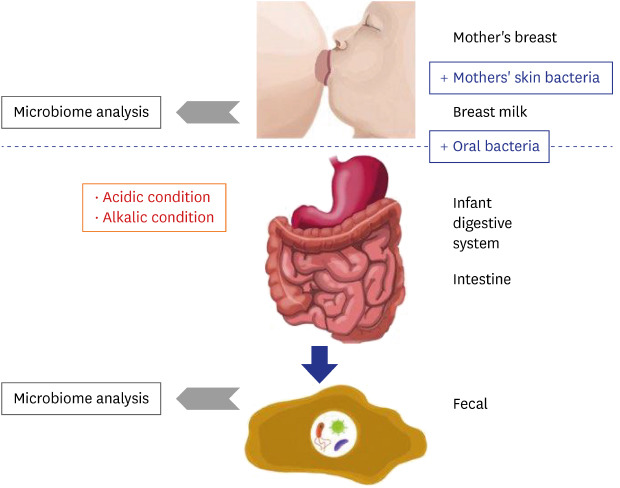
As shown in Fig. 4, microbiome changes in the HBM samples were more dramatic than those in IF samples, indicating that the HBM microbiome may be affected by both, various environmental factors and individual genetic background. For example, the HBM microbiome may be easily affected by the skin microbiome, which is directly exposed to the environment. On the other hand, the microbiome in IF first passes the infant digestive system, resulting in relatively less dynamic changes as in Fig. 7. This trend is an important point for understanding the differences in HBM and IF sample microbiomes.
Fig. 7. Proposed model for the establishment of infant fecal and human breast milk microbiomes. Blue boxes indicate factors that promote microbiome expansion and red boxes indicate environmental conditions that are detrimental to the microbiome. Grey arrow point to next generation sequencing microbiome analysis.

Administration of probiotics during the lactation period changed the composition of HBM and IF microbiomes in various ways. Two possible mechanisms explain these changes: 1) dynamic changes in the microbiome could be triggered indirectly, by extra-intestinal changes in physiological conditions17,18,19; and 2) the changes could be elicited directly, by a series of events. As an example of the latter, pregnancy triggers an increase of progesterone levels, leading to increased intestinal permeability, and causing microbiome inflow from the intestine to the bloodstream, to be finally transferred to the mammary gland.20,21,22,23,24,25
Twenty-one and 15 species were only detected in the L. rhamnosus and S. boulardii groups, respectively. Their abundance in the microbiome might hence increase in response to probiotic intake. In the L. rhamnosus group, three Bacteroides species were detected: Bacteroides nordii, which was detected in the human feces48; Bacteroides xylanisolvens, which was isolated from the human intestine; and Bacteroides caecimuris, which was detected in the mouse intestine.49 This suggested that these species might have specific mechanisms to L. rhamnosus enrichment. In S. boulardii group, three Ruminococcus species, which are typically detected among the human gut microbiota, were detected.
The HBM microbiome plays an important role in the primary colonization of a newborn's gut. Two hypotheses explain how the colonization could occur: 1) HBM microbes directly colonize an infant's gut44,50,51,52; and 2) HBM microbes act as pre-biotics, with HBM oligosaccharides indirectly affecting the intestine and extra-intestinal environment.44,50,51 In our study, we did not observe consistent differences or changes in the individual HBM and IF microbiomes, indicating that the HBM microbiota may directly colonize the infant's gut. However, as mentioned above, as yet unidentified processes of microbial transfer between the mother and infant could affect the observed complex changes in the HBM and IF microbiomes.
Based on the findings of the current and former studies, we propose a model of the interaction between the mother's and infant's microbiomes. The microbiome of the mother's breast is transferred to the infant's mouth, intermixed with the mother's skin bacteria and infant's oral bacteria. Many bacterial species are subsequently eliminated under the generally unpermissive acidic and alkaline conditions of the infant digestive system (Fig. 7). After removal of these species, some of the remaining microbes settle in the intestine together with the pre-existing bacteria (Fig. 7). Consequently, the microbiome remnants are detected in the IF samples (Fig. 7).
Based on the detected bacterial genera or species, a matrix of bacterial characteristics can be generated to test whether the model explains the existence of each genus or species or not. For example, Lactobacillus genus is found in both HBM and IF samples, but is proportionally more abundant in the former. Because Lactobacillus genus is resistant to acidic conditions,52 these bacteria can be transferred from the mother to the infant. A reduced relative abundance can also be explained by loss through transfer from the mother to the infant. Even though each species in the same genus may exhibit different characteristics, e.g., resistance to acidic or alkaline conditions, this model can be used to gain insight into the HBM and IF microbiomes, and into the mechanism of their initial formation and dynamics.
This study had several limitations; first, we only focused on the microbial species. However, one of the probiotics tested herein was S. boulardii (Bioflor), a eukaryote. Therefore, changes in the population of eukaryotic species, especially fungal species, should also be investigated. Such analysis may provide additional detailed insight into the mechanisms that drive microbiome changes upon probiotic treatment. Second, the intestinal microbiome may depends on several factors, such as the delivery type, maternal age, antibiotics treatment in infants and external environments, but this differences were not reflected.26,27,28,29,30
In conclusion, this is the first Korean microbiome study in which the effect of different probiotic intake by the mother on the microbiota in HBM and IF samples were investigated. We showed that oral probiotic supplementation in mothers during the lactation period increases the HBM Lactobacillus levels, and changes the microbial composition and diversity in HBM and IF. These changes may have important consequences for infant health. The maintenance of a healthy and balanced intestinal microbiome during the lactation period, possibly via administration of validated probiotic products, should be considered as an important approach positively influencing the HBM microbiome.
ACKNOWLEDGMENTS
The authors thank to the lactating mothers for their participation in this study, and thank Macrogen (Seoul, Korea) for help in data analysis. We also gratefully acknowledge the support of Editage (www.editage.co.kr) for English language editing.
Footnotes
Funding: This research was funded by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT, No. NRF-2019R1F1A1059569).
Disclosure: The authors have no potential conflicts of interest to disclose.
- Conceptualization: Yi DY.
- Data curation: Park J, Shin DY.
- Formal analysis: Park J.
- Writing - original draft: Shin DY, Park J.
- Writing - review & editing: Yi DY, Park J.
SUPPLEMENTARY MATERIALS
List of participants and samples for this microbiome analysis
List of raw data statistics and alpha diversity test results
Top 20 abundant genera detected in 43 HBM samples
Top 20 frequent genera detected in 43 HBM samples
Top 20 abundant genera detected in 49 IF samples
Top 20 frequent genera detected in 49 IF samples
List of 51 species only identified only in HBM samples
List of 85 species identified only in IF samples
List of 43 species identified in both HBM and IF samples
List of 115 species identified only in HBM samples
List of 59 bacterial species identified only in IF samples
List of 82 species identified in both HBM and IF samples
List of 135 species identified only in HBM samples
List of 70 species identified only in IF samples
List of 73 species identified in both HBM and IF samples
References
- 1.McDowell MM, Wang CY, Kennedy-Stephenson J. Breastfeeding in the United States: findings from the national health and nutrition examination surveys, 1999–2006. NCHS Data Brief. 2008;(5):1–8. [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention. Breastfeeding among US children. [Updated 2020]. [Accessed May 28, 2020]. https://www.cdc.gov/breastfeeding/about-breastfeeding/index.html.
- 3.Centers for Disease Control and Prevention. CDC national survey of maternity practices in infant nutrition and care (mPINC) [Updated 2020]. [Accessed July 2, 2020]. https://www.cdc.gov/breastfeeding/data/mpinc.
- 4.Arora T, Singh S, Sharma RK. Probiotics: interaction with gut microbiome and antiobesity potential. Nutrition. 2013;29(4):591–596. doi: 10.1016/j.nut.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Gao Z, Guo B, Gao R, Zhu Q, Wu W, Qin H. Probiotics modify human intestinal mucosa-associated microbiota in patients with colorectal cancer. Mol Med Rep. 2015;12(4):6119–6127. doi: 10.3892/mmr.2015.4124. [DOI] [PubMed] [Google Scholar]
- 6.Singh RK, Chang HW, Yan D, Lee KM, Ucmak D, Wong K, et al. Influence of diet on the gut microbiome and implications for human health. J Transl Med. 2017;15(1):73. doi: 10.1186/s12967-017-1175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vitetta L, Coulson S, Linnane AW, Butt H. The gastrointestinal microbiome and musculoskeletal diseases: a beneficial role for probiotics and prebiotics. Pathogens. 2013;2(4):606–626. doi: 10.3390/pathogens2040606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SY, Yu J, Ahn KM, Kim KW, Shin YH, Lee KS, et al. Additive effect between IL-13 polymorphism and cesarean section delivery/prenatal antibiotics use on atopic dermatitis: a birth cohort study (COCOA) PLoS One. 2014;9(5):e96603. doi: 10.1371/journal.pone.0096603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JY, Seo JH, Kwon JW, Yu J, Kim BJ, Lee SY, et al. Exposure to gene-environment interactions before 1 year of age may favor the development of atopic dermatitis. Int Arch Allergy Immunol. 2012;157(4):363–371. doi: 10.1159/000328778. [DOI] [PubMed] [Google Scholar]
- 10.Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med. 2016;22(10):1187–1191. doi: 10.1038/nm.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alam C, Bittoun E, Bhagwat D, Valkonen S, Saari A, Jaakkola U, et al. Effects of a germ-free environment on gut immune regulation and diabetes progression in non-obese diabetic (NOD) mice. Diabetologia. 2011;54(6):1398–1406. doi: 10.1007/s00125-011-2097-5. [DOI] [PubMed] [Google Scholar]
- 12.Xu WT, Nie YZ, Yang Z, Lu NH. The crosstalk between gut microbiota and obesity and related metabolic disorders. Future Microbiol. 2016;11(6):825–836. doi: 10.2217/fmb-2015-0024. [DOI] [PubMed] [Google Scholar]
- 13.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med. 2015;7(307):307ra152. doi: 10.1126/scitranslmed.aab2271. [DOI] [PubMed] [Google Scholar]
- 15.Xuan C, Shamonki JM, Chung A, Dinome ML, Chung M, Sieling PA, et al. Microbial dysbiosis is associated with human breast cancer. PLoS One. 2014;9(1):e83744. doi: 10.1371/journal.pone.0083744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cremon C, Barbaro MR, Ventura M, Barbara G. Pre- and probiotic overview. Curr Opin Pharmacol. 2018;43:87–92. doi: 10.1016/j.coph.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Alberda C, Gramlich L, Meddings J, Field C, McCargar L, Kutsogiannis D, et al. Effects of probiotic therapy in critically ill patients: a randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2007;85(3):816–823. doi: 10.1093/ajcn/85.3.816. [DOI] [PubMed] [Google Scholar]
- 18.Sanaie S, Ebrahimi-Mameghani M, Mahmoodpoor A, Shadvar K, Golzari SE. Effect of a probiotic preparation (VSL#3) on cardiovascularrisk parameters in critically-ill patients. J Cardiovasc Thorac Res. 2013;5(2):67–70. doi: 10.5681/jcvtr.2013.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta N, Kumar A, Sharma P, Garg V, Sharma BC, Sarin SK. Effects of the adjunctive probiotic VSL#3 on portal haemodynamics in patients with cirrhosis and large varices: a randomized trial. Liver Int. 2013;33(8):1148–1157. doi: 10.1111/liv.12172. [DOI] [PubMed] [Google Scholar]
- 20.Perez PF, Doré J, Leclerc M, Levenez F, Benyacoub J, Serrant P, et al. Bacterial imprinting of the neonatal immune system: lessons from maternal cells? Pediatrics. 2007;119(3):e724–32. doi: 10.1542/peds.2006-1649. [DOI] [PubMed] [Google Scholar]
- 21.Jiménez E, Fernández L, Maldonado A, Martín R, Olivares M, Xaus J, et al. Oral administration of Lactobacillus strains isolated from breast milk as an alternative for the treatment of infectious mastitis during lactation. Appl Environ Microbiol. 2008;74(15):4650–4655. doi: 10.1128/AEM.02599-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arroyo R, Martín V, Maldonado A, Jiménez E, Fernández L, Rodríguez JM. Treatment of infectious mastitis during lactation: antibiotics versus oral administration of Lactobacilli isolated from breast milk. Clin Infect Dis. 2010;50(12):1551–1558. doi: 10.1086/652763. [DOI] [PubMed] [Google Scholar]
- 23.Jost T, Lacroix C, Braegger CP, Rochat F, Chassard C. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ Microbiol. 2014;16(9):2891–2904. doi: 10.1111/1462-2920.12238. [DOI] [PubMed] [Google Scholar]
- 24.Abrahamsson TR, Sinkiewicz G, Jakobsson T, Fredrikson M, Björkstén B. Probiotic lactobacilli in breast milk and infant stool in relation to oral intake during the first year of life. J Pediatr Gastroenterol Nutr. 2009;49(3):349–354. doi: 10.1097/MPG.0b013e31818f091b. [DOI] [PubMed] [Google Scholar]
- 25.Fernández L, Langa S, Martín V, Maldonado A, Jiménez E, Martín R, et al. The human milk microbiota: origin and potential roles in health and disease. Pharmacol Res. 2013;69(1):1–10. doi: 10.1016/j.phrs.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 26.La Rosa PS, Warner BB, Zhou Y, Weinstock GM, Sodergren E, Hall-Moore CM, et al. Patterned progression of bacterial populations in the premature infant gut. Proc Natl Acad Sci U S A. 2014;111(34):12522–12527. doi: 10.1073/pnas.1409497111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azad MB, Konya T, Maughan H, Guttman DS, Field CJ, Chari RS, et al. Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. CMAJ. 2013;185(5):385–394. doi: 10.1503/cmaj.121189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim G, Bae J, Kim MJ, Kwon H, Park G, Kim SJ, et al. Delayed establishment of gut microbiota in infants delivered by cesarean section. Front Microbiol. 2020;11:2099. doi: 10.3389/fmicb.2020.02099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madan JC, Hoen AG, Lundgren SN, Farzan SF, Cottingham KL, Morrison HG, et al. Association of cesarean delivery and formula supplementation with the intestinal microbiome of 6-week-old infants. JAMA Pediatr. 2016;170(3):212–219. doi: 10.1001/jamapediatrics.2015.3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345(6200):1048–1052. doi: 10.1126/science.1254529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez-Llorente C, Plaza-Diaz J, Aguilera M, Muñoz-Quezada S, Bermudez-Brito M, Peso-Echarri P, et al. Three main factors define changes in fecal microbiota associated with feeding modality in infants. J Pediatr Gastroenterol Nutr. 2013;57(4):461–466. doi: 10.1097/MPG.0b013e31829d519a. [DOI] [PubMed] [Google Scholar]
- 32.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 34.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 35.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabrera-Rubio R, Collado MC, Laitinen K, Salminen S, Isolauri E, Mira A. The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am J Clin Nutr. 2012;96(3):544–551. doi: 10.3945/ajcn.112.037382. [DOI] [PubMed] [Google Scholar]
- 37.Murphy K, Curley D, O'Callaghan TF, O'Shea CA, Dempsey EM, O’Toole PW, et al. The composition of human milk and infant faecal microbiota over the first three months of life: a pilot study. Sci Rep. 2017;7(1):40597. doi: 10.1038/srep40597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heikkilä MP, Saris PE. Inhibition of Staphylococcus aureus by the commensal bacteria of human milk. J Appl Microbiol. 2003;95(3):471–478. doi: 10.1046/j.1365-2672.2003.02002.x. [DOI] [PubMed] [Google Scholar]
- 39.Jost T, Lacroix C, Braegger C, Chassard C. Assessment of bacterial diversity in breast milk using culture-dependent and culture-independent approaches. Br J Nutr. 2013;110(7):1253–1262. doi: 10.1017/S0007114513000597. [DOI] [PubMed] [Google Scholar]
- 40.Martín R, Heilig HG, Zoetendal EG, Jiménez E, Fernández L, Smidt H, et al. Cultivation-independent assessment of the bacterial diversity of breast milk among healthy women. Res Microbiol. 2007;158(1):31–37. doi: 10.1016/j.resmic.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 41.Turroni F, Foroni E, Pizzetti P, Giubellini V, Ribbera A, Merusi P, et al. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl Environ Microbiol. 2009;75(6):1534–1545. doi: 10.1128/AEM.02216-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fanaro S, Vigi V, Chierici R, Boehm G. Fecal flora measurements of breastfed infants using an integrated transport and culturing system. Acta Paediatr. 2003;92(5):634–635. doi: 10.1080/08035350310011632. [DOI] [PubMed] [Google Scholar]
- 43.Favier CF, Vaughan EE, De Vos WM, Akkermans AD. Molecular monitoring of succession of bacterial communities in human neonates. Appl Environ Microbiol. 2002;68(1):219–226. doi: 10.1128/AEM.68.1.219-226.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martín V, Maldonado-Barragán A, Moles L, Rodriguez-Baños M, Campo RD, Fernández L, et al. Sharing of bacterial strains between breast milk and infant feces. J Hum Lact. 2012;28(1):36–44. doi: 10.1177/0890334411424729. [DOI] [PubMed] [Google Scholar]
- 45.Hunt KM, Foster JA, Forney LJ, Schütte UM, Beck DL, Abdo Z, et al. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS One. 2011;6(6):e21313. doi: 10.1371/journal.pone.0021313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ward TL, Hosid S, Ioshikhes I, Altosaar I. Human milk metagenome: a functional capacity analysis. BMC Microbiol. 2013;13(1):116. doi: 10.1186/1471-2180-13-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martín R, Langa S, Reviriego C, Jimínez E, Marín ML, Xaus J, et al. Human milk is a source of lactic acid bacteria for the infant gut. J Pediatr. 2003;143(6):754–758. doi: 10.1016/j.jpeds.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 48.Song YL, Liu CX, McTeague M, Finegold SM. “Bacteroides nordii” sp. nov. and “Bacteroides salyersae” sp. nov. isolated from clinical specimens of human intestinal origin. J Clin Microbiol. 2004;42(12):5565–5570. doi: 10.1128/JCM.42.12.5565-5570.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lagkouvardos I, Pukall R, Abt B, Foesel BU, Meier-Kolthoff JP, Kumar N, et al. The mouse intestinal bacterial collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat Microbiol. 2016;1(10):16131. doi: 10.1038/nmicrobiol.2016.131. [DOI] [PubMed] [Google Scholar]
- 50.Coppa GV, Bruni S, Morelli L, Soldi S, Gabrielli O. The first prebiotics in humans: human milk oligosaccharides. J Clin Gastroenterol. 2004;38(6) Suppl:S80–3. doi: 10.1097/01.mcg.0000128926.14285.25. [DOI] [PubMed] [Google Scholar]
- 51.Oozeer R, van Limpt K, Ludwig T, Ben Amor K, Martin R, Wind RD, et al. Intestinal microbiology in early life: specific prebiotics can have similar functionalities as human-milk oligosaccharides. Am J Clin Nutr. 2013;98(2):561S–71S. doi: 10.3945/ajcn.112.038893. [DOI] [PubMed] [Google Scholar]
- 52.Tannock GW. A special fondness for lactobacilli. Appl Environ Microbiol. 2004;70(6):3189–3194. doi: 10.1128/AEM.70.6.3189-3194.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of participants and samples for this microbiome analysis
List of raw data statistics and alpha diversity test results
Top 20 abundant genera detected in 43 HBM samples
Top 20 frequent genera detected in 43 HBM samples
Top 20 abundant genera detected in 49 IF samples
Top 20 frequent genera detected in 49 IF samples
List of 51 species only identified only in HBM samples
List of 85 species identified only in IF samples
List of 43 species identified in both HBM and IF samples
List of 115 species identified only in HBM samples
List of 59 bacterial species identified only in IF samples
List of 82 species identified in both HBM and IF samples
List of 135 species identified only in HBM samples
List of 70 species identified only in IF samples
List of 73 species identified in both HBM and IF samples