

Abstract
Conventional dendritic cells (cDCs) play a central role in T-cell anti-tumor responses. We studied the significance of Notch-regulated DC immune responses in a mouse model of colitis-associated colorectal cancer (CRC) in which there is epithelial down-regulation of Notch/Hes1 signaling. This defect phenocopies that caused by GMDS (GDP-mannose 4,6-dehydratase) mutation in human CRCs. We found that, although wild-type immune cells restrained dysplasia progression and decreased incidence of adenocarcinoma in chimeric mice, the immune system with Notch2 deleted in all blood lineages or in only DCs promoted inflammation-associated transformation. Notch2 signaling deficiency not only impaired cDC terminal differentiation, but also down-regulated CCR7 expression, reduced DC migration and suppressed antigen cross-presentation to CD8+ T cells. Transfer of Notch-primed DCs restrained inflammation-associated dysplasia progression. Consistent with the mouse data, we observed a correlation between infiltrating cDC1 and Notch2 signaling in human CRCs and found that GMDS mutant CRCs showed decreased CCR7 expression and suppressed cDC1 signature gene expression. Suppressed cDC1 gene signature expression in human CRC was associated with a poor prognosis. In summary, our study supports an important role for Notch2 signaling in cDC1-mediated anti-tumor immunity and indicates that Notch2-controlled DCs restrain inflammation-associated colon cancer development in mice.
Keywords: Notch, dendritic cells, anti-tumor immunity, inflammation-associated carcinogenesis
Introduction
Inflammation increases risk for cancer (1). Colorectal cancer (CRC) is a multifactorial disease that progresses from transformed epithelial cells in a complex microenvironment composed of immune cells, cytokines, and gut bacteria. The immune system can affect all aspects of CRC development by suppressing tumor initiation and progression as well as promoting proliferation and metastasis. The importance of the anti-tumor immune response is underscored by the rapid development of immune-based cancer therapies over the last 30 years (2). Although the effect of cancer immunotherapy and checkpoint blockade has centered largely around cytotoxic CD8+ T cells, emerging work has begun to reveal a critical role for dendritic cells (DCs) in promoting T-cell anti-tumor immunity (3–8). However, exactly how DCs in a complex tumor immune microenvironment impact inflammation-associated CRC carcinogenesis has not yet been defined.
Previously we reported that genetic disruption of the GDP-4-keto-6-deoxy-mannose-3,5,epimerase-4-reductase (Fx) gene results in spontaneous colitis, colitis-associated dysplasia, and ultimately adenocarcinoma (9). In this animal model, loss of GDP-4-keto-6-deoxy-mannose-3,5,epimerase-4-reductase disrupts the conversion of GDP-mannose to GDP-fucose and leads to fucosylglycan deficiency (10), a condition phenocopied by deletion of the GDP-mannose 4,6-dehydratase (GMDS) gene (11), which is seen in 6-13% of CRCs (9,12). Notch signaling transactivation following Notch–ligand interaction requires the post-translation modification of Notch with O-fucose addition to the consensus Ser/Thr residue present on the core ligand-binding EGF-like repeats and subsequent modification by Fringe, an N-acetyl-glucosaminyl transferase (13–15). As a result, fucosylglycan deficiency in the gut epithelium disrupts Notch and downstream Hes1 signaling, resulting in aberrant crypt proliferation and goblet-cell expansion accompanied by profound inflammation and serrated-like lesions (SSLs) (16,17). Importantly, loss of Hes1 expression is commonly observed in sessile serrated adenomas/polyps (SSA/p) and CRC in the SSA pathway (9,18). Fx deletion also alters blood-cell homeostasis by affecting Notch-dependent T-cell differentiation and myelopoiesis (13,15). A role for hematopoietic fucosyglycan-deficiency in the gut inflammation and transformation seen in Fx−/− mice was suggested by bone marrow chimeric studies in which wild-type (WT) bone marrow cells have a colitis-limiting and tumor-suppressing effect (9). This finding prompted our current investigation of whether fucosyglycan-deficiency in the context of aberrant Notch signaling alters immune surveillance in one or more blood lineages and promotes inflammation and progression of colitis-associated carcinogenesis.
Notch signaling is evolutionarily conserved to determine cell fate and has emerged as a critical regulator of lymphoid-cell development and T-cell function (19). Less elucidated is the significance of Notch in innate-immune cell development and function. Several groups reported that Notch2 regulates terminal differentiation of splenic and lamina propria conventional dendritic cell (cDC) (20–22). Additionally, Notch activation in DCs interacts with signaling triggered by various TLR agonists to modulate inflammatory cytokine expression (23). Furthermore, Notch signaling in DCs is critical for evoking anti-tumor responses to mouse melanoma cells (24). However, the role of Notch-regulated cDC immune responses in CRC and particularly in the development of inflammation-associated CRC is unknown. Here, by using Fx−/− mice as a model of colitis-associated CRC, we identified that Notch2-controlled DCs play a critical role in limiting the development of inflammation-associated colon cancer in mice. Through database analysis, we identified a link between Notch2 and cDC1 in human CRC and showed that fucosylation-deficient CRCs have decreased CCR7 expression and suppressed cDC1 signature gene expression.
Materials and Methods
Mice
The animal research was approved by Case Western Reserve University Institutional Animal Care and Use Committee. C57Bl/6 (Ly5.2) and B6.SJL-Ptrca Pep3b/BoyJ (B6.BoyJ:Ly5.1) mice were maintained in the lab (9,13). Rbpjflox/flox (RbpjF/F) mice were a gift from Dr Tasuku Honjo. Notch1F/F mice were a gift from Dr Ralph Kopan. VavCre/Notch1F/F and VavCre/Notch2F/F mice were generated by crossing Vav-Cre mice (#008610; Jackson Laboratory) with Notch1F/F and Notch2F/F mice (#010525; Jackson Laboratory), respectively. Mx-Cre/RbpjF/F, CD4-Cre/RbpjF/F, and Lys-Cre/RbpjF/F mice were generated by crossing Mx-Cre (#003556; Jackson Laboratory), CD4-Cre (#022071; Jackson Laboratory) and Lys-Cre (#004781; Jackson Laboratory), with RbpjF/F mice, respectively. CD11c-Cre/Notch2F/F mice were generated by crossing CD11c-Cre mice (#008068; Jackson Laboratory) with Notch2F/F mice. OT-1 mice (003831) were obtained from Jackson Laboratory. Fx−/− mice (Ly5.2 and Ly5.1) were generated by our lab and maintained on fucose-supplemented chow diet (0.5% L-fucose) from weaning as described (9,13, 25). Briefly, in experiments where fucose-deficient Fx−/− mice were used, mice were maintained on a fucose-supplemented diet until 12 weeks, and then on standard chow for 4 weeks before use. For bone marrow transplantation, all Fx−/− recipient mice were maintained on fucose-supplemented diet before transplantation and for 14 days after transplantation at which time they were switched to the standard diet. In some experiments, mice received antibiotics containing amoxicillin (0.06%), clarithromycin (0.01%), metronidazole (0.02%) and omeprazole (0.0004%) for 8 weeks (9).
Bone marrow transplantation and mouse histology
Bone marrow transplantation was performed in lethally-irradiated (950 cG) 8-12 week old mice (Ly5.1) by i.v. transfer of 2 X106 donor cells (Ly5.2) (13). Mouse colitis scoring and dysplasia scoring were performed as described (9). Briefly, the final histological score is the sum of the indices of active inflammation, chronic inflammation, transmural inflammation, as well as the ulceration (scale 0-3) and regeneration (scale 0-4). The index of inflammation (active, chronic and transmural) is the product of the intensity of the inflammation (scale 0-3) and the area of involvement (scale 0-4). Dysplastic scores were determined by the percentage of epithelial cells showing cytological dysplastic changes.
DC isolation, immunophenotyping, differentiation, and priming with Notch ligand
Mouse spleens were crushed on a sterile 40 μm cell strainer using the flat end of the plunger from a sterile 1 ml syringe. Dissociated cells were passed through the strainer using 1 ml of FACS buffer (HBSS with 0.5% BSA and 1mM EDTA). The cell suspension was subjected to red cell lysis using the Red Blood Cell Lysis Buffer (11814389001, Sigma) for 5 minutes at room temperature. cDC1 enriched cells were isolated from the dissociated splenocytes by negative selection with biotinylated B220 (RA3-6B2, #103204, BioLegend) and CD11b (M1/70, #101204, BioLegend) antibody and anti-Biotin MicroBeads (#130-090-485, Miltenyi Biotec) followed by positive selection with CD11c MicroBeads (#130-125-835, Miltenyi Biotec).
To prepare bone marrow-derived DCs (BMDCs), bone marrow cells were flushed from femurs and tibias using a 21–gauge needle with FACS buffer followed by red cell lysis and filtered through a sterile 40 μm cell strainer. Lysed bone marrow cells were suspended in RPMI (#SH30027.01, Hyclone) supplemented with 10% FBS (# SH30088.03, Hyclone), 1% L-glutamine (#25030081, Thermo Fisher Scientifc), 1% sodium pyruvate (# 11360070, Thermo Fisher Scientifc), 1% MEM-NEAA (# 11140050, Thermo Fisher Scientifc), 55 μM 2-mercaptoethanol (#21985023, Thermo Fisher Scientifc), and 100 ng/ml Flt3L (# 250-31L, PeproTech). Cell were plated in 6-well plates at 4 x 106/well and cultured at 37°C in a humidified atmosphere at 5% CO2 for 7 days.
To prime DCs with DLL1, BMDCs were prepared as described previously and cultured first with Flt3L for 3 days. Cells were then co-cultured with OP9 cells transduced with retroviruses encoding green fluorescent protein (GFP) or Notch ligand DLL1 starting on day 3 and co-cultured for 4 more days. OP9 and OP9-DLL1 cells were gifts from Dr John Lowe and authenticated by genotyping and FACS analysis for GFP expression (13). These cells were authenticated and tested for mycoplasma yearly. These cells were cultured in MEM alpha medium (# 12561072, Thermo Fisher Scientifc) supplemented with 20% FBS (#SH30088.03, Hyclone). Prior to co-culture with DCs, OP9 cells were treated with mitomycin C (10 mg/mL) for 2 hours. BMDCs primed with DLL1 were transferred into Fx−/− mice via i.v. injection (1 x 106/mouse) weekly for 6 weeks.
FACS analysis was performed on BD FACSAria I and BD CytoFLEX and analyzed using BD FACSDiva software version 4.1. and CytExpert software. Briefly, 0.2 – 1 x 106 cells were incubated with 1:100 dilutions of appropriate antibodies in 0.2 ml FACS buffer on ice for 20 minutes followed by 2 washes with FACS buffer (26). Antibodies used included: CD4 (RM4-5, #100508, Biolegend), CD8α (53-6.7, #100712, Biolegend), B220 (RA3-6B2, #103204, Biolegend), CD11b (M1/70, #101216, Biolegend), Gr-1 (RB6-8C5, #108404, Biolegend), MHCII (I-A/I-E; M5/114.15.2, #107606, Biolegend), TER119 (TER-119, #116204, Biolegend), CD103 (M290, #557495, BD), NK1.1 (PK136, #108704, Biolegend), ESAM (1G8, #136204, Biolegend), c-kit (2B8, #105812, Biolegend), Sca1 (D7, #108114, Biolegend), CD11c (HL3, #561241, BD), Flt3 (A2F10, #135306, Biolegend), CD24 (M1/69, #138506, Biolegend), XCR1 (ZET, #148206, Biolegned), CD115 (AFS98, #135532, Biolegend), and SIRPα (P84, #144028, Biolegend).
DC migration and antigen presentation analysis
Transwell assay was performed using 24-well plates with 6.5 mm transwells with 5.0μm pore polycarbonate membrane (#3421, Corning). Briefly, 100μl of cDC1-enriched splenocyte suspension (2x105/ml) in RPMI medium supplemented with 2% FBS, 1% L-glutamine, 1% sodium pyruvate, 1% MEM-NEAA (migration medium) were added to the top chamber and 600μl migration medium containing chemokines [100ng/ml of CCL2, #250-10; CCL5, #250-07; CCL19, #250-17B; CCL21, #250-013 (all from PeproTech); XCL1, #783502, Biolegend] was added to the bottom chamber. Migrated cells at the bottom chamber were recovered 3 hours later and enumerated by flow cytometry by gating on the viable cDC1 cells.
For cross presentation assays, endotoxin-free chicken ovalbumin (OVA) (#S7951-1MG, Sigma Aldrich) was added (10 ng/ml) to enriched cDC1. CD8+ T cells were isolated from spleens from OT-1 mice by negative selection using biotinylated antibodies (B220, Ter119, CD11b, Gr1, CD11c, NK1.1) and streptavidin microbeads (#130-048-101, Miltenyi Biotech). CD8+ T cells were stained with carboxyfluorescein succinimidyl ester (CFSE, 1 μM) (65-0850-84, Invitrogen) in RPMI with 10% FBS at 37°C in a humidified atmosphere at 5% CO2 for 15 minutes and washed twice with the same staining buffer. A 96-well plate was pre-coated with 1:1000 anti-CD3 (145-2C11, #557306, BD) and anti-CD28 (D665, #566883, BD) in PBS for 90 minutes. 10x104 cells/well were co-cultured in the pre-coated 96-well plate with 2x104/well CD11c+ cells in RPMI with 10% FBS for 3 days at 37°C in a humidified atmosphere at 5% CO2. T-cell proliferation was assessed for CFSE dilution using the FACS buffer indicated by the FITC fluorescence signal on the FACSAria I.
Chromosome Immunoprecipitation (ChIP)
The upstream 5000 bp promoter sequence (−5000 nt to −1 nt) of mouse Ccr7 gene was searched for the RBPJ binding motif TG(G/A)GAA in both positive and negative complement strands by using BioEdit 7.2 software. BMDCs primed with Flt3L for 6 days as described above (see DC isolation, immunophenotyping, differentiation, and priming with Notch ligand) were transferred to 6-well plates pre-coated with recombinant mouse DLL1-Fc (2.5μg/ml, #5026-DL-050, R&D Systems) and cultured for 17 hours. ChIP assay was performed using an EZ-ChIP Assay Kit (#17-295, Millipore) according to the manufacturer’s instructions. Briefly, 3x106 harvested BMDCs were fixed with formaldehyde with a final concentration of 1% and incubated for 10 minutes at 37°C. Crosslinking was stopped by glycine (0.125M) for 5 minutes. Cells were resuspended in 600 μl of SDS Lysis Buffer with protease inhibitors (#20-163, Millipore) and incubated for 10 minutes on ice. Cell lysate was sonicated for 60 cycles, each with a 10-second pulse at 50% of maximal power, followed by a 20-second cooling period on ice to shear DNA to lengths of about 200 base pairs. Samples were subjected to immunoprecipitation using rabbit polyclonal anti-RBP-JK antibody (# 25949, Abcam) overnight at 4°C followed by adding 60 μl of Protein A Agarose (Cat # 16-157C, Millipore) for one hour at 4°C. The immunoprecipitated chromatin was analyzed by qRT-PCR as described below (see Affymetrix GeneChip array and qRT-PCR analysis). The primers used were listed in Supplementary Table 1.
Affymetrix GeneChip array and qRT-PCR analysis
Total RNA was extracted from spleen cDC1 cells using the RNeasy Mini Kit (#74104, Qiagen). RNA was reverse transcribed and labeled according to the instruction of Affymetrix WT Pico protocol by using 9 ng total RNA (GeneChip WT Pico Reagent Kit, #902622, Thermo Fisher Scientific, Inc), and 5.5ug of cDNAs were hybridized to Affymetrix Gene Chip Mouse Gene 2.0 ST Array (#902118, Thermo Fisher Scientific, Inc). GeneChips were scanned using Affymetrix GeneChip Scanner 3000. Normalized robust multi-array average (RMA) values were calculated and used to calculate fold change for each gene and sorted by log2 fold change (GSE163958). A total of 327 genes were identified with a fold change greater than 2 (log2FC > 1) between cDC1 enriched cells isolated from on-fucose and off-fucose mice. A heatmap of the top 50 genes with the greatest fold change was generated using Clustvis. The 327 genes were cross referenced against the Molecular Signature Database (MSigDB, Broad Institute) to identify gene sets enriched in differentially expressed genes.
For qRT-PCR, total RNA was prepared from freshly isolated cDC1 cells as described above. 500 ng of total RNA was reverse transcribed using the Bio-Rad iScript™ Select cDNA Synthesis Kit (#170-8897, Bio-Rad) in 20 µl reactions following the manufacturer’s instructions. 2 μl cDNA was amplified with the BIO-RAD iQ™ SYBR Green Supermix (# 1708880, Bio-Rad) in 10 μl PCR reactions and the primer sets specific for mouse genes in triplicates with the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). The relative gene expression was calculated based on ΔΔCt method and normalized to β-Actin. The sequences of the primer sets are listed in the Supplemental Table 2.
TCGA gene expression analysis and CRC survival analysis
CRC data from TCGA PanCancer Atlas (27) was accessed using cBioPortal. The PanCancer Atlas includes 592 CRC patients who have mRNA expression data and 120 individuals were available for survival analysis. The mRNA co-expressions were reported and graphed for genes of interest based on RSEM values batch normalized from Illumina HiSeq RNASeq data. Normalized expression values were transformed into Z scores and ranked by the mean expression value of signature genes. Spearman correlation between each expressed gene was reported along with the p-value and multiple testing corrected q-value using cBioPortal. Overall survival for patients relative to genes of interest was analyzed using the Gene Expression Profiling Interactive Analysis (GEPIA) web server by categorizing samples as “High” or “Low” based on the 75% quantile of the expression values (28). Mutation profiling and Copy Number Variation (CNV) were assembled to classify samples into GMDS wild-type and mutant groups separately. Samples with either CNV score less than 0 or detected mutation on gene of interest were classified as mutant samples, while the rest are considered as wild type. Data processing and statistical analysis were implemented in R scripts. R package DESeq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html) was applied to perform differential analysis, and R package fgsea (https://bioconductor.org/packages/release/bioc/html/fgsea.html) used for Gene Set Enrichment analysis (GSEA).
Screening of GMDS mutation
Study of archived human CRC was approved by the institute review board of the University Hospitals Case Medical Center (UHCMC). A total of 90 de-identified human CRC specimens were screened for GMDS mutation using primers for exons 1-2, 2-4, and 5-7, respectively, as described (29). GMDS-WT (n=9) and GMDS-mutant CRCs (n=7) were Included in the study.
Statistics
Data are presented as means ± SD, unless otherwise stated. Differences in variables were assessed by Student t test for two groups or one-way ANOVA for at least three groups using SPSS version 20 software.
Results
Pan-Notch- or Notch2-deficient hematopoietic cells promote colitis and CRC development
Reconstituting Fx−/− mice with WT hematopoietic cells results in decreased intestinal inflammation and more than 50% reduction of CRC (9). We first studied if defective Notch function in hematopoietic cells of Fx−/− mice contributes to the progression of colitis-associated cancer development in these mice by reconstituting the hematopoietic compartment of Fx−/− mice with Notch-deficient bone marrow cells from Mx1-Cre/RbpjF/F (RBP-Jk−/−or R−/−) mice since the recombination signal binding protein-Jk (RBP-Jk or RBPJ) is the critical transcription factor downstream of all four mammalian Notch receptors. Bone marrow cells from Mx1-Cre/Rbpj+/+ or RbpjF/F (WT) and Mx1-Cre/RbpjF/+ (RBP-Jk+/− or R+/−) were used as control donors. WT mice were used as recipient controls. All Fx−/− recipient mice were maintained on fucose-supplemented diet (on-fucose diet) before transplantation and for 14 days after transplantation to allow mice to acquire a fucosylation-replete phenotype through a fucosylation salvage pathway (25). Mice were then switched to regular diet (off-fucose diet) and studied for histology and immune-cell function two months after diet switch; this time point was selected based on our previous studies where Fx−/− recipients showed defined inflammation and dysplasia histology after receiving WT bone marrow transplantation (9).
Colonic histology analysis revealed that Fx−/− mice receiving WT cells or cells carrying a single copy of Rbpj (Rbpj+/−) displayed variable colitis and dysplasia (8.8 and 28 are inflammation and dysplasia indexes for RBP-J+/− while 6.1 and 19.4 are for WT, respectively). Invasive adenocarcinoma developed in 40% of Fx−/− mice receiving RBP-Jk+/− cells but only developed in 2 out of 12 (16.7%) mice receiving WT cells (Table 1). In comparison, Fx−/− mice receiving RBP-Jk−/− cells displayed more severe inflammation and dysplasia, showing statistically significant higher mean scores reaching 13 and 50, respectively, when compared with mice receiving WT bone marrow (Fig. 1A–D; Table 1), and 7 out of 9 mice developed adenocarcinoma (Table 1). In WT recipients, neither RBP-Jk+/− nor RBP-Jk−/− cells caused inflammation or dysplasia. Accordingly, we found that most inflammatory cytokine expression by WT recipients were not affected by the type of bone marrow cells transplanted. In contrast, consistent with our previous report that Fx−/− gut mucosa was sufficient to induce inflammation and dysplasia (9), most of the inflammatory cytokines expressed by the colonic epithelium were increased in Fx−/− mice receiving control RBP-Jk+/− cells. Inflammatory cytokine expression was further enhanced in Fx−/− mice receiving RBP-Jk−/− cells (Fig. 1E). These findings suggest that Notch signaling deficiency in hematopoietic cells promotes inflammation-associated carcinogenesis of the intestinal epithelium of Fx−/− mice in a dose-dependent manner. Consistent with our previous findings that the microbiome is critical for promoting inflammation-associated carcinogenesis, antibiotic treatment completely eliminated dysplasia and significantly decreased inflammation in Fx−/− mice receiving RBP-Jk−/− cells (Fig. 1A–B, two rightmost columns). None of the mice treated with antibiotics developed adenocarcinoma.
Table 1.
Inflammation score, dysplasia score, and CRC frequency in transplanted Fx−/− mice
| Donor | Mean Inflammation Score | Mean Dysplastic Score | Number of mice | CRC Frequency |
|---|---|---|---|---|
| WT | 7.04 | 17.9 | 12 | 2/12 |
| Fx | 11.06 | 42.2 | 9 | • * |
| Mx-Cre/Rbpjk+/+ or RbpjkF/F | 6.1 | 19.4 | 8 | 3/8 |
| Mx-Cre/RbpjkF/+ | 8 | 28 | 10 | 4/10 |
| Mx-Cre/RbpjkF/F | 12.8 | 50 | 9 | 7/9 |
| Vav-Cre/N1F/+ | 9.4 | 16 | 5 | 0/5 |
| Vav-Cre/N1F/F | 9 | 13 | 5 | 0/5 |
| Vav-Cre/N2F/+ | 7.6 | 30 | 5 | 1/5 |
| Vav-Cre/N2F/F | 12.3 | 46.7 | 6 | 3/6 |
| CD4-Cre/RbpjkF/+ | 5.3 | 21.2 | 8 | 2/8 |
| CD4-Cre/RbpjkF/F | 4.8 | 14 | 10 | 0/10 |
| Lys-Cre/RbpjkF/+ | 7.1 | 15.7 | 7 | 1/7 |
| Lys-Cre/RbpjkF/F | 8.5 | 25 | 6 | 0/6 |
| CD11c-Cre/N2+/+ or N2F/F | 7 | 17.9 | 12 | 3/12 |
| CD11c-Cre/N2F/+ | 7.7 | 28 | 15 | 6/15 |
| CD11c-Cre/N2F/F | 9.6 | 38.4 | 16 | 7/16 |
Mice all died less than 2 months after bone marrow transfer.
Fig. 1. Notch-deficient hematopoietic cells promote colitis and CRC development.
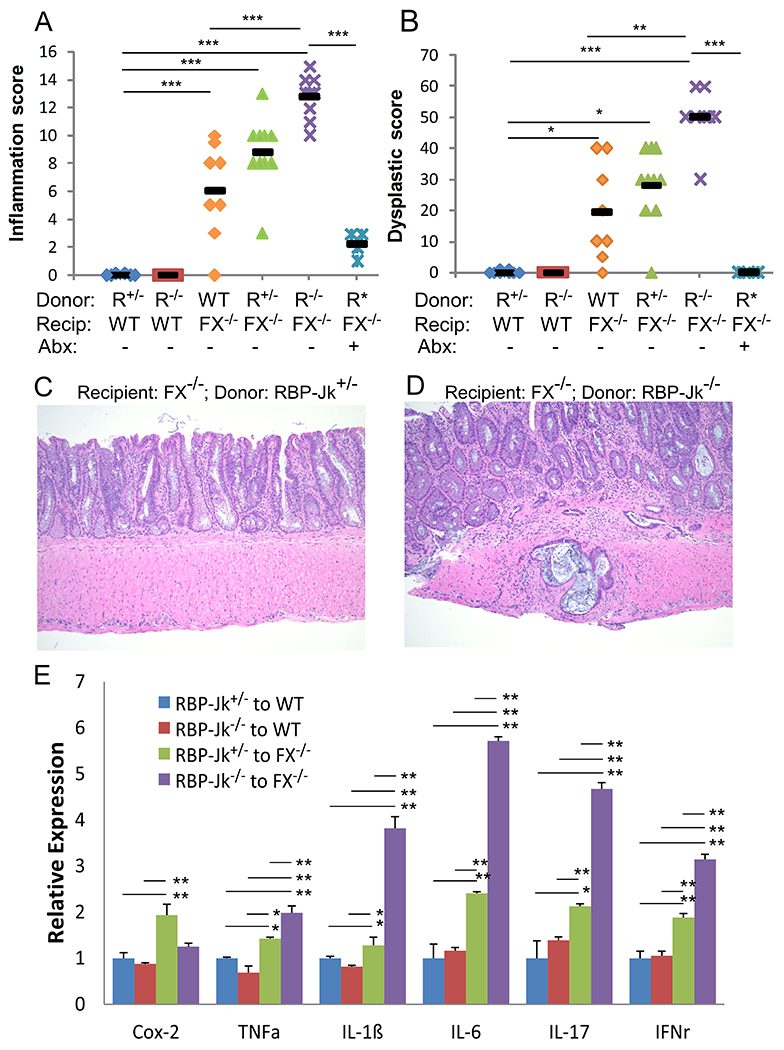
Reciprocal transplantation was performed in wild-type (WT) mice receiving total bone marrow cells from Mx-Cre/RbpjF/+ (RBP-Jk+/−; R+/−) (n=6) or Mx-Cre/RbpjF/F (RBP-Jk−/−; R−/−) mice (n=6), and in Fx−/− mice receiving WT (Mx1-Cre/Rbpj+/+ or RbpjF/F) (n=8), Mx-Cre/RbpjF/+(RBP-Jk+/−; R+/−) (n=10) or Mx-Cre/RbpjF/F (RBP-Jk−/−; R−/−) bone marrow cells, either non-treated (n=9) or treated with antibiotics (R*) (60d) (n=9). Data were pooled from 3 experiments. Colon inflammation (A) and dysplastic scores (B) were recorded 2 months after transplantation. (C-D) Representative H&E staining of colonic tissues from Fx−/− mice transplanted with RBP-Jk+/−or RBP-Jk−/−bone marrow cells, showing worsening inflammation and CRC development in the latter. Images were taken under 20X magnification. (E) Inflammatory cytokine mRNA expression in colon tissue from recipients in 4 transplant settings was measured by qRT-PCR and normalized to the expression in WT mice receiving RBP-Jk+/−cells (n=6-9/group from 3 experiments). Black bars (A-D) indicate means. Results shown in E are mean ± SD. Student t-test was performed; *p<0.05, **p<0.01
To discern which Notch receptor (Notch 1-4) was required for limiting inflammation-associated transformation in colon epithelium, we reconstituted lethally-irradiated Fx−/− mice with Notch1- or Notch2-deficient bone marrow cells (Vav-Cre/Notch1F/F or Vav-Cre/Notch2F/F) or their controls (Vav-Cre/Notch1F/+ or Vav-Cre/Notch2F/+) and compared the gut pathology after transplantation. Only mice reconstituted with Notch2-deficient (Supplementary Fig. S1A–D) but not Notch1-deficient (Supplementary Fig. S1E–H) hematopoietic cells developed significant colitis and dysplasia. The inflammatory and dysplastic indexes in these mice were similar to those recorded for Fx−/− mice reconstituted with the RBP-Jk−/− cells (Supplementary Fig. S1A–B). Accordingly, inflammatory cytokine expression was further enhanced in Fx−/− mice receiving Notch2-deficient bone marrow cells (Supplementary Fig. S1I). Half of the mice receiving Notch2-deficient bone marrow cells developed CRC, while none of the mice receiving Notch1-deficient cells had cancer (Table 1).
Notch2-dependent DCs are essential for limiting colitis-associated CRC development
Since Notch1 is critical for T-cell differentiation and adaptive immune function (19), and RBP-Jk-dependent signaling has also been shown to promote T-cell cytotoxicity (30), we asked if defective Notch function in T cells promotes inflammation-associated cancer development. Surprisingly, we found that loss-of-Notch signaling in T cells does not enhance inflammation and dysplastic progression in Fx−/− mice (Supplementary Fig. S2). None of the recipient mice developed CRC after receiving CD4-Cre/RbpjF/F cells (Table 1). Therefore, lack of Notch signaling in hematopoietic cells promoted inflammation-associated transformation in this model through a Notch2-controled and T cell–independent mechanism. Furthermore, we excluded a role of Notch-deficient myeloid cells in this process (Supplementary Fig. S3).
Because Notch2 regulates terminal DC differentiation (20–22), we sought to determine if the phenotype associated with Notch2-deficient bone marrow reconstitution of Fx−/− mice can be recapitulated by Notch2-deficient DC bone marrow chimerism. We transplanted CD11c-Cre/Notch2F/F marrow cells to WT or Fx−/− mice. CD11c-Cre/Notch2F/+ or WT cells were used in control transplantation. Compared with Fx−/− mice receiving WT cells, Fx−/− mice receiving CD11c-Cre/Notch2F/F cells displayed significantly higher inflammation (9.6 and 7.0 for recipients of CD11c-Cre/Notch2F/F and WT bone marrow, respectively, p=0.025) (Fig. 2A) and dysplasia scores (38.4 and 17.9 for recipients of CD11c-Cre/Notch2F/F and WT bone marrow, respectively, p=0.01) (Fig. 2B). These findings were consistent with the histologic analysis (Fig 2C and 2D). In addition, around 45% of mice developed cancer in the CD11c-Cre/Notch2F/F group. These inflammation and dysplasia scores are similar to those recorded for Fx−/− mice receiving Fx−/− bone marrow cells. Notably, mice receiving DCs carrying a single copy of Notch2 had moderately increased levels of inflammation and dysplasia (Table 1), where 40% of mice also developed adenocarcinoma, doubling the incidence compared with Fx−/− mice receiving WT DCs. These findings suggested that Notch2-regulated DCs attenuated inflammation-associated carcinogenesis in this colitis-associated CRC model. Like RBP-Jk-deficient hematopoietic cells, Notch2-deficient DCs promoted colitis-associated carcinogenesis in a dose-dependent manner.
Fig. 2. Notch2-dependent DCs are essential for limiting colitis-associated CRC development.
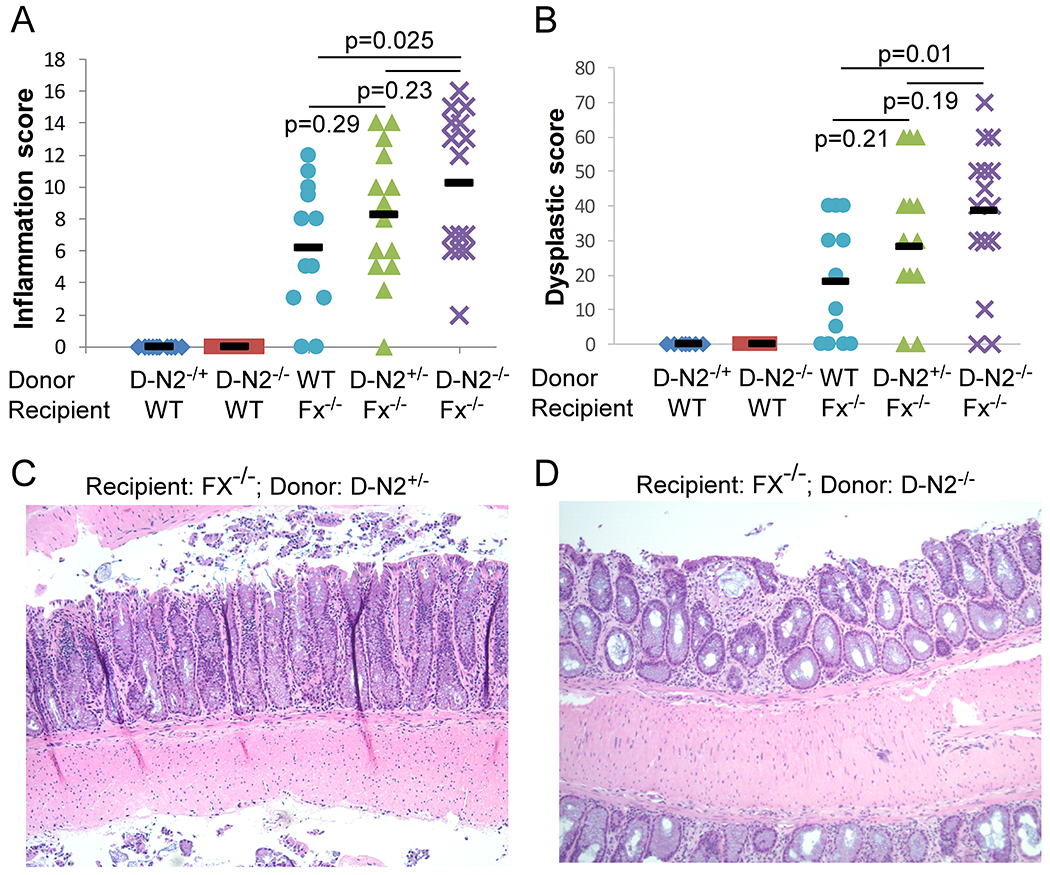
WT mice were transplanted with CD11c-Cre/Notch2F/+ (D-N2+/−) (n=10) or CD11c-Cre/Notch2F/F (D-N2−/−) marrow (n=10), and Fx−/− mice received WT (CD11c-Cre/Notch2+/+ or Notch2F/F) (n=12), CD11c-Cre/Notch2F/+ (D-N2+/−) (n=14) or CD11c-Cre/Notch2F/F (D-N2−/−) bone marrow cells (n=15). Data were pooled from 4 experiments. Colon inflammation (A) and dysplastic scores (B) were recorded 2 months after transplantation. (C-D) Representative H&E staining of colonic tissues from Fx−/− mice transplanted with D-N2+/− or D-N2−/− cells show worse inflammation in D than C (20X magnification). Black bars (A-B) indicate means. Student t-test was performed; p-values were shown.
Conventional DC differentiation is impaired in Fx−/− mice due to Notch2 dysregulation
We next examined how DC frequency and function are altered in Fx−/− mice. We characterized the DCs in Fx−/− mice by the markers expressed on cDC1 and cDC2. We found that the numbers of splenic, mesenteric lymph node (mLN), and lamina propria (LP) DCs were decreased in Fx−/− mice maintained on regular diet compared with those maintained on fucose-supplemented diet (Fig. 3A). In addition, cDC1 (CD11c+MHCII+CD8+CD11b−) and cDC2 (CD11c+MHCII+CD8−CD11b+) (Fig. 3B) frequencies were decreased by 92% and 68% respectively in the spleen. The expression of XCR1 on cDC1s was markedly decreased in off-fucose Fx−/− mice (Fig. 3C). The analysis of bone marrow DC progenitors revealed a mild reduction in the frequencies of committed precursors of cDCs (pre-cDCs) but similar frequencies of the macrophage and DC precursors (MDPs) and the common DC precursors (CDPs) (31) in off-fucose Fx−/− mice compared with controls (Supplementary Fig. S4A–B). In mLN, cDC1 frequencies were also decreased by 63%, while cDC2 remained unchanged (Supplementary Fig. S4C). LP and mLN migratory CD103+ DCs were decreased by 34% and 44% in frequencies, respectively, in off-fucose Fx−/− mice (Fig. 3D). Because similar levels of inflammation and dysplasia were observed in non-transplanted Fx−/− mice and Fx−/− mice reconstituted by CD11c-Cre/Notch2F/F bone marrow cells, we suspected that Notch2 signaling loss in the hematopoietic compartment could similarly alter the frequencies of DC subsets. Analysis of DCs derived from CD11c-cre/Notch2F/F donors in chimeric recipients revealed that total splenic DC numbers were decreased in both WT and Fx−/− recipients compared to recipients receiving control (CD11c-cre/Notch2F/+) cells. In addition, decreased cDC1s and cDC2s derived from CD11c-cre/Notch2F/F donors in both WT and Fx−/− recipient mice recapitulated the altered DC differentiation observed in Fx−/− mice (Fig. 3E). However, MDPs, CDPs and pre-cDCs were not changed in the CD11c-cre/Notch2F/F mice even though XCR1 expression was decreased in CD8+ DCs (Supplementary Fig. S5A–E).
Fig. 3. Conventional DC differentiation was impaired in Fx−/− mice due to Notch2 signaling loss.
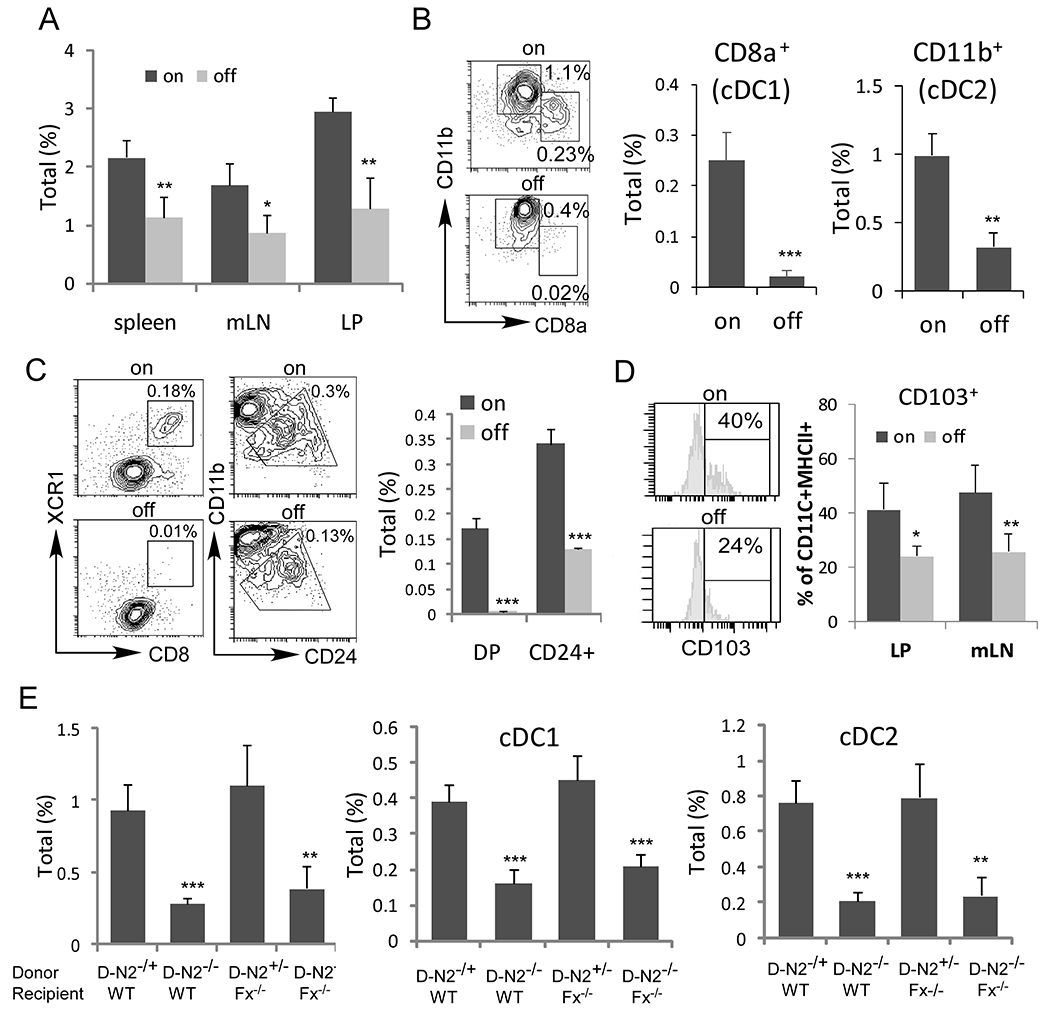
(A) Total DCs (CD11c+MHC II+) among live cells from spleen, LN and LP in Fx−/− mice maintained with fucose-supplemented (on) (n=6) or regular diet (off) (n=6) were determined by FACS analysis. (B) Representative FACS profile of splenic cDC1s (CD8α+CD11b−) and cDC2s (CD8α−CD11b+) after gating on CD11c+MHC II+ DCs were shown. Frequencies were calculated based on the total viable splenocytes. (C) Representative FACS profile of CD8α+XCR1+ (DP) and CD24+ cDC1 after gating on CD11c+MHC II+ DC cells were shown. Frequencies were calculated based on the total viable splenocytes. (D) Representative FACS profile and frequencies of CD103+ DCs in mLN and LP after gating on CD11c+MHC II+ DC cells were shown. (E) Bone marrow chimeric studies were performed in WT mice, which received CD11c-Cre/Notch2F/+ (D-N2+/−) (n=6) or CD11c-Cre/Notch2F/F (D-N2−/−) bone marrow (n=6), and in Fx−/− mice, which received CD11c-Cre/Notch2F/+ (D-N2+/−) (n=6) or CD11c-Cre/Notch2F/F (D-N2−/−) bone marrow cells (n=6). Total DCs and the fraction of the indicated DC subsets from spleen (E) were determined by FACS analysis. Results shown are mean ± SD. Student t-test was performed; *p<0.05, **p<0.01, *** p<0.001
Conventional DC migration and cross-presentation are suppressed by Notch2 dysregulation
Although decreased numbers of cDC1 would limit their function, the impaired Notch2 signaling in DC subsets may directly impair anti-tumor activity. To investigate this possibility, we assessed the migration of DCs toward chemokine ligands in transwell migration assays. We found that cDC1-enriched cells isolated from the spleens of off-fucose Fx−/− mice (off-fucose DC) displayed decreased migration toward CCL19 (Fig. 4A) and CCL21 (Fig. 4B) compared with cDC1-enriched cells from on-fucose Fx−/− mice (on-fucose DC). Consistent with the altered expression of XCR1 in cDC1 of Fx−/− mice, these cells showed a decrease in migration toward XCL1 (Fig. 4C). In comparison, both off-fucose and on-fucose DCs showed minimal migration toward CCL2 and CCL5 (Fig. 4B). Consistently, expression of CCR7, which is the receptor for CCL19 and CCL21, was decreased in DCs from off-fucose Fx−/− mice, while expression of CCR2 and CCR5, which are the receptors for CCL2 and CCL5, respectively, remained unchanged (Fig. 4D). We then analyzed the ability of Fx−/− DCs to cross-prime T cells. Using a co-culture assay with CFSE-labeled CD8+ T cells from OVA-specific OT-I mice and OVA-pulsed cDC1 cells, we found decreased cross-priming of CD8+T cells by freshly isolated cDC1 (Fig. 4E) from off-fucose Fx−/− mice. We asked if the decreased cross-priming is associated with aberrant IL12 expression. Although IL12 expression did not differ significantly between off-fucose DCs and control DCs, its expression was much lower in off-fucose DCs after LPS stimulation (Fig. 4F). To determine if impaired migration and cross-priming of off-fucose DC from Fx−/− mice is caused by the suppressed Notch2 signaling, we examined the migration and T-cell priming of CD11c-cre/Notch2F/F DCs. We found similar reduction in migration of Notch2-deficient cDC1s and suppression of cross-priming of CD8+ T cells by Notch2-deficient DCs (Fig. 4G–K). Expression of CCR7 and IL12 was also decreased in Notch2-deficient DCs (Fig. 4J & L).
Fig. 4. Conventional DC migration and antigen cross-presentation are impaired in Fx−/−mice due to Notch2 signaling loss.
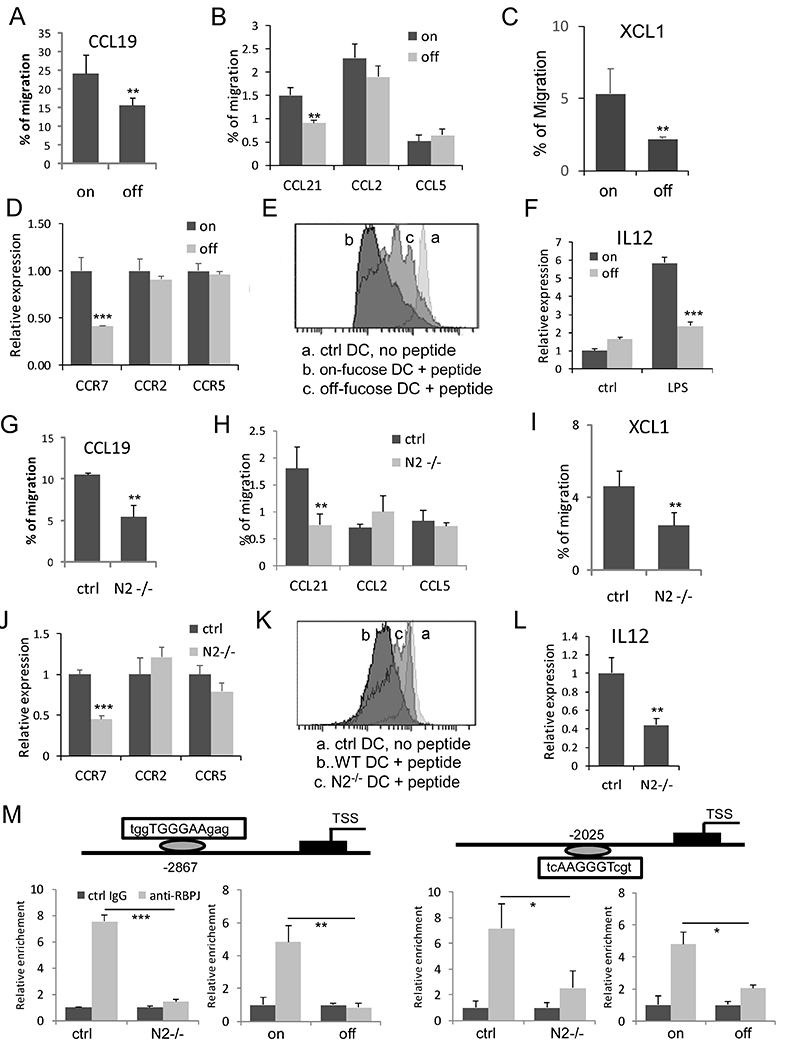
(A-C) Enriched cDC1 cells isolated from the spleens of Fx−/− mice maintained with fucose-supplemented diet (on) or regular diet (off) were assessed in transwell assays for ability to migrate toward CCL19 (A); CCL21, CCL2, and CCL5 (B); and XCL1 (C). Expression of chemokine receptors was determined by qRT-PCR (D). (E) Cross-priming of CD8+ T cells in vitro. Bone marrow-derived DCs on day 7 were incubated with OVA and were incubated with CFSE-labeled OT-I CD8+T cells at a 1:5 ratio for 3 days. The levels of CFSE were assessed by FACS in gated CD8+ T cells, with the individual peaks of CFSE dilution quantified. No CFSE dilution observed in the absence of OVA. Representative FACS profile from 3 similar experiments was shown. (F) IL12 expression was determined in bone marrow-derived total DCs with and without LPS stimulation. (G-I) Enriched cDC1s isolated from the spleen of CD11c-Cre/Notch2F/F (N2−/−) and control CD11c-Cre/Notch2F/+ mice were assessed for migration toward CCR9 (G); CCL21, CCL2, and CCL5 (H); and XCL1 (I). Expression of chemokine receptors in N2−/− DCs and control DCs was determined by qRT-PCR (J). (K) Cross-priming of CD8+ T cells by N2−/− and control DCs. Representative FACS profile from 3 similar experiments was shown. (L) IL12 expression was determined by qRT-PCR in bone marrow-derived total DCs. (M) ChIP analysis with control rabbit IgG (ctrl IgG) and anti-RBPJ showing CCR7 promoter region contains two RBPJ/CSL sites approximately 2.8 kb (positive strand) and 2.0 kb (negative strand) upstream that were bound by RBPJ in WT BMDCs and BMDC cells from Fx−/− mice maintained with fucose-supplemented diet (on), but not in N2−/− DCs or BMDC cells from Fx−/− mice maintained with regular diet (off). Results shown in A-C, E-G and J-K are mean ± SD. Student t-test was performed; * p<0.05, **p<0.01, *** p<0.001
Notch regulates gene expression by forming a transcriptional complex with the DNA binding protein RBPJ/CSL. To investigate if Notch2 directly regulates CCR7 expression, we searched the promoter region of CCR7 and found several potential RBPJ/CSL binding motifs (TGGGAA). Chromosome immunoprecipitation (ChIP) analysis of BMDCs stimulated by DLL1 showed RBPJ binds strongly with two RBPJ/CSL sites (~2.8 kb and 2.0 kb) upstream of the CCR7 promoter. In comparison, none of the sites were bound by RBPJ in Notch2-deficient or Fx−/− cells (Fig. 4M). These findings indicate that CCR7 is directly regulated by Notch2 signaling and its suppressed expression contributes to the aberrant migration of Fx−/− and Notch2-deficient DCs.
To gain a comprehensive understanding of how fucose deficiency impacts DC maturation and function, we performed gene expression profiling of cDC1 cells from Fx−/− mice maintained with on-fucose or off-fucose diet. A total of 275 genes were up-regulated and 52 genes were down-regulated in response to fucose treatment (Supplementary Fig. S6A). Analysis of the top differentially regulated genes revealed that these genes were enriched in pathways such as immune-effector processes, myeloid leukocyte activation, exocytosis, and secretion. Additionally, some genes regulate molecular functions such as signaling receptor binding and endopeptidase activity (Supplementary Fig. S6B). The divergent gene expression implicated in altered immune-effector functions was validated by qRT-PCR, which showed up-regulation of genes encoding the myeloid inflammatory proteins and the leukocyte immunoglobulin-like receptor subfamily B member 4 (Lilrb4) by fucose-deficient cDC1s. Importantly, expression of genes encoding chemokine receptors (Ccr7), migration (Fscn1 and Hspa8), antigen processing (H2-m2), and immune defense molecules such as guanylate-binding proteins (Gbp9 and Gbp10) was down-regulated in fucose-deficient cDC1s (Supplementary Fig. S6C).
cDC1 abundance correlates with Notch2 signaling and GMDS mutation status in patients with CRC
To establish the relevance of Notch2 regulation of cDC1 in human CRC, we stratified patients in the CRC dataset of TCGA by expression of cDC1-associated genes (CCR7, XCR1, FLT3, CLEC9A, and THBD) (32, 33) and found that a higher cDC1 signature in tumors was positively associated with survival (Fig. 5A). The correlation between CCR7 and representative cDC1 signature genes (BATF3, FLT3, CLEC9A, XCR1, and THBD) indicates that CCR7 serves as a marker of tumor-infiltrating cDC1s for human CRC (Supplementary Fig. S7A). We then compared the cDC1 signature gene expression and the expression NOTCH2 and DLL1, which encodes the primary Notch ligand that drives Notch2-dependent cDC differentiation (34), and found they have a significant positive correlation (Fig. 5B). Consistent with our finding that Notch2 regulates cDC1 function, we found NOTCH2 gene expression in CRC correlates with the cDC1 gene THBD (Fig. 5C) and FLT3 as well as XCR1 (Supplementary Fig. S7B). Furthermore, we found that expression of the cDC1 signature gene (BATF3, FLT3, CLEC9A, XCR1, and THBD) correlated with the CD8+ T-cell genes CD8A and CD3E in human CRC (Fig. 5D).
Fig 5. Association between Notch signaling and cDC1 signature genes and altered cDC1 gene expression in human GMDS mutant CRCs.
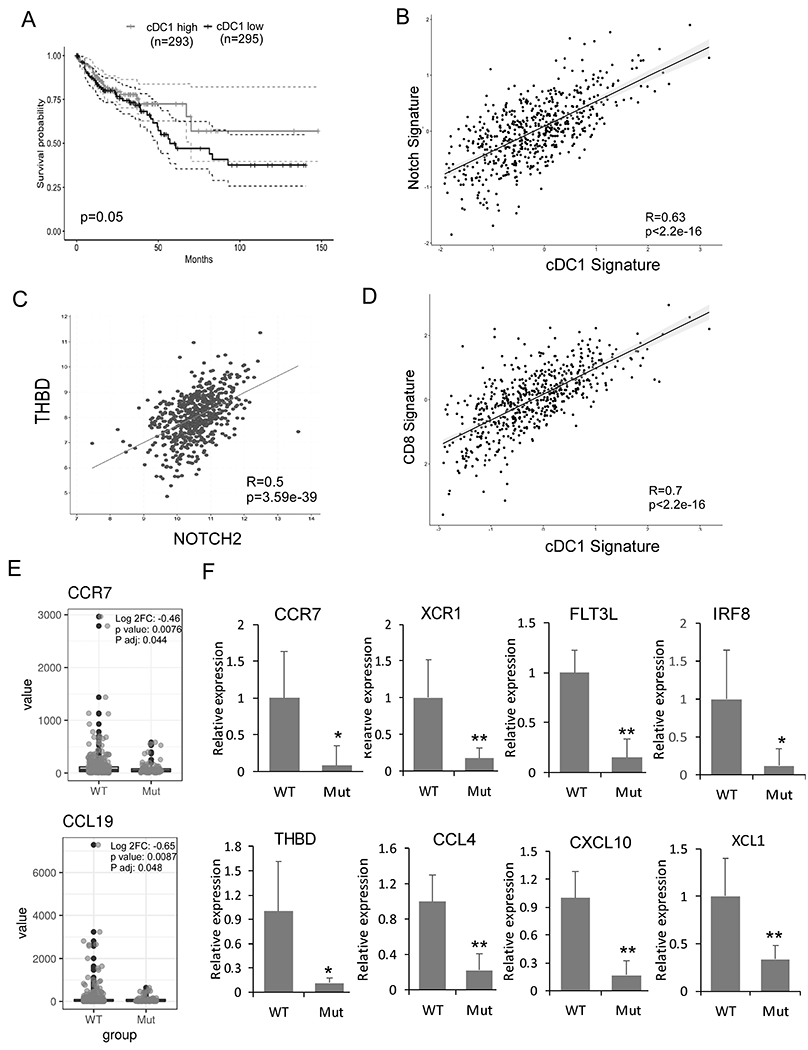
The following data analysis was performed on TCGA data base. (A) Prognostic value of the cDC1 signature (CCR7, XCR1, FLT3, CLEC9A, and THBD) for overall survival of human CRC patients comparing top and bottom quartiles. (B-D) Scatterplots showing correlation analyses of the cDC1 gene expression signature (BATF3, FLT3, CLEC9A, XCR1, and THBD) with the Notch gene expression signature (NOTCH2, DLL1) (B); of NOTCH2 and THBD (C); and the cDC1 gene expression signature (BATF3, FLT3, CLEC9A, XCR1, and THBD) with a CD8+ T-cell signature (CD8A and CD3E) (D). Spearman correlation coefficient and p values are shown. (E) CRCs from TCGA were stratified based on GMDS mutation. GSEA was performed using DC genes CCR7 and CCL19. (F) mRNA expression in archived tissues from GMDS WT (n=9) and GMDS mutant CRC (n=7) by qRT-PCR and normalized to the expression in GMDS WT CRC. Results shown are mean ± SD. Student t-test was performed; *p<0.05, **p<0.01
We then determined the impact of fucosylation deficiency in human CRC by performing GSEA after stratifying CRC patient data in TCGA on the basis of GMDS mutation. Supporting our findings in Fx−/− mice, we found a reduction of CCR7 and CCL19 expression in human GMDS-mutated CRCs compared with CRCs with no GMDS mutation (Fig. 5E). In addition, GMDS-mutant CRCs showed down-regulation of FLT3 and down-regulation of genes encoding proteins involved in MHC class I antigen processing and presentation (TREML4) and in cytoskeletal assembly (LDB3) (Supplementary Fig. S7C), but up-regulation of genes encoding proteins involved in releasing and responding to inflammatory cytokines (TNFAIP2, LILAR2, and NUCB2) and the inhibition of cell migration (BST2) (Supplementary Fig. S7D). Further, we found that reduced GMDS expression is associated with poor disease-free survival (Supplementary Fig. S8). Finally, we verified that representative cDC1 signature genes (CCR7, XCR1, THBD, IRF8 and FLT3L) and chemokine ligands (XCL1, CXCL10, CCL4) were suppressed in our cohort of GMDS-mutant CRCs compared with GMDS-WT CRCs (Fig. 5F) (5,35,36). In summary, these analyses support the hypothesis that Notch2 signaling regulates the tumor-associated cDC1 population and that fucosylation deficient CRCs have similar suppression of cDC1 signature genes as observed in our mouse model.
Notch-primed DCs suppresses inflammation-associated dysplasia progression
Finally, to demonstrate that Notch signaling in DCs restrains colitis-associated transformation, we adoptively transferred WT or Notch-primed DC cells to Fx−/− mice. Unprimed or Notch-primed DCs were both derived from bone marrow progenitors with Flt3L. Expanded DCs were transferred into Fx−/− mice weekly for 6 weeks (Fig. 6A). The inflammation and dysplastic indices of Fx−/− mice receiving unprimed DC cells remain unchanged when compared with control PBS treated mice (15.8 and 44 are inflammation and dysplasia indexes for DC-treated while 15.6 and 46 are for PBS-treated, respectively). However, we found that mice receiving Notch-primed DCs had significantly decreased inflammation and dysplasia (Fig. 6B and 6C). The incidence of CRC in mice receiving Notch-primed DCs also decreased to 20% compared with 38% in the control group (p<0.05). The histological improvement was accompanied with decreased expression of inflammatory cytokines, including IL1β, IL6, Cox2, and TNFα, although expression of IFNγ was increased by around 2-fold, suggesting attenuated inflammation and improved cytotoxicity in response to DC infusion (Fig. 6D). In summary, these findings indicate that ex vivo Notch-primed DCs were able to restrain colitis-associated CRC development.
Fig 6. Adoptive transfer of Notch-primed DCs limited inflammation associated transformation.
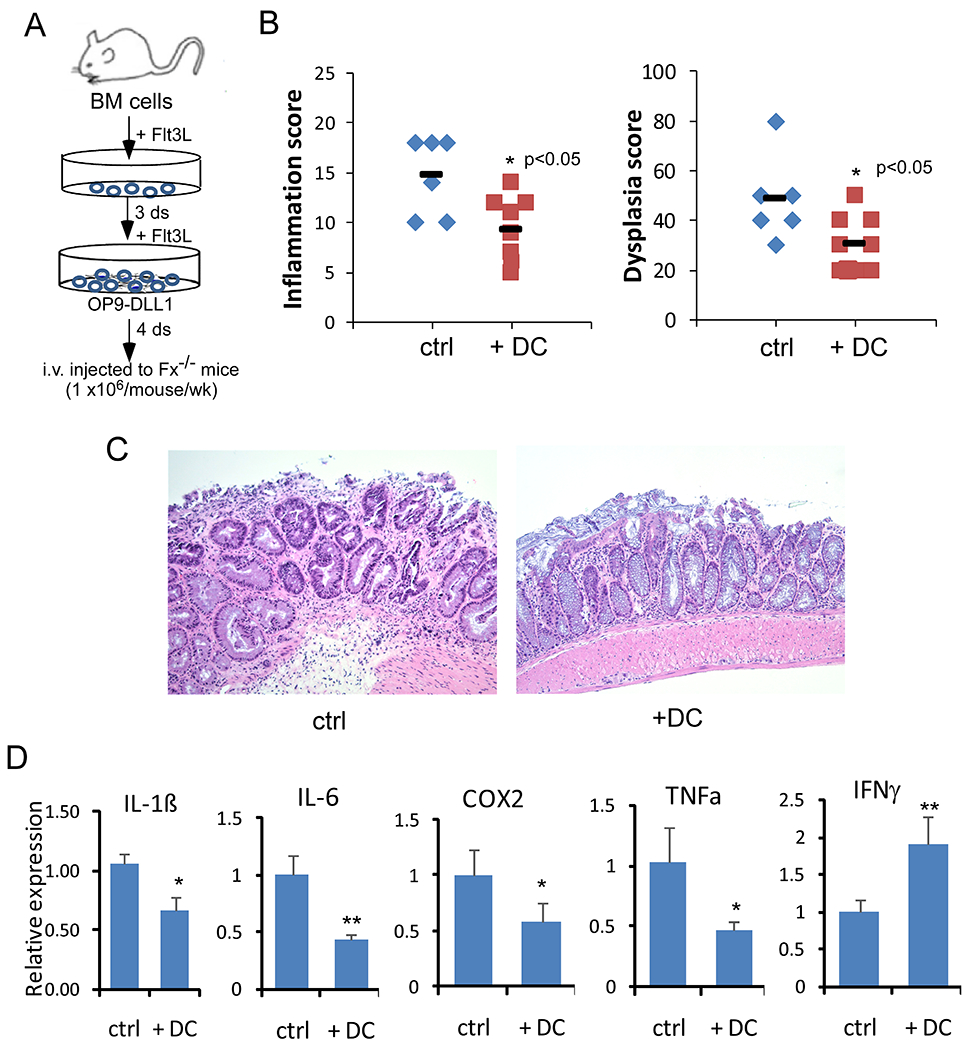
(A) Schematic of in vitro priming of bone marrow-derived DCs with OP9-DLL1 in the presence of Flt3L and adoptive transfer. (B) Colon inflammation and dysplastic scores were recorded 5 days after the last dose of DC transfer (n=9) or PBS injection (control; n=6). Data were pooled from 3 experiments. (C) Representative H&E staining of colonic tissues from Fx−/− mice received PBS (control) or primed DCs. (D) Inflammatory cytokine mRNA expression in colon tissue from Fx−/− mice receiving PBS or DC injection was measured by qRT-PCR and normalized to the expression in Fx−/− mice receiving PBS. Results shown in B and D are mean ± SD. Student t-test was performed; *p<0.05, **p<0.01
Discussion
We have uncovered a novel mechanism implicating defective Notch-dependent DC function in promoting inflammation-associated dysplasia and progression to colon cancer transformation. We found more progression of inflammation-associated dysplasia to CRC in Fx−/− mice reconstituted with Notch2-defective DCs while adoptive transfer of Notch-primed DCs attenuated transformation. Additionally, we revealed a direct correlation between Notch2 signaling and infiltrating cDC1 and the association of the suppressed cDC1 signature with a poor prognosis in human CRC. Our findings thus reveal a critical role for Notch2-dependent cDC1s in restraining inflammation-associated transformation in our mouse model and tumor progression in human CRCs.
Our previous studies show that in our model of colitis-associated CRC, the intestinal epithelium of Fx−/− mice is chronically inflamed and displays aberrant proliferation and progression of dysplasia to adenocarcinoma in a defined temporal and histopathological sequence (9). Here, we revealed that fucose deficiency impairs DC function in a manner that is regulated by Notch2 signaling. The differentiation of cDCs depends on a few key transcription factors (for cDC1s, it is IRF8, BATF3, and ID2; for cDC2s, it is IRF4 and ZEB2) (37–39). It is also known that Notch2 is involved in the differentiation of terminal cDC1, CD11b+ESAMhi cDC2 and CD103+CD11b+ intestinal DCs (20–22). In Fx−/− mice, Fx locus deletion impairs Notch signaling by abolishing fucosylation and Fringe-mediated modification of Notch ligand binding EGF-like repeats, affecting the development of multiple hematopoietic lineages through dysregulated Notch1 or Notch2 signaling (13,15). Here, we showed that decreased numbers cDC1 and cDC2 cells in Fx−/− mice phenocopied altered cDC differentiation in mice with Notch2 deleted in CD11c cells (CD11c-Cre/Notch2F/F). This indicates that lack of fucosylation impairs Notch2-dependent signaling in final DC differentiation as we found that the cDC precursors including MDP and CDP frequencies were not altered. Further, the functional defects of Fx-null cDC1 were shared by Notch2-null cDC1. Defective anti-tumor activity in the absence of Notch2 signaling was illustrated in vivo by bone marrow chimeric studies showing that gut inflammation and dysplasia were worsened by Notch signaling-deficient (Mx1-Cre/RBPJkF/F) total bone marrow cells or Notch2-deficient (Vav-Cre/Notch2F/F) cells. The crippled anti-transforming activity of Notch2-deficient cells was attributed to DCs but not T cells or myeloid lineage cells in a dosage-dependent manner.
Migration and cross presentation to cytotoxic T cells were major functional defects identified in Notch2-deficient and Fx−/− DCs, consistent with qRT-PCR and array-based transcriptome analysis showing down-regulation of migration regulating genes and antigen processing genes. Particularly, we found that Notch signaling directly regulates CCR7 expression and that down-regulated CCR7 is accompanied with decreased migration toward CCL19 and CCL21 in both Fx-null and Notch2-null DCs. This is consistent with reports that CCR7 is critical for cDC1 to present tumor antigens and that Notch priming promotes cDC1 development and up-regulates CCR7 (40–42). Although terminal differentiation of cDC1 and cDC2 were both affected in Fx−/− mice, numbers of cDC1s were more prominently decreased in the draining lymph nodes while the numbers of migratory CD103+ DCs were decreased in both mLN and LP. This further supports that defective DC migration is a prominent feature in this animal model. CD103+DCs in LP and mLN are uniquely capable of generating gut-tropic CD8+ effector T cells (43). Indeed, we observed decreased cross-presentation to CD8+ T cells by Fx−/− and Notch2-deficient cDC1s but unchanged cDC2 cross-presentation to CD4+ T cells. Supporting this notion, human CRCs with GMDS mutation not only had decreased CCR7 expression and cDC1 signature gene expression but also show decreased expression of cDC1-recruiting CCL4 (35) and decreased XCL1, which is expressed by NK cells to recruit XCR1+ cDC1 to form cDC1/NK clusters within tumor tissues (44). It is possible that cDC1 development is compromised in the fucosylglycan-deficient environment of GMDS mutant tumors, and/or cDC1 recruitment is suppressed in GMDS mutant tumors by the altered cytokine milieu. Others have reported that GMDS mutant CRCs escape NK-mediated surveillance by TRAIL-induced apoptosis. The underlying mechanism remains elusive but has been found to be independent of O-glycosylation (29). Considering that Fx−/− and Notch2-deficient cDC1s show reduced XCR1 expression, defective cDC1 recruitment through XCL1 may further compromise cDC1 migration and represent another mechanism of escaped tumor surveillance associated with fucosylation deficiency.
We extended our findings beyond fucosylation-deficient tumors by showing a direct link between infiltrating cDC1 and Notch2 signaling in human CRC. Further, consistent with the finding that cDC1s recruit CD8+ T cells into the tumor microenvironment, we found that the cDC1 gene expression signature correlated with CD8A and CD3E expression in human CRC (5). Decreased intra-tumoral cDC1s and circulating cDC1s in CRC patients relate to disease stage (45,46). Our TCGA analysis extends these findings by revealing that lower cDC1 biomarker expression is associated with worse survival among CRC patients. Unlike the Fx−/− mice, human cancer-cell-extrinsic mechanisms of suppressed cDC1 signature associated with inhibited Notch activity may involve fucosylation-independent mechanisms. The correlation between the Notch2/DLL1 gene signature and the cDC1 gene signature suggests that down-regulation of DLL1 in the tumor microenvironment could potentially account for the suppressed Notch2-dependent cDC1 homeostasis. Chronic inflammation regulates specific inflammatory conditions that can control activation of the Notch pathway in DCs. For example, DLL1 is down-regulated while JAG1 is up-regulated in the intestinal epithelium of off-fucose Fx−/− mice. JAG1 is up-regulated in other cell types under various chronic inflammation conditions (47, 48). Both Notch ligands JAG1 and DLL1 can activate Notch signaling but show opposite effects on DC differentiation (34). While DLL1 induces DC differentiation, JAG1 stimulation limits full DC differentiation by inducing an altered Hes1 activation. How inflammation alters Notch ligand expression and induces aberrant Notch signaling to suppress DC anti-tumor immunity needs to be explored.
Chronic inflammation induces epigenetic modification and DNA modification in intestinal epithelial cells, promoting dysplasia and contributing to initiation and progression of CRC. During this process, innate immune cells and adaptive immune cells are recruited, which generates an environment with pro-inflammatory cytokines and highly genotoxic oxygen/nitrogen reactive species (49). Indeed, up-regulation of an array of pro-inflammatory cytokines was a prominent feature of colonic epithelium of the Fx−/− mice and up-regulation of cytokines was further enhanced when mice were reconstituted with Notch2 signaling-deficient whole bone marrow cells or DCs. In contrast, adoptive transfer of Notch-primed DCs decreased expression levels of inflammatory cytokines, including IL1β, IL6 and TNFα. Thus, Notch2-dependent DCs appear to limit pro-tumorigenic inflammation in this model. It is interesting to note that Notch-deficient T cells appear to have a mild protective effect and decrease inflammation-associated dysplasia progression. However, the role of Notch signaling dictated by different Notch isoforms and the mechanism by which they coordinate immune responses in DC cells and other immune cells to promote an inflammation-limiting and anti-tumor microenvironment remains to be further defined.
Enhancing DC numbers and function within the pre-cancerous and early cancer lesions is paramount for cancer prevention (50). For immunotherapy, cDC1s are of particular interest in cellular vaccination strategies as they traffic tumor antigens to the draining lymph nodes and cross-present cell-associated neoantigens to cytotoxic T cells (4,41). Our findings reveal a mechanism whereby Notch signaling plays a critical role in the regulation of DC migration and antigen presentation. These findings suggest a strategy of enhancing DC anti-tumor activity by stimulating Notch signaling to eliminate transformed cells and to prevent tumor progression. This approach potentially promotes DC migration and increases antigen delivery without patient-specific antigen targeting. Future work is required to identify molecules and pathways that are regulated by Notch or Notch-suppressing factors in the tumor microenvironment, which are potentially important adjuvants to enhance the efficiency of antigen presentation by DCs and the stimulatory capacity of these cells in the context of anti-tumor activity.
Supplementary Material
Synopsis:
Conventional dendritic cells (DCs) play a key role in the anti-tumor immune response. The data show that Notch2 deletion in all cells or in only DCs promotes inflammation-associated colorectal carcinogenesis; targeting Notch2-controlled DCs could restrain colon cancer.
Acknowledgement
We thank Dr Kenneth Murphy and Ivaylo Ivanov for providing bone marrow cells from CD11c-Cre/Notch2F/F mice for part of this study. We thank Ms Alison W. Xin for editing this manuscript.
Financial support: This work was supported in part by research funding from NCI CA222064, NIH HL103827, Case GI SPORE Research Development Award, and NIDDK DDRCC Pilot/Feasibility Award (to LZ), and by the Department of Pathology Case Western Reserve University faculty startup fund to WX and LZ.
Footnotes
Disclosure of Potential Conflicts of Interest: The authors have declared that no conflict of interest exists.
References
- 1.Grivennikov SI, Greten FR, and Karin M (2010) Immunity, inflammation, and cancer. Cell 140, 883–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen DS, and Mellman I (2017) Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 [DOI] [PubMed] [Google Scholar]
- 3.Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, et al. (2018) Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 49, 1148–1161.e1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. (2016) Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spranger S, Dai D, Horton B, and Gajewski TF (2017) Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31, 711–723.e714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. (2008) Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 322, 1097–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spranger S, Bao R, and Gajewski T (2015) Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523. [DOI] [PubMed] [Google Scholar]
- 8.Sánchez-Paulete AR, Cueto FJ, Martínez-López M, Labiano S, Morales-Kastresana A, Rodríguez-Ruiz ME, et al. (2016) Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer discovery 6, 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Huang D, Chen KY, Cui M, Wang W, Huang X, et al. (2017) Fucosylation Deficiency in Mice Leads to Colitis and Adenocarcinoma. Gastroenterology 152, 193–205 e110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tonetti M, Sturla L, Bisso A, Benatti U, and De Flora A (1996) Synthesis of GDP-L-fucose by the human FX protein. J Biol Chem 271, 27274–27279 [DOI] [PubMed] [Google Scholar]
- 11.Ohyama C, Smith PL, Angata K, Fukuda MN, Lowe JB, and Fukuda M (1998) Molecular cloning and expression of GDP-D-mannose-4,6-dehydratase, a key enzyme for fucose metabolism defective in Lec13 cells. J Biol Chem 273, 14582–14587 [DOI] [PubMed] [Google Scholar]
- 12.Nakayama K, Moriwaki K, Imai T, Shinzaki S, Kamada Y, Murata K, et al. (2013) Mutation of GDP-mannose-4,6-dehydratase in colorectal cancer metastasis. PLoS One 8, e70298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou L, Li LW, Yan Q, Petryniak B, Man Y, Su C, et al. (2008) Notch-dependent control of myelopoiesis is regulated by fucosylation. Blood 112, 308–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan Q, Yao D, Wei LL, Huang Y, Myers J, Zhang L, et al. (2010) O-fucose modulates notch-controlled blood lineage commitment. Am J Pathol 176, 2921–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao D, Huang Y, Huang X, Wang W, Yan Q, Wei L, et al. (2011) Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood 117, 5652–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. (2005) Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435, 959–963 [DOI] [PubMed] [Google Scholar]
- 17.Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, and Artavanis-Tsakonas S (2005) Notch signals control the fate of immature progenitor cells in the intestine. Nature 435, 964–968 [DOI] [PubMed] [Google Scholar]
- 18.Cui M, Awadallah A, Liu W, Zhou L, and Xin W (2016) Loss of Hes1 Differentiates Sessile Serrated Adenoma/Polyp From Hyperplastic Polyp. Am J Surg Pathol 40, 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radtke F, MacDonald HR, and Tacchini-Cottier F (2013) Regulation of innate and adaptive immunity by Notch. Nature reviews. Immunology 13, 427–437 [DOI] [PubMed] [Google Scholar]
- 20.Caton ML, Smith-Raska MR, and Reizis B (2007) Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med 204, 1653–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, et al. (2011) Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35, 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, et al. (2013) Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nature immunology 14, 937–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gentle ME, Rose A, Bugeon L, and Dallman MJ (2012) Noncanonical Notch signaling modulates cytokine responses of dendritic cells to inflammatory stimuli. Journal of immunology (Baltimore, Md. : 1950) 189, 1274–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng F, Wang YC, Hu XB, Liu XW, Ji G, Chen YR, et al. (2010) The transcription factor RBP-J-mediated signaling is essential for dendritic cells to evoke efficient anti-tumor immune responses in mice. Molecular cancer 9, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith PL, Myers JT, Rogers CE, Zhou L, Petryniak B, Becker DJ, et al. (2002) Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol 158, 801–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, Yu S, Zimmerman G, Wang Y, Myers J, Yu VW, et al. (2015) Notch Receptor-Ligand Engagement Maintains Hematopoietic Stem Cell Quiescence and Niche Retention. Stem cells (Dayton, Ohio) 33, 2280–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang K, Creighton CJ, Davis C, Donehower L, Drummond J, Wheeler D, et al. (2013) The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics 45, 1113–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang Z, Li C, Kang B, Gao G, Li C, and Zhang Z (2017) GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research 45, W98–w102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moriwaki K, Noda K, Furukawa Y, Ohshima K, Uchiyama A, Nakagawa T, et al. (2009) Deficiency of GMDS leads to escape from NK cell-mediated tumor surveillance through modulation of TRAIL signaling. Gastroenterology 137, 188–198, 198, e181–182 [DOI] [PubMed] [Google Scholar]
- 30.Maekawa Y, Minato Y, Ishifune C, Kurihara T, Kitamura A, Kojima H, et al. (2008) Notch2 integrates signaling by the transcription factors RBP-J and CREB1 to promote T cell cytotoxicity. Nature immunology 9, 1140–1147 [DOI] [PubMed] [Google Scholar]
- 31.Yáñez A, Coetzee SG, Olsson A, Muench DE, Berman BP, Hazelett DJ, et al. (2017) Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 47, 890–902.e894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. (2017) Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell reports 18, 248–262 [DOI] [PubMed] [Google Scholar]
- 33.Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, et al. (2012) Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 37, 60–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng P, Nefedova Y, Corzo CA, and Gabrilovich DI (2007) Regulation of dendritic-cell differentiation by bone marrow stroma via different Notch ligands. Blood 109, 507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spranger S, Bao R, and Gajewski TF (2015) Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 [DOI] [PubMed] [Google Scholar]
- 36.Aliberti J, Reis e Sousa C, Schito M, Hieny S, Wells T, Huffnagle GB, et al. (2000) CCR5 provides a signal for microbial induced production of IL-12 by CD8α+ dendritic cells. Nature immunology 1, 83–87 [DOI] [PubMed] [Google Scholar]
- 37.Mildner A, and Jung S (2014) Development and function of dendritic cell subsets. Immunity 40, 642–656 [DOI] [PubMed] [Google Scholar]
- 38.Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, et al. (2016) Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 45, 669–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. (2012) Deciphering the transcriptional network of the dendritic cell lineage. Nature immunology 13, 888–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. (2016) Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirkling ME, Cytlak U, Lau CM, Lewis KL, Resteu A, Khodadadi-Jamayran A, et al. (2018) Notch Signaling Facilitates In Vitro Generation of Cross-Presenting Classical Dendritic Cells. Cell reports 23, 3658–3672.e3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balan S, Arnold-Schrauf C, Abbas A, Couespel N, Savoret J, Imperatore F, A. C, et al. (2018) Large-Scale Human Dendritic Cell Differentiation Revealing Notch-Dependent Lineage Bifurcation and Heterogeneity. Cell reports. 24:1902–1915.e1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Forster R, et al. (2005) Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med 202, 1063–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. (2018) NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 172, 1022–1037.e1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwaab T, Weiss JE, Schned AR, and Barth RJ Jr. (2001) Dendritic cell infiltration in colon cancer. Journal of immunotherapy (Hagerstown, Md. :1997) 24, 130–137 [PubMed] [Google Scholar]
- 46.Della Porta M, Danova M, Rigolin GM, Brugnatelli S, Rovati B, Tronconi C, et al. (2005) Dendritic cells and vascular endothelial growth factor in colorectal cancer: correlations with clinicobiological findings. Oncology 68, 276–284 [DOI] [PubMed] [Google Scholar]
- 47.Morga E, Mouad-Amazzal L, Felten P, Heurtaux T, Moro M, Michelucci A, et al. (2009) Jagged1 regulates the activation of astrocytes via modulation of NFkappaB and JAK/STAT/SOCS pathways. Glia 57, 1741–1753 [DOI] [PubMed] [Google Scholar]
- 48.Wang W, Zimmerman G, Huang X, Yu S, Myers J, Wang Y, et al. (2016) Aberrant Notch Signaling in the Bone Marrow Microenvironment of Acute Lymphoid Leukemia Suppresses Osteoblast-Mediated Support of Hematopoietic Niche Function. Cancer research 76, 1641–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, et al. (2008) DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. The Journal of clinical investigation 118, 2516–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Z, Han C, and Fu YX (2020) Targeting innate sensing in the tumor microenvironment to improve immunotherapy. Cellular & molecular immunology 17, 13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.